

Abstract
Purpose.
To investigate the role of the lipid mediator, resolvin E1 (RvE1), in corneal inflammation.
Methods.
The effect of RvE1 on stimulated human corneal epithelial cells (HCECs) and neutrophils, and mouse macrophage was assessed. C57BL/6 mouse corneas were abraded and treated with RvE1 either before or after stimulation with lipopolysaccharide (LPS) and antibiotic-killed Pseudomonas aeruginosa and Staphylococcus aureus. The levels of CXC chemokines in the cornea were quantified, and the presence of neutrophils in corneal infiltrates was detected by immunohistochemistry and by in vivo confocal microscopy. The effect of RvE1 on apoptosis in the corneal epithelium was assessed using the TUNEL assay.
Results.
RvE1 significantly inhibited cytokine production in HCECs and neutrophils, and mouse macrophages and cornea. The development of corneal infiltrates, specifically neutrophils, in response to stimulation with LPS, P. aeruginosa, and S. aureus was also significantly reduced. There was no apoptotic effect of RvE1 on mouse corneal epithelial cells.
Conclusions.
RvE1 inhibits corneal inflammation induced by LPS, Gram negative (P. aeruginosa) and Gram positive (S. aureus) bacteria. These findings indicate that RvE1 as a potential anti-inflammatory therapy for patients with corneal inflammation and also, when given together with antibiotics, for bacterial keratitis.
Keywords: cornea, keratitis, bacteria LPS, Pseudomonas aeruginosa, Staphylococcus aureus, resolvin E1 (RvE1)
The results presented in this manuscript demonstrate RvE1 inhibition of corneal inflammation induced by LPS, Gram negative (P. aeruginosa) and Gram positive (S. aureus) bacteria.
Acute inflammation is an essential process in defending the host against infection. Following infection, circulating neutrophils are generally the first cells to infiltrate the inflamed tissue to eliminate the infecting microbes; however, these cells also release proteinases that cause tissue damage prior to undergoing cell death by apoptosis. Proinflammatory lipid mediators such as prostaglandin E2 (PGE2) and leukotriene B4 (LTB4) are generated during the acute stage of inflammation and contribute to the inflammatory process by enhancing recruitment of neutrophils.1–3 Once the organisms have been killed, the lipid profile at the site changes to lipid mediators that counter inflammation, including resolvins and lipoxins, and contribute to resolution of the inflammatory response, including macrophage clearance of apoptotic neutrophils.2
Resolvins belong to a novel class of endogenous proresolving specialized mediators (SPA) that are derived via lipoxygenation from the essential dietary omega-3 polyunsaturated fatty acids eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), and include the E-series resolvins (RvE) derived from EPA, and the D-series resolvins (RvD), neuroprotectins/protectins (NPD1/PD1), maresins, and aspirin triggered epimers that are derived from DHA.2–4 Among the E-series resolvins, the biological functions of RvE1 include blocking expression of proinflammatory cytokines and trans-endothelial neutrophil migration.5 RvE1 is also a potent counter-regulator of superoxide anion generation, thereby preventing tissue destruction resulting from inflammation.6 In addition to these anti-inflammatory effects, RvE1 attracts noninflammatory monocytes and macrophages, and promotes neutrophil clearance from the inflamed site through macrophage-mediated efferocytosis.2
We have used murine models to identify potential causes of corneal inflammation, and demonstrated that activation of toll-like receptors (TLR), including TLR2 and TLR4 by Staphylococcus aureus and Pseudomonas aeruginosa or LPS, respectively, leads to production of proinflammatory and chemotactic cytokines in the cornea, neutrophil infiltration, and loss of corneal clarity demonstrated by in vivo confocal imaging.7–10 Therefore, the purpose of the current study was to determine if RvE1 inhibits proinflammatory and chemotactic cytokine production by human corneal epithelial cells and neutrophils, and in murine models of corneal inflammation.
Methods
Mouse Strains
C57BL/6 mice (6–8 weeks old) were obtained from the Jackson Laboratory. All mice were maintained in pathogen-free conditions in microisolator cages, and were treated in accordance with the guidelines provided in the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. The animal protocol was also approved by the Case Western Reserve University IACUC.
Drug Preparation
Lipopolysaccharide (LPS) was obtained from Invivogen (San Diego, CA, USA). RvE1 was kindly provided by Resolvyx Pharmaceuticals, Inc. (Bedford, MA, USA) and prepared in PBS.
Source of Bacteria
P. aeruginosa strain ATCC 19660 was obtained from ATCC, and S. aureus strain 8325-4 was a gift from Richard O'Callaghan (University of Mississippi). Bacteria were grown overnight (18 hours) in a brain–heart infusion broth, and aliquots from these stationary cultures were diluted 1:100, and grown until OD650 = 0.2, which we found to be 1 × 108 bacteria/mL. For antibiotic treatment, bacteria were centrifuged, washed once with phosphate buffered saline (PBS), and resuspended at 2 × 109 bacteria/mL in 0.3% Tobramycin in PBS (Sigma-Aldrich Corp., St. Louis, MO, USA). Bacterial killing was confirmed by the absence of growth on tryptic soy broth (TSB) agar plates.
Cell Lines and In Vitro Stimulation
The SV-40-transfected human corneal epithelial cell line (HCE-T) was obtained from ATCC (American Type Culture Collection, Manassas, VA, USA), and was grown to 80% confluency in keratinocyte serum-free medium (KSFM) containing 0.05 mg/mL bovine pituitary extract and 5 ng/mL epidermal growth factor in collagen-coated plates. Before stimulation, the cells underwent epidermal growth factor starvation overnight, as described.10,11 The human neutrophil promyelocyte cell line (HL-60) was maintained in RPMI with 10% FBS. Cells were and incubated 5 days in 1.2% DMSO to induce differentiation to the neutrophil phenotype. Both cell types were incubated with RvE1, followed by stimulation with tobramycin-killed S. aureus or P. aeruginosa or LPS. HCE cell lines were incubated for 2 hours with IFN-γ to induce expression of the MD-2 coreceptor of TLR4 before the addition of LPS.12 After 3 hours, cell-free supernatants were collected, and IL-8, IL-6, and CXCL1 were measured by ELISA according to the manufacturer's directions (R&D Systems, Minneapolis, MN, USA). Absorption was measured at 450 nm on a VersaMax microplate reader using SoftMax Pro software 5.2 (Molecular Devices, Sunnyvale, CA, USA).
Peritoneal Macrophage Isolation and In Vitro Stimulation
For macrophages, mice were injected intraperitoneally with 3% thioglycolate, and cells were recovered by peritoneal lavage after 3 days. Cell purity was > 97%. All cells were incubated with RvE1, followed by stimulation with tobramycin-killed S. aureus or P. aeruginosa or LPS. After 3 hours, cell-free supernatants were collected, and IL-1β, TNF-α, and CXCL1/KC were measured by sandwich ELISA according to the manufacturer's directions (R&D Systems) as before.
Murine Models of Corneal Inflammation
Six- to 10-week-old C57BL/6 mice from The Jackson Laboratory were anesthetized by intraperitoneal injection of 0.4 mL 2,2,2-tribromoethanol (1.2%), and the central corneal epithelium was abraded with three contiguous scratches made with a 26-gauge needle. Histologic examination of the abraded cornea showed that the wound was limited to the epithelial layer. For corneal inflammation, 1 × 107 antibiotic-killed S. aureus or P. aeruginosa in 2.5 μL PBS, 20 μg of LPS in 2 μL sterile endotoxin-free water was placed on the ocular surface as described.12–15 RvE1 prepared as 2 μg in 2 μL PBS was placed on the corneal surface either before or after inflammation, and after 24 hours, mice were euthanized.
Cytokine Analysis in the Cornea
To measure cytokine production in the corneas, individual corneas were dissected and homogenized in 0.2 mL PBS using a Mixer Mill MM300 (Retsch; Qiagen, Germantown, MD, USA) for 4 minutes at 33 Hz. A 25-μL aliquot was assayed by ELISA according to the manufacturer's directions (R&D Systems).
Detection of Neutrophils in the Cornea by Immunohistochemistry
Eyes were enucleated and snap frozen in liquid nitrogen, and 5-μm sections were fixed in 4% formaldehyde for 30 minutes. The slides were then washed with 0.05 M Tris buffered saline (TBS; pH 7.6), and the sections were incubated for 2 hours with antineutrophil antibody NIMP-R14 diluted 1:100 in 1% fetal calf serum/TBS (1% FCS/TBS). After washing, sections were incubated for 45 minutes with FITC-conjugated rabbit anti-rat antibody (Vector Laboratories, Burlingame, CA, USA) diluted 1:200 in 1% FCS/TBS, mounted in antifade medium containing 4′, 6-diamidino-phenylindole (DAPI; Vectashield; Vector Laboratories), and the number of neutrophils per section was counted (from limbus to limbus at 400×) by fluorescence microscopy (Olympus Optical Co. Ltd., Tokyo, Japan). This approach has been used extensively in our laboratory for studies of corneal inflammation.12–14,16
Examination of Stromal Thickness and Reflectivity
Analysis of cellular infiltration in the corneal stroma was accomplished by in vivo confocal microscopy (Confoscan; Nidek Technologies America, New Orleans, LA, USA). Briefly, mice were immobilized on a secure platform. A 40× objective was maneuvered into place on the corneal surface using transparent gel (Genteal; Novartis Ophthalmics, Duluth, GA, USA) as a medium between the corneal surface and the objective, and the software (NAVIS; Lucent Technologies, Murray Hill, NJ, USA) captured images every 1 μm and stored them as a stack for analysis of corneal thickness and haze. Stromal thickness was determined as the area between basal epithelium and corneal endothelium, and stromal haze was defined as stromal thickness X combined light intensity of each image of the corneal stroma. To obtain this, the series of intensity values for each corneal stroma was saved on a computer spreadsheet (Excel; Microsoft, Redmond, WA, USA), and exported into another program (Prism; Graph Pad Software, San Diego, CA, USA) to generate a curve using the “curves and regression” function. The total area under the curve was then calculated by using this software as previously described.12,14,16
Apoptosis Assay
Cell viability was measured in vitro measured by trypan blue exclusion. The corneal sections were incubated with terminal deoxynucleotidyltransferase–mediated dUTP nick end labeling (TUNEL) reagents according to the manufacturer's directions (Roche, Penzberg, Germany), counterstained with DAPI to show the nuclei. TUNEL-positive cells were detected and quantified by fluorescence microscopy.
Statistics
Statistical analysis was performed using one-way ANOVA with Tukey post hoc analyses (Prism; GraphPad Software). P < 0.05 was considered significant.
Results
RvE1 Inhibits Bacteria and LPS-Induced Cytokine Production by Human Corneal Epithelial Cells, Human Neutrophils, and Mouse Macrophages
Bacterial keratitis is treated using antibiotics; therefore, human corneal epithelial and neutrophil cell lines and murine peritoneal macrophages were stimulated with tobramycin-killed S. aureus or P. aeruginosa, or with LPS in the presence of resolvin E1 (RvE1).
As shown in Figure 1, upper panels, tobramycin killed S. aureus, P. aeruginosa, and LPS stimulated IL-8 production by human neutrophils and corneal epithelial cells. S. aureus- and P. aeruginosa-induced IL-8 was almost completely inhibited by the lowest concentration of RvE1 (100 pg/mL), whereas IL-8 production induced by LPS was ablated in neutrophils and in corneal epithelial cells. Similarly, IL-6 and CXCL-1 production in response to S. aureus or P. aeruginosa or LPS was significantly and dose-dependently inhibited by RvE1. Murine peritoneal macrophages also produced IL-1β, TNF-α, and CXCL1 in response to these stimuli, which was inhibited by 0.1 to 1 ng/mL RvE1 (Fig. 2).
Figure 1.

Effect of RvE1 on LPS-, P. aeruginosa-, and S. aureus-stimulated human neutrophil and corneal epithelial cell lines. Human neutrophil (HL-60) and corneal epithelial (HCE-T) cell lines were stimulated with LPS or tobramycin-killed P. aeruginosa or S. aureus in the presence of RvE1. After 3 hours, cytokines in the cells' supernatants were quantified by ELISA. Data shown are the mean ± SEM of three wells per sample in a representative experiment. Experiments were repeated three times with similar results. The overall p value by one-way ANOVA is < 0.0001 for all cytokines produced by neutrophils and HCECs stimulated with LPS, P. aeruginosa or S. aureus. The P values on the graphs are from the Tukey post hoc analyses.
Figure 2.

Effect of RvE1 on LPS-, P. aeruginosa-, and S. aureus-stimulated primary murine macrophages. Mouse peritoneal macrophages were stimulated with LPS or tobramycin-killed P. aeruginosa or S. aureus in the presence of RvE1. After 3 hours, IL-1β, TNF-α, and CXCL1 in culture supernatants were quantified by ELISA. Data shown are the mean ±SEM of three wells per sample in a representative experiment. Experiments were repeated three times with similar results. The overall P value by one-way ANOVA is < 0.0001 for all cytokines produced by neutrophils and HCECs following each stimulus and shown as median and range. *P < 0.05 derived from Tukey post hoc analyses.
We therefore conclude that RvE1 inhibits TLR2 and TLR4 activation by resident and infiltrating cells of the cornea.
RvE1 Inhibits Bacteria- and LPS-Induced CXCL1/KC Production in the Cornea
Neutrophils are the predominant cell type recruited to the cornea following infectious or inflammatory stimulation,13 and degranulation and release of proteases cause changes in corneal thickness and haze in animal models of S. aureus, P. aeruginosa, and LPS-induced corneas disease.9,10 As neutrophil recruitment to the corneal stroma is mediated by CXCL1/KC, and is elevated early in S. aureus, P. aeruginosa, or LPS-induced corneal inflammation, we examined CXCL1 production 6 hours after corneal abrasion and topical application of RvE1 and exposure to S. aureus, P. aeruginosa, or LPS.
Figure 3 shows that CXCL1 was elevated in corneas treated with tobramycin killed S. aureus, P. aeruginosa, or LPS, compared with trauma control corneas. However, chemokine production in RvE1 treated corneas was significantly lower, demonstrating the inhibitory effect of RvE1 in corneal inflammation.
Figure 3.
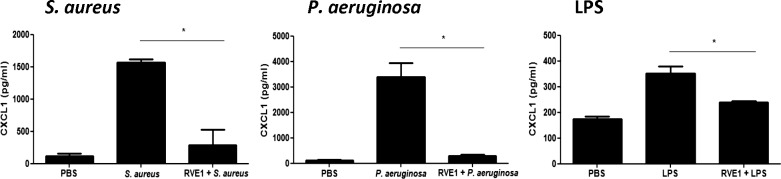
Effect of RvE1 on CXC chemokine production in murine corneas. Corneas of C57BL/6 mice were abraded with three parallel, superficial scratches, and RvE1 (2 μg in 2 μL) was added topically. After 2 hours, 2 μL LPS (20 μg) or 2.5 μL tobramycin-killed P. aeruginosa or S. aureus (1 × 107) was added topically, and 3 hours later, corneas were dissected and homogenized, and CXCL1 was measured by ELISA. This experiment is representative of two repeat studies with three mice per group. RvE1-treated corneas reduced chemokine production significantly. The overall P value by one-way ANOVA is < 0.0001 for all cytokines. *P < 0.05 derived from Tukey post hoc analyses. Data are shown as median and range.
RvE1 Inhibits Neutrophil Recruitment to the Corneal Stroma in Bacteria- and LPS-Induced Inflammation
To determine if there is an inhibitory effect of RvE1 on neutrophil, infiltration was measured by directly counting neutrophils per 5-μm corneal section. As shown in Figure 4, corneas exposed to killed bacteria or LPS had significantly more neutrophils per corneal section than trauma control corneas (abraded and given topical PBS). Mice given topical application of RvE1 either before or after stimulation had significantly less neutrophils than in the absence of the inhibitor (Fig. 4). Further, there was no significant difference between pre- and posttreatment groups, demonstrating the inhibitory effect of RvE1 even after the inflammatory response has been initiated.
Figure 4.

Effect of RvE1 on neutrophil recruitment to the corneal stroma in mouse models of LPS-, P. aeruginosa-, and S. aureus-induced corneal inflammation. Mouse corneas were abraded and treated with 2 μg of RvE1 before or after inducing inflammation by LPS, tobramycin-killed P. aeruginosa or S. aureus. After 24 hours, neutrophil numbers were assessed by direct counting of 5-μm sections. Data points represent individual corneas from groups of four to six mice, and data are shown as median and range. The overall P value by one-way ANOVA is < 0.0001 for all cytokines. *P < 0.05 derived from Tukey post hoc analyses, and the experiment was repeated twice with similar results.
RvE1 Inhibits Bacteria- and LPS-Induced Elevated Corneal Thickness and Haze
The corneal stroma is composed of antiparallel rows of collagen fibrils, regularly spaced by intermediate proteoglycans, primarily keratan sulfate. The predominant cell types present are keratocytes, which are quiescent, translucent cells organized in the stroma to form a regular array, and macrophages and dendritic cells.17,18 When examined by vivo confocal microscopy, which scans the entire corneal stroma (∼80 μm), the normal transparent corneal stroma shows no infiltrating cells, whereas our previous findings showed that topical exposure to LPS or P. aeruginosa stimulates a pronounced cellular infiltrate detected by their high reflectivity in the cornea.9,10
To assess the effect of RvE1 on corneal thickness and haze, corneas were abraded as before, and 2 μg RvE1 was added before or after stimulation with tobramycin-killed P. aeruginosa or S. aureus, or with LPS. After 24 hours, corneal thickness and haze were measured using in vivo confocal microscopy (Confoscan, Nidek, Fremont, CA, USA).
Images of the central corneal stroma were captured, and as shown in Figure 5, upper panels, there were few infiltrating cells in corneas treated with PBS (trauma control), whereas mice given S. aureus exhibit an intense cellular infiltrate in the central corneal stroma (detected as small, light reflective cells). Further, corneas pre- or posttreated with RvE1 showed very few reflective cells in the central corneal stroma. Quantification shows significantly less stromal thickness and haze in corneas given topical RvE1 that was administered either prior to or following stimulation with killed S. aureus. Similar results were found in mice stimulated with either P. aeruginosa or LPS (Fig. 5, center and lower panels).
Figure 5.

Effect of RvE1 on LPS-, P. aeruginosa-, and S. aureus-induced increased corneal thickness and reflectivity. Mouse corneas were abraded and exposed to LPS- or tobramycin-killed P. aeruginosa and S. aureus as described in the legend to Figure 4. After 24 hours, corneas were examined by in vivo confocal microscopy. Left panels: representative confocal images of the central corneal stroma showing infiltrates as small reflective cells. Center and right panels: Stromal thickness and reflectivity were quantified as described in Methods. Data points represent individual corneas from groups of four to six mice, and the experiment was repeated twice with similar results. The overall P value by one-way ANOVA is < 0.0001 for all cytokines. *P < 0.05 derived from Tukey post hoc analyses. Median and range are indicated.
Collectively, these data show that RvE1 inhibits changes to the cornea induced by Gram-negative and Gram-positive bacteria and bacterial products such as LPS.
RvE1 Does Not Induce Apoptosis in Corneal Epithelial Cells
To determine whether there is any cytotoxic effect of RvE1 on corneal epithelial cells in vivo, corneas were abraded, and topical RvE1 was added before or after stimulation with LPS or with tobramycin-killed P. aeruginosa or S. aureus as described above. After 24 hours, eyes were processed for histology, and 5-μm corneal sections were incubated with TUNEL reagents to identify apoptotic cells.
As shown in Figure 6, right panels, following LPS activation, TUNEL-positive cells were predominantly seen in the corneal stroma, and correspond with the location of NIMPR14+ neutrophils. There were very few TUNEL-positive cells in the corneal epithelium either with LPS alone, or after RvE1 was applied topically either before or after stimulation. Similarly, corneas stimulated with killed P. aeruginosa or S. aureus had very few TUNEL-positive cells in the epithelium. These observations indicate that RvE1 does not induce apoptosis of corneal epithelial cells.
Figure 6.
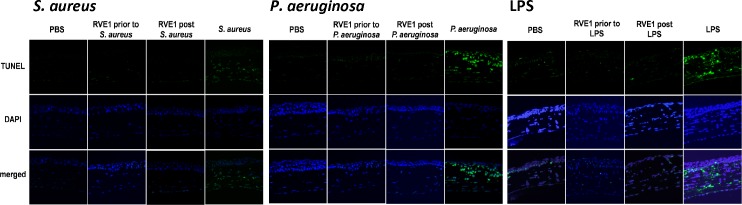
Effect of RvE1 on apoptosis of cells in inflamed corneas. Corneas were treated as described in the legend to Figure 4. After 24 hours, eyes were processed for histology, and 5-mm sections were stained with TUNEL reagents, and counterstained with DAPI to identify individual cells. Images from LPS-, P. aeruginosa-, and S. aureus-induced corneal inflammation are representative of three mice per group. Note that the only TUNEL-positive cells are in the corneal stroma and correspond to the presence of neutrophils.
Discussion
We and others have shown that neutrophils are the major cell type in corneal ulcers caused by Gram-positive and Gram-negative bacteria,16 and in sterile corneal infiltrates of patients with adverse effects to contact lens wear.14 Further, our studies using murine models of infection with live bacteria, bacterial products, or antibiotic-killed bacteria have focused on anti-inflammatory therapies that block Toll-Like receptor activation and neutrophil recruitment in the corneal stroma.10,19–21 However, anti-inflammatory lipid mediators such as lipoxins and resolvins have both anti-inflammatory and proresolving activities, and 15-lipoxygenase and heme-oxygenase inhibit inflammation of cornea neovascularization and inflammation in the uvea and retina.22,15,23
Results of our current in vitro and in vivo studies support an anti-inflammatory role for RvE1 in corneal inflammation as we demonstrate inhibitory activity of RvE1 on cytokine production by corneal epithelial cells, neutrophils, and macrophages, which each contribute to inducing corneal inflammation.12,20 Similar concentrations of RvE1 block bacteria and LPS-induced CXCL8/IL-8 and IL-6 production in the human HL-60 neutrophil cell line. As neutrophils express most TLRs and respond to S. aureus, P. aeruginosa, and LPS,24,25 these findings extend our understanding of this inflammatory process by demonstrating that RvE1 can function directly on neutrophils by inhibiting CXCL8/IL-8 and IL-6. The profound effect of RvE1 on IL-8 production by human corneal epithelial cells and neutrophils indicates a potential role for this inhibitor to impair recruitment of neutrophils to the corneal stroma, and neutrophil-mediated tissue damage and loss of corneal transparency and visual impairment.
We and others showed that the inability of epithelial cells from the cornea and other tissues to respond to LPS is due to the absence of the MD-2 coreceptor of TLR42,26; however, IFN-γ induces MD-2 expression and confers LPS responsiveness by corneal epithelial cells.12 CD14 also mediates TLR4 activation of the MyD88-independent, TRIF dependent pathway to produce other inflammatory mediators, including CCL5, which recruits monocytes.27 Our current studies indicate that RvE1 inhibits LPS keratitis by reducing activation and production of CXC chemokine production by corneal epithelial cells, neutrophils, and macrophages and thereby limits recruitment of neutrophils to the corneal stroma. Further, we demonstrate that RvE1 has potent inhibitory activity in murine models of S. aureus, P. aeruginosa, and LPS-induced corneal inflammation. This model, while clearly limited by short-term exposure at the corneal surface, is an important first step in examining anti-inflammatory agents in the context of bacteria-associated corneal inflammation. Our previous studies demonstrated that S. aureus, P. aeruginosa and LPS can activate the host innate immune response by activation of the TLR2 and TLR4/MD-2 signaling cascade, respectively, in resident myeloid and corneal epithelial cells, resulting in production of chemotactic and proinflammatory cytokines, which then mediate recruitment of neutrophils from limbal vessels to the corneal stroma, resulting in corneal inflammation.9,10 This understanding of the sequence of events leading to corneal inflammation allows for a more targeted approach to blocking inflammation. One approach is to target TLR activation, and we reported that killed P. aeruginosa- and LPS-induced corneal inflammation can be blocked by the MD-2 antagonist Eritoran tetrasodium (E5564); however, this reagent is specific for Gram-negative bacteria and does not affect corneal inflammation induced by Gram-positive bacteria or TLR2 agonists.10 We also demonstrated that blocking TLR signaling by short-chain ceramide in response to antibiotic killed S. aureus and LPS stimulation inhibits corneal inflammation.11 Blockade of extravasation using the lymphocyte functional antigen-1 (LFA-1) antagonist lifitegrast (SAR 1118) also inhibits corneal inflammation induced by exposure to S. aureus and P. aeruginosa.21 In the current study, RvE1 clearly inhibits corneal inflammation through blocking neutrophil recruitment to the cornea as well as cytokine production in response to stimulation with antibiotic killed S. aureus (TLR2), and P. aeruginosa or LPS (TLR4). TLRs are therefore a feasible target for therapeutic intervention when combined with antibiotics. Future studies will examine the combined effect of antibiotics and RvE1 following infection with live bacteria.
The pre- and posttreatment anti-inflammatory activity of RvE1 demonstrated here indicates that RvE1 may have broader effectiveness by inhibiting other clinical manifestations of inflammation. Our findings are consistent with recent studies showing an inhibitory effect of RvE1 in a murine model of dry eye, preventing disruption of the corneal epithelial barrier and protecting against goblet cell loss.28 RvD1 and RvE1 also inhibited corneal neovascularization induced by suture,29 and the RvE1 analog RX-10045 reduced corneal stromal haze following photorefractive keratectomy.30 RvE1 also blocked angiogenesis in Herpes simplex keratitis, in part by inhibiting cytokine production neutrophil infiltration to the cornea31 and increased corneal epithelial cell migration by activating EGF receptor signaling and enhancing the wound healing response.32 These findings are consistent with our observations that topical application of RvE1 does not cause apoptosis of corneal epithelial cells, and may also promote wound healing. Further, RvE1 induces neutrophil apoptosis, which likely contributes to resolution of the inflammatory response.33
The chemokine-related receptor ChemR23 is widely expressed in human tissues including dendritic cells and macrophages,34 and ChemR23 siRNA knock-down experiments reduced RvE1 regulation of cytokine production by human dendritic cells.32 Similarly, knockdown of ChemR23 in human corneal epithelial cells prevented RvE1 dependent inhibition of inflammation induced by hyperosmolar stress (Yuan J, et al. IOVS 2012:ARVO E-Abstract 5242). Therefore, in the current study, RvE1 likely activates ChemR23 on resident corneal macrophages and epithelial cells to inhibit chemokine production.
In summary, results presented in the current study demonstrate that RvE1 inhibits corneal inflammation induced by antibiotic-treated S. aureus and P. aeruginosa, which are two common causes of corneal ulcers worldwide. Furthermore, topical application of RvE1 before or after inflammation significantly reduces this immune response without inducing epithelial cell apoptosis, thereby demonstrating proof of concept for the use of RvE1 in combination with antibiotics in prophylactic and possibly therapeutic applications for bacterial keratitis. RVE1 may also have a therapeutic effect on sterile corneal infiltrates associated with contact lens wear.
Acknowledgments
We thank Catherine Doller, Scott Howell, and Dawn Smith in the Visual Sciences Research Center core facility for outstanding technical assistance.
Supported by the National Institutes of Health Grants R01 EY14362 (EP) and P30 EY11373 and Visual Sciences Research Center Core grant (EP). Additional support for this work was provided by the Research to Prevent Blindness Foundation and the Ohio Lions Eye Research Foundation, and EP is the recipient of an Alcon Research Institute award.
Disclosure: J.-E. Lee, Resolvyx Pharmaceuticals, Inc. (E); Y. Sun, Resolvyx Pharmaceuticals, Inc. (E); P. Gjorstrup, Resolvyx Pharmaceuticals, Inc. (I, E, S); E. Pearlman, Resolvyx Pharmaceuticals, Inc. (E)
JL, YS, and EP have no financial interest in material presented herein.
PG is an officer of Resolvyx and holds equity.
References
- 1.Buckley CD,, Gilroy DW,, Serhan CN.Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity. 2014; 40: 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Serhan CN.Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014; 510: 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Serhan CN,, Clish CB,, Brannon J,, Colgan SP,, Chiang N,, Gronert K.Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med. 2000; 192: 1197–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gronert K,, Kantarci A,, Levy BD, et al. A molecular defect in intracellular lipid signaling in human neutrophils in localized aggressive periodontal tissue damage. J Immunol. 2004; 172: 1856–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwab JM,, Serhan CN.Lipoxins and new lipid mediators in the resolution of inflammation. Curr Opin Pharma. 2006; 6: 414–420. [DOI] [PubMed] [Google Scholar]
- 6.Hasturk H,, Kantarci A,, Ohira T, et al. RvE1 protects from local inflammation and osteoclast-mediated bone destruction in periodontitis. FASEB J. 2006; 20: 401–403. [DOI] [PubMed] [Google Scholar]
- 7.Johnson AC,, Heinzel FP,, Diaconu E,, et al. Activation of toll-like receptor (TLR)2, TLR4, and TLR9 in the mammalian cornea induces MyD88-dependent corneal inflammation. Invest Ophthalmol Vis Sci. 2005; 46: 589–595. [DOI] [PubMed] [Google Scholar]
- 8.Khatri S,, Lass JH,, Heinzel FP, et al. Regulation of endotoxin-induced keratitis by PECAM-1, MIP-2, and toll-like receptor 4. Invest Ophthalmol Vis Sci. 2002; 43: 2278–2284. [PubMed] [Google Scholar]
- 9.Sun Y,, Hise AG,, Kalsow CM,, Pearlman E.Staphylococcus aureus-induced corneal inflammation is dependent on Toll-like receptor 2 and myeloid differentiation factor 88. Infect Immun. 2006; 74: 5325–5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun Y,, Pearlman E.Inhibition of corneal inflammation by the TLR4 antagonist Eritoran tetrasodium (E5564). Invest Ophthalmol Vis Sci. 2009; 50: 1247–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun Y,, Fox T,, Adhikary G,, Kester M,, Pearlman E.Inhibition of corneal inflammation by liposomal delivery of short-chain C-6 ceramide. J Leukoc Biol. 2008; 83: 1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roy S,, Sun Y,, Pearlman E.Interferon-gamma-induced MD-2 protein expression and lipopolysaccharide (LPS) responsiveness in corneal epithelial cells is mediated by Janus tyrosine kinase-2 activation and direct binding of STAT1 protein to the MD-2 promoter. J Biol Chem. 2011; 286: 23753–23762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pearlman E,, Sun Y,, Roy S, et al. Host defense at the ocular surface. Int Rev Immunol. 2013; 32: 4–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holden BA,, Reddy MK,, Sankaridurg PR,, et al. Contact lens-induced peripheral ulcers with extended wear of disposable hydrogel lenses: histopathologic observations on the nature and type of corneal infiltrate. Cornea. 1999; 18: 538–543. [PubMed] [Google Scholar]
- 15.Leedom AJ,, Sullivan AB,, Dong B,, Lau D,, Gronert K.Endogenous LXA4 circuits are determinants of pathological angiogenesis in response to chronic injury. Am J Pathol. 2010; 176: 74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karthikeyan RS,, Priya JL,, Leal SM JR,, et al. Host response and bacterial virulence factor expression in Pseudomonas aeruginosa and Streptococcus pneumoniae corneal ulcers. PLoS One. 2013; 8: e64867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamrah P,, Liu Y,, Zhang Q,, Dana MR.The corneal stroma is endowed with a significant number of resident dendritic cells. Invest Ophthalmol Vis Sci. 2003; 44: 581–589. [DOI] [PubMed] [Google Scholar]
- 18.Knickelbein JE,, Watkins SC,, McMenamin PG,, Hendricks RL.Stratification of antigen-presenting cells within the normal cornea. Ophthalmol Eye Dis. 2009; 1: 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adhikary G,, Sun Y,, Pearlman E.C-Jun NH2 terminal kinase (JNK) is an essential mediator of Toll-like receptor 2-induced corneal inflammation. J Leukoc Biol. 2008; 83: 991–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun Y,, Karmakar M,, Roy S, et al. TLR4 and TLR5 on corneal macrophages regulate Pseudomonas aeruginosa keratitis by signaling through MyD88-dependent and -independent pathways. J Immunol. 2010; 185: 4272–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun Y,, Zhang R,, Gadek TR,, O'Neill CA,, Pearlman E.Corneal inflammation is inhibited by the LFA-1 antagonist, lifitegrast (SAR 1118). J Ocul Pharmacol Ther. 2013; 29: 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gronert K.Resolution the grail for healthy ocular inflammation. Exp Eye Res. 2010; 91: 478–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Medeiros R,, Rodrigues GB,, Figueiredo CP,, et al. Molecular mechanisms of topical anti-inflammatory effects of lipoxin A(4) in endotoxin-induced uveitis. Mol Pharma. 2008; 74: 154–161. [DOI] [PubMed] [Google Scholar]
- 24.Hayashi F,, Means TK,, Luster AD.Toll-like receptors stimulate human neutrophil function. Blood. 2003; 102: 2660–2669. [DOI] [PubMed] [Google Scholar]
- 25.Mantovani A,, Cassatella MA,, Costantini C,, Jaillon S.Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011; 11: 519–531. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J,, Kumar A,, Wheater M,, Yu FS.Lack of MD-2 expression in human corneal epithelial cells is an underlying mechanism of lipopolysaccharide (LPS) unresponsiveness. Immun Cell Biol. 2009; 87: 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roy S,, Karmakar M,, Pearlman E.CD14 mediates Toll-like receptor 4 (TLR4) endocytosis and spleen tyrosine kinase (Syk) and interferon regulatory transcription factor 3 (IRF3) activation in epithelial cells and impairs neutrophil infiltration and Pseudomonas aeruginosa killing in vivo. J Biol Chem. 2014; 289: 1174–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Paiva CS,, Schwartz CE,, Gjorstrup P,, Pflugfelder SC.Resolvin E1 (RX-10001) reduces corneal epithelial barrier disruption and protects against goblet cell loss in a murine model of dry eye. Cornea. 2012; 31: 1299–1303. [DOI] [PubMed] [Google Scholar]
- 29.Jin Y,, Arita M,, Zhang Q, et al. Anti-angiogenesis effect of the novel anti-inflammatory and pro-resolving lipid mediators. Invest Ophthalmol Vis Sci. 2009; 50: 4743–4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Torricelli AA,, Santhanam A,, Agrawal V,, Wilson SE.Resolvin E1 analog RX-10045 0.1% reduces corneal stromal haze in rabbits when applied topically after PRK. Mol Vis. 2014; 20: 1710–1716. [PMC free article] [PubMed] [Google Scholar]
- 31.Rajasagi NK,, Reddy PB,, Suryawanshi A,, Mulik S,, Gjorstrup P,, Rouse BT.Controlling herpes simplex virus-induced ocular inflammatory lesions with the lipid-derived mediator resolvin E1. J Immunol. 2011; 186: 1735–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arita M,, Bianchini F,, Aliberti J, et al. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J Exp Med. 2005; 201: 713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.El Kebir D,, Gjorstrup P,, Filep JG.Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc Natl Acad Sci U S A. 2012; 109: 14983–14988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wittamer V,, Franssen JD,, Vulcano M,, et al. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J Exp Med. 2003; 198: 977–985. [DOI] [PMC free article] [PubMed] [Google Scholar]