

Abstract
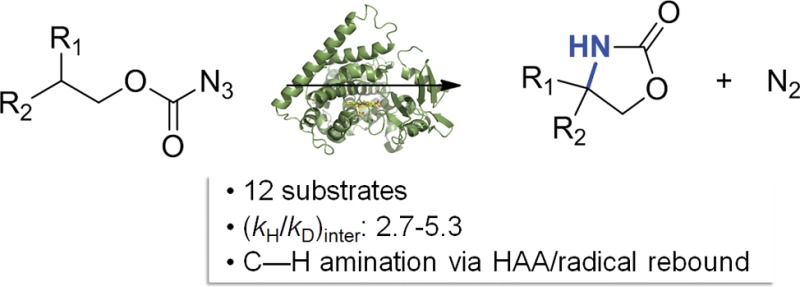
Cytochrome P450 enzymes can effectively promote the activation and cyclization of carbonazidate substrates to yield oxazolidinones via an intramolecular nitrene C–H insertion reaction. Investigation of the substrate scope shows that while benzylic/allylic C–H bonds are most readily aminated by these biocatalysts, stronger, secondary C–H bonds are also accessible to functionalization. Leveraging this “non-native” reactivity and assisted by fingerprint-based predictions, improved active-site variants of the bacterial P450 CYP102A1 could be identified to mediate the aminofunctionalization of two terpene natural products with high regio- and stereoselectivity. Mechanistic studies and KIE experiments show that the C–H activation step in these reactions is rate-limiting and proceeds in a stepwise manner, namely, via hydrogen atom abstraction followed by radical recombination. This study expands the reactivity scope of P450-based catalysts in the context of nitrene transfer transformations and provides first-time insights into the mechanism of P450-catalyzed C–H amination reactions.
Keywords: C−H amination, cytochrome P450, biocatalysis, carbonazidate, oxazolidinones
1. Introduction
Expanding the scope of protein-based catalysts beyond the reaction space encompassed by natural enzymes constitutes an important, and increasingly sought-after goal in biocatalysis.1−11 A synthetically most valuable transformation is the direct functionalization of aliphatic C–H bonds, which occur ubiquitously in man-made and natural compounds. Of particular interest are methods for the direct conversion of aliphatic C–H bonds into C–N bonds, owing to the abundance of nitrogen functionalities in biologically active molecules.12,13 Accordingly, significant efforts have been devoted over the past several years to the development of synthetic transition metal catalysts to promote C(sp3)—H amination transformations, especially via nitrene C–H insertion.14−18 In stark contrast, natural enzymes capable of supporting C(sp3)—H amination chemistry via direct C–H bond functionalization are extremely rare (e.g., SAM-dependent lysine 2,3-aminomutase)19 and none is known to catalyze nitrene C–H insertion reactions. This “gap” prompted us and others to develop engineered and artificial biocatalytic systems for promoting this transformation.7,20−22 In independent studies, our laboratory and the Arnold group demonstrated that engineered cytochrome P450 enzymes containing either a cysteine-7 or a serine-ligated heme20 can catalyze the intramolecular C–H amination of arylsulfonyl azides with high catalytic efficiency and selectivity. More recently, activation of arylsulfonyl azide substrates for intramolecular C–H amination using alternative hemoprotein-based catalysts21 as well as for intermolecular sulfimidation23 was reported.
Based on the promising nitrene transfer reactivity exhibited by engineered variants of CYP102A1 (P450BM3)24 with arylsulfonyl azides,7 we envisioned the possibility to exploit P450 catalysts for engaging alternative nitrene donors in intramolecular C–H amination processes. In this regard, the construction of oxalidinone rings via cyclative nitrene C–H insertion25−27 is of particular relevance as these compounds can provide rapid access, upon hydrolysis, to valuable (chiral) 1,2-aminoalcohols, key functional motifs in many bioactive molecules28 and chiral auxiliaries.29 To date, this type of transformation has been achieved with rhodium or silver-based catalysts in the presence of carbamate-derived iminoiodinanes25,26 or N-tosyloxycarbamates27 as the substrates. Compared to the latter, organic azides represent considerably more attractive nitrene precursors, owing to their high atom economy and the release of environmentally benign N2 gas as the byproduct of the amination reaction.18,30 Here, we report that engineered cytochrome P450 enzymes can effectively catalyze the cyclization of carbonazidate substrates to yield oxazolidinones via an intramolecular C–H amination reaction.
2. Experimental Section
2.1. Chemical Synthesis
Synthentic procedures and characterization data for the acyloxycarbamates 1–3, carbonazidates 4, 6–14, 23–24, Z-12, deuterated probe compounds D-6, D1-11, D2-11, and the corresponding oxazolidinone and carbamate products are provided as Supporting Information.
2.2. Protein Expression and Purification
Wild-type CYP102A1 and its engineered variants were expressed from pCWori-based plasmids containing the P450 gene under the control of an IPTG-inducible promoter (BamHI/EcoRI cassette) according to procedures described previously.31 Typically, cultures of recombinant DH5α cells were grown at 37 °C (200 rpm) in Terrific Broth (TB) medium supplemented with ampicillin (100 mg/L) until OD600 reached 1.0 and then induced with 0.25 mM β-d-1-thiogalactopyranoside (IPTG) and 0.3 mM δ-aminolevulinic acid (ALA). After induction, cultures were shaken at 150 rpm and 27 °C and harvested after 20 h by centrifugation at 4000 rpm at 4 °C. Cell lysates were prepared by sonication and loaded on Q Sepharose resin. P450s were eluted using 20 mM Tris, 340 mM NaCl, pH 8.0. After buffer exchange (50 mM potassium phosphate buffer, pH 8.0), the enzymes were stored at −80 °C. P450 concentration was determined from CO-binding difference spectra (ε450–490 = 91 000 M–1 cm–1). The vector encoding for the thermostable phosphite dehydrogenase (PTDH) variant Opt13 was kindly provided by the Zhao group.32 PTDH was overexpressed from pET-15b-based vector in BL21(DE3) cells and purified using Ni-affinity chromatography according to the published procedure.32
2.3. P450 Reactions
The enzymatic reactions were carried out at a 400 μL-scale using 5–20 μM purified P450 and 10 mM substrate in the presence of a PTDH-based cofactor regeneration system (1 μM PTDH, 100 μM NADP+, 50 mM sodium phosphite). In a typical procedure, a solution containing the cofactor regeneration system in potassium phosphate buffer (50 mM, pH 8.0) was degassed by bubbling argon into the mixture for 5 min in a sealed vial. A buffered solution containing the P450 enzyme was carefully degassed in a similar manner in a separate vial. The two solutions were then mixed together via cannulation. Reactions were initiated by addition of 8 μL of azide (from a 0.5 M stock solution in methanol) with a syringe, and the reaction mixture was stirred for 16 h at room temperature under positive argon pressure. Control reactions with hemin were conducted following an identical procedure with the difference that the P450 was replaced by 80 μL of a hemin solution (100 μM in DMSO:H2O, 1:1) and sodium dithionite (10 mM) was used as the reductant. Prior to chromatographic analysis (see Analytical Methods below), the reaction mixtures were extracted with 400 μL CH2Cl2, followed by evaporation of the organic layer and dissolution of the residue in 100 μL MeOH. Calibration curves for quantification of the different oxazolidinones (Figure S4) were generated using authentic standards produced synthetically (see Synthetic Procedures in SI). All measurements were performed in triplicate. For each experiment, negative control samples containing either no enzyme or no reductant were included. For the CO-inactivation experiment, carbon monoxide was bubbled through the reaction mixture containing the P450 enzyme and sodium dithionite prior to addition of the azide substrate. For the natural product substrates (menthol- and borneol-derived carbonazidates), the reaction was quenched by adding excess dithionite, followed by stirring for 24 h before the addition of internal standard and extraction with CH2Cl2. The organic layer was then analyzed by GC-MS.
2.4. Analytical Methods
GC/MS analyses were performed on a Shimadzu GCMS-QP2010 equipped with a RTX-XLB column (30 m × 0.25 mm × 0.28 μm) and a quadrupole mass analyzer. Separation method: 1 μL injection, inj. temp.: 250 °C, detector temp.: 220 °C. Gradient: column temperature set at 50 °C for 1 min, then to 280 °C at 30 °C/min, then to280 °C for 4 min. Total run time was 13.67 min. Enantiomeric excess was determined by chiral gas chromatography (GC) using a Shimadzu GC-2010 gas chromatograph equipped with a FID detector and a Agilent CYCLOSIL-B column (30 m × 0.25 mm × 0.25 μm film). Separation method for 4-methyl-4-(p-tolyl)oxazolidin-2-one (15): 1 μL injection, injector temp.: 200 °C, detector temp: 300 °C. Gradient: column temperature set at 150 °C for 0 min, then to 200 °C at 4.5 °C/min, then to 230 °C at 1.5 °C/min, then to 245 °C at 15 °C/min for 8 min. Separation method for 4-phenyloxazolidin-2-one (20): 1 μL injection, injector temp.: 200 °C, detector temp: 300 °C. Gradient: column temperature set at 150 °C for 0 min, then to 200 °C at 4.5 °C/min, then to 220 °C at 1 °C/min, then to 245 °C at 15 °C/min for 8 min. HPLC analyses were performed using a Shimadzu LC-2010A-HT equipped with a VisionHT-C18 reverse-phase column and a multidiode UV–VIS detector. Injection volume: 10 uL. Flow rate: 1 mL/min. Gradient: 10% acetonitrile in water (0.1% TFA) for 3 min, then increased to 90% over 24 min.
2.5. Kinetic Isotope Effect (KIE) Experiments
P450 reactions were carried out at a 400 μL-scale using 20 μM P450, 10 mM NADPH and stopped after 1 h. For KIE determination with 6 and D-6, parallel reactions with either of the carbonazidate substrates were carried out at varying substrate concentration (100 μM to 2 mM). The reactions were analyzed by HPLC and KIE values were calculated from the resulting plots of initial rate against substrate concentration. For the intra- and intermolecular KIE analyses with D1-11 and with equimolar mixtures of 11 and D2-11, respectively, the reactions were carried out as described above using 10 mM total substrate concentration, followed by GC-MS analysis. KIE values were determined based on the ratios between the relative abundances of the characteristic fragments with m/z of 105 (nondeuterated) and m/z of 106 (deuterated) (Figure S5). Authentic standards of product 20 and D-20 injected individually and in equimolar mixture were used as references (Figure S5).
3. Results and Discussion
In preliminary experiments, a series of oxycarbonyl-based nitrene donors were prepared using 2-phenylpropanol derivatives as model substrates (Table 1). These compounds comprised acetoxy- (1) and aryloxy-carbamate (2, 3) derivatives, which have been previously investigated in the context of rhodium-catalyzed nitrene transfer reactions.27 Given the ability of P450s to activate sulfonyl azides,7 the carbonazidate derivative 4 was also synthesized. In addition to being readily accessible from a synthetic standpoint, carbonazidates are most desirable precursors for oxazolidinone formation via nitrene C–H insertion because of their high atom economy, with N2 being produced as byproduct as opposed to AcOH or ArOH with 2 or 3. Despite these favorable features and the successful application of carbonazidates in the context of sulfimidation,33,34 olefin aziridination,35,36 and aminohalogenation,33,37 catalytic intramolecular C–H amination reactions involving these compounds have not been reported to our knowledge. CYP102A1 variant FL#62 was chosen for the initial scouting experiments. Compared to wild-type CYP102A1, FL#62 contains five active site mutations (V78A, F81S, A82 V, F87A, A184 V), which contribute to expand its active site and make it accessible to large and bulky substrates.31,38,39 In addition, FL#62 exhibited the highest C–H amination activity in the presence of arylsulfonyl azides in our previous studies.7
Table 1. C–H Amination Reactivity of Wild-Type CYP102A1 and Variant FL#62 on Different Carbonyl-Based Nitrene Donors[a].
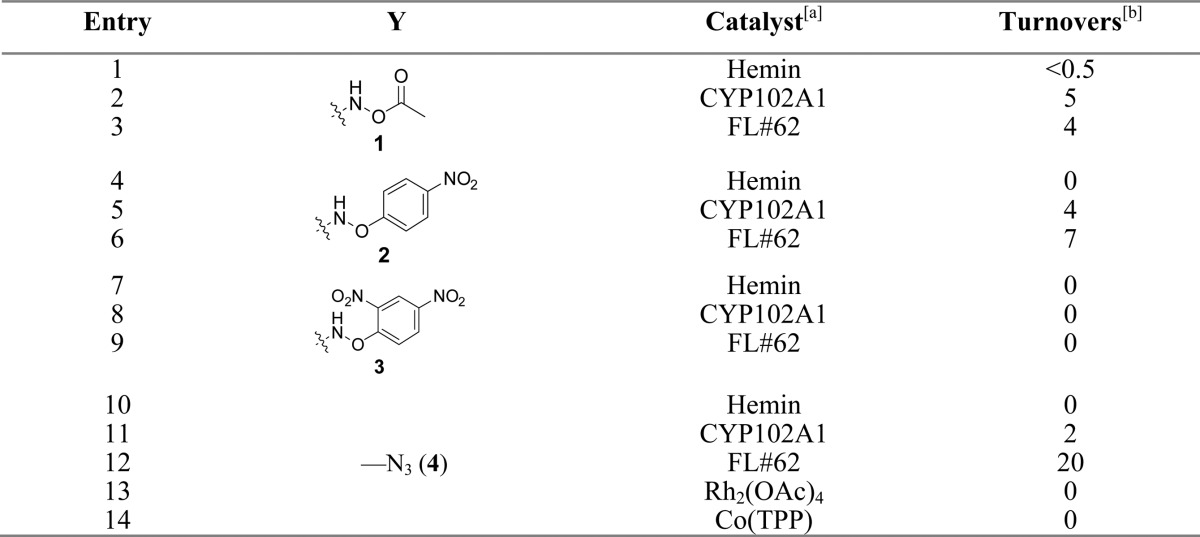
Reaction conditions: 20 μM P450, 10 mM substrate, 10 mM NADPH. Hemin reactions: 200 μM hemin, 10 mM substrate, 10 mM Na2S2O4. Rh2(OAc)4/Co(TPP) reactions: 5 mol % catalyst, 200 mM 4 in dry toluene.
As determined by HPLC (SD within 15%).
P450 reactions with substrates 1–4 were performed in the presence of NADPH, to reduce the enzyme to its ferrous state, and anaerobically, both of which conditions were previously found to be critical for supporting P450-dependent nitrene transfer reactivity.7 Gratifyingly, both wild-type CYP102A1 and FL#62 were found to yield appreciable amounts of the desired oxazolidinone 5 in the presence of the acetoxy- and aryloxy-carbamates 1–3 (turnover number (TON): 4–7, Table 1). Importantly, significantly better conversion to the oxazolidinone product could be achieved from the carbonazidate substrate 4 (Table 1) with FL#62 as the catalyst. In contrast, little or no cyclic product was formed in the presence of wild-type CYP102A1 or free hemin. Notably, no reaction was observed in the presence of 4 under standard amination conditions in the presence of Rh2(OAc)4, as noted previously,27 or Co-(TPP) (TPP = tetraphenylporphyrin), two prominent organometallic catalysts investigated in the context of C–H amination reactions.16,40
Toward optimization of the reaction conditions, the importance of the source of reducing equivalents in the P450-catalyzed conversion of carbonazidate 4 was investigated. These experiments showed that cyclization of 4 could be achieved by replacing NADPH with the inexpensive dithionite (Na2S2O4), albeit only with lower efficiency (5 vs 20 TON). This difference could be attributed to the kinetically competitive reduction of the carbonazidate substrate to the corresponding carbamate by action of the inorganic reductant as compared to the enzymatic C–H amination reaction (0.2 turnovers/min). More promisingly, comparable number of turnovers as observed in the presence of an excess of NADPH (10 mM) could be obtained using catalytic amounts of NADPH (100 μM) in combination with a phosphite dehydrogenase32 based cofactor regeneration system. Thus, under optimized conditions, over 70 catalytic turnovers were achieved for the FL#62-catalyzed transformation of 4 into 5 (Entry 1, Table 2). These improvements notwithstanding, the yield of the C–H amination product remained moderate (∼4%), with the remainder of the carbonazidate substrate being converted to a carbamate and alcohol products as discussed in more details later.
Table 2. Scope of P450-Catalyzed C–H Amination with Carbonazidate Substrates[a].
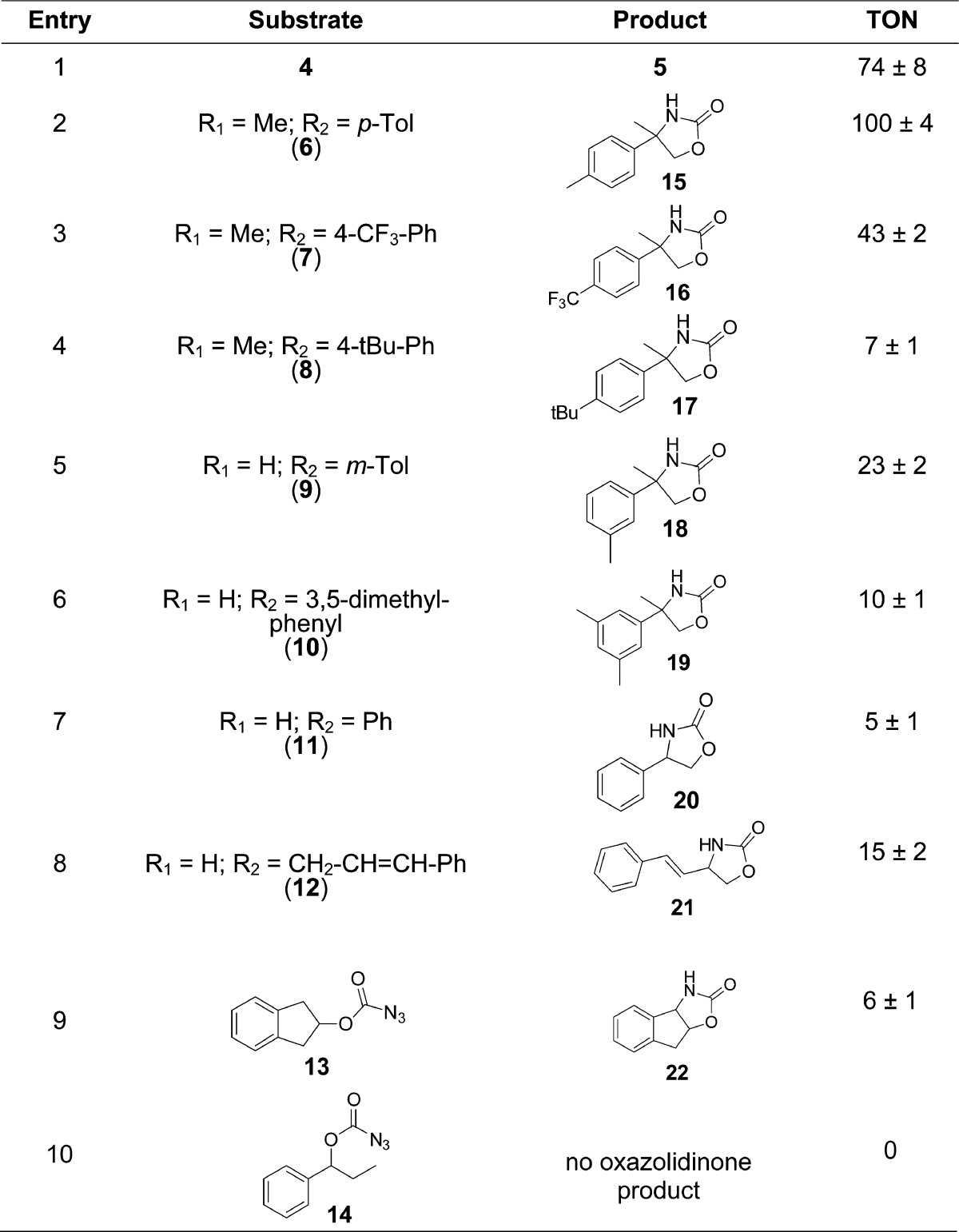
Reactions conditions: 5 μM P450, 10 mM carbonazidate, cofactor regeneration system (100 μM NADP+, 1 μM phosphite dehydrogenase, 50 mM Na2PO3), 16 h. Turnover numbers (TON) were measured by HPLC from triplicate experiments.
3.1. Investigation of Substrate Scope
To gain insights into the influence of electronic and steric effects on the P450-catalyzed C–H amination reaction, a range of carbonazidates (6–10) were then prepared and tested for cyclization in the presence of FL#62. As shown in Table 2, various substitutions at the level of the aromatic ring of the substrate were tolerated by the enzyme, resulting in turnover numbers ranging from 10 to 100 (Entries 1–6, Table 2). Interestingly, an increase in TON was observed upon substitution of the para position with a methyl group (6; 100 vs 74 TON), whereas a reduction in C–H amination activity resulted from substitution of this site with an isosteric but electronwithdrawing trifluoromethyl group (7; 43 TON). This trend in reactivity is consistent with the electrophilic character of the reactive species envisioned to mediate these reactions (vide infra). Notably, derivatives with a bulky substituent (tert-butyl) at the para position (8) as well as double substitutions on the aromatic ring (10) were also processed by the enzyme.
With compounds 11–14, the influence of the relative strength of the target C–H bond on the efficiency of the reaction was investigated. Interestingly, FL#62-mediated amination of the secondary, benzylic C–H bond in 11 as well as in the more conformationally rigid 13 was found to occur with significantly lower efficiency (10-fold) as compared to the tertiary, benzylic C–H site in 4 (5–6 vs 74 turnovers). These results suggest a clear dependence of the P450-dependent amination activity on the C–H bond strength, a conclusion further supported by the comparatively higher activity of the enzyme on 12 (15 TON), and lack of activity on 14, in which the C–H bonds in β to the carbonazidate group have estimated bond dissociation energies (BDE) of 86 and 98 kcal/mol, respectively.41 This bias notwithstanding, the catalytic turnovers achieved by these P450 catalysts with carbonazidates compare very favorably with those reported for rhodium or silver catalysts in the context of oxalidinone synthesis from carbamate-iminoiodinane or N-tosyloxy-carbamate precursors (5–25 turnovers).25−27 Furthermore, the absence of aziridine formation in the reaction with 12 supports the chemoselectivity of the P450 catalyst toward nitrene C–H insertion over the potentially competing aziridination reaction.27,42
Chiral analysis of the oxazolidinone products generated from 4, 8, and 11 in the presence of FL#62 showed only low levels of enantioinduction (2–5% ee). These results were rather surprising as moderate to high degree of stereo- or enantioselectivity (40–91% ee) were observed in the cyclization of arylsulfonyl azides by means of FL#62 and related CYP102A1 variants.7 Screening of a panel of 15 FL#62-derived active site variants against carbonazidates 4, 8, and 11 gave similar results, suggesting that the limited chiral induction observed with these substrates is not dependent upon the active site configuration of the enzyme but it is rather linked to the mechanism of the C–H activation step operating in these processes as discussed further below.
3.2. Natural Product Substrates
To further investigate the scope and potential utility of the P450 catalysts in the context of oxazolidinone synthesis, carbonazidate derivatives of two natural product terpenes, menthol (23) and borneol (24), were subjected to FL#62 catalysis (Figure 1). Notably, both compounds could be converted to a single five-membered cyclic product, namely, the trans-oxazolidinone 25 and the cis-oxazolidinone 26, respectively, with high regio- and stereoselectivity. The high degree of site-selectivity obtained in the reactions with these substrates contrasts with the unselective nitrene C–H insertion events observed upon pyrolysis or photolysis of related compounds.43,44
Figure 1.
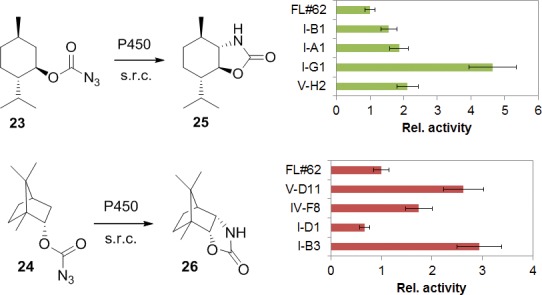
P450-catalyzed C–H amination in terpene natural products. The graphs illustrates the relative activity, based on TON, for the FL#62-derived active-site variants selected by fingerprint analysis (SCA). The amino acid mutations in the P450 variants can be found in Table S1. “s.r.c.” = standard reaction conditions, as indicated in Table 2.
Despite the successful cyclization of 23 and 24, we noted that the catalytic efficiency of FL#62 on these substrates was only modest (TON ∼ 4). Recently, we reported a fingerprint-based method for predicting the substrate reactivity of engineered P450s on target molecules that are structurally related to the probe compounds used for their fingerprinting (referred to as “fingerprint single component analysis” or SCA).38,39 Substrates 23 and 24 share significant scaffold similarity with two of said fingerprint probes, namely, the cyclohexane- and the bornane-based probes (Figure S1). Using SCA, four candidate P450 catalysts with potentially improved activity for the cyclization of these substrates were identified from our previously assembled collection of FL#62-based variants, which were derived from this P450 via simultaneous mutagenesis of 2 to 6 active site positions.31,39 Gratifyingly, these P450s were found capable of processing 23 and 24 supporting 3- to 5-fold higher catalytic turnovers than FL#62 (Figure 2). Altogether, these studies demonstrate the possibility to apply these P450 catalysts for C–H amination transformations in the context of structurally and stereochemically complex molecules. Furthermore, these results support the functionality of our fingerprint-based methods for P450 substrate reactivity prediction also in the context of P450-mediated C–H aminations.
Figure 2.
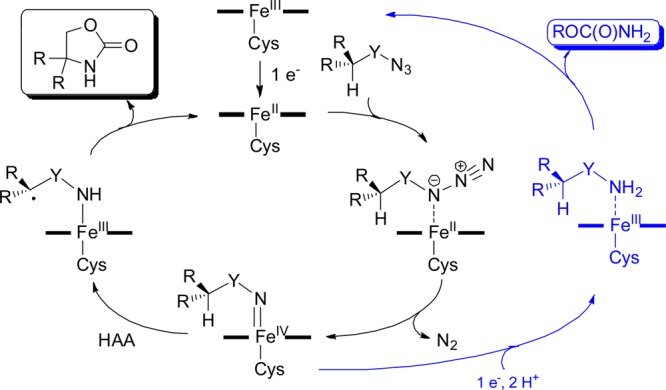
Proposed mechanism for P450-catalyzed conversion of carbonazidates to oxazolidinones. The competing pathway leading to the carbamate byproduct is indicated in blue. Y = —OC(O)—.
3.3. Mechanistic Studies
Further experiments were conducted to shed light into the mechanism of these reactions. The requirement for a reductant (i.e., dithionite or NADPH) and complete loss of carbonazidate C–H amination activity upon complexation of the reduced P450 with CO provided clear evidence in support of the involvement of the ferrous heme in catalysis. Accordingly and building upon our investigations with arylsulfonyl azides7 and previous studies with metalloporphyrin systems,45−48 we envision the catalytic cycle to initiate with the binding of the carbonazidate substrate to the ferrous heme to form an azido-iron(II) complex, which would then undergo nitrogen loss to give a putative imido-iron(IV) intermediate (Figure 2). Reasonably, the subsequent C–H activation event could then proceed through (a) a stepwise hydrogen atom abstraction (HAA)/radical rebound mechanism, analogous to that mediated by oxo-iron(IV)porphyrin π cation radical (Compound I) in P450-catalyzed C–H oxidations,49,50 or (b) a concerted nitrene C–H insertion, analogous to that operating in rhodium-catalyzed C–H aminations.51 To probe this step, the cis isomer of carbonazidate 12 ((Z)-12, Figure 3) was subjected to FL#62-catalyzed amination conditions, with scrambling of the double bond geometry upon cyclization being revealing of a radical mechanism. Interestingly, only formation of the rearranged trans oxazolidinone (E)-21 was observed (TON: 16), a product identical to that generated starting from (E)-12 (Figure 3). In contrast, rhodium-catalyzed cyclization of carbamate (Z)-27 activated in situ with PhI(OAc)2 yielded the cis oxazolidinone product (Z)-21. Thus, these results unambiguously revealed that a stepwise mechanism, involving HAA followed by radical recombination, is operative in the P450 amination reaction (Figure 2). Furthermore, the complete Z → E rearrangement of the olefinic bond in (Z)-12 is suggestive of the formation of a relatively long-lived carbon-centered radical, which could account for the observed lack of stereo/enantioselectivity upon amination of the benzylic C–H sites in 4, 8, and 11 by FL#62 and its variants. On the other hand, the high stereoselectivity observed in the case of the menthol and borneol derivatives (Figure 1) could stem from the faster rebound step expected to occur for the less stabilized secondary alkyl radical intermediate formed during the intramolecular C–H amination of these substrates.
Figure 3.

Rearrangement studies. (a) FL#62-catalyzed cyclization of (E)-12 (or its isomer (Z)-12) leads to formation of trans oxazolidinone (E)-21, as determined by HPLC (Figure S3 in SI) and GC-MS analysis. The Z → E rearrangement in case of (Z)-12 is indicative of the formation radical intermediate via HAA. (b) Rhodium-catalyzed cyclization of carbamate Z-27 is not accompanied by scrambling of the double bond configuration, in agreement with the concerted nitrene C–H insertion mechanism proposed for these catalysts.51
3.4. Kinetic Isotope Effect (KIE) Experiments
According to the mechanistic model of Figure 2, cleavage of the C–H bond was expected to be rate-limiting during oxazolidinone formation. To examine this aspect, the effect of H/D substitution on the C–H amination rate of substrate 6 was determined (Figure 4a). The positive primary kinetic isotope effect observed in these experiments (kH/kD: 2.7 ± 0.3 at 20 °C) is consistent with the involvement of C–H bond cleavage in the rate-determining step of the reaction. At the same time, this value is considerably smaller than and comparable to, respectively, that observed for C–H amination reactions with Ru-porphyrins (kH/kD: 6–12)46,52 and rhodium catalysts (kH/kD: 1.8–4.5),51,53 which were established to operate via a stepwise and concerted C–H activation pathway, respectively. In light of the large KIE difference compared to the Ru-porphyrin system, we wondered whether other steps prior to C–H bond cleavage (e.g., activation of the azide via N2 extrusion; Figure 2), may be also kinetically relevant in the P450 reaction, thus resulting in partial “masking” of the actual KIE associated with the C–H activation step. A powerful approach to examine this aspect is through comparison of KIEintra versus KIEinter.54,55 Accordingly, the mono- (D1-11) and gem-dideuterated (D2-11) analogues of compound 11 (Figure 3b) were synthesized (see SI for details) and utilized in intra- and intermolecular competition experiments, respectively. Expectedly, a KIEintra > KIEinter would reveal that azide activation is also rate-limiting, whereas KIEintra ≈ KIEinter would indicate that C–H bond cleavage is the only rate-determining step in the reaction. Analysis of the FL#62-catalyzed cyclization of D1-11 yielded a KIEintra = 4.7 ± 0.3, whereas the competition experiment with a 1:1 mixture of 11 and D2-11 yielded a KIEinter value of 5.3 ± 0.4 (Figure 3b), as determined by mass spectrometry (Figure S5). The close similarity between these values (within experimental error) thus supports a scenario in which C–H bond cleavage is likely to be the predominant rate-determining step in these P450-catalyzed C–H amination reactions. At the same time, it is possible that in the presence of weaker C–H bonds (e.g., tertiary vs secondary C–H bond in 6 vs 11) other steps may become kinetically important, thereby affecting the extent of the KIE associated with the H/D substitution at the level of the aminated C–H bond.
Figure 4.
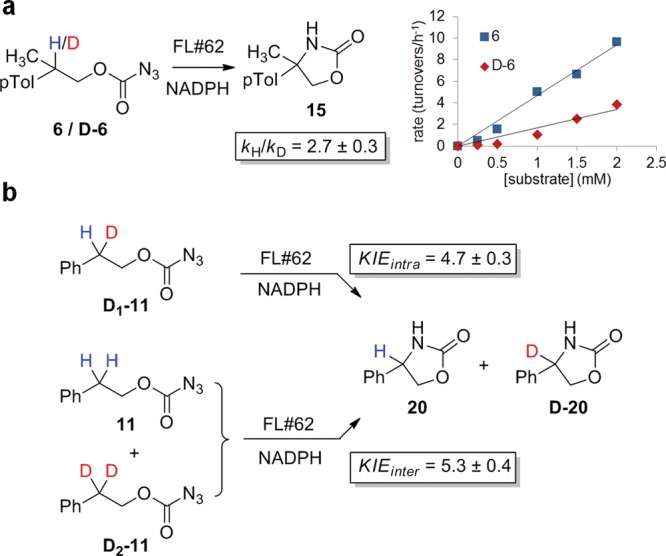
Kinetic isotope effect (KIE) experiments. (a) KIE value for FL#62-catalyzed amination of 6 and its deuterated analogue, D-6. The graph reports a plot of initial rate versus substrate concentration for the two reactions. (b) Comparison of KIEintra and KIEinter values as determined from intramolecular (D1-11) and intermolecular (1:1 mixture of 11 and D2-11) competition experiments, followed by GC-MS analysis (Figure S5).
3.5. Competing Pathways
Whereas the studies above provided insights into the productive pathway leading to oxazolidinone formation, evidence for the occurrence of competing, unproductive pathways was also gathered. Indeed, in addition to the oxazolidinone product, a considerable amount of the acyclic carbamate and of the corresponding alcohol was observed in the P450 reactions with all the substrates tested. Whereas the carbonazidate is completely consumed in these reactions, the carbamate and alcohol products were found to account for the majority of the consumed substrate (∼90%). Nearly complete (>95%) suppression of either byproduct in the presence of CO-saturated buffer proved that they are both generated at the level of the heme center in the enzyme and not from reaction with the flavins in the reductase domain or components of the cofactor regeneration system. As proposed in the context of our previous studies with arylsulfonyl azides, where a similar reduction byproduct (i.e., sulfonamide) was observed,7 we suspect the acyclic carbamate to originate from overreduction and protonation of the imido-iron intermediate (blue path, Figure 2), a process potentially favored by the native two single-electron/proton transfer mechanism operating in P450s as required for the reductive activation of O2 to Compound I.50 Interestingly, a similar “uncoupling” phenomenon was observed during P450-catalyzed nitrene transfer to sulfides recently investigated by Arnold and co-workers.23 Possible pathways leading to the alcohol byproduct could involve hydrolysis of the heme bound carbonyl-nitrenoid species or radical decarboxylation during catalysis. Although these uncoupling mechanisms currently limit the synthetic utility of these P450-catalyzed transformations, we anticipate that targeting structural elements mediating the native electron/proton transfer pathway in these enzymes could provide a future avenue to further increase their efficiency as C–H amination catalysts.
4. Conclusions
In conclusion, we have reported the successful synthesis of oxazolidinones via the P450-catalyzed cyclization of carbonazidates. In addition to expanding the reaction scope of cytochrome P450 enzymes, this report describes the first example in which these atom-economical and readily accessible nitrene donors are successfully employed in catalytic intramolecular C–H amination reactions. Our structure–reactivity analyses indicate that whereas benzylic/allylic C–H bonds with a BDE < 90 kcal/mol are most readily aminated by these biocatalysts, activation of stronger C–H bonds (i.e., nonbenzylic/nonallylic 2 °C–H) is also feasible. Our mechanistic and KIE analyses indicate that the C–H activation step is rate-limiting and proceeds in a stepwise manner (i.e., via HAA/radical rebound), reminiscent of that involved in P450 hydroxylations. Overall, these studies provide first-time insights into the mechanism of P450-mediated C–H amination as well as a basis for further optimization of these catalysts. The experiments with the terpene substrates demonstrate the feasibility of applying these P450-based amination catalysts for the transformation of complex natural products, with active site mutagenesis providing a way to improve the catalytic activity toward a particular target substrate. Because carbonazidates can be readily synthesized from alcohols, the reactivity reported here could open exciting opportunities for the application of these enzymes in synthesis, which may include, for example, coupling selective P450 hydroxylation31,39,56 with P450-catalyzed carbonazidate cyclization to install 1,2-aminoalcohol functionalities at remote C(sp3)—H positions in organic molecules.
Supporting Information Available
The following file is available free of charge on the ACS Publications website at DOI: 10.1021/cs5018612.
Synthetic procedures, compound characterization data and NMR spectra, additional figures and tables (PDF)
This work was supported by the U.S. National Institute of Health (R01 grant GM098628). MS instrumentation was supported by the U.S. NSF grant CHE-0946653.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Wilson M. E.; Whitesides G. M. J. Am. Chem. Soc. 1978, 100, 306–307 10.1021/ja00469a064. [DOI] [Google Scholar]
- Collot J.; Gradinaru J.; Humbert N.; Skander M.; Zocchi A.; Ward T. R. J. Am. Chem. Soc. 2003, 125, 9030–9031 10.1021/ja035545i. [DOI] [PubMed] [Google Scholar]
- Reetz M. T.; Peyralans J. J. P.; Maichele A.; Fu Y.; Maywald M. Chem. Commun. 2006, 4318–4320 10.1039/b610461d. [DOI] [PubMed] [Google Scholar]
- Abe S.; Niemeyer J.; Abe M.; Takezawa Y.; Ueno T.; Hikage T.; Erker G.; Watanabe Y. J. Am. Chem. Soc. 2008, 130, 10512–10514 10.1021/ja802463a. [DOI] [PubMed] [Google Scholar]
- Mayer C.; Gillingham D. G.; Ward T. R.; Hilvert D. Chem. Commun. 2011, 47, 12068–12070 10.1039/c1cc15005g. [DOI] [PubMed] [Google Scholar]
- Coelho P. S.; Brustad E. M.; Kannan A.; Arnold F. H. Science 2013, 339, 307–310 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]
- Singh R.; Bordeaux M.; Fasan R. ACS Catal. 2014, 4, 546–552 10.1021/cs400893n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Srivastava P.; Zhang C.; Lewis J. C. ChemBioChem 2014, 15, 223–227 10.1002/cbic.201300661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J. C. ACS Catal. 2013, 3, 2954–2975 10.1021/cs400806a. [DOI] [Google Scholar]
- Sreenilayam G.; Fasan R. Chem. Commun. 2015, 51, 1532–1534 10.1039/C4CC08753D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordeaux M.; Tyagi V.; Fasan R. Angew. Chem., Int. Ed. 2014, 54, 1744–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick A. R.; Sanford M. S. Tetrahedron 2006, 62, 2439–2463 10.1016/j.tet.2005.11.027. [DOI] [Google Scholar]
- Davies H. M. L.; Manning J. R. Nature 2008, 451, 417–424 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller P.; Fruit C. Chem. Rev. 2003, 103, 2905–2919 10.1021/cr020043t. [DOI] [PubMed] [Google Scholar]
- Collet F.; Dodd R. H.; Dauban P. Chem. Commun. 2009, 5061–5074 10.1039/b905820f. [DOI] [PubMed] [Google Scholar]
- Roizen J. L.; Harvey M. E.; Du Bois J. Acc. Chem. Res. 2012, 45, 911–922 10.1021/ar200318q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey J. L.; Sarpong R. Chem. Sci. 2013, 4, 4092–4106 10.1039/c3sc51420j. [DOI] [Google Scholar]
- Driver T. G. Org. Biomol. Chem. 2010, 8, 3831–3846 10.1039/c005219c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey P. A.; Magnusson O. T. Chem. Rev. 2003, 103, 2129–2148 10.1021/cr020422m. [DOI] [PubMed] [Google Scholar]
- McIntosh J. A.; Coelho P. S.; Farwell C. C.; Wang Z. J.; Lewis J. C.; Brown T. R.; Arnold F. H. Angew. Chem., Int. Ed. 2013, 52, 9309–9312 10.1002/anie.201304401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordeaux M.; Singh R.; Fasan R. Bioorg. Med. Chem. 2014, 22, 5697–5704 10.1016/j.bmc.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyster T. K.; Farwell C. C.; Buller A. R.; McIntosh J. A.; Arnold F. H. J. Am. Chem. Soc. 2014, 136, 15505–15508 10.1021/ja509308v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farwell C. C.; McIntosh J. A.; Hyster T. K.; Wang Z. J.; Arnold F. H. J. Am. Chem. Soc. 2014, 136, 8766–8771 10.1021/ja503593n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narhi L. O.; Fulco A. J. J. Biol. Chem. 1987, 262, 6683–6690 10.1103/PhysRevB.35.6683. [DOI] [PubMed] [Google Scholar]
- Espino C. G.; Du Bois J. Angew. Chem., Int. Ed. 2001, 40, 598–600. [DOI] [PubMed] [Google Scholar]
- Cui Y.; He C. Angew. Chem., Int. Ed. 2004, 43, 4210–4212 10.1002/anie.200454243. [DOI] [PubMed] [Google Scholar]
- Lebel H.; Huard K.; Lectard S. J. Am. Chem. Soc. 2005, 127, 14198–14199 10.1021/ja0552850. [DOI] [PubMed] [Google Scholar]
- Kotti S. R. S. S.; Timmons C.; Li G. G. Chem. Biol. Drug Des. 2006, 67, 101–114 10.1111/j.1747-0285.2006.00347.x. [DOI] [PubMed] [Google Scholar]
- Ager D. J.; Prakash I.; Schaad D. R. Chem. Rev. 1996, 96, 835–875 10.1021/cr9500038. [DOI] [PubMed] [Google Scholar]
- Intrieri D.; Zardi P.; Caselli A.; Gallo E. Chem. Commun. 2014, 50, 11961. 10.1039/C4CC90355B. [DOI] [PubMed] [Google Scholar]
- Zhang K. D.; Shafer B. M.; Demars M. D.; Stern H. A.; Fasan R. J. Am. Chem. Soc. 2012, 134, 18695–18704 10.1021/ja3073462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan M. J.; Johannes T. W.; Zhao H. Biotechnol. Bioeng. 2008, 99, 268–274 10.1002/bit.21546. [DOI] [PubMed] [Google Scholar]
- Bach T.; Schlummer B.; Harms K. Chemistry 2001, 7, 2581–2594. [DOI] [PubMed] [Google Scholar]
- Tamura Y.; Uchida T.; Katsuki T. Tetrahedron Lett. 2003, 44, 3301–3303 10.1016/S0040-4039(03)00609-9. [DOI] [Google Scholar]
- Bergmeier S. C.; Stanchina D. M. J. Org. Chem. 1999, 64, 2852–2859 10.1021/jo9823893. [DOI] [PubMed] [Google Scholar]
- Bergmeier S. C.; Stanchina D. M. J. Org. Chem. 1997, 62, 4449–4456 10.1021/jo970473x. [DOI] [PubMed] [Google Scholar]
- Kamon T.; Shigeoka D.; Tanaka T.; Yoshimitsu T. Org. Biomol. Chem. 2012, 10, 2363–2365 10.1039/c2ob07190h. [DOI] [PubMed] [Google Scholar]
- Zhang K.; El Damaty S.; Fasan R. J. Am. Chem. Soc. 2011, 133, 3242–3245 10.1021/ja109590h. [DOI] [PubMed] [Google Scholar]
- Kolev J. N.; O’Dwyer K. M.; Jordan C. T.; Fasan R. ACS Chem. Biol. 2014, 9, 164–173 10.1021/cb400626w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H.; Zhang X. P. Chem. Soc. Rev. 2011, 40, 1899–1909 10.1039/c0cs00070a. [DOI] [PubMed] [Google Scholar]
- Luo Y.Handbook of Bond Dissociation Energies in Organic Compounds; CRC Press LLC: Boca Raton, FL, 2003. [Google Scholar]
- Padwa A.; Flick A. C.; Leverett C. A.; Stengel T. J. Org. Chem. 2004, 69, 6377–6386 10.1021/jo048990k. [DOI] [PubMed] [Google Scholar]
- Banks M. R.; Blake A. J.; Cadogan J. I. G.; Dawson I. M.; Gosney I.; Grant K. J.; Gaur S.; Hodgson P. K. G.; Knight K. S.; Smith G. W.; Stevenson D. E. Tetrahedron 1992, 48, 7979–8006 10.1016/S0040-4020(01)80472-5. [DOI] [Google Scholar]
- Meth-Cohn O. Acc. Chem. Res. 1987, 20, 18–27 10.1021/ar00133a003. [DOI] [Google Scholar]
- Mansuy D.; Battioni P.; Mahy J. P. J. Am. Chem. Soc. 1982, 104, 4487–4489 10.1021/ja00380a031. [DOI] [Google Scholar]
- Au S. M.; Huang J. S.; Yu W. Y.; Fung W. H.; Che C. M. J. Am. Chem. Soc. 1999, 121, 9120–9132 10.1021/ja9913481. [DOI] [Google Scholar]
- Fantauzzi S.; Gallo E.; Caselli A.; Ragaini F.; Casati N.; Macchi P.; Cenini S. Chem. Commun. 2009, 3952–3954 10.1039/b903238j. [DOI] [PubMed] [Google Scholar]
- Lyaskovskyy V.; Suarez A. I. O.; Lu H. J.; Jiang H. L.; Zhang X. P.; de Bruin B. J. Am. Chem. Soc. 2011, 133, 12264–12273 10.1021/ja204800a. [DOI] [PubMed] [Google Scholar]
- Groves J. T.; Mcclusky G. A.; White R. E.; Coon M. J. Biochem. Biophys. Res. Commun. 1978, 81, 154–160 10.1016/0006-291X(78)91643-1. [DOI] [PubMed] [Google Scholar]
- Denisov I. G.; Makris T. M.; Sligar S. G.; Schlichting I. Chem. Rev. 2005, 105, 2253–2277 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- Fiori K. W.; Espino C. G.; Brodsky B. H.; Du Bois J. Tetrahedron 2009, 65, 3042–3051 10.1016/j.tet.2008.11.073. [DOI] [Google Scholar]
- Leung S. K.; Tsui W. M.; Huang J. S.; Che C. M.; Liang J. L.; Zhu N. J. Am. Chem. Soc. 2005, 127, 16629–16640 10.1021/ja0542789. [DOI] [PubMed] [Google Scholar]
- Collet F.; Lescot C.; Liang C.; Dauban P. Dalton Trans. 2010, 39, 10401–10413 10.1039/c0dt00283f. [DOI] [PubMed] [Google Scholar]
- Shearer J.; Zhang C. X.; Hatcher L. Q.; Karlin K. D. J. Am. Chem. Soc. 2003, 125, 12670–12671 10.1021/ja0359409. [DOI] [PubMed] [Google Scholar]
- Jung H. H.; Floreancig P. E. Tetrahedron 2009, 65, 10830–10836 10.1016/j.tet.2009.10.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolev J. N.; Zaengle J. M.; Ravikumar R.; Fasan R. ChemBioChem 2014, 15, 1001–1010 10.1002/cbic.201400060. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.