

Abstract
Although there is general consensus that deep brain stimulation (DBS) yields substantial clinical benefit in patients with Parkinson's disease (PD), the therapeutic mechanism of DBS remains a matter of debate. Recent studies demonstrate that DBS targeting the globus pallidus internus (GPi-DBS) suppresses pathological oscillations in firing rate and between-cell spike synchrony in the vicinity of the electrode but has negligible effects on population-level firing rate or the prevalence of burst firing. The present investigation examines the downstream consequences of GPi-DBS at the level of the primary motor cortex (M1). Multielectrode, single cell recordings were conducted in the M1 of two parkinsonian nonhuman primates (Macaca fasicularis). GPi-DBS that induced significant reductions in muscular rigidity also reduced the prevalence of both beta (12–30 Hz) oscillations in single unit firing rates and of coherent spiking between pairs of M1 neurons. In individual neurons, GPi-DBS-induced increases in mean firing rate were three times more common than decreases; however, averaged across the population of M1 neurons, GPi-DBS induced no net change in mean firing rate. The population-level prevalence of burst firing was also not affected by GPi-DBS. The results are consistent with the hypothesis that suppression of both pathological, beta oscillations and synchronous activity throughout the cortico-basal ganglia network is a major therapeutic mechanism of GPi-DBS.
Keywords: deep brain stimulation, MPTP, nonhuman primate, Parkinson's disease, primary motor cortex, globus pallidus
deep brain stimulation (DBS), an established neurosurgical intervention for Parkinson's disease (PD), involves the delivery of continuous, high-frequency (150 Hz) electrical stimulation to a target nucleus [i.e., the globus pallidus internus (GPi) or the subthalamic nucleus (STN)] via a chronically implanted macroelectrode (Cooper et al. 1980; Benabid et al. 1987; Agid 1999; Starr et al. 1998). Although the antiparkinsonian efficacy of DBS is well recognized (Rodriguez-Oroz et al. 2005; Bronstein et al. 2011), there remains much debate as to the underlying therapeutic mechanism (Perlmutter and Mink 2006; Lozano and Lipsman 2013).
Historically, pathological changes in tonic firing rate at critical nodes of the cortico- basal ganglia (BG) network were thought to underlie parkinsonism (Albin et al. 1989; DeLong 1990). This hypothesis is challenged by other studies suggesting instead the importance of increased burst firing, excessive oscillatory and synchronous activity in the beta-frequency range, and a breakdown of functional segregation in the cortico-BG network (Filion and Tremblay 1991; Raz et al. 2000; Goldberg et al. 2002, 2004; Hutchison et al. 1994, 2004; Wichmann et al. 1999; Soares et al. 2004). Excessive beta-oscillatory activity is hypothesized to induce an antikinetic state, which inhibits voluntary movement in downstream BG-recipient motor centers [e.g., primary motor cortex (M1) (Marsden et al. 2001b; Brown et al. 2004b)]. Therefore, M1 activity is believed to play a critical role in the expression of PD symptoms.
Early studies hypothesized that DBS works by inhibiting neuronal activity in the vicinity of the stimulating electrode, analogous to a reversible pallidotomy (Benazzouz et al. 1995; Boraud et al. 1996; Dostrovsky et al. 2000). It was suggested that this inhibition of BG output reduces the excessive inhibitory tone impinging on the motor cortex.
We have recently reported that therapeutic GPi-DBS in parkinsonian nonhuman primates (NHPs) induces a complex combination of changes in activity in the pallidum (McCairn and Turner 2009). At the single unit level, this includes increases or decreases in tonic firing rate, plus phasic and dynamic entrainment by the stimulation pulse. Across the population, however, we observed only a negligible reduction in mean firing rate (−6.9 sp/s) and no change in burstiness. In contrast, beta oscillations and synchrony were reduced dramatically. Similar observations have been made by others during high-frequency microstimulation of the GPi (Bar-Gad et al. 2004; Erez et al. 2009; Chiken and Nambu 2013) or the STN (Meissner et al. 2005; Moran et al. 2011).
These findings suggest that suppression of system-wide pathological oscillations and synchrony are important components of the mechanism of action of GPi-DBS. To test that hypothesis, we studied the effects of GPi-DBS on the activity of multiple single units in the M1 of macaque monkeys that were rendered parkinsonian using 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP).
Antidromic-driven high-frequency resonance in cortex has been postulated to play key a role in the therapeutic mechanism of STN-DBS (Li et al. 2007; Dejean et al. 2009; Gradinaru et al. 2009; Li et al. 2012) and perhaps GPi-DBS as well (Johnson et al. 2009). Our experimental methods allowed us to assay directly the prevalence of antidromic-like driving and thus the potential involvement of high-frequency resonance of M1 during therapeutic GPi-DBS.
METHODS
Two mature female (3 and 4 kg) cynomolgus macaques (Macaca fasicularis) were used for this study (monkey C and monkey E). All aspects of animal use were approved by the University of California, San Francisco Animal Care and Use Committee and were in accord with the Guide for the Care and Use of Laboratory Animals (National Research Council 1996).
Surgery.
After the animal was acclimatized to the recording environment, the animal underwent aseptic surgery under isoflurane anesthesia, following ketamine induction (10 mg/kg im). A cylindrical titanium recording chamber (19-mm inner diameter) was implanted over a craniotomy to allow access to the right GP and M1 via a coronal approach (Szabo and Cowan 1984) (35° from vertical; Horsley-Clark anterior: 15 mm, lateral: 13 mm, depth: 18 mm) using an Alpha Omega four channel microdrive. The chamber and head fixation hardware were fixed to the skull using bone screws and methyl-methacrylate cement. Prophylactic antibiotics and analgesics were administered postsurgically.
MPTP administration.
Experimental parkinsonism was induced using the overlesioned hemi-parkinsonian model (Oiwa et al. 2003; McCairn and Turner 2009). Animals received initial infusions of MPTP via the right intracarotid artery (0.55 mg/kg), followed by supplemental systemic doses (0.4 mg/kg iv, 0.35–0.75 mg/kg im) over the course of several weeks, until a stable, moderate to severe parkinsonism was induced. Symptom severity was rated regularly according to a scale developed by Schneider et al. (2003).
Indwelling GPi-DBS macroelectrode.
Standard microelectrode mapping was used to identify the chamber coordinates for DBS electrode placement, which allowed targeting of the GPi. A custom DBS electrode was assembled from two Teflon-coated Pt-Ir microwires (50-μm diameter) glued inside a 5-mm length of 28-gauge stainless steel cannula (Turner and DeLong 2000; McCairn and Turner 2009). The cut ends of the microwires extended below the cannula tip by 1.0 and 1.5 mm. Insulation was stripped from ∼0.5 mm of the distal ends of each microwire (surface areas: ∼0.08 mm2, impedance: 5–10 kΩ at 1 kHz, charge density: 100 μC·cm−2·phase−1). The electrode assembly was implanted transdurally, via the chronic recording chamber using a protective guide cannula (26 gauge) and stylus mounted in the microdrive. Upon reaching the target location for implantation, the guide tube and stylus were withdrawn, and the electrode assembly was left floating in the brain with only the proximal ends of the microwires exiting the dura. The proximal ends were led through a port in the side of the recording chamber (which was subsequently sealed with cyanoacrylate glue) and soldered to a head-mounted connector.
Clinical testing.
To measure the therapeutic effects of GPi-DBS, transient changes in posture and rigidity were measured at the elbow joint using a servo-controlled torque motor (Aerotech, Pittsburgh, PA). During torque testing, the animal's left arm (contralateral to the DBS electrode) was held in the horizontal plane and secured in a padded splint, with the elbow joint aligned with the vertical axis of rotation of the motor. The motor moved the elbow through a sinusoidal displacement of ±20° around the elbow's neutral position at a cycle rate of 1 Hz, with reactive torque sampled from the motor at a rate of 1 KHz (see Fig. 2Bi). Significant changes in the work required to move the elbow were detected by converting the torque transients per sinusoid into total “work” required expressed as Newton meters per degree (McCairn and Turner 2009). These data were then tested for significance by constructing histograms of the mean work on- and off-stimulation, with confidence limits set at P = 0.01 (see Fig. 2Bii).
Fig. 2.

Stimulation artifact rejection and behavioral response to deep brain stimulation (DBS). Ai: neuronal signal from M1 acquired just before and during GPi-DBS, but with artifact subtraction disabled. Note the presence of large voltage transients during DBS (black) preventing discrimination of single unit spikes. Aii: expanded view of a short time period during DBS showing the large voltage transients that occurred time locked to the delivery of the stimulation pulse (vertical dashed lines; 150-Hz, 1-mA, 200-μs pulse width). Aiii: same microelectrode signal acquired on a parallel acquisition channel with artifact subtraction working shows no evidence of shock artifacts while action potentials and recording noise are preserved. (Note the different voltage scales in Aii and Aiii.) Action potentials that were completely obscured by artifacts in the unsubtracted data stream were easily detected in the processed data stream. Aiv: peristimulus sweeps (n = 4,500) of the recorded signal (top trace) demonstrate that it was possible to record and discriminate action potentials across the entire peristimulus period, including times close to stimulus pulse delivery (time zero). The bottom traces show statistical tests of the efficacy of the artifact rejection. The mean waveforms of spikes discriminated during DBS (thick black lines; ±SE indicated by light gray shading) were required to fall within the 95% confidence interval for all spikes discriminate during nonstimulation periods (dark gray shading). The test was conducted separately for four 2-ms epochs, defined by the vertical dashed lines. Bi: clinical testing using the torque motor. The figure shows one unprocessed record of constant angular displacement (gray trace) ±20° at 1 Hz and corresponding torque trace (black trace) off- and on-stimulation. Bii: measure of elbow rigidity calculated as cycle-by-cycle work derived from the integrated resistive torque (i.e., “work”) required to move the elbow joint through a ± 20° cycle at 1 Hz from one recording session that consisted of ten 30-s long periods of stimulation. Note the large reduction of work during application of GPi-DBS and the persistence of the effect after stimulation has stopped. Biii: postural transients (*) increased in frequency during GPi-DBS. This particular example shows raw torque (black traces) for 7 successive blocks of GPi-DBS (gray shading) sampled from monkey C.
Data acquisition and artifact subtraction.
The extracellular activity of multiple single neurons in M1 was sampled using a four-electrode microdrive and glass-insulated tungsten microelectrodes (0.5–1.0 MΩ impedance; MT; Alpha Omega Engineering, Nazareth, Israel). Data were passed through a low-gain headstage (gain = 4×, 2 Hz to 7.5 kHz bandpass) and then digitized at 24 kHz (16-bit resolution; Tucker Davis Technologies, Alachua, FL). The digitized signals were filtered (300 Hz to 6 kHz; First order Butterworth) and saved to disk either as continuous data or as 38 sample-long snippets of the continuous data stream.
During periods of GPi-DBS, online signal processing (Tucker Davis Technologies) was used to eliminate the large stimulation-induced electrical artifacts in the neuronal recordings (see Fig. 2, Ai-Aiii). This was accomplished through use of a low gain headstage and preamplifier and 16-bit A/D conversion to prevent artifact-induced saturation of the analog electronics, synchronization of stimulus delivery, analog-to-digital conversion, and artifact subtraction using the same microsecond-accurate clock, thereby eliminating shock-to-shock variation in the shape of the artifact caused by temporal jitter, and subtraction of an artifact “template” from the data stream at the time of every shock. The template was computed as a 6.1-ms-long moving average of the previous 100 shocks. Thus the template was allowed to change over time in response to gradual changes in artifact shape (e.g., due to changes in an animal's posture). This artifact subtraction algorithm was applied in real-time during simultaneous data acquisition across four channels so that its efficacy could be monitored. Valid operation of the subtraction method was readily apparent during data collection in that stimulation-related voltage transients were absent from the signals played back on oscilloscope and audio monitors. Offline analyses, using custom Matlab scripts, were used to verify that stimulation-related voltage transients were not included in the action potentials of sorted spikes and that the subtraction method did not distort the shapes of spike waveforms. Additional quality testing of spike waveforms during stimulation was achieved through constructing peristimulus sweeps of the recorded signal aligned to each stimulus pulse. The mean waveform shape was analyzed in 2-ms epochs pre- and poststimulus delivery to determine that action potentials were of a consistent size and shape with those recorded outside the stimulus period (see Fig. 2Biv). Interspike interval (ISI) histograms were then constructed to determine if there were significant increases or decreases in activity equal to the stimulation rate. Further details on the efficacy of the artifact subtraction methods can be found in McCairn and Turner (2009).
Recording and stimulation protocol.
The animal was seated in a primate chair and placed in a sound attenuating booth. The primary motor cortex was identified before experimental recording sessions using sensory examination and intracortical microstimulation (5–50 μA, 20 biphasic pulses, 200 μs each phase, 300 Hz; Fig. 1B). During recording experiments, up to four microelectrodes were lowered transdurally into the primary motor cortex via the implanted recording chamber. As soon as the action potentials of one or more single neurons were isolated on the recording electrode(s), high-frequency stimulation was delivered for <5 s to train artifact templates into the artifact rejection system. Stimulation was delivered to the GPi using an isolated constant current stimulator (model 2100; A-M Systems, Carlsborg, WA), with symmetric biphasic pulses (cathodal followed by anodal) 200 μs in duration, delivered at 150 Hz. Data were then collected during an initial 1-min off-stimulation (“baseline” or “control”) period, followed by multiple 30-s blocks of stimulation (DBSon) each separated by >30 s of no stimulation (DBSoff). Stimulation currents ranged from 200 to 1,000 μA (mean current 400 μA, SD = 120 μA). All of the recordings were obtained during rigidity testing. During recording, the animal's left (contralateral) arm was secured in the torque motor's manipulandum and the elbow was rotated through a sinusoidal displacement of ± 20° at 1 Hz.
Fig. 1.
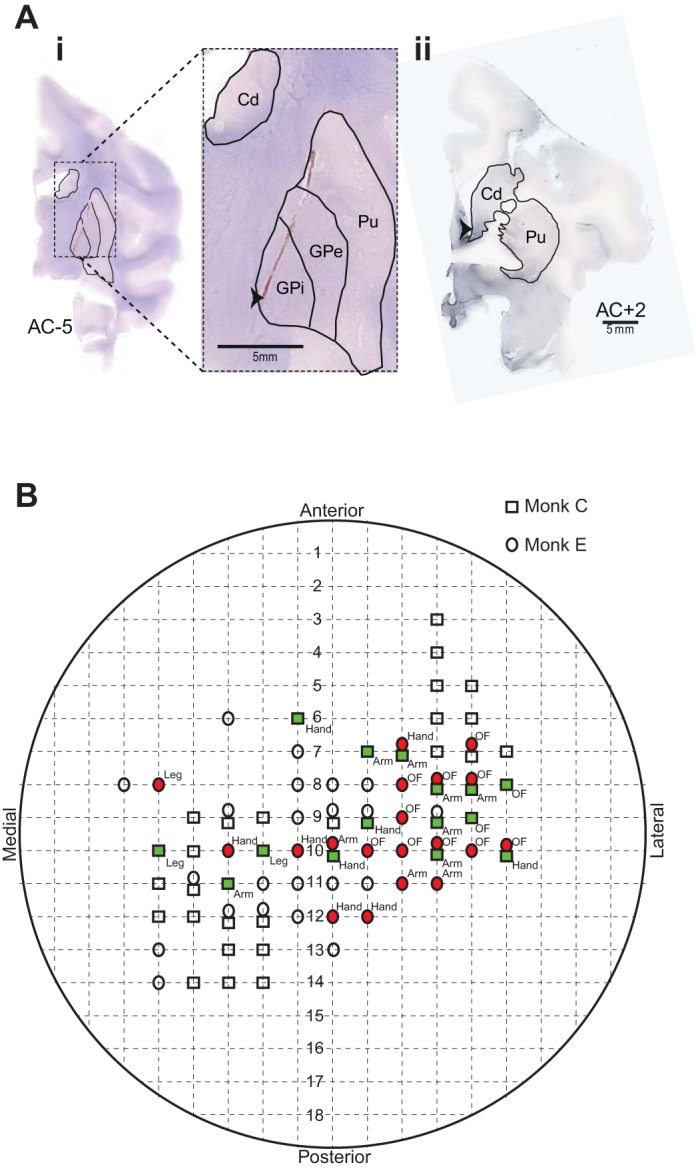
Histology and intracortical mapping. Ai: cresyl violet stained slide, showing a coronal section through the electrode implanted hemisphere, which highlights the gliosis mark produced by placement of the stimulating electrode (black arrow). Aii: tyrosine hydroxylase was depleted throughout the dorsal caudate (Cd) and putamen (Pu) but preserved in the limbic regions of the ventral caudate (black arrow). GPi, globus pallidus internus; GPe, globus pallidus externus. B: chamber schematic showing the areas of the chamber sampled for primary motor cortex (M1) recording and microexcitable sites at which intracortical microstimulation elicited movement of specific body parts (filled shapes). OF, orofacial.
Offline analysis of neuronal activity.
The action potentials of individual neurons were sorted offline by drawing contours around waveform clusters in principal component space (Offline Sorter, Plexon, Plano, TX). Neurons were accepted for further analysis if they met the following criteria: 1) the recording was from a location within the primary motor cortex (defined by microelectrode mapping, sensitivity to sensory driving, and microstimulation); 2) a unit's action potentials were of a consistent shape and could be separated with a high degree of certainty from the background noise and other recorded units; 3) no more than 0.1% of the unit's ISIs were less than 1.5 ms; 4) stable unit isolation was present for a minimum of three stimulation periods with corresponding control periods, and 5) there was no evidence that single unit isolation was corrupted by the appearance of voltage transients caused by GPi-DBS. Following sorting, each neuron's autocorrelation function was inspected to determine that each unit displayed a clear refractory period. This precluded the possibility that the identified unit was corrupted by shock artifacts.
The primary tests for long-duration effects of DBS on a neuron's firing were performed on a histogram constructed around DBSon [i.e., a peri-DBS histogram (PdbsH), bin size = 1 s]. Significant responses to GPi-DBS were initially determined via 99% confidence interval threshold crossings. The latency of any significant response was taken as the first bin that crossed the 99% confidence interval relative to the start of DBS. To calculate population-scale mean firing rate changes during GPi-DBS, the data were normalized by subtracting the mean of the 30-s prestimulation data just before DBSon to give a measure of neural activity around a baseline of zero. To assess the stability of any rate changes across the stimulation period, the peristimulus histogram was divided into equal epochs covering the pre-, actual, and poststimulation period, each consisting of 10 s. The data for each stimulation period (epochs 4–6) were compared with control periods (epochs 1–3) and poststimulation periods (epochs 7–9) using multicomparison ANOVA with Tukey's honestly significant difference error correction (see Fig. 3).
Fig. 3.

Stimulation induced modulation of firing rates in M1. Exemplar raw data from 2 neurons whose firing rate increased (A) and decrease (B) with GPi-DBS. C and D: peri-DBS histograms (PdbsHs) and raster plots for the units shown in A and B. Vertical dashed line: time of onset of GPi-DBS. Gray shading: the period DBS was active. Horizontal solid and dashed lines: mean and 99% confidence level for firing rates from the pre-DBS control period. E, top: color plot of PdbsHs for all cells recorded in the study (1 horizontal row for each cell) aligned to stimulation (DBSon). Firing rates have been normalized by subtracting the mean firing rate computed from the 30 s before DBSon. Vertical dashed lines: 10 s-long analysis epochs. The on-stimulation period corresponded to epochs 4–6. E, bottom: box and whisker plot for each analysis epoch showing median and range of firing rates. The horizontal ends of each box indicate the upper and lower quartile range. The whiskers extend to 1.5× the interquartile range, while outliers are displayed as +'s.
DBS-induced changes to the pattern of firing were investigated by building ISI histograms and testing the coefficient of variation (CV). The different experimental states (DBSon and DBSoff) were concatenated, and only sessions with >1,000 spikes were used for further analysis. The data from each neuron were then pooled and differences in mean CV between the experimental states were determined using Student's t-test.
Burst discharges were detected using the Legendy surprise method (Legendy and Salcman 1985), which is described in detail by Wichmann and Soares (2006). Bursts were defined as groups of three or more spikes whose ISIs were unusually short compared with other ISIs of a spike train. The parameters for analysis used a surprise threshold of 5, which equates to an alpha <0.001 that the candidate burst would occur as part of a Poisson-distributed sequence of spikes. Spike trains were concatenated across all off- and on-stimulation periods. The concatenated control and stimulation trains were analyzed separately. Three fundamental measures for burst analysis were used: 1) the number of identified bursts in the 30 s before stimulation was compared with the number of bursts in the 30 s of stimulation; 2) the fraction of time within a spike train spent in bursts; and 3) the fraction of total spikes contained within bursts. A change in a single neuron's propensity to fire in bursts between different experimental states was tested using a Wilcoxon rank-sum test. In addition, effects of DBS on burst morphology were analyzed by constructing burst-triggered averages and 95% confidence intervals for a cell's periburst instantaneous frequency of firing (McCairn and Turner 2009; Pasquereau and Turner 2011). Separate averages were computed around the onset of bursts detected in control and stimulation trains. The mean preburst firing rate (500 ms immediately before burst onset) was subtracted from each burst-triggered average to aid comparison between stimulation conditions. Significant differences between the two states were tested for by performing a t-test across the concatenated pre- and post-DBS matrices, which gave a P value for each analyzed bin. Comparing burst-triggered averages provided a way to detect potential effects of DBS on the general structure of bursts without extracting an unreasonably large number of burst parameters.
Oscillations in neuronal activity were detected using methods adapted from previous studies (McCairn and Turner 2009; Pasquereau and Turner 2011). To detect oscillations, a discrete Fourier transform was applied to nonoverlapping 1,024-point segments of the spike delta function (1-kHz resolution) and smoothed with a Hanning window of the same length. This yielded power spectral density estimates for frequencies between 0 and 500 Hz, with a resolution of 1 Hz. Peaks in the normalized spectra between 3 and 200 Hz were tested for significance relative to the SD of the normalized spectra in the 340- to 500-Hz “control” range. Frequencies up to 200 Hz were included to detect the fundamental spectral correlates of phasic responses to stimulation. The threshold for significance applied to each peak in a spectrum was corrected for multiple comparisons (Bonferroni correction based on n = 161 spectral points in the tested range 3–160 Hz) to yield an omnibus alpha of 0.01. Autocorrelograms were inspected to verify the qualitative accuracy of the spectral analysis (1-ms resolution, maximum lag = 1 s, smoothed with a σ = 3 ms Gaussian). Note that we did not perform shuffled normalization of the computed spectra (i.e., following Rivlin-Etzion et al. 2006), consistent with our previous study of M1 activity in parkinsonism (Pasquereau and Turner 2011). Thus, in the current study, spectral peaks could reflect either rhythmic oscillations in firing rate or the rhythmic generation of individual spikes. A full justification for this approach is provided in Pasquereau and Turner (2011).
Functional connectivity and the presence of correlated activity between pairs of neurons in the spectral domain were determined using coherence analysis. Coherence was calculated by dividing the cross spectral density of the two cells being analyzed by the square root of the product of the autospectrum of each signal (1-ms resolution, maximum lag = 1 s, smoothed with a σ = 3 ms Gaussian).
Histology.
At the completion of the study, animals were anesthetized deeply and perfused transcardially using phosphate-buffered saline with 10% formalin. Stimulating electrodes were left in place until after perfusion. The tissue was blocked in the coronal plane, cut in 40-μm sections, mounted, and stained with cresyl violet (Fig. 1Ai). To verify depletion of dopaminergic terminals in the striatum, selected sections were stained using tyrosine hydroxylase (TH) immunohistochemistry. These sections were incubated for 24 h with a mouse anti-TH monoclonal antibody, followed by 1 h of incubation with a biotinylated horse anti-mouse antibody (Chemicon International, Temecula, CA). Sections were then incubated with streptavidin for 1 h and revealed using DAB-Vector SK-400 (Vector Laboratories, Burlingame, CA; Fig. 1Aii). The locations of stimulating electrodes within the GPi were clearly visible based on the locations of defects left in the histologic sections by the electrode shaft.
RESULTS
Clinical status and reconstruction of the stimulation site.
After MPTP administration, both animals showed moderate to severe parkinsonian signs bilaterally. Daily symptom ratings using the Schneider scale for parkinsonism (Schneider et al. 2003) were 37 and 38 for the two animals, compared with a mean of 8 before MPTP treatment. Observable symptoms included decreased spontaneous movement, limb rigidity, bradykinesia, action tremor, and stooped posture. Both animals had very low rates of spontaneous movement in their home cages (mean 9.0 movements/min), compared with the >50 movements/min rate typical for neurologically intact macaques.
Postmortem histological examination confirmed that the stimulating electrode targeted the GPi in both animals, an example of which is shown in Fig. 1Ai. In the figure, gliosis caused by the macroelectrode assembly is visible; the tip of the stimulating probe and actual site of stimulation (indicated by an arrow) is located in the dorsomedial regions of the GPi, adjacent to the internal capsule. The animals also showed widespread depletion of tyrosine hydroxylase, the rate-limiting enzyme for conversion of tyrosine to dopamine (Fig. 1Aii). Preserved fibers in the example shown here (indicated by arrows) can be seen at the level of the ventral caudate (Fig. 1Aii), a common feature observed with MPTP toxicity (Jan et al. 2003).
GPi-DBS was associated with a reduction in muscular rigidity, which persisted for >10 s following the cessation of GPi-DBS (Fig. 2Bii). GPi-DBS was also associated with an increase in transient postural shifts; these changes in body posture were readily detectable in the torque traces as the animal moved against the arm that was fixed in the splint of the torque motor (Fig. 2Biii). The behavioral results with respect to muscular rigidity and postural transients have been published previously, and a more detailed description of the experimental findings can be found in McCairn and Turner (2009).
Electrophysiological data and responses to GPi-DBS.
A total of 132 cells [monkey C (n = 78) and monkey E (n = 54)] were sampled from the arm area of M1 during GPi-DBS. All of the cells were recorded while a torque motor moved the contralateral elbow through 20° sinusoidal displacements. Of the 132 cells sampled, 36% [monkey C (n = 22) and monkey E (n = 25), total n = 47] showed a significant change in mean firing rate during GPi-DBS. The changes in firing rate manifested as either increases in activity [27%, n = 35, monkey C (n = 17) and monkey E (n = 18); Fig. 3, A and C] or decreases [9%, n = 12, monkey C (n = 5) and monkey E (n = 7); Fig. 3, B and D]. Increases in firing were more common than decreases (χ2 = 9.4; P < 0.01). There was, however, no significant difference in the latency of firing rate changes for the two response types. The mean onset latency was 2.7 s (SD ± 2.9) for increases in firing rate and 2.0 s (SD ± 3.4; P = 0.4, t-test) for decreased firing rates. Across the population, GPi-DBS had no net effect on the population mean firing rate (Fig. 3E). The population mean firing rate was slightly elevated during the stimulation period (+0.46 ± 0.17 sp/s), but this increase was nonsignificant (P = 0.05, ANOVA).
Minimal antidromic-like driving in M1 during GPi-DBS.
Previous studies, using GPi-DBS in MPTP monkeys (Johnson et al. 2009), have suggested that stimulation of the output nucleus of the BG leads to an antidromic activation motor cortexes, specifically M1. We examined our recordings for evidence of short-latency antidromic-like activation of M1 by searching for driving of a neuron's action potentials at a fixed latency following individual stimuli delivered to the GPi. Our ability to record continuously throughout the peristimulus interval, unimpeded by shock artifacts, allowed us to determine the prevalence of short-latency driving of M1 during GPi-DBS and thereby assess the idea that antidromic induction of high-frequency resonance in the cortex plays a critical role in the therapeutic mechanism of DBS (Li et al. 2007; Dejean et al. 2009; Johnson et al. 2009; Li et al. 2012).
Among the 132 cells in the database, short fixed-latency driving was found in only one neuron (<1% of all neurons studied). For this one neuron, GPi stimulation at 400 μA (150 Hz) induced action potentials at a constant short latency (2.0 ± 0.4 ms) in response to a small fraction of the stimuli delivered (fraction of stimuli that yielded an antidromic spike = 6.4%). The temporally locked activity was present for short periods (mean 0.18 ± 0.04 s) separated by long periods of silence (5.3 ± 14.3 s). The mean firing rate during the periods of antidromic-like driving was 101.3 ± 15.6 Hz. The low fraction of cells showing any indication of antidromic-like driving runs contrary to the idea that antidromic-induced cortical resonance is a primary component of therapeutic GPi-DBS.
GPi-DBS did not suppress burst firing.
Although one therapeutic mechanism hypothesized for GPi-DBS is suppression of burst firing (Shi et al. 2006; Vitek 2002), our previous work found no evidence for a decrease in burst firing in the pallidum during GPi-DBS (McCairn and Turner 2009). Similarly, we found here that few cells (11%, n = 15) showed a change in the prevalence of burst firing between the two states and those cells were split evenly between increased and decreased burstiness with stimulation (n = 7 and n = 8, respectively). Figure 4Ai shows an example of decreased burst firing following the onset of stimulation. In the figure, the rasters show spike times relative to DBSon (black tic marks) and each red tick mark corresponds to the start of an identified burst (DBSoff burst/s median = 12, DBSon burst/s median = 0; P = 0.0003, Wilcoxon rank sum). Figure 4Aii shows an example of a cell that increased its propensity to fire in bursts following the onset of stimulation (DBSoff burst/s median = 0, DBSon burst/s median = 5; P = 0.0006, Wilcoxon rank sum). The low number of cells with modified burst firing between the two states and roughly equal propensity to decrease and increase burstiness led to no significant change in measures of burst firing across the population (P > 0.2, paired t-test; Fig. 4, Bi and Bii). Similarly, the population-averaged burst structure was indistinguishable on- and off-stimulation (Fig. 4Biii).
Fig. 4.

Burst properties and firing patterns in M1 during stimulation. Raster plots of spike times (black tics) and burst events (red arrows) for 2 neurons in which GPi-DBS decreased the rate of burst firing (Ai) and increased burst firing (Aii). B: cell-by-cell measures of burst firing during DBSon were well predicted by the same measures from the no stimulation (DBSoff) condition. This was true both for the number of bursts/s and the %spikes in burst. Biii: GPi-DBS did not alter the time-magnitude structure of bursts. Population mean firing rates aligned on the time of burst onset under DBSoff (black) and DBSon (gray) conditions (dashed lines: SE). Inset: P values from t-tests comparing DBSoff vs. DBSon population mean periburst firing rates. D: for most cells, firing rate variability [coefficient of variation (CV) of interspike intervals] was also highly similar under DBSon and DBSoff conditions.
CV of ISIs also showed no difference between the off- and on-stimulation conditions (DBSoff CV = 1.29, SD ± 0.5 vs. DBSon CV = 1.3, SD ± 0.7; P = 0.7, paired t-test; Fig. 4Biv). Therefore, although GPi-DBS affected the prevalence and structure of bursts in some individual neurons, these effects yielded no net change in the amount or pattern of burst firing or regularity of firing when averaged across the population.
GPi-DBS suppressed beta oscillatory activity.
A subset of M1 neurons (20%, n = 27) displayed significant rhythmic firing in the beta (12–30 Hz)-frequency band (Fig. 5). The mean oscillation frequency for these cells was 17.25 ± 4.0 Hz. Figure 5A shows an example of an M1 neuron's autocorrelation function off- and on-stimulation. In the off-stimulation condition (black), the autocorrelation function shows substantial peaks and valleys indicative of oscillatory firing in the beta-frequency range. This motif transitioned to a flatter profile in the on-stimulation condition (Fig. 5A, gray trace). Spectral analysis of this neuron's spike train showed a marked peak at 14.8 Hz in the off-stimulation condition that was attenuated significantly during GPi-DBS. Consistent with the observation that short-latency entrainment and high-frequency resonance were rare in our data, the reduction in beta oscillations was achieved without the appearance of peaks at the frequency of stimulation (150 Hz) or its lower harmonics (Fig. 5B). GPi-DBS reduced oscillatory power in all beta-rhythmic cells but one, causing a significant reduction in the overall prevalence of oscillatory activity (χ2 = 27.01; P < 0.0001; Fig. 5C). In contrast, the prevalence of high-frequency oscillations did not differ significantly between DBSon and DBSoff conditions (i.e., the gamma band and oscillations between gamma and 200 Hz).
Fig. 5.
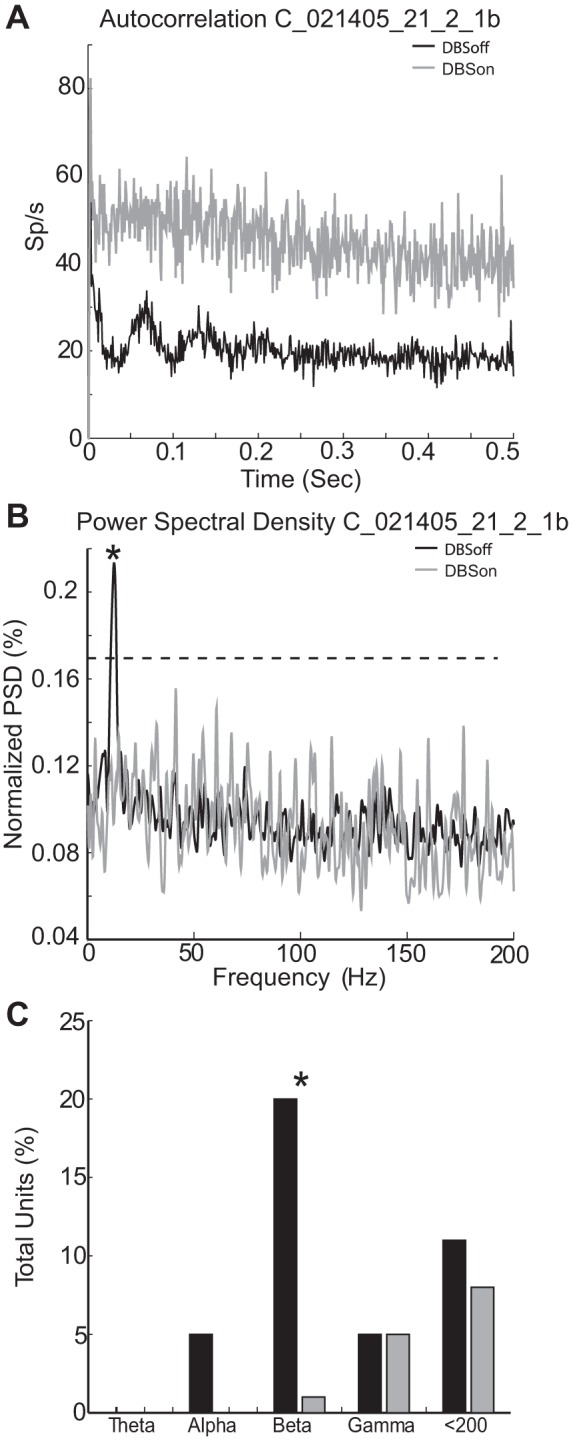
Firing pattern and spectral properties in M1 during stimulation. A: exemplar autocorrelation function and its change during GPi-DBS. In the DBSoff condition (black trace), the cell tended to spike at regular intervals, revealed as significant peaks and valleys in its autocorrelation function, within the beta-frequency range. During stimulation (gray trace), the cell's autocorrelation function flattened and lost the characteristic peaks and valleys observed off-stimulation. B: power spectra for the same exemplar data used in A under DBSoff and DBSon conditions. A prominent spectral peak at 14.8 Hz (*) was abolished during stimulation. Dashed horizontal line: threshold for significance. C: fraction of cells with one or more significant spectral peak under DBSoff and DBSon conditions (black and gray bins, respectively). Note that the prevalence of cells showing oscillatory activity in the beta-frequency range was reduced significantly during stimulation (*).
GPi-DBS also reduced oscillations that were coherent (i.e., synchronized) between neuron pairs in M1. A total of 207 neuronal pairs were sampled simultaneously from different electrodes. Coherent activity <30 Hz was found in 18% of the recorded pairs. Figure 6A shows an example in which, before stimulation, the firing of a pair of neurons was highly synchronized at 15.6 Hz (R2 = 0.15; Fig. 6A, black trace). It can be seen that GPi-DBS completely abolished this synchronized oscillation (Fig. 6A, gray trace). In agreement with this example, stimulation reduced the overall prevalence of synchronized firing across the population of cell pairs studied (Fig. 6B; χ2 = 28.13; P < 0.0001). In summary, GPi-DBS reduced the prevalence of pathological low-frequency oscillations (<30 Hz), both in the firing of individual neurons and in the synchronized firing of neuronal pairs.
Fig. 6.

Suppression of synchronized spiking during GPi-DBS. A: spike activity of a pair of simultaneously recorded M1 neurons was synchronized at 15.6 Hz (*) in the DBSoff condition (black trace). That coherent activity was suppressed completely during GPi-DBS (gray trace). B: GPi-DBS reduced the prevalence of low-frequency synchronized rhythmic firing in the M1 population. The bar plot indicates the fraction of cell pairs with 1 or more significant coherence peak in the indicated frequency ranges during control periods (DBSoff) and stimulation (DBSon). Note the elevated prevalence of significant coherent firing in the beta frequency band in the absence of stimulation (black bins) and the significant reduction in that prevalence during stimulation (marked by an *).
DISCUSSION
Our results demonstrate that GPi-DBS induces both increases and decreases in the firing rates of individual neurons within M1, with excitation being the predominant response among the population of cells that were directly affected by stimulation. However, population-scale mean firing rates were only marginally affected between experimental states (DBSoff vs. DBSon). GPi-DBS also had a minimal impact on burst statistics and firing variability measured at the population level. The most obvious neurophysiological correlate of reduced parkinsonism was a suppression of rhythmic firing and synchronous activity in the beta (12–30 Hz) range. Antidromic-driven activity and associated high-frequency resonant activity were uncommon in this study. It is, therefore, unlikely that high-frequency resonance in the cortex (e.g., at the 150-Hz stimulation frequency or harmonics) played an important role as a therapeutic mechanism during GPi-DBS.
Potential confounds and limitations.
Although we used low-impedance macroelectrodes and clinically relevant currents and frequencies, the experimental electrodes did not replicate the exact geometry of clinical electrodes. Additionally, the pulse width used in this study was 0.2 ms (vs. <0.1 ms used clinically). Another limitation is that we were unable to confidently reconstruct the laminar position or the efferent targets of the M1 neurons studied. Recent work has shown that subpopulations within M1 are affected differently by the parkinsonian state (Pasquereau and Turner 2011). It is also important to recognize that the present results, collected while an animal's arm was being moved passively via a torque motor, may have limited relevance for other behavioral contexts [e.g., in animals at rest or during performance of a behavioral task (Montgomery and Gale 2008)].
We observed two primary effects of GPi-DBS on the animal's behavior: a reduction in limb rigidity and an increase in the frequency of postural transients. The latter presumably reflected movements executed by the animal to achieve a more comfortable resting position. A potential confound is that the observed suppression of beta activity may be secondary to the increased frequency of movement and not a direct effect of stimulation. It should be pointed out, however, that although GPi-DBS often increased the frequency of spontaneous movement, these movements appear sporadically. The suppression of low-frequency oscillatory activity was still present in the absence of any detectable movement by the animal. A detailed analysis and discussion of the clinical effects of GPi-DBS can be found in McCairn and Turner (2009).
Modulation of rate firing.
Supporting evidence for GPi-DBS-induced modulation of the motor cortexes of PD patients comes from a number of studies. These reports have relied on noninvasive PET and MRI measures, and although clear changes have been observed in pre- and supplementary motor areas, there are as yet no conclusive reports of gross changes in M1 activity from GPi-DBS (Davis et al. 1997; Limousin et al. 1997; Devos et al. 2002; Fukuda et al. 2002; Valalik et al. 2009). We report both increases and decreases in the firing rate of individual cells during GPi-DBS, with no net change in the population mean firing rates. This observation would appear to support the previous imaging studies.
Can current understanding of the network effects of DBS explain our observations? The local effects of GPi-DBS on firing rates within the pallidum appear to be mixed, with some neurons showing dramatic reductions in firing rate (Benazzouz et al. 1995; Boraud et al. 1996; Dostrovsky et al. 2000; Wu et al. 2001; McCairn and Turner 2009; Bar-Gad et al. 2004; Erez et al. 2009; Liu et al. 2012; Chiken and Nambu 2013) and others showing increases (McCairn and Turner 2009; Bar-Gad et al. 2004; Erez et al. 2009; Liu et al. 2012; Chiken and Nambu 2013). These observations may explain our observation of a mixture of increases and decreases in M1 firing rates, by way of DBS-induced disinhibition and inhibition (respectively) of GPi-receiving thalamo-cortical projections (Montgomery 2006; Anderson et al. 2003).
Direct electrophysiological support for pallidal stimulation modulating M1 has recently been demonstrated in NHPs (Johnson et al. 2009). GPi-DBS was found to reduce the mean firing rates of cortical neurons, often in a phasic stimulus-locked manner. The latency of inhibition was on the order of ∼3 ms following delivery of the stimulation pulse; the short-latency timing of these responses appears to rule out mediation via the known multisynaptic pathways from pallidum to cortex (Anderson and Turner 1991; Nambu et al. 1988; Buford et al. 1996; Inase et al. 1996). Methodological differences (artifact rejection techniques) and differences in the stimulating probe most likely explain the difference between this and our study. The macroelectrode used in the previous study was significantly larger than the one used here and may have been more effective at exciting fibers in the internal capsule. It remains unclear, however, how stimulation in the GPi is able to elicit short-latency inhibition, not excitation as would be predicted (Li et al. 2007; Dejean et al. 2009; Gradinaru et al. 2009; Li et al. 2012).
Claims of direct activation of the cortex following GPi-DBS have also recently been published from studies in dystonic patients (Tisch et al. 2008; Bhanpuri et al. 2014). These studies used EEG recording with relatively slow rates of stimulation (∼9–10 Hz). It was reported that any changes in EEG amplitude were detectable at ∼25 ms after pulse delivery, which is close to the latencies expected for orthodromic pallido-thalamo-cortical transmission (Herrojo et al. 2014). As our study did not use EEG or LFP measurement in M1, it is difficult to make a direct comparison with the results reported here.
Antidromic-driven high-frequency resonance in M1 was minimal.
Recent work using DBS in the STN of rodents has suggested that antidromic modulation of the cortex (and induction of “high-frequency resonance”), through activation of fibers in the internal capsule, may underlie the mechanism of STN-DBS (Li et al. 2007; Dejean et al. 2009; Li et al. 2012; Gradinaru et al. 2009). The short-latency inhibition of M1 during GPi-DBS reported by Johnson et al. (2009) may also be seen as consistent with a high-frequency resonance mechanism for DBS.
Our artifact subtraction method makes it possible to identify short-latency responses (0–3 ms) in close spatial proximity to the stimulating electrode (McCairn and Turner 2009; McCairn et al. 2013). Using this technique, we found only one M1 cell (out of 132 cells studied) that responded at a short fixed latency as would be expected of antidromic driving. The precise pathway responsible for this effect is a matter of speculation. Observations that high-frequency microstimulation of the GPi is not transmitted reliably to M1 (Rivlin-Etzion et al. 2008) makes an orthodromic pathway (i.e., GPi→thalamus→cortex) unlikely. It appears more likely that GPi-DBS in our animals spread to the internal capsule where it excited a small number of corticofugal fibers. Alternatively, there is some evidence for a sparse but direct cortical innervation of the pallidum (Naito and Kita 1994; Smith and Wichmann 2014). The low fidelity of driving observed here (spikes elicited by ∼6% of shocks) is consistent with a previous study of antidromic activation of cortical neurons during high-frequency stimulation (Iremonger et al. 2006). The paucity of stimulation driven high-frequency resonance in our recordings, combined with pronounced and persistent therapeutic effects of GPi-DBS, is inconsistent with the idea that antidromic-driven resonance in cortex is a primary therapeutic mechanism of action for GPi-DBS.
GPi-DBS did not suppress burst firing.
We found no stimulation-induced change in the overall prevalence of burst discharges or in the characteristics of their structure. This finding corresponds with our observations from the GP of MPTP-treated NHPs (McCairn and Turner 2009) and is supported by the recent work of Hahn and colleagues (Hahn et al. 2008). This finding brings into question the concept that GPi-DBS reduces parkinsonism by suppressing burst firing (Vitek 2002; Shi et al. 2006). The concept that DBS does not alleviate parkinsonism through abolition of burst firing may, however, only apply to GPi-DBS. Recent work in rodents has suggested that the activation of the fibers surrounding the STN and subsequent antidromic activation of cortical structures can lead to destabilization of synchronized bursting activity within M1 (Li et al. 2012).
GPi-DBS suppressed beta-oscillatory activity.
Parkinsonian symptoms have been associated with the presence of synchronized beta oscillations in the BG-thalamo-cortical network both in animal models (Goldberg et al. 2004; Mallet et al. 2008; McCairn and Turner 2009; Pasquereau and Turner 2011) and in human patients (Hutchison et al. 2004; Kuhn et al. 2004, 2009; Brown and Williams 2005; Hammond et al. 2007; Eusebio et al. 2011). It is important to note, however, that some parkinsonian signs may appear before excessive beta firing emerges (Leblois et al. 2007; Mallet et al. 2005), and the assumption that increased beta oscillations play a causal role in parkinsonism has been questioned (Montgomery 2012). We demonstrated previously that, at the level of the GP, the most consistent features associated with DBS-induced reductions in parkinsonism were reductions in oscillations and coherent oscillations between pairs of pallidal neurons (McCairn and Turner 2009).
Similarly, in the present study, we found a subset of M1 neurons that showed beta-oscillatory activity and pairs of simultaneously recorded neurons whose firing rates were modulated coherently at beta frequencies. These presumed pathological oscillations were reduced in strength or eliminated completely by DBS. Similar reductions in beta activity have been observed in the STN in NHPs (Meissner et al. 2005; Moran et al. 2012) and have been inferred from studies in PD patients (Marsden et al. 2001a; Brown et al. 2004a; Hammond et al. 2007; Eusebio et al. 2011). Our results suggest that the oscillation-suppressive effects of DBS extend beyond the local neuronal circuits that are directly modulated by the stimulation pulse.
The mechanism for suppression of oscillatory activity in M1 is most likely dependent on the specific nucleus being stimulated. In the case of the STN, it appears that much of the suppressive effect in M1 is achieved through “sporadic” antidromic activation of the cortex via the internal capsule (Li et al. 2007; Dejean et al. 2009; Gradinaru et al. 2009; Li et al. 2012). Data from the present study suggest that it is sufficient to suppress oscillatory activity in the output nucleus of the BG (i.e., the GPi) without significantly activating the fibers of the internal capsule to cause a reduction in M1 oscillatory activity.
Therapeutic GPi-DBS also suppressed beta-range coherence. The role that coherence/synchrony plays in the pathophysiology of parkinsonism has yet to be elucidated. It has been suggested that the increase in coherence/synchrony may reflect a breakdown in the functional segregation of discrete cortico-BG neural circuits (Filion and Tremblay 1991; Raz et al. 2000; Goldberg et al. 2002, 2004; Hutchison et al. 2004; Kuhn et al. 2004; Brown and Williams 2005; Bergman et al. 1998). Presumably, as with beta oscillations, the mechanism that mediates a DBS-induced reduction in synchrony depends on the nucleus being stimulated. In the case of GPi-DBS, it is probable that a reduction in synchrony local to the site of stimulation in the GPi (McCairn and Turner 2009) is then communicated to downstream circuits including M1.
Conclusion.
In summary, the effects of GPi-DBS on M1 activity closely resembled those reported for the pallidum. GPi-DBS caused significant reductions in the prevalence of oscillatory and synchronous activity in single and pairs of neurons but no gross alteration in population-level measures of baseline firing rate or burst firing. These results reinforce the concept that reductions of pathological oscillations and synchrony play a major role in the therapeutic mechanism of GPi-DBS.
The observations and conclusions presented here, however, should be placed in the context of the established clinical effects of GPi-DBS, which are known to evolve over multiple time scales from minutes, hours, days, and months (Temperli et al. 2003). The changes to the prevalence of oscillatory and synchronous activity reported here were observed at short time intervals relative to the typical duration of clinical DBS treatment. Therefore, our results may not reflect the totality of the therapeutic mechanisms of GPi-DBS and further studies are needed to test for more long-term effects.
GRANTS
This work was supported by National Institute of Neurological Disorders and Stroke Grants NS-044551, NS-070865, and 1P30-NS0-76405-01A1 (to R. S. Turner).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: K.W.M. and R.S.T. conception and design of research; K.W.M. and R.S.T. performed experiments; K.W.M. and R.S.T. analyzed data; K.W.M. and R.S.T. interpreted results of experiments; K.W.M. and R.S.T. prepared figures; K.W.M. and R.S.T. drafted manuscript; K.W.M. and R.S.T. edited and revised manuscript; K.W.M. and R.S.T. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Eric Schaible, who designed and built the data acquisition system used in this study, and Masaki Isoda, Matthew Panico, Andrew Zimnik, and Benjamin Pasquereau for helpful comments on the manuscript.
REFERENCES
- Agid Y. Continuous high frequency stimulation of deep brain structures in brain pathology. Brain Res Bull 50: 475, 1999. [DOI] [PubMed] [Google Scholar]
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci 12: 366–375, 1989. [DOI] [PubMed] [Google Scholar]
- Anderson ME, Postupna N, Ruffo M. Effects of high-frequency stimulation in the internal globus pallidus on the activity of thalamic neurons in the awake monkey. J Neurophysiol 89: 1150–1160, 2003. [DOI] [PubMed] [Google Scholar]
- Anderson ME, Turner RS. Activity of neurons in cerebellar-receiving, and pallidal-receiving areas of the thalamus of the behaving monkey. J Neurophysiol 66: 879–893, 1991. [DOI] [PubMed] [Google Scholar]
- Bar-Gad I, Elias S, Vaadia E, Bergman H. Complex locking rather than complete cessation of neuronal activity in the globus pallidus of a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated primate in response to pallidal microstimulation. J Neurosci 24: 9410–9419, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benabid AL, Pollak P, Louveau A, Henry S, de RJ. Combined (thalamotomy and stimulation) stereotactic surgery of the VIM thalamic nucleus for bilateral Parkinson disease. Appl Neurophysiol 50: 344–346, 1987. [DOI] [PubMed] [Google Scholar]
- Benazzouz A, Piallat B, Pollak P, Benabid AL. Responses of substantia nigra pars reticulata, and globus pallidus complex to high frequency stimulation of the subthalamic nucleus in rats: electrophysiological data. Neurosci Lett 189: 77–80, 1995. [DOI] [PubMed] [Google Scholar]
- Bergman H, Feingold A, Nini A, Raz A, Slovin H, Abeles M, Vaadia E. Physiological aspects of information processing in the basal ganglia of normal, and parkinsonian primates. Trends Neurosci 21: 32–38, 1998. [DOI] [PubMed] [Google Scholar]
- Bhanpuri NH, Bertucco M, Ferman D, Young SJ, Liker MA, Krieger MD, Sanger TD. Deep brain stimulation evoked potentials may relate to clinical benefit in childhood dystonia. Brain Stimul 7: 718–726, 2014. [DOI] [PubMed] [Google Scholar]
- Boraud T, Bezard E, Bioulac B, Gross C. High frequency stimulation of the internal Globus Pallidus (GPi) simultaneously improves parkinsonian symptoms and reduces the firing frequency of GPi neurons in the MPTP-treated monkey. Neurosci Lett 215: 17–20, 1996. [DOI] [PubMed] [Google Scholar]
- Bronstein JM, Tagliati M, Alterman RL, Lozano AM, Volkmann J, Stefani A, Horak FB, Okun MS, Foote KD, Krack P, Pahwa R, Henderson JM, Hariz MI, Bakay RA, Rezai A, Marks WJ Jr, Moro E, Vitek JL, Weaver FM, Gross RE, DeLong MR. Deep brain stimulation for Parkinson disease: an expert consensus and review of key issues. Arch Neurol 68: 165, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown P, Mazzone P, Oliviero A, Altibrandi MG, Pilato F, Tonali PA, Di L, V. Effects of stimulation of the subthalamic area on oscillatory pallidal activity in Parkinson's disease. Exp Neurol 188: 480–490, 2004a. [DOI] [PubMed] [Google Scholar]
- Brown P, Williams D. Basal ganglia local field potential activity: character, and functional significance in the human. Clin Neurophysiol 116: 2510–2519, 2005. [DOI] [PubMed] [Google Scholar]
- Brown P, Mazzone P, Oliviero A, Altibrandi MG, Pilato F, Tonali PA, Di Lazzaro V. Effects of stimulation of the subthalamic area on oscillatory pallidal activity in Parkinson's disease. Exp Neurol 188: 480–490, 2004b. [DOI] [PubMed] [Google Scholar]
- Buford JA, Inase M, Anderson ME. Contrasting locations of pallidal-receiving neurons, and microexcitable zones in primate thalamus. J Neurophysiol 75: 1105–1116, 1996. [DOI] [PubMed] [Google Scholar]
- Chiken S, Nambu A. High-frequency pallidal stimulation disrupts information flow through the pallidum by GABAergic inhibition. J Neurosci 33: 2268–2280, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper IS, Upton AR, Amin I. Reversibility of chronic neurologic deficits. Some effects of electrical stimulation of the thalamus and internal capsule in man. Appl Neurophysiol 43: 244–258, 1980. [DOI] [PubMed] [Google Scholar]
- Davis KD, Taub E, Houle S, Lang AE, Dostrovsky JO, Tasker RR, Lozano AM. Globus pallidus stimulation activates the cortical motor system during alleviation of parkinsonian symptoms. Nat Med 3: 671–674, 1997. [DOI] [PubMed] [Google Scholar]
- Dejean C, Hyland B, Arbuthnott G. Cortical effects of subthalamic stimulation correlate with behavioral recovery from dopamine antagonist induced akinesia. Cereb Cortex 19: 1055–1063, 2009. [DOI] [PubMed] [Google Scholar]
- DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci 13: 281–285, 1990. [DOI] [PubMed] [Google Scholar]
- Devos D, Derambure P, Bourriez JL, Cassim DF, Blond S, Guieu JD, Destee A, Defebvre L. Influence of internal globus pallidus stimulation on motor cortex activation pattern in Parkinson's disease. Clin Neurophysiol 113: 1110–1120, 2002. [DOI] [PubMed] [Google Scholar]
- Dostrovsky JO, Levy R, Wu JP, Hutchison WD, Tasker RR, Lozano AM. Microstimulation-induced inhibition of neuronal firing in human globus pallidus. J Neurophysiol 84: 570–574, 2000. [DOI] [PubMed] [Google Scholar]
- Erez Y, Czitron H, McCairn K, Belelovsky K, Bar-Gad I. Short-term depression of synaptic transmission during stimulation in the globus pallidus of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated primates. J Neurosci 29: 7797–7802, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eusebio A, Thevathasan W, Doyle GL, Pogosyan A, Bye E, Foltynie T, Zrinzo L, Ashkan K, Aziz T, Brown P. Deep brain stimulation can suppress pathological synchronisation in parkinsonian patients. J Neurol Neurosurg Psychiatry 82: 569–573, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filion M, Tremblay L. Abnormal spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res 547: 142–151, 1991. [PubMed] [Google Scholar]
- Fukuda M, Ghilardi MF, Carbon M, Dhawan V, Ma Y, Feigin A, Mentis MJ, Ghez C, Eidelberg D. Pallidal stimulation for parkinsonism: improved brain activation during sequence learning. Ann Neurol 52: 144–152, 2002. [DOI] [PubMed] [Google Scholar]
- Goldberg JA, Boraud T, Maraton S, Haber SN, Vaadia E, Bergman H. Enhanced synchrony among primary motor cortex neurons in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine primate model of Parkinson's disease. J Neurosci 22: 4639–4653, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Rokni U, Boraud T, Vaadia E, Bergman H. Spike synchronization in the cortex/basal-ganglia networks of Parkinsonian primates reflects global dynamics of the local field potentials. J Neurosci 24: 6003–6010, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradinaru V, Mogri M, Thompson KR, Henderson JM, Deisseroth K. Optical deconstruction of parkinsonian neural circuitry. Science 324: 354–359, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn PJ, Russo GS, Hashimoto T, Miocinovic S, Xu W, McIntyre CC, Vitek JL. Pallidal burst activity during therapeutic deep brain stimulation. Exp Neurol 211: 243–251, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C, Bergman H, Brown P. Pathological synchronization in Parkinson's disease: networks, models, and treatments. Trends Neurosci 30: 357–364, 2007. [DOI] [PubMed] [Google Scholar]
- Herrojo RM, Huebl J, Schonecker T, Kupsch A, Yarrow K, Krauss JK, Schneider GH, Kuhn AA. Involvement of human internal globus pallidus in the early modulation of cortical error-related activity. Cereb Cortex 24: 1502–1517, 2014. [DOI] [PubMed] [Google Scholar]
- Hutchison WD, Dostrovsky JO, Walters JR, Courtemanche R, Boraud T, Goldberg J, Brown P. Neuronal oscillations in the basal ganglia, and movement disorders: evidence from whole animal and human recordings. J Neurosci 24: 9240–9243, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison WD, Lozano AM, Davis KD, Saint-Cyr JA, Lang AE, Dostrovsky JO. Differential neuronal activity in segments of globus pallidus in Parkinson's disease patients. Neuroreport 5: 1533–1537, 1994. [DOI] [PubMed] [Google Scholar]
- Inase M, Buford JA, Anderson ME. Changes in the control of arm position, movement, and thalamic discharge during local inactivation in the globus pallidus of the monkey. J Neurophysiol 75: 1087–1104, 1996. [DOI] [PubMed] [Google Scholar]
- Iremonger KJ, Anderson TR, Hu B, Kiss ZH. Cellular mechanisms preventing sustained activation of cortex during subcortical high-frequency stimulation. J Neurophysiol 96: 613–621, 2006. [DOI] [PubMed] [Google Scholar]
- Jan C, Pessiglione M, Tremblay L, Tande D, Hirsch EC, Francois C. Quantitative analysis of dopaminergic loss in relation to functional territories in MPTP-treated monkeys. Eur J Neurosci 18: 2082–2086, 2003. [DOI] [PubMed] [Google Scholar]
- Johnson MD, Vitek JL, McIntyre CC. Pallidal stimulation that improves parkinsonian motor symptoms also modulates neuronal firing patterns in primary motor cortex in the MPTP-treated monkey. Exp Neurol 219: 359–362, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn AA, Tsui A, Aziz T, Ray N, Brucke C, Kupsch A, Schneider GH, Brown P. Pathological synchronisation in the subthalamic nucleus of patients with Parkinson's disease relates to both bradykinesia, and rigidity. Exp Neurol 215: 380–387, 2009. [DOI] [PubMed] [Google Scholar]
- Kuhn AA, Williams D, Kupsch A, Limousin P, Hariz M, Schneider GH, Yarrow K, Brown P. Event-related beta desynchronization in human subthalamic nucleus correlates with motor performance. Brain 127: 735–746, 2004. [DOI] [PubMed] [Google Scholar]
- Leblois A, Meissner W, Bioulac B, Gross CE, Hansel D, Boraud T. Late emergence of synchronized oscillatory activity in the pallidum during progressive Parkinsonism. Eur J Neurosci 26: 1701–1713, 2007. [DOI] [PubMed] [Google Scholar]
- Legendy CR, Salcman M. Bursts, and recurrences of bursts in the spike trains of spontaneously active striate cortex neurons. J Neurophysiol 53: 926–939, 1985. [DOI] [PubMed] [Google Scholar]
- Li Q, Ke Y, Chan DC, Qian ZM, Yung KK, Ko H, Arbuthnott GW, Yung WH. Therapeutic deep brain stimulation in Parkinsonian rats directly influences motor cortex. Neuron 76: 1030–1041, 2012. [DOI] [PubMed] [Google Scholar]
- Li S, Arbuthnott GW, Jutras MJ, Goldberg JA, Jaeger D. Resonant antidromic cortical circuit activation as a consequence of high-frequency subthalamic deep-brain stimulation. J Neurophysiol 98: 3525–3537, 2007. [DOI] [PubMed] [Google Scholar]
- Limousin P, Greene J, Pollak P, Rothwell J, Benabid AL, Frackowiak R. Changes in cerebral activity pattern due to subthalamic nucleus or internal pallidum stimulation in Parkinson's disease. Ann Neurol 42: 283–291, 1997. [DOI] [PubMed] [Google Scholar]
- Liu LD, Prescott IA, Dostrovsky JO, Hodaie M, Lozano AM, Hutchison WD. Frequency-dependent effects of electrical stimulation in the globus pallidus of dystonia patients. J Neurophysiol 108: 5–17, 2012. [DOI] [PubMed] [Google Scholar]
- Lozano AM, Lipsman N. Probing, and regulating dysfunctional circuits using deep brain stimulation. Neuron 77: 406–424, 2013. [DOI] [PubMed] [Google Scholar]
- Mallet N, Le MC, Charpier S, Gonon F. Feedforward inhibition of projection neurons by fast-spiking GABA interneurons in the rat striatum in vivo. J Neurosci 25: 3857–3869, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet N, Pogosyan A, Sharott A, Csicsvari J, Bolam JP, Brown P, Magill PJ. Disrupted dopamine transmission, and the emergence of exaggerated beta oscillations in subthalamic nucleus and cerebral cortex. J Neurosci 28: 4795–4806, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden J, Limousin-Dowsey P, Fraix V, Pollak P, Odin P, Brown P. Intermuscular coherence in Parkinson's disease: effects of subthalamic nucleus stimulation. Neuroreport 12: 1113–1117, 2001a. [DOI] [PubMed] [Google Scholar]
- Marsden JF, Limousin-Dowsey P, Ashby P, Pollak P, Brown P. Subthalamic nucleus, sensorimotor cortex, and muscle interrelationships in Parkinson's disease. Brain 124: 378–388, 2001b. [DOI] [PubMed] [Google Scholar]
- McCairn KW, Iriki A, Isoda M. Deep brain stimulation reduces tic-related neural activity via temporal locking with stimulus pulses. J Neurosci 33: 6581–6593, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCairn KW, Turner RS. Deep brain stimulation of the globus pallidus internus in the parkinsonian primate: local entrainment, and suppression of low-frequency oscillations. J Neurophysiol 101: 1941–1960, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner W, Leblois A, Hansel D, Bioulac B, Gross CE, Benazzouz A, Boraud T. Subthalamic high frequency stimulation resets subthalamic firing, and reduces abnormal oscillations. Brain 128: 2372–2382, 2005. [DOI] [PubMed] [Google Scholar]
- Montgomery EB. Effects of GPi stimulation on human thalamic neuronal activity. Clin Neurophysiol 117: 2691–2702, 2006. [DOI] [PubMed] [Google Scholar]
- Montgomery EB. The epistemology of Deep Brain Stimulation and neuronal pathophysiology. Front Integr Neurosci 6: 78, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery EB, Gale JT. Mechanisms of action of deep brain stimulation (DBS). Neurosci Biobehav Rev 32: 388–407, 2008. [DOI] [PubMed] [Google Scholar]
- Moran A, Stein E, Tischler H, Bar-Gad I. Decoupling neuronal oscillations during subthalamic nucleus stimulation in the parkinsonian primate. Neurobiol Dis 45: 583–590, 2012. [DOI] [PubMed] [Google Scholar]
- Moran A, Stein E, Tischler H, Belelovsky K, Bar-Gad I. Dynamic stereotypic responses of Basal Ganglia neurons to subthalamic nucleus high-frequency stimulation in the parkinsonian primate. Front Syst Neurosci 5: 21, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito A, Kita H. The cortico-pallidal projection in the rat: an anterograde tracing study with biotinylated dextran amine. Brain Res 653: 251–257, 1994. [DOI] [PubMed] [Google Scholar]
- Nambu A, Yoshida S, Jinnai K. Projection on the motor cortex of thalamic neurons with pallidal input in the monkey. Exp Brain Res 71: 658–662, 1988. [DOI] [PubMed] [Google Scholar]
- Oiwa Y, Eberling JL, Nagy D, Pivirotto P, Emborg ME, Bankiewicz KS. Overlesioned hemiparkinsonian non human primate model: correlation between clinical, neurochemical, and histochemical changes. Front Biosci 8: a155–a166, 2003. [DOI] [PubMed] [Google Scholar]
- Pasquereau B, Turner RS. Primary motor cortex of the parkinsonian monkey: differential effects on the spontaneous activity of pyramidal tract-type neurons. Cereb Cortex 21: 1362–1378, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmutter JS, Mink JW. Deep brain stimulation. Annu Rev Neurosci 29: 229–257, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz A, Vaadia E, Bergman H. Firing patterns, and correlations of spontaneous discharge of pallidal neurons in the normal and the tremulous 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine vervet model of parkinsonism. J Neurosci 20: 8559–8571, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivlin-Etzion M, Marmor O, Saban G, Rosin B, Haber SN, Vaadia E, Prut Y, Bergman H. Low-pass filter properties of basal ganglia cortical muscle loops in the normal, and MPTP primate model of parkinsonism. J Neurosci 28: 633–649, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivlin-Etzion M, Ritov Y, Heimer G, Bergman H, Bar-Gad I. Local shuffling of spike trains boosts the accuracy of spike train spectral analysis. J Neurophysiol 95: 3245–3256, 2006. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Oroz MC, Obeso JA, Lang AE, Houeto JL, Pollak P, Rehncrona S, Kulisevsky J, Albanese A, Volkmann J, Hariz MI, Quinn NP, Speelman JD, Guridi J, Zamarbide I, Gironell A, Molet J, Pascual-Sedano B, Pidoux B, Bonnet AM, Agid Y, Xie J, Benabid AL, Lozano AM, Saint-Cyr J, Romito L, Contarino MF, Scerrati M, Fraix V, Van Blercom N. Bilateral deep brain stimulation in Parkinson's disease: a multicentre study with 4 years follow-up. Brain 128: 2240–2249, 2005. [DOI] [PubMed] [Google Scholar]
- Schneider JS, Gonczi H, Decamp E. Development of levodopa-induced dyskinesias in parkinsonian monkeys may depend upon rate of symptom onset and/or duration of symptoms. Brain Res 990: 38–44, 2003. [DOI] [PubMed] [Google Scholar]
- Shi LH, Luo F, Woodward DJ, Chang JY. Basal ganglia neural responses during behaviorally effective deep brain stimulation of the subthalamic nucleus in rats performing a treadmill locomotion test. Synapse 59: 445–457, 2006. [DOI] [PubMed] [Google Scholar]
- Smith Y, Wichmann T. The cortico-pallidal projection: an additional route for cortical regulation of the basal ganglia circuitry. Mov Disord 2014. December 5 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares J, Kliem MA, Betarbet R, Greenamyre JT, Yamamoto B, Wichmann T. Role of external pallidal segment in primate parkinsonism: comparison of the effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism, and lesions of the external pallidal segment. J Neurosci 24: 6417–6426, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr PA, Vitek JL, Bakay RA. Ablative surgery, and deep brain stimulation for Parkinson's disease. Neurosurgery 43: 989–1013, 1998. [DOI] [PubMed] [Google Scholar]
- Szabo J, Cowan WM. A stereotaxic atlas of the brain of the cynomolgus monkey (Macaca fascicularis). J Comp Neurol 222: 265–300, 1984. [DOI] [PubMed] [Google Scholar]
- Temperli P, Ghika J, Villemure JG, Burkhard PR, Bogousslavsky J, Vingerhoets FJ. How do parkinsonian signs return after discontinuation of subthalamic DBS? Neurology 60: 78–81, 2003. [DOI] [PubMed] [Google Scholar]
- Tisch S, Rothwell JC, Zrinzo L, Bhatia KP, Hariz M, Limousin P. Cortical evoked potentials from pallidal stimulation in patients with primary generalized dystonia. Mov Disord 23: 265–273, 2008. [DOI] [PubMed] [Google Scholar]
- Turner RS, DeLong MR. Corticostriatal activity in primary motor cortex of the macaque. J Neurosci 20: 7096–7108, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valalik I, Emri M, Lengyel Z, Mikecz P, Tron L, Csokay A, Marian T. Pallidal deep brain stimulation and L-dopa effect on PET motor activation in advanced Parkinson's disease. J Neuroimaging 19: 253–258, 2009. [DOI] [PubMed] [Google Scholar]
- Vitek JL. Mechanisms of deep brain stimulation: excitation or inhibition. Mov Disord 17, Suppl 3: S69–S72, 2002. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Bergman H, Starr PA, Subramanian T, Watts RL, DeLong MR. Comparison of MPTP induced changes in spontaneous neuronal discharge in the internal pallidal segment, and in the substantia nigra pars reticulata in primates. Exp Brain Res 125: 397–409, 1999. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Soares J. Neuronal firing before, and after burst discharges in the monkey basal ganglia is predictably patterned in the normal state and altered in parkinsonism. J Neurophysiol 95: 2120–2133, 2006. [DOI] [PubMed] [Google Scholar]
- Wu YR, Levy R, Ashby P, Tasker RR, Dostrovsky JO. Does stimulation of the GPi control dyskinesia by activating inhibitory axons? Mov Disord 16: 208–216, 2001. [DOI] [PubMed] [Google Scholar]