

Abstract
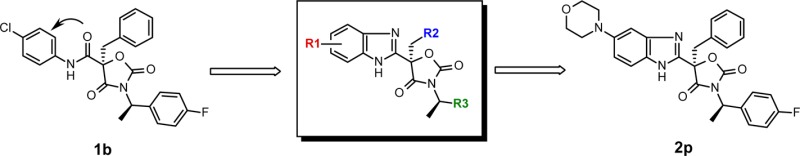
Elaboration of the oxazolidinedione series led to replacement of the exocyclic amides with substituted benzimidazoles. The structure–activity relationship (SAR) exploration resulted in the discovery of potent and selective nonsteroidal mineralocorticoid receptor (MR) antagonists with significantly improved microsomal stability and pharmacokinetic (PK) profile relative to the HTS hit 1a. One compound 2p possessed comparable efficacy as spironolactone (SPL) at 100 mg/kg (p.o.) in the rat natriuresis model. As such, this series was validated as a lead series for further optimization.
Keywords: mineralocorticoid receptor, antagonist, amide replacement, oxazolidinedione, natriuresis effect
The mineralocorticoid receptor (MR) is a nuclear hormone receptor located in the sodium ion-transporting epithelia (e.g., kidney, colon) and nonepithelia tissues including heart, blood vessels, brain, and salivary and sweat glands.1 The major endogenous ligand for MR is aldosterone (A in Figure 1), which plays an essential role in salt and water homeostasis by retaining sodium and water. Aldosterone binding in MR triggers a cascade of events including modulation of gene expression and protein synthesis, which then promote renal sodium reabsorption and potassium excretion in the distal nephron. Patients with primary hyperaldosteronism frequently suffer from hypertension, which may be driven by high MR activity leading to sodium reabsorption and fluid retention.2
Figure 1.

Structures of MR antagonists SPL, SPL, and EPL.
The inhibition of the renin–angiotensin–aldosterone system by angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) has been established as a successful approach in treating hypertension and congestive heart failure (CHF).3 Unfortunately, ACE inhibitors or ARBs fail to reach blood pressure lowering goals in a significant number of hypertensive patients treated with diuretics.3 This result is often in part due to both an abnormal activation of MR by elevated levels of aldosterone and salt imbalance, which may cause hypertension and other detrimental effects to the vasculature. Hence, direct MR antagonism has been utilized as an alternative or adjunct therapy for cardiovascular diseases. For example, spironolactone4 (SPL) and eplerenone5 (EPL) are two marketed MR antagonists (Figure 1) used successfully to treat hypertension and heart failure. However, use of SPL is limited due to sexual adverse effects including gynecomastia and menstrual irregularities, which mainly result from the off-target activity related to the androgen receptor (AR). While EPL is significantly more selective than SPL, resulting in a lower incidence of gynecomastia, it is less efficacious than SPL, as reflected by EPL’s inferior potency and pharmacokinetic (PK) profile.6
In light of the current limitations of the marketed MR antagonists, a potent and selective small molecule MR antagonist may provide clinicians with an additional valuable tool for treating hypertension and CHF. Toward this end, several classes of nonsteroidal MR antagonists have emerged over the past few years.7−15 Herein, we report our preliminary efforts on the discovery of benzimidazole oxazolidinediones as a novel class of potent MR antagonists.
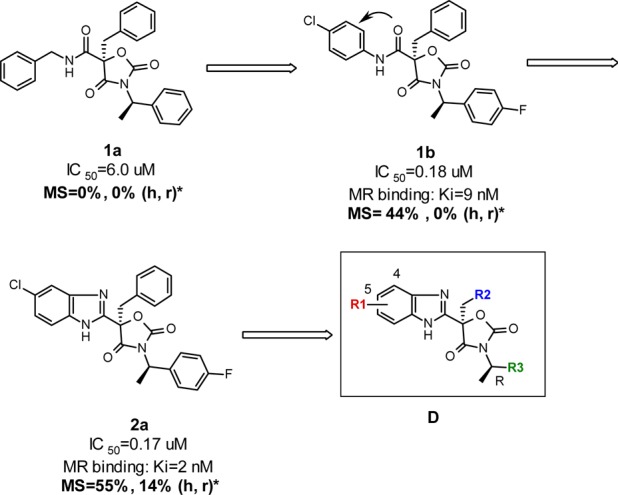
Starting from the high-throughput screening hit 1a (Figure 2), a preliminary study provided a series of nonsteroidal, potent, and selective oxazolidinedione MR antagonists.16 However, these compounds, in general, had poor liver microsomal stability; they were almost completely metabolized in 30 min upon incubation in the presence of human or rat liver microsomes. The in vitro metabolite identification study revealed that the primary metabolic pathways of these compounds originated from the hydrolysis of the amide, dealkylation of the benzylamine moiety, and extensive oxidation of the three aromatic groups. In light of these findings, aniline-derived amide 1b was prepared in order to minimize the N-dealkylation of the benzylamine. In addition, fluorine and chlorine atoms were introduced to two of the three aryl groups in order to reduce their propensity toward oxidative metabolism. While these additions did not improve rat liver microsomal stability (MS, 0%), measured by the remaining parent molecule at 30 min postincubation, they did increase the human liver microsomal stability (44%). Furthermore, the MR antagonist potency of 1b was significantly increased relative to 1a. It was further envisaged that the issue of amide hydrolysis could be solved by replacing the amide group with a heterocyclic bioisostere, such as monocyclic heteroaryls including oxadiazole, triazole, or imidazole, as described previously.17 Alternatively, we envisage that a bicyclic heteroaryl such as benzimidazole might provide distinct structure–activity relationships (SARs) and different properties. Gratifyingly, analogue 2a, the first benzimidazole analogue prepared in this series, not only displayed similar MR potency as 1b but also demonstrated improved rat liver microsomal stability. Thus, a new benzimidazole-based oxazolidinedione class emerged.
Figure 2.
Evolutions leading to benzimidazole oxazolidinedione analogues.
Compounds generated during the hit-to-lead process were screened for in vitro activity against human MR in a cell-based functional assay (hMR-NH-Pro),18 which is a commercially available PathHunter protein–protein interaction assay that measures the ability of compounds to antagonize full-length human MR binding to a coactivator peptide. In this assay, the average IC50 values for SPL and EPL were 11 and 240 nM, respectively. In addition, most compounds were also characterized in human and rat liver microsome stability assays.
The exploration of the SAR with respect to the R2 and R3 regions of scaffold D (Figure 2) indicates that phenyl and 4-fluorophenyl, respectively, afforded the best activity against MR. Tables 1 and 2 summarize the efforts in optimizing the R1 moiety of the scaffold, namely, the 5- and 4-positions of the benzimidazole, respectively.
Table 1. In Vitro SAR of 5-Substituted Benzimidazole Analoguesa.

Values are the average of two experiments, each in 10-point titrations.
Percent remaining at 0.5 h and 1 μM incubation in human (h) and rat (r) liver microsomes (LM).
Table 2. In Vitro SAR of 4-Substituted Benzimidazole Analoguesa.

Values are the average of two experiments, each in 10-point titrations.
Percent remaining at 0.5 h and 1 μM incubation in human (h) and rat (r) liver microsomes (LM).
Analogues containing small halides (2a, 2c, 3b), trifluoromethyl (2d, 3c), alkoxide (2e), nitrile (2i), amide (2h), and sulfonamide (2f, 3e) provided modest MR activity with IC50 values ranging from 100 to 500 nM in the functional assay. Substituting benzimidazole with heteroaryls or heterocycles at the 4-position led to several compounds (2k, 2l, 2n, 2o, 2p, 2q, 2s, 2t, 2u) with improved IC50 values below 100 nM. In contrast, the analogues bearing 4-subsituted heteroaryls (3f–3k) were, in general, less active than the 5-substituted counterparts (2j–2o, 2q–2u).
Most compounds depicted in Table 1 had acceptable human microsomal stability (>50% of parent remaining after 30 min of incubation). The rat microsomal stability varied significantly depending on the substituents of the benzimidazole. Two types of modifications were adopted to successfully improve the rat liver microsomal stability of compounds. The first was to introduce sulfonamide functionality (2f), and the second was to place a heteroaryl group (2j, 2k, 2m, 2o, 2p) at the 5-position of the benzimidazole moiety.
Substitution at the 4-position of benzimidazole (Table 2) also had a profound impact on the rat liver microsomal stability. The trifluoromethyl analogue 3c had the highest rat microsomal stability followed by amide 3d and sulfonamide 3e.
Related to MR are other nuclear hormone receptors, such as androgen receptor (AR), glucocorticoid receptor (GR), estrogen receptor (ER), and progesterone receptor (PR). It is critical to develop MR antagonists with excellent selectivity to avoid off-target mediated adverse effects. As such, cell-based reporter gene assays were performed with a panel of nuclear hormone receptors including human AR, GR, ERα, ERβ, and PRβ (Table 3). As described in Table 3, SPL (B) had modest selectivity for MR over AR (∼60-fold) and some residual activity against GR and PRβ. In contrast, EPL (C) displayed remarkable selectivity against other nuclear hormone receptors examined. While SPL, EPL, and benzimidazole oxazolidinediones were typically completely inactive in the agonist mode of the cell-based counterscreening assays, the selectivity in the antagonist mode continued to be a challenge. Most oxazolidinediones appeared to have some GR, AR, ERα, ERβ, and PRβ antagonist activity. Several compounds, such as 2o, 2p, and 2q, displayed over 40-fold selectivity profile across the board, which represents a significant improvement over the previously described compounds (>5-fold).17 Compared to SPL, which had a 60-fold selectivity against AR, compounds 2o and 2q had a selectivity of 90- and 110-fold, respectively.
Table 3. Nuclear Hormone Receptor Selectivity of Representative Analogues (NH_PRO Antagonist Mode Assay).
| compd | MR IC50 (μM) | GR IC50 (μM) | AR IC50 (μM) | ERα IC50 (μM) | ERβ IC50 (μM) | PRβ IC50 (μM) |
|---|---|---|---|---|---|---|
| B | 0.011 | 4.1 | 0.67 | >20 | >20 | 4.0 |
| C | 0.24 | 18 | >20 | >20 | >20 | >20 |
| 2a | 0.16 | 3.8 | 3.1 | 3.3 | 2.4 | 2.4 |
| 2d | 0.12 | 3.4 | 3.0 | 3.0 | 2.7 | 3.2 |
| 2f | 0.22 | >20 | 13 | 17 | 12 | 4.9 |
| 2m | 0.12 | 3.0 | 3.1 | 6.3 | 2.6 | 2.9 |
| 2o | 0.039 | 2.9 | 3.4 | 4.9 | 3.0 | 2.8 |
| 2p | 0.040 | 4.9 | 7.7 | 6.1 | 5.3 | 1.9 |
| 2q | 0.034 | 3.1 | 3.7 | 7.9 | 2.7 | 2.9 |
| 3e | 0.21 | 17 | 11 | >20 | 7.3 | 8.2 |
The PK profiles of a subset of compounds in rats are shown in Table 4. A range of oral bioavailability (6–54%) was observed for these compounds, of which 2d, 2o, 2p, and 3c had modest to good bioavailability. It is worth noting that several compounds (2a, 2d, 2q, and 3c) displayed high plasma protein binding (>99%) in whole plasma, and thus, free fraction was typically measured in the presence of 10% plasma and presumed to be correlated to the free fraction in total plasma. By introducing polar substituents to benzimidazole such as a sulfonamide (2f, 3e), heteroaryl (2m), or heterocycle (2p), higher plasma free fraction was observed, and generally rat liver microsomal stability was improved as well (Tables 1 and 2).
Table 4. Rat PK Profiles and Plasma Protein Binding Data in Human and Rat Plasma (10%)a.
| compd | F% | Clp (mL/min/kg) | Vdss (L/kg) | T1/2 (h) | AUCNpo (μM·h·kg/mg) | free fraction in 10% plasma (%, human, rat) |
|---|---|---|---|---|---|---|
| 2a | 15 | 47 | 4.6 | 1.5 | 0.11 | 0.2, 1 |
| 2d | 36 | 33 | 3.4 | 1.7 | 0.33 | 0.2, 0.7 |
| 2f | 6 | 32 | 1.8 | 1.6 | 0.067 | 3.4, 6.4 |
| 2m | 27 | 19 | 1.6 | 1.8 | 0.49 | 4, 1.7 |
| 2o | 54 | 20 | 1.9 | 1.2 | 0.79 | - |
| 2p | 44 | 67 | 2.3 | 0.69 | 0.07 | 9.3, 6.5 |
| 2q | 11 | 80 | 5.0 | 0.94 | 0.046 | 0.2, 0.8 |
| 3c | 44 | 23 | 3.4 | 2.4 | 0.067 | 0.2, 0.5 |
| 3e | 15 | 55 | 3.2 | 1.2 | 0.085 | 5.8, 6.4 |
Values are an average of two wistar han rats for IV dose and three rats for p.o. dose. Formulation: The IV doses were formulated at 1 mg/kg in DMSO/PEG200/water (10:60:30), and the oral doses were formulated at 2 mg/kg in imwitor/tween (1:1) and given by oral gavage. F%, oral bioavailability; Clp, plasma clearance; Vdss, volume of distribution; Cmax, observed maximal plasma concentration following oral dosing; T1/2, terminal half-life; AUCNpo; normalized area under the curve for oral dosing.
Potent MR antagonists with reasonable rat PK were further evaluated in an acute rat pharmacodynamic model, which measured the amount of urinary sodium excretion (natriuresis) over 6 h after a single oral dose, using SPL as a positive control. It should be noted that rat and human MRs have 90% identity, and the two proteins share the exactly same DNA and ligand binding domains. Therefore, compounds are presumed to have similar human and rat MR activities.19 Of several compounds being tested (2a, 2f, 2j, 2p, and 2q) in the natriuresis rat model, 2a, 2f, and 2q exhibited a modest but statistically significant PD effect (p < 0.05). Furthermore, compound 2p had the most pronounced dose-dependent natriuretic effect as shown in Figure 3, and the effect at 100 mg/kg was comparable to SPL at 30 or 100 mg/kg. Interestingly, previous amide replacement by monocyclic heteroaryls17 exhibited much weaker efficacy at the same doses in the same model (data not shown). Taking in vivo efficacy and selectivity into consideration, there are unique advantages of using substituted benzimidazoles as replacement of amide.
Figure 3.
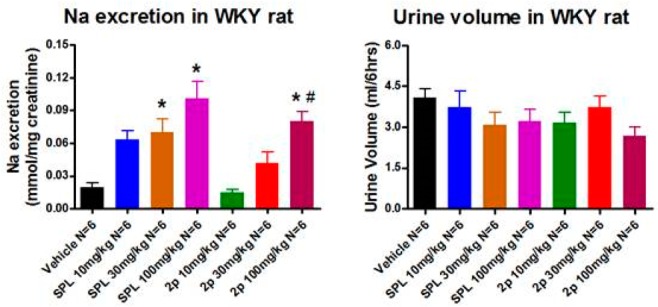
Dose-dependent natriuretic effect of SPL and 2p in WKY rats. Vehicle: inwitor/tween, 2 mL/kg. Animals fed with low salt diet; jugular vein bleedings were performed 6 h postdose for drug exposure (mpk, mg/kg; *, p < 0.05 vs vehicle; #, p < 0.05 vs 2p 10 mg/kg).
A representative synthesis of oxazolidinedione analogues is illustrated in Scheme 1. The α-hydroxylation of malonate 4, applying vigorous air bubbling over 3 days, afforded the hydroxylated intermediate 5, which then underwent a monohydrolysis to yield monocarboxylic acid 6. Subsequent chiral supercritical fluid chromatography (SFC) separation provided the chiral acid 7, which was then coupled with 1-chloro-3,4-benzenediamine to generate amide intermediate 8, followed by heating in acetic acid to yield the benzimidazole intermediate 9. Finally, the reaction of the benzimidazole 9 with isocyanate 10 in the presence of NaOH provided compound 2a. The cross-coupling reaction of 11, the 5-Br analogue of 2a, with various boronic acids/esters generated the corresponding heteroaryl-substituted analogues. For example, the Suzuki coupling of compound 11 and pyrimidine boronic acid 12 afforded 2u in good yield.
Scheme 1.

Reagents and conditions: (a) CsF, DMF, 40 °C to rt, 3 d, 71%; (b) KOH, EtOH, 46%, rt, overnight; (c) chiral SFC (OJ-H, 4.6 × 100 mm, 5% MeOH/0.1% TFA/CO2, 2.5 mL/min, 100 bar); (d) HATU, iPr2NEt, 1-chloro-3,4-benzenediamine, DMF, overnight; (e) AcOH, 75 °C, 2 h, 58% over 2 steps; (f) NaOH, THF, 70 °C, 25 min, 77%; (g) Pd(OAc)2, dppf, K3PO4, EtOH, 120 °C, microwave, 5 min, 76%.
In summary, oxazolidinediones derived from the HTS hit 1a represent a novel class of MR antagonists. SAR studies were directed to enhance potency and human and rat liver microsomal stability, as well as rat PK in order to elicit PD effects in rats. Improvements in liver microsomal stability and PK were realized by replacing the central amide functionality with benzimidazole. The endeavor of optimizing the benzimidazole series by adding a polar substituent led to several MR antagonists with acceptable rat PK, potency against MR, and selectivity against other nuclear hormone receptors. Ultimately, analogue 2p demonstrated similar natriuretic effect in rats as SPL at 100 mg/kg. As such, the benzimidazole oxazolidinedione class became a validated lead series for further optimization, which will be reported in due course.
Supporting Information Available
Experimental procedures, NMR, LC/MS data, and chiral separation. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Chai W.; Danser A. H. Why are mineralocorticoid receptor antagonists cardioprotective?. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2006, 374, 153–162. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funder J. W. Glucocorticoid and mineralocorticoid receptors: Biology and clinical relevance. Annu. Rev. Med. 1997, 48, 231–240. and references therein.. [DOI] [PubMed] [Google Scholar]
- McKelvie R. S.; Yusuf S.; Pericak D.; Avezum A.; Burns R. J.; Probstfield J.; Tsuyuki R. T.; White M.; Rouleau J.; Latini R.; Maggioni A.; Young J.; Pogue J. Comparison of candesartan, enalapril, and their combination in congestive heart failure, randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study, the RESOLVD pilot study investigators. Circulation 1999, 100, 1056–1064. [DOI] [PubMed] [Google Scholar]
- Ouzan J.; Perault C.; Lincoff A. M.; Carre E.; Mertes M. The role of spironolactone in the treatment of patients with refractory hypertension. Am. J. Hypertens. 2002, 15, 333–339. [DOI] [PubMed] [Google Scholar]
- Krum H.; Nolly H.; Workman D.; He W.; Roniker B.; Krause S.; Fakouhi K. Efficacy of eplerenone added to renin-angiotensin blockade in hypertensive patients. Hypertension 2002, 40, 117–123. [DOI] [PubMed] [Google Scholar]
- Parthasarathy H. K.; Ménard J.; White W. B.; Young W. F. Jr; Williams G. H.; Williams B.; Ruilope L. M.; McInnes G. T.; Connell J. M.; MacDonald T. M. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J. Hypertens. 2011, 29, 980–990. [DOI] [PubMed] [Google Scholar]
- Neel D. A.; Brown M. L.; Lander P. A.; Grese T. A.; Defauw J. M.; Doti R. A.; Fileds T.; Kelley S. A.; Smith S.; Zimmerman K. M.; Steinberg M. I.; Jadhav P. K. 3,3-Bisaryloxindoles as mineralocorticoid receptor antagonists. Bioorg. Med. Chem. Lett. 2005, 15, 2553–2557. [DOI] [PubMed] [Google Scholar]
- Meyers M. J.; Hu X. Non-steroidal mineralocorticoid receptor antagonists. Expert Opin. Ther. Pat. 2007, 17, 17–23. [Google Scholar]
- Bell M. D.; Gernert D. L.; Grese T. A.; Belvo M. D.; Borromeo P. S.; Kelley S. A.; Kennedy J. H.; Kolis S. P.; Lander P. A.; Richey R.; Sharp V. S.; Stephenson G. A.; Williams J. D.; Yu H.; Zimmerman K. M.; Steinberg M. I.; Jadhav P. K. (S)-N-{3-[1-Cyclopropyl-1-(2,4-difluoro-phenyl)-ethyl]-1H-indol-7-yl}-methanesulfonamide: A potent, nonsteroidal, functional antagonist of the mineralocorticoid receptor. J. Med. Chem. 2007, 50, 6443–6445. [DOI] [PubMed] [Google Scholar]
- Arhancet G. B.; Woodard S. S.; Dietz J. D.; Garland D. J.; Wagner G. M.; Iyanar K.; Collins J. T.; Blinn J. R.; Numann R. E.; Hu X.; Huang H.-C. Stereochemical requirements for the mineralocorticoid receptor antagonist activity of dihydropyridines. J. Med. Chem. 2010, 53, 4300–4304. [DOI] [PubMed] [Google Scholar]
- Arhancet G. B.; Woodard S. S.; Iyanar K.; Case B. L.; Woerndle R.; Dietz J. D.; Garland D. J.; Collins J. T.; Payne M. A.; Blinn J. R.; Pomposiello S. I.; Hu X.; Heron M. I.; Huang H. C.; Lee L. F. Discovery of novel cyanodihydropyridines as potent mineralocorticoid receptor antagonists. J. Med. Chem. 2010, 53, 5970–5978. [DOI] [PubMed] [Google Scholar]
- Meyers M. J.; Arhancet G. B.; Hockerman S. L.; Chen X.; Long S. A.; Mahoney M. W.; Rico J. R.; Garland D. J.; Blinn J. R.; Collins J. T.; Yang S.; Huang H.-C.; McGee K. F.; Wendling J. M.; Dietz J. D.; Payne M. A.; Homer B. L.; Heron M. I.; Reitz D. B.; Hu X. Discovery of (3S, 3aR)-2-(3-chloro-4-cynaophenyl)-3-cyclopentyl-3, 3a, 4, 5-tetrahydro-2H-benzo[g]indazole-7-carboxylic acid (PF3883845), an orally efficacious mineralocorticoid receptor (MR) antagonist for hypertension and nephropathy. J. Med. Chem. 2010, 53, 5979–6002. [DOI] [PubMed] [Google Scholar]
- Hasui T.; Matsunaga N.; Ora T.; Ohyabu N.; Nishigaki N.; Imura Y.; Igata Y.; Matsui H.; Motoyaji T.; Tanaka T.; Habuka N.; Sogabe S.; Ono M.; Siedem C. S.; Tang T. P.; Gauthier C.; De Meese L. A.; Boyd S. A.; Fukumoto S. Identification of benzoxazin-3-one derivatives as novel, potent, and selective nonsteroidal mineralocorticoid receptor antagonists. J. Med. Chem. 2011, 54, 8616–8631. [DOI] [PubMed] [Google Scholar]
- Piotrowski D. W. Mineroalocorticoid receptor antagonists for the treatment of hypertension and diabetic nephropathy. J. Med. Chem. 2012, 55, 7957–7966. [DOI] [PubMed] [Google Scholar]
- Casimiro-Garcia A.; Piotrowski D. W.; Ambler C.; Arhancet G. B.; Banker M. E.; Banks T.; Boustany C. M.; Cai C.; Chen X.; Eudy R.; Hepworth D.; Hulford C. A.; Jennings S. M.; Loria P. M.; Meyers M. J.; Petersen D. N.; Raheja N. K.; Sammons M.; She L.; Song K.; Vrieze D.; Wei L. Identification of (R)-6-(1-(4-cyano-3methylphenyl)-5-cyclopentyl-4,5-dihydro-1H-pyrazol-3-yl-2–15methoxynicotinic acid, a highly potent and selective nonsteroidal mineralocorticoid receptor antagonist. J. Med. Chem. 2014, 57, 4273–4288. [DOI] [PubMed] [Google Scholar]
- Yang C.; Shen H. C.; Wu Z.; Chu H. D.; Cox J. M.; Balsells J.; Crespo A.; Brown P.; Zamlynny B.; Wiltsie J.; Clemas J.; Gibson J.; Contino L.; Lisnock J.; Zhou G.; Garcia-Calvo M.; Bateman T.; Xu L.; Tong X.; Crook M.; Roy S.; Tata J. R.; Sinclair P. Discovery of novel oxazolidinedione derivatives as potent and selective mineralocorticoid receptor antagonists. Bioorg. Med. Chem. Lett. 2013, 23, 4388–4392. [DOI] [PubMed] [Google Scholar]
- Cox J. M.; Chu H. D.; Yang C.; Shen H. C.; Wu Z.; Balsells J.; Crespo A.; Brown P.; Zamlynny B.; Wiltsie J.; Clemas J.; Gibson J.; Contino L.; Lisnock J.; Zhou G.; Garcia-Calvo M.; Bateman T.; Xu L.; Tong X.; Crook M.; Sinclair P. Mineralocorticoid receptor antagonists: identification of heterocyclic amide replacements in the oxazolidinedione series. Bioorg. Med. Chem. Lett. 2014, 24, 1681–1684. [DOI] [PubMed] [Google Scholar]
- Nuclear Hormone Receptors. http://www.discoverx.com/nhrs/prod-nhrs.php.
- UniProt. http://www.uniprot.org.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.