

Abstract
The neuromodulator acetylcholine (ACh) shapes neocortical function during sensory perception, motor control, arousal, attention, learning, and memory. Here we investigate the mechanisms by which ACh affects neocortical pyramidal neurons in adult mice. Stimulation of cholinergic axons activated muscarinic and nicotinic ACh receptors on pyramidal neurons in all cortical layers and in multiple cortical areas. Nicotinic receptor activation evoked short-latency, depolarizing postsynaptic potentials (PSPs) in many pyramidal neurons. Nicotinic receptor-mediated PSPs promoted spiking of pyramidal neurons. The duration of the increase in spiking was membrane potential dependent, with nicotinic receptor activation triggering persistent spiking lasting many seconds in neurons close to threshold. Persistent spiking was blocked by intracellular BAPTA, indicating that nicotinic ACh receptor activation evoked persistent spiking via a long-lasting calcium-activated depolarizing current. We compared nicotinic PSPs in primary motor cortex (M1), prefrontal cortex (PFC), and visual cortex. The laminar pattern of nicotinic excitation was not uniform but was broadly similar across areas, with stronger modulation in deep than superficial layers. Superimposed on this broad pattern were local differences, with nicotinic PSPs being particularly large and common in layer 5 of M1 but not layer 5 of PFC or primary visual cortex (V1). Hence, in addition to modulating the excitability of pyramidal neurons in all layers via muscarinic receptors, synaptically released ACh preferentially increases the activity of deep-layer neocortical pyramidal neurons via nicotinic receptors, thereby adding laminar selectivity to the widespread enhancement of excitability mediated by muscarinic ACh receptors.
Keywords: acetylcholine, nicotinic acetylcholine receptor, neocortical pyramidal neurons
the neuromodulator acetylcholine (ACh) shapes neocortical function during sensory perception (Disney et al. 2007; Metherate 2004), motor control (Berg et al. 2005), arousal (Jones 2008; Steriade 2004), attention (Herrero 2008; Parikh and Sarter 2008), learning (Kilgard 2003; Ramanathan et al. 2009), and memory (Winkler et al. 1995). A decline in neocortical ACh has been tied to conditions such as depression (Dilsaver 1986), schizophrenia (Raedler and Tandon 2006), Alzheimer's disease (Whitehouse et al. 1982), and Parkinson's disease (Whitehouse et al. 1983).
Most cholinergic axons in neocortex arise from nucleus basalis and other basal forebrain nuclei such as substantia innominata (Wainer and Mesulam 1990). The cholinergic projection from basal forebrain plays a central role in shaping neocortical components of arousal, attention, learning, memory, sensory perception, and motor control. For example, stimulation of vibrissal primary motor cortex (M1) evokes whisker movements that are enhanced by activation of basal forebrain (Berg et al. 2005) and basal forebrain lesions impair motor control (Gharbawie and Whishaw 2003) and motor map rearrangement during motor learning (Conner et al. 2003).
ACh affects neocortical networks, in part by modulating the activity of pyramidal neurons. Pyramidal neurons express nicotinic and muscarinic ACh receptors (nAChRs and mAChRs) on their plasma membranes (Mrzljak et al. 1993; van der Zee et al. 1992), but ACh is thought to act on pyramidal neurons primarily via mAChRs. Activation of mAChRs evokes an initial hyperpolarization and subsequent slow depolarization of many cortical pyramidal neurons. The hyperpolarization results from activation of an SK-type potassium current, whereas the slow depolarization has been linked to a number of currents, including M-, AHP-, and inward rectifier-type potassium currents and a nonspecific cation current (Carr and Surmeier 2007; Delmas and Brown 2005; Gulledge and Stuart 2005; Haj-Dahmane and Andrade 1996; Krnjevic 1971; McCormick and Prince 1985, 1986; McCormick and Williamson 1989; Zhang and Séguéla 2010). There are reports of ACh activating nAChRs on pyramidal neurons (Chu et al. 2000; Guillem et al. 2011; Kassam et al. 2008; Poorthuis et al. 2013a; Roerig et al. 1997; Zolles et al. 2009; but see also Gil et al. 1997; Porter et al. 1999; Vidal and Changeux 1993). Few authors have studied nicotinic postsynaptic currents in neocortical pyramidal neurons (Chu et al. 2000; Roerig et al. 1997). Hence the functional roles of nAChRs on pyramidal neurons remain obscure.
Here we studied how ACh affects pyramidal neurons, focusing on the role of nAChRs. To drive synaptic release of ACh, we expressed the light-activated protein channelrhodopsin-2 (ChR2) in cholinergic neurons in the basal forebrain, allowing us to selectively stimulate cholinergic axons in neocortex (Kalmbach et al. 2012). In contrast to many previous reports, we find that ACh excites pyramidal neurons via both mAChRs and nAChRs. Activation of nAChRs occurs with short latency, consistent with nAChRs being located at synapses between cholinergic axons and pyramidal neurons, and can evoke persistent spiking via a calcium-activated conductance. Direct nAChR-mediated effects occurred in pyramidal neurons in several neocortical areas and in all neocortical layers, indicating that direct excitation of pyramidal neurons via nAChRs can occur across neocortical layers and areas. However, laminar and regional differences in both the incidence and amplitude of the nAChR-mediated depolarization suggest regional differences in the modulation of neocortical networks by nAChRs.
METHODS
All experiments and procedures were approved by the Northwestern University Institutional Animal Care and Use Committee (IACUC).
Two approaches were employed to selectively express ChR2 in cholinergic neurons: 1) stereotaxic injection of a floxed viral vector into the basal forebrain of ChAT-Cre mice and 2) crossing ChAT-Cre and floxed ChR2 mouse lines.
For experiments using virally delivered ChR2, we used Tg(ChAT-Cre)60Gsat mice (GENSAT), which express Cre-recombinase on a choline acetyltransferase (ChAT) promoter, resulting in Cre expression in cholinergic neurons throughout the brain. Into this mouse we injected adeno-associated virus with a double-floxed inverse open reading frame [EF1a-DIO-hChR2(H134R)eYFP, Virus Vector Core, University of North Carolina], which drives expression of ChR2-yellow fluorescent protein (ChR2-YFP) in infected neurons containing Cre. Four hundred nanoliters of virus was injected into the basal forebrain at postnatal day 21 with the use of stereotaxic coordinates (0.2 mm A-P, 1.7 mm M-L, 4.5 mm D-V). Three weeks after injection, ChR2 is expressed almost exclusively in cholinergic neurons and their axons in neocortex, with no adverse effects (Porter et al. 1999).
In some experiments, selective expression of ChR2 in cholinergic neurons was achieved without the use of viral vectors by crossing ChAT-Cre [B6;129S6-Chattm1(cre)Lowl/J, Jax 006410] and floxed ChR2 [129S-Gt(ROSA)26Sortm32(CAG-COP4*H134R/EYFP)Hze/J, Jax 012569, Ai32 (Madisen et al. 2012)] mouse lines to produce ChAT-ChR2(Ai32) mice. Cre+ mice were crossed with ChR2+/+ mice to yield Cre+ ChR2+/− offspring. These offspring were crossed to generate Cre+ ChR2+/+ mice, which were used for experiments. Results from ChAT-ChR2(Ai32) mice were similar to those from viral infections, and results from these two approaches were pooled, unless otherwise noted.
Somatic and axonal labeling with ChR2-YFP was examined in fixed sections. Tissue was fixed by transcardial perfusion with 4% paraformaldehyde in phosphate buffer, and 100- to 200-μm-thick coronal sections were cut with a vibrating microtome. In some sections, the YFP signal was enhanced with an anti-green fluorescent protein (GFP) primary (ab13970, 1:10,000, Abcam) and a fluorescent secondary antibody (613111, 1:750, Invitrogen). Images were acquired by widefield or two-photon microscopy. Widefield images were acquired with a GFP filter set and a Hamamatsu Orca-285 camera. Two-photon images were acquired with 880-nm illumination from a Coherent Chameleon Ultra II Ti:sapphire laser and a 505- to 545-nm emission filter.
Whole cell recordings were obtained from pyramidal neurons in 300-μm-thick acute parasagittal slices of M1 and coronal slices of prefrontal cortex (PFC) and primary visual cortex (V1) from postnatal day 33–61 mice of either sex. In virally infected mice, recordings were obtained ∼3 wk after viral injection. Slices were prepared in ice-cold artificial cerebrospinal fluid (ACSF; in mM: 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 20 NaHCO3, 5 HEPES, 25 glucose, 2 CaCl2, and 1 MgCl2, pH 7.3, oxygenated with 95% O2-5% CO2). Whole cell recording pipettes were 4–8 MΩ when filled with intracellular solution [in mM: 135 K gluconate, 4 KCl, 10 HEPES, 10 Na2-phosphocreatine, 4 Mg-ATP, 0.3 Na2-GTP, with 0.2% (wt/vol) biocytin and 10 μM Alexa Fluor 594, pH 7.3].
Recordings were obtained at 35–37°C with a feedback circuit and temperature controller (TC-324B, Warner Instruments, Hamden, CT). In experiments using calcium-free ACSF, 2 mM CaCl2 was replaced with 4 mM MgCl2 to give a final MgCl2 concentration of 5 mM. ChR2 was activated by widefield illumination through a microscope objective (Olympus, ×20/0.95 NA or ×40/0.8 NA) using a blue light-emitting diode (LED; Thorlabs LEDC5 and LEDD1 or DC2100 driver). The maximum steady-state intensity was 20 mW/mm2. In some experiments (see Fig. 8, A and B), illumination was restricted by closure or partial closure of the fluorescence field stop. The illumination area was measured off-line by bleaching a thin, immobilized film of fluorescence on a microscope slide. Closure of the field stop had no effect on illumination intensity per unit area. In restricted-illumination experiments, the soma was positioned in the center of the field of illumination unless noted otherwise.
Fig. 8.
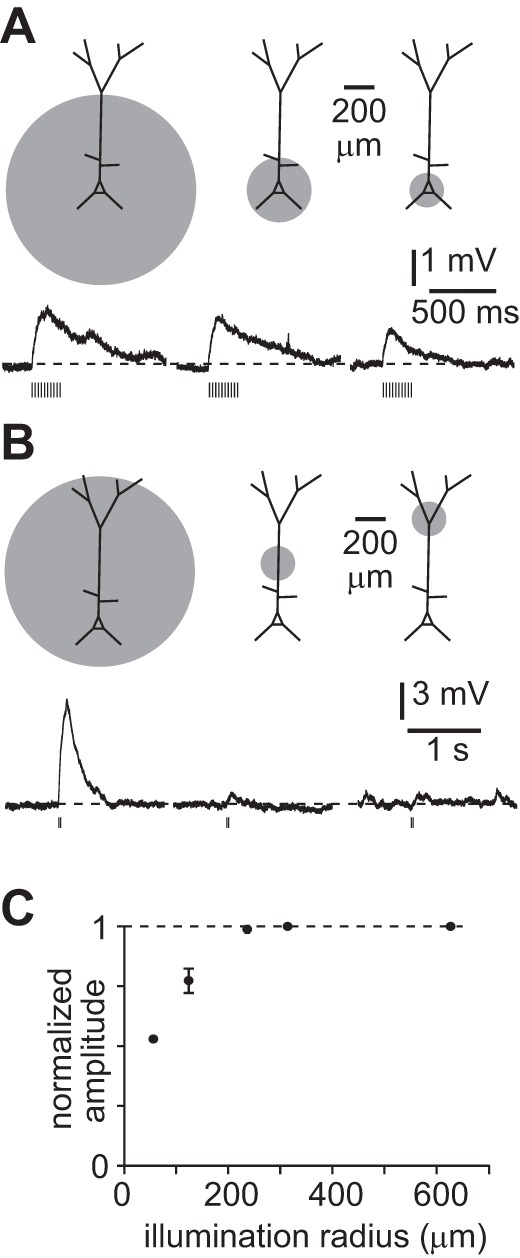
Fast depolarization evoked by activation of perisomatic nAChRs. A: examples of nAChR PSPs during localized illumination centered on the soma, with schematic illustrating approximate illumination areas; 10 × 5-ms illumination at 20 Hz. ChAT-ChR2(Ai32) mouse. Measured at rest. B: examples of nAChR PSPs during localized illumination of different regions of the dendritic arbor, with schematic illustrating approximate illumination areas; 2 × 5-ms illumination at 20 Hz. Virus-injected mouse. C: summary plot showing peak amplitude of depolarization with illumination areas of different radii, centered on the soma, in 7 recordings. Points represent mean ± SE peak amplitude normalized to average amplitude with full field of illumination. Illumination radii: 55 μm, 7 neurons; 110–118 μm, 7 neurons; 235 μm, 2 neurons; 312.5 μm, 7 neurons; 625 μm, 2 neurons.
The amplitudes of voltage responses were calculated by subtracting the average baseline membrane potential for ≥1 s before illumination from the peak of the response. Voltage responses that failed to exceed 3 standard deviations of the baseline membrane potential were assigned an amplitude of 0 mV. During brief bursts of nAChR postsynaptic potentials (PSPs), the peak amplitude was calculated by subtracting the preburst membrane potential from the most depolarized potential during the burst.
Local application of ACh was by pressure ejection of 100 μM ACh in ACSF from a glass pipette ∼50 μm from the soma (30 psi; Toohey Spritzer IIe, Toohey, Fairfield, NJ).
Mecamylamine hydrochloride and atropine were obtained from Sigma-Aldrich. Dihydro-β-erythroidine hydrobromide (DHβE), methyllycaconitine citrate (MLA), galantamine hydrobromide, and physostigmine hemisulfate were obtained from Tocris Bioscience. NBQX disodium salt, (R)-CPP, gabazine (SR95531), CGP 52432, tetrodotoxin citrate, and (R,S)-MCPG were obtained from Ascent Scientific. In some experiments, we used (2R)-amino-5-phosphonopentanoic acid (AP5) to block NMDA receptors, instead of CPP. AP5 was obtained from Tocris.
Statistical analyses were performed with the Graphpad QuickCalcs online tool (http://www.graphpad.com/quickcalcs/) or in Graphpad Instat 3.06 (GraphPad Software, La Jolla, CA). Continuous data (such as pharmacology of the nAChR PSP) were analyzed with a two-tailed t-test, Kruskal-Wallis test, or Mann-Whitney test and categorical data with Fisher's exact test.
The kinetics and latency of nAChR PSPs were measured by fitting a sum of two exponentials to the mean of 10 trials. In each trial the PSP was evoked with a single 2-ms or (occasionally) 5-ms blue light illumination. The signal-to-noise ratio of our recordings was not sufficient to permit accurate measurement of the timing of the initial rise of the membrane potential of a nAChR PSP, which is smaller and slower than a glutamatergic PSP. To accurately estimate the point of initial rise we therefore measured the time from the start of illumination to 5% of the peak amplitude of the PSP. We would expect this method of latency measurement to overestimate the latency of each PSP by ∼1 ms, and we therefore corrected the latency of each PSP by back-extrapolating the fit to the resting membrane potential preceding the stimulus.
Pyramidal neurons were identified by their somatic shape and spiking pattern and by their large apical dendrites, which were visible by fluorescence microscopy once filled with indicator. Neurons were routinely filled with indicator and their dendrites examined by fluorescence microscopy. Neurons with truncated primary apical dendrites, lacking apical dendrites, and with spiking patterns more typical of interneurons (narrow spikes, large afterhyperpolarization) were excluded from further analysis.
Pyramidal cells were classified according to the laminar locations of their somata. Laminar borders were based on the Allen Brain Atlas and consistent with laminar variation in opacity of slices, which was visible under brightfield illumination. Distances from the pial surface of the slice were measured perpendicular to the pia. In M1, cortical layers were layer 2/3 150–500 μm, layer 5A 600–750 μm, layer 5B 800-1,200 μm, and layer 6 >1,300 μm. In PFC, cortical layers were layer 2/3 100–300 μm, layer 5 330–550 μm, and layer 6 >550–800 μm (Guillem et al. 2011). In visual cortex, cortical layers were layer 2/3 100–275 μm, layer 5 450–750 μm, and layer 6 >750 μm (Olivas et al. 2012; Petrof et al. 2012).
Pyramidal neurons projecting to defined postsynaptic target tissues were labeled with fluorescent microspheres (RetroBeads, Lumafluor). Beads were injected into dorsolateral striatum (0 mm A-P, −2 mm M-L, 2.5 mm D-V), cervical spinal cord (between approximately −0.5 mm M-L and the midline, approximately 1 mm D-V, at the level of the C2 vertebra), or ventral posteromedial thalamus (two sites: 1.6 mm A-P, 1.2 mm M-L, 3.25 mm D-V and 1.6 mm A-P, 1.7 mm M-L, 3.5 mm D-V). Recordings were obtained 5–11 days after bead injection from M1 contralateral to striatal and spinal injections and ipsilateral to thalamic injections.
RESULTS
To stimulate cholinergic axons entering neocortex, we used the blue light-activated membrane protein ChR2 (Nagel et al. 2003). We expressed ChR2 in cholinergic neurons in the basal forebrain (Fig. 1, A–C), obtaining selectivity for cholinergic neurons with a ChAT-Cre mouse line and floxed virus or by crossing ChAT-Cre and floxed ChR2 mouse lines. We obtained whole cell recordings from layer 5B pyramidal neurons in acute slices of M1 containing ChR2-labeled axons (Fig. 1D). Mean resting potential was −62 ± 1 mV (51 neurons). Widefield illumination of the slice with a blue LED evoked one or more of four voltage responses: a slow depolarization, a hyperpolarization, a medium depolarization, or a fast depolarization (Fig. 1F). All responses were absent in tetrodotoxin (TTX, 500 nM; 17 neurons) and after removal of extracellular calcium (10 neurons), indicating that all four voltage responses were evoked by spikes, leading to vesicular release (Fig. 2).
Fig. 1.

Stimulation of cholinergic axons evokes 4 responses in pyramidal neurons. A: schematic of a coronal slice illustrating the expression of channelrhodopsin-2 (ChR2)-yellow fluorescent protein (YFP) in cholinergic neurons in the basal forebrain and their axonal projections to neocortex. CP, caudate putamen; LV, lateral ventricle. B: image of ChR2-YFP-labeled neurons in nucleus basalis (NB) in a ChAT-ChR2(Ai32) mouse: maximum-intensity projection from a 2-photon z-stack through a fixed section. C: image of ChR2-YFP-labeled axons in primary motor cortex (M1) in a ChAT-ChR2(Ai32) mouse: maximum-intensity projection from a 2-photon z-stack through a fixed section. D and E: pyramidal neuron in layer 5B of M1 3 wk after virus injection. Neuron was filled with Alexa Fluor 594 during whole cell recording (D) and surrounding cholinergic axons expressing ChR2-YFP (E). Both images are maximum-intensity projections from 2-photon z-stacks. F: examples of voltage recordings from 4 layer 5B pyramidal neurons at rest. Each voltage response was evoked by stimulation of cholinergic axons by widefield illumination of the slice with a blue LED (stimulus: 10 × 5 ms at 20 Hz; blue bars). Example of slow depolarization includes a preceding hyperpolarization. Examples of slow depolarization and fast hyperpolarization were obtained from virus-injected mice. Examples of medium depolarization and fast depolarization were obtained from ChAT-ChR2(Ai32) mice.
Fig. 2.
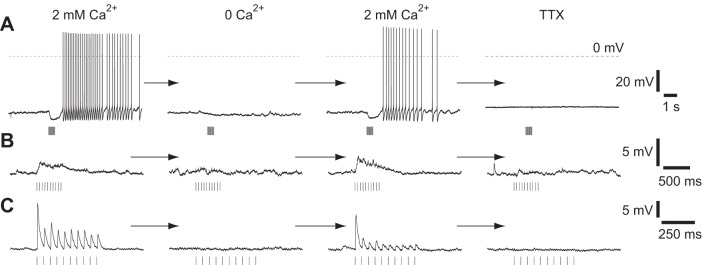
Light-evoked responses are eliminated by tetrodotoxin (TTX) and by removal of extracellular calcium: examples of the slow depolarization and hyperpolarization (A, neuron depolarized by DC current injection), medium depolarization (B, neuron at rest), and fast depolarization (C, neuron at rest) from 3 neurons. All responses were reversibly eliminated by removal of external calcium and by addition of 500 nM TTX. Stimulus: 10 × 5-ms illumination at 20 Hz. All examples were obtained from ChAT-ChR2(Ai32) mice.
We used pharmacology to identify the receptors underlying each of the four voltage responses. The hyperpolarization and slow depolarization were mediated by mAChRs: both hyperpolarization and slow depolarization were eliminated by the mAChR antagonist atropine (1 μM, 140 of 141 neurons; Fig. 3) but not by the nAChR antagonist mecamylamine (100 μM, 11 of 13 neurons; Fig. 3) or by glutamate receptor (GluR) or GABA receptor (GABAR) antagonists (10 μM NBQX and 10 μM CPP; 1 μM gabazine and 3 μM CGP 52432; 14 of 16 neurons).
Fig. 3.
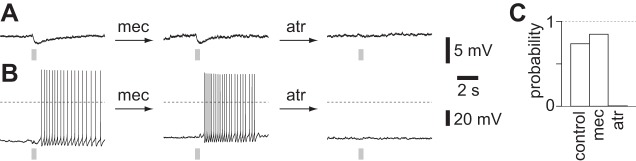
Pharmacology of hyperpolarization and slow depolarization. A: example of sequential addition of 100 μM mecamylamine (mec) and 1 μM atropine (atr) to a neuron displaying a light-evoked hyperpolarization. Stimulus: 10 × 5-ms illumination at 20 Hz. Neuron held close to threshold by DC current injection. Recording from a virus-injected mouse. B: example of sequential addition of 100 μM mecamylamine and 1 μM atropine to a neuron displaying a light-evoked slow depolarization. Stimulus: 10 × 5-ms illumination at 20 Hz. Neuron held close to threshold by DC current injection. Recording from a ChAT-ChR2(Ai32) mouse. C: probability of evoking a muscarinic ACh receptor (mAChR)-mediated potential during constant depolarization by DC current injection. Control, 38 neurons; mec, 100 μM mecamylamine, 13 neurons; atr, 1 μM atropine, 141 neurons.
The medium depolarization was mediated by nAChRs. It was eliminated by mecamylamine (100 μM, 12 of 12 neurons, P < 0.05, paired 2-tailed t-test) and enhanced 73 ± 21% by the nAChR allosteric potentiator and cholinesterase antagonist galantamine (1 μM, 23 neurons, P < 0.05, paired 2-tailed t-test) and 27 ± 15% by the ACh esterase inhibitor physostigmine (0.5 μM, 4 neurons, P < 0.05, paired 2-tailed t-test), and insensitive to antagonists of mAChRs, GluRs, and GABARs (5 of 5, 7 of 7, and 8 of 8 neurons, respectively; Fig. 4). The medium depolarization was unaffected by the α7 nAChR antagonist MLA (10 nM, 12 of 12 neurons) and eliminated by DHβE (10 μM, 16 of 16 neurons, P < 0.05, paired 2-tailed t-test; Fig. 4), which has high affinity for α4β2-containing nAChRs. Hence the medium depolarization is mediated by non-α7 nAChRs, probably α4β2-containing nAChRs.
Fig. 4.

Pharmacology of medium depolarization. Example recordings from 6 neurons. Stimulus: 2–3 × 5-ms illumination at 20 Hz. physo, 0.5 μM physostigmine; atr, 1 μM atropine; mec, 100 μM mecamylamine; gal, 1 μM galantamine; NBQX/CPP, 10 μM NBQX and 10 μM CPP; GBZ/CGP, 1 μM gabazine and 3 μM CGP 52432; MLA, 10 nM methyllycaconitine; DHβE, 10 μM dihydro-β-erythroidine hydrobromide. All examples at resting membrane potential and from virus-injected mice. Summary: means ± SE, normalized to predrug amplitude. TTX, 7 neurons; 0 Ca2+, 7 neurons; NBQX/CPP, 7 neurons; GBZ/CGP, 8 neurons; physostigmine, 4 neurons; atropine, 5 neurons; mecamylamine, 12 neurons; galantamine, 23 neurons; MLA, 12 neurons; DHβE, 16 neurons. *Significant effects (P < 0.05, paired 2-tailed t-test).
The fast depolarization was mediated by ionotropic GluRs: the fast depolarization was not inhibited by nAChR, mAChR, or GABAR antagonists (each P < 0.05, paired 2-tailed t-test) but was eliminated by ionotropic GluR antagonists (Fig. 5A). GluR-mediated fast depolarizations were observed only in a subset of ChAT-ChR2(Ai32) mice and not in virally infected mice. Furthermore, for each ChAT-ChR2(Ai32) mouse we found that GluR-mediated fast depolarizations were obtained in either all or no neurons. We recorded from multiple neurons in slices from 25 mice. In 13 of these 25 mice, we observed GluR-mediated fast depolarizations in every neuron; 12 of 25 mice exhibited no GluR-mediated fast depolarization in any recording. Hence it is likely that the GluR-mediated fast depolarization results from expression of ChR2 in noncholinergic neurons in a subset of ChAT-ChR2(Ai32) mice. To eliminate GluR-mediated responses, we included GluR antagonists in all remaining experiments with ChAT-ChR2(Ai32) mice. The presence of GluR-mediated responses in some ChAT-ChR2(Ai32) mice is the only difference we observed between genetic and viral methods of driving ChR2 expression in cholinergic neurons; in other respects, the two methods of driving ChR2 expression were equivalent [no difference in amplitudes of nAChR PSPs, P > 0.05, Mann-Whitney test; 37 and 22 neurons from virally infected and ChAT-ChR2(Ai32) mice, respectively]. We therefore pooled results from the two techniques.
Fig. 5.
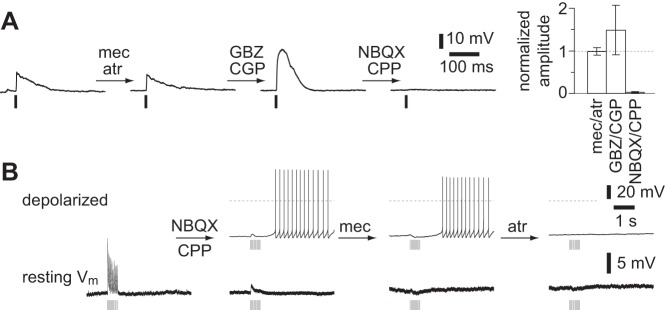
Pharmacology of fast depolarization and pharmacological separation of multiple responses. A: example and summary of the pharmacology of the fast depolarization in a ChAT-ChR2(Ai32) mouse. Example: sequential addition of mec/atr, 100 μM mecamylamine and 1 μM atropine; GBZ/CGP, 1 μM gabazine and 3 μM CGP 52432; NBQX/CPP, 10 μM NBQX and 10 μM CPP; 2-ms illumination. Neuron at rest. Summary: means ± SE, normalized to predrug amplitude. mec/atr, GBZ/CGP, and NBQX/CPP, 5, 3, and 3 neurons, respectively. Antagonists were added sequentially in each recording. B: example of the effect of sequential application of drugs to a neuron from a ChAT-ChR2(Ai32) mouse with a combination of all 4 responses. Bottom: voltage responses at resting membrane potential (Vm). Top: voltage responses during depolarizing somatic current injection.
All four postsynaptic responses could occur individually, but most pyramidal neurons exhibited a combination of responses mediated by two or more receptors (Fig. 5B). Hence synaptically released ACh activates pyramidal neurons via nAChRs and mAChRs, and often via both types of ACh receptor in the same neuron.
Activation of pyramidal neuron mAChRs by synaptically released ACh.
The peak amplitude of the hyperpolarization, evoked by brief illumination (≤10 × 5 ms at 20 Hz), was 1.1 ± 0.5 mV, and the decay time constant was 774 ± 73 ms (10 neurons). The slow depolarization displayed a peak amplitude of 2.0 ± 0.6 mV (6 neurons) and lasted several seconds (Table 1). At resting membrane potentials, the hyperpolarization and slow depolarization were rare, occurring in only 7% (8 of 112) and 19% (21 of 112) of layer 5B pyramidal neurons, respectively.
Table 1.
Amplitude and kinetics of cholinergic responses evoked in pyramidal neurons in layer 5 of primary motor cortex
| Amplitude | Decay Time Constant | |
|---|---|---|
| Medium Depolarization (nAChR) | 4.2 ± 1.1 mV(22 neurons) | 180 ± 23 ms (14 neurons) |
| Hyperpolarization (mAChR) | 1.2 ± 0.5 mV (10 neurons) | 774 ± 73 ms (10 neurons) |
| Slow depolarization (mAChR) | 2.0 ± 0.6 mV (6 neurons) |
Values are means ± SE. Measurements at resting membrane potential. For muscarinic ACh receptor (mAChR)-mediated responses, amplitude and decay time constant were measured after 10 × 5-ms illuminations at 20 Hz. For the nicotinic ACh receptor (nAChR) postsynaptic potential, amplitudes were measured after 10 × 5-ms illuminations at 20 Hz and the decay time constant after a single 2- to 5-ms illumination.
Many previous authors have reported activation of pyramidal neurons via mAChRs upon application of ACh to the soma, by pressure ejection from a nearby pipette (Gulledge et al. 2007; Gulledge and Stuart 2005; McCormick and Prince 1985; McCormick and Williamson 1989), with relatively few authors reporting nAChR-mediated depolarization of pyramidal neurons (Chu et al. 2000; Guillem et al. 2011; Kassam et al. 2008; Poorthuis et al. 2013a; Roerig et al. 1997; Zolles et al. 2009). Similar to many previous studies, we found that pressure ejection of ACh onto the soma evoked a mAChR-mediated hyperpolarization and slow depolarization (5 of 6 and 6 of 6 neurons, respectively; Fig. 6A) but no nAChR-mediated depolarization.
Fig. 6.
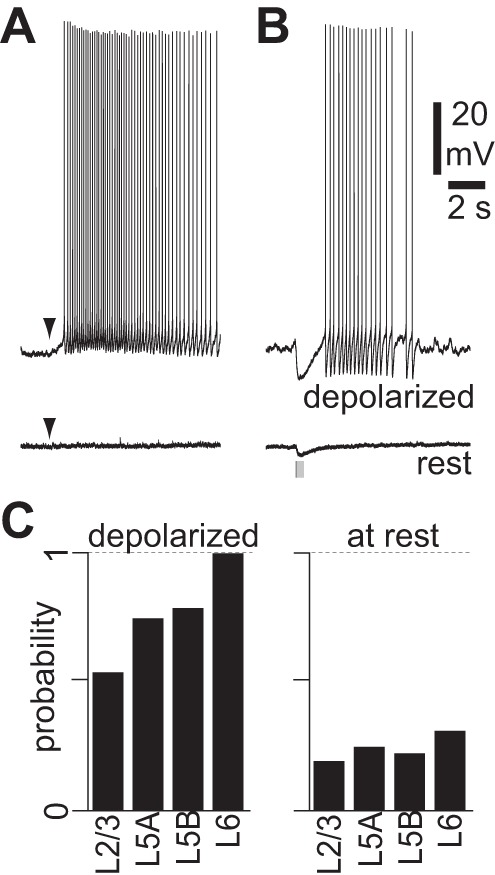
mAChR-mediated hyperpolarization and slow depolarization. A: example of pressure application of ACh (100 μM, 10 ms, arrowheads) at rest and during constant depolarization in a neuron displaying a mAChR-mediated slow depolarization. NBQX, CPP, gabazine, and CGP 52432 were present to prevent network effects. B: example of blue light stimulus at rest and during constant depolarization in a neuron from a virus-injected mouse displaying a mAChR-mediated hyperpolarization and slow depolarization. Stimulus: 10 × 5-ms illumination at 20 Hz. NBQX, CPP, gabazine, and CGP 52432 present throughout. C: incidence of light-evoked mAChR-mediated responses in pyramidal neurons in different cortical layers at rest and during depolarization by somatic current injection. Probability was calculated as the number of neurons in which we observed mAChR responses divided by the total number of recordings. Laminar locations determined by distance from pia (see methods). Depolarized: layer(L)2/3, 8 neurons; L5A, 8 neurons; L5B, 48 neurons; L6, 7 neurons. At rest: L2/3, 22 neurons; L5A, 8 neurons; L5B, 84 neurons; L6, 32 neurons.
One possible interpretation of our results might be that synaptically released ACh activates primarily nAChRs and usually fails to activate mAChRs, whereas pressure ejection onto the soma activates a different population of receptors, primarily mAChRs. mAChR activation modulates primarily potassium conductances (McCormick 1992), and the reversal potential for potassium is approximately −90 mV. mAChR activation may therefore exert little effect on the membrane potential at rest: both mAChR-mediated hyperpolarization and slow depolarization are larger when the neuron is depolarized (Gulledge and Stuart 2005; McCormick and Prince 1986). To maximize the effects of mAChR activation on the membrane potential and, therefore, the probability that we would observe activation of mAChRs, we depolarized neurons by somatic current injection (Fig. 6B). Brief stimuli (≤10 × 5 ms illumination at 20 Hz) evoked mAChR-mediated voltage responses in 79% of depolarized layer 5B pyramidal neurons (Fig. 6C), indicating that synaptically released ACh activates mAChRs in the majority of layer 5B pyramidal neurons but that the resulting hyperpolarization or depolarization from rest is often too small to be observed.
Previous authors have also reported that ACh, applied by pressure application, more readily evokes postsynaptic mAChR-mediated responses in pyramidal neurons in deep than superficial layers of the neocortex (Gulledge et al. 2007; McCormick and Prince 1986; McCormick and Williamson 1989). Similarly, we found that synaptically released ACh more readily excites pyramidal neurons via mAChRs in deep than superficial layers of M1, with the proportion of pyramidal neurons that exhibited a mAChR-mediated slow depolarization or hyperpolarization ranging from 53% in layer 2/3 to 91% in layer 6 pyramidal neurons (Fig. 6C).
We conclude that ACh released by a brief burst of cholinergic activity activates mAChRs on the majority of pyramidal neurons throughout M1. Compared with pressure application of ACh, activation of cholinergic synapses with brief bursts of stimuli provides relatively weak activation of mAChRs that often fails to affect the somatic membrane potential at rest.
ACh release evokes a nicotinic PSP.
Brief illumination (≤10 × 5 ms at 20 Hz) in mAChR, GluR, and GABAR antagonists evoked a nAChR-mediated medium depolarization in almost all (82 of 90) layer 5B pyramidal neurons at rest. We stimulated cholinergic axons with brief bursts of stimuli at 20 Hz, a stimulus designed to mimic the spiking of cholinergic basal forebrain neurons during paradoxical sleep and awake states, when cholinergic basal forebrain neurons spike in bursts, each of ∼4–6 spikes at ∼25 Hz (Lee et al. 2005; Manns et al. 2000). During such bursts, summation occurred at stimulus frequencies greater than ∼5 Hz. In our experiments summation may result, in part, from accumulation of calcium entering through ChR2 channels in the presynaptic terminal (Zhang and Oertner 2007) and may therefore be an artifact of the optogenetic stimulus. Nonetheless, summation permitted us to evoke a substantial depolarization via nAChRs: a burst of stimuli at 20 Hz frequently evoked a peak depolarization of 5–10 mV (Fig. 7, A and B).
Fig. 7.
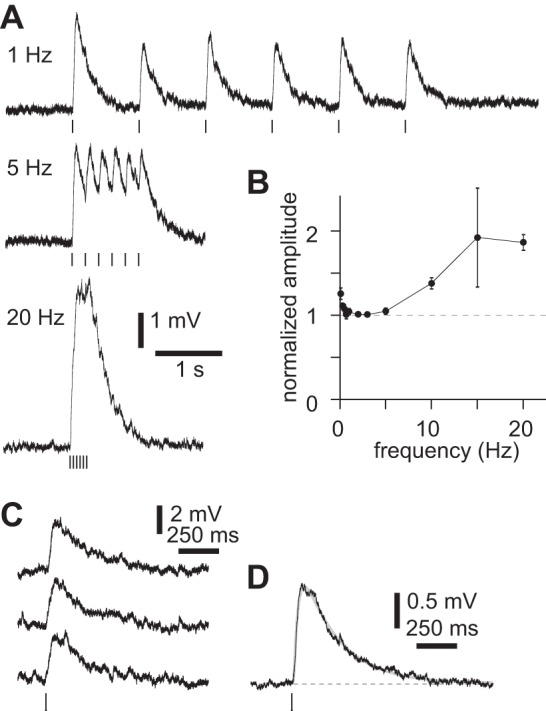
Latency and kinetics of nicotinic ACh receptor (nAChR)-mediated medium depolarization. A: example of summation of nAChR postsynaptic potentials (PSPs) in a neuron from a virus-injected mouse. Six 2-ms illuminations at 1, 5, and 20 Hz in the presence of atropine, NBQX, CPP, gabazine, and CGP 52432. Measured at rest. B: summary plot showing peak amplitudes after six 2-ms illuminations at up to 20 Hz. In each trial, peak amplitude was normalized to that of the first PSP. Points represent means ± SE. 0.033 Hz, 11 neurons; 0.1 Hz, 13 neurons; 0.5 Hz, 14 neurons; 0.7 Hz, 14 neurons; 1 Hz, 14 neurons; 2 Hz, 14 neurons; 3 Hz, 2 neurons; 5 Hz, 7 neurons; 10 Hz, 11 neurons; 15 Hz, 2 neurons; 20 Hz, 14 neurons. C: nAChR PSPs in a neuron from a virus-infected mouse, evoked by 2-ms illumination in atropine, NBQX, CPP, gabazine, and CGP 52432. Measured at rest. D: mean response to 2-ms illumination, with fit (sum of 2 exponentials; gray). Rise and decay time constants 27.5 and 231 ms. Virus-infected mouse.
We measured the latency and kinetics of the nAChR-mediated depolarization evoked by a single 2-ms illumination in 1 μM atropine to inhibit mAChRs and GluR and GABAR antagonists to eliminate any indirect effects. In 15 of 15 neurons, 2-ms illumination evoked reproducible depolarizations (Fig. 7C), with a mean amplitude of 1.4 ± 0.2 mV (11 neurons). To the mean response we fit a sum of two exponentials (Fig. 7D) with mean rise and decay time constants of 28 ± 4 ms and 180 ± 23 ms (14 neurons; Table 1). These relatively slow kinetics are comparable to those of PSPs mediated by α4β2-containing nAChRs in interneurons (Bell et al. 2011) but slower than PSPs mediated by α7 nAChRs (Fedorov et al. 2012; Frazier et al. 1998). These kinetics suggest that the medium depolarization is a PSP mediated by non-α7 nAChRs, consistent with the pharmacology of the medium depolarization presented above.
The latency of the nAChR PSP, measured from the start of illumination, was 5.9 ± 0.8 ms (12 neurons). Compared with electrical stimulation, ChR2 drives relatively slow depolarization of the neuronal membrane, typically with rise and decay time constants of at least several milliseconds (Lin et al. 2009; Yizhar et al. 2011), which may be further elongated by the time constant of the membrane. The latency of the fast depolarization was 5.3 ± 0.5 ms (3 neurons). Hence the latency of the nicotinic PSP was only ∼0.5 ms longer than that of a monosynaptic glutamatergic PSP, consistent with the medium depolarization being a monosynaptic PSP generated at synapses between cholinergic axons and pyramidal neurons.
nAChRs appear to be distributed throughout the dendritic trees of cortical pyramidal neurons (Nakayama et al. 1995; van der Zee et al. 1992), but the locations of cholinergic synapses are unknown. To determine whether nAChR PSPs were evoked primarily by cholinergic synapses in the proximal or distal dendrites of layer 5B pyramidal neurons, we measured nAChR PSPs during restricted illumination of the slice (Fig. 8, A and B). Restricting illumination to a radius of less than ∼300 μm around the soma was necessary to reduce the amplitude of the nAChR PSP, and the amplitude was reduced by 50% when the radius of illumination was ∼50 μm (Fig. 8C; 7 neurons). Illumination of the tuft dendrites failed to evoke a nAChR PSP at the soma (Fig. 8B; 3 neurons). Hence the nAChRs that contribute to the somatic depolarization in our experiments are likely to be within 300 μm of the soma and many are probably located in the proximal 50 μm of the apical and basal arbor.
nAChR activation can evoke persistent spiking.
We next addressed the functional consequences of nAChR activation. nAChR PSPs increased the spike rate of layer 5B pyramidal neurons in M1, depolarized beyond threshold by somatic current injection (Fig. 9A). The increase was independent of initial spike rate (Fig. 9B), with 2- to 5-ms illumination increasing spike rate by 2.2 ± 0.1 Hz (6 neurons) and a brief burst (10 × 5 ms at 20 Hz) increasing spike rate by 3.1 ± 0.2 Hz (4 neurons). Hence during spiking nAChR activation causes a linear, additive change in spike rate with no change in the slope of the input-output relationship. The elevated spike rate was maintained during repetitive stimulation and declined after cessation of the cholinergic stimulus with a decay time constant of 185 ± 21 ms (Fig. 9C; 2- or 5-ms illumination,; 6 neurons), which matches the decay of the underlying nAChR PSP.
Fig. 9.
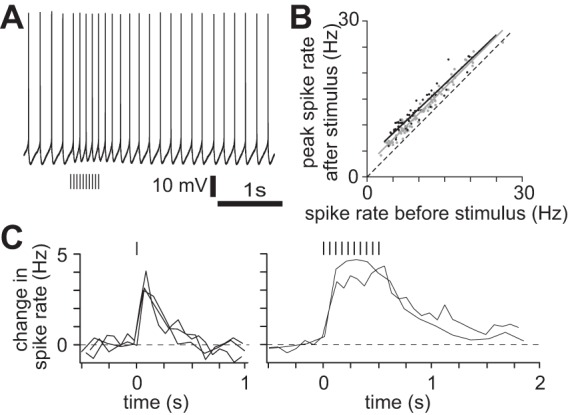
nAChR activation enhances spiking of layer 5 pyramidal neurons. A: example of effect of cholinergic stimulation on spike rate during ongoing spiking; 400-pA constant current injection at soma. 10 × 5-ms illumination at 20 Hz. NBQX, CPP, gabazine, CGP 52432, and atropine present throughout. Virus-injected mouse. B: summary of increase in spike rate during ongoing spiking. Each point represents 1 trial. Lines, best linear fit. Grey, single 2- to 5-ms illumination, increase in spike rate 2.2 ± 0.1 Hz, 6 neurons; black: 10 × 5-ms illumination at 20 Hz, increase in spike rate 3.1 ± 0.2 Hz, 4 neurons. C: kinetics of change in spike rate with one 2-ms illumination and burst of 10 × 5 ms at 20 Hz. Each line represents a single trial.
With the membrane depolarized from rest but subthreshold, cholinergic stimulation evoked persistent spiking. We evoked nAChR PSPs during long step current injections of increasing amplitude until the nAChR PSP evoked one or more spikes. A nAChR PSP reduced rheobase by 11.6 ± 3.7 pA (from 344 ± 26 pA to 337 ± 26 pA, 18 neurons, 5-ms illumination in the presence of atropine). However, under these conditions, a nAChR PSP typically evoked multiple spikes. The spike rate during the first second after stimulus onset was 3.0 ± 0.2 Hz (6 neurons) for a single 2- to 5-ms illumination and 7.1 ± 1.2 Hz for a brief burst (10 × 5 ms at 20 Hz, 7 neurons). Spike rate declined slowly after the last nAChR PSP (Fig. 10), but spiking typically continued until the holding current was removed, which was up to 4 s after the initial spike (Fig. 10).
Fig. 10.
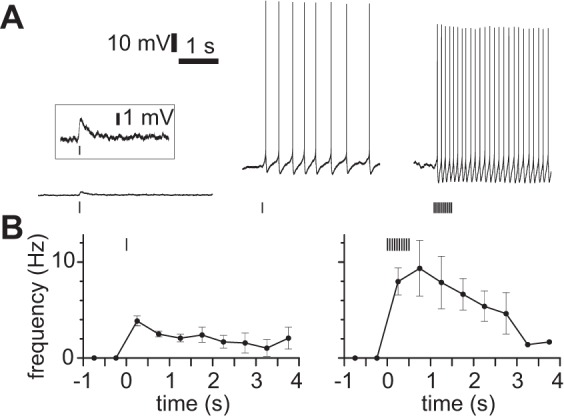
Persistent spiking after nAChR activation. A: examples from virus-injected mice of persistent spiking in atropine, NBQX, CPP, gabazine, and CGP 52432. Left: single 5-ms illumination at resting membrane potential. Inset: expanded view of same response, same timescale. Center: persistent spiking after single 2-ms illumination, 350-pA constant current injection at soma. Right: persistent spiking after 10 × 5-ms illumination at 20 Hz, 400-pA constant current injection at soma. B: mean spike rate during persistent spiking. Stimulus starting at 0 ms. Left: single 2- to 5-ms illumination, 7 neurons. Right: 10 × 5 ms at 20 Hz, 7 neurons.
We defined persistent spiking as spiking that continued for at least 500 ms after the end of the cholinergic stimulus. By this definition, persistent spiking occurred in every neuron (13 of 13 neurons), but in 6 of 13 neurons persistent spiking occurred on some but not all trials. In trials in which persistent spiking failed to occur, the nAChR PSP evoked only a single spike (mean 1.0 ± 0, 14 trials from 4 neurons, single 2- to 5-ms illumination), whereas in trials in which persistent spiking occurred the nAChR PSP evoked 7.5 ± 1.6 spikes (10 trials in the same 4 neurons with the same stimulus). Comparing trials with and without persistent spiking, the resting membrane potential at the start of the trial (−62.3 ± 1.9 and −63.2 ± 1.5 mV, respectively), the current injected to depolarize the neuron (209 ± 20 and 268 ± 23 pA, respectively), the membrane potential immediately before the nAChR PSP (−48.0 ± 2.0 and −48.5 ± 1.8 mV, respectively), and the threshold of the first spike (−35.2 ± 1.9 and −39.1 ± 1.9 mV, respectively) were similar (18 and 14 trials, respectively; each P > 0.05, paired 2-tailed t-test). These measurements indicate that trial-to-trial variability in perisomatic membrane potential, input resistance, and spike threshold do not account for the variability in persistent spiking, although they do not exclude a role for such changes in the distal dendrite, as a result of ongoing synaptic activity, for example.
Persistent spiking required activation of nAChRs on pyramidal neurons but not GluRs, GABARs, or mAChRs (Fig. 11). Persistent spiking occurred in the presence of 10 μM NBQX, 10 μM CPP, 1 μM gabazine, 3 μM CGP, and 1–10 μM atropine (13 of 13 neurons). Subsequent addition of 100 μM mecamylamine (in the continued presence of GluR and GABAR antagonists and of atropine) blocked persistent spiking (3 of 3 neurons; Fig. 11A), but 100 μM MCPG did not (3 of 3 neurons).
Fig. 11.
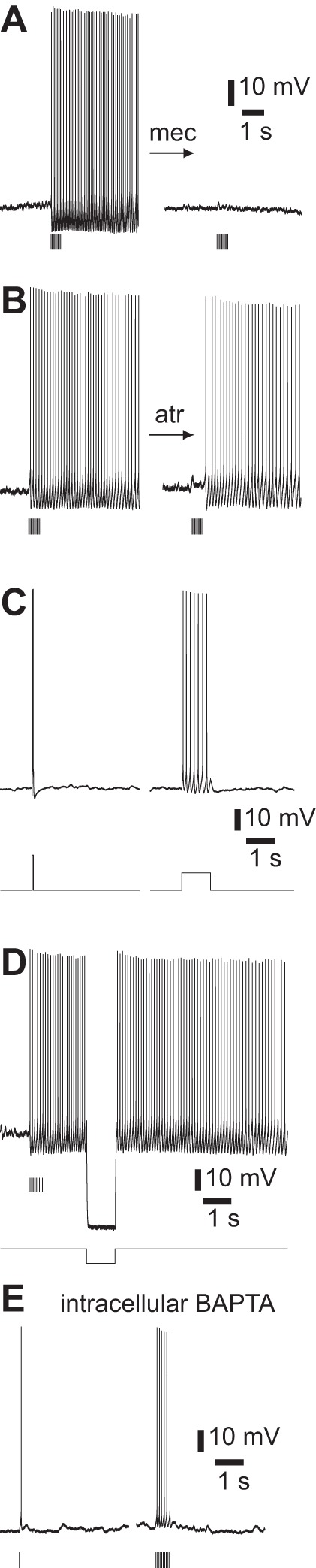
Pharmacology of persistent spiking. A: inhibition of persistent spiking by 100 μM mecamylamine; 10 × 5-ms illumination at 20 Hz. Atropine, NBQX, CPP, gabazine, and CGP 52432 present throughout. Measured near threshold. Virus-injected mouse. B: failure of 1 μM atropine to inhibit persistent spiking; 10 × 5-ms illumination at 20 Hz. NBQX, CPP, gabazine, and CGP 52432 present throughout. ChAT-ChR2(Ai32) mouse. C: examples of failure to evoke persistent spiking with somatic current injection; 400-pA constant current injection to bring cell close to threshold. Left: baseline membrane potential −49 mV, additional 100 pA for 50 ms, resulting in 2 spikes. Right: baseline membrane potential −47 mV, additional 50 pA for 1 s. NBQX, CPP, gabazine, CGP 52432, and atropine present throughout. Virus-injected mouse. D: hyperpolarization failed to end persistent spiking; 10 × 5-ms illumination at 20 Hz, −300-pA hyperpolarizing current injection. Atropine, NBQX, CPP, gabazine, and CGP 52432 present throughout. Virus-injected mouse. E: inhibition of persistent spiking by 10 mM intracellular BAPTA in a ChAT-ChR2(Ai32) mouse. Left: 5-ms illumination. Right: 10 × 5-ms illumination at 20 Hz. Atropine, NBQX, CPP, gabazine, and CGP 52432 present throughout.
nAChR activation provides more than just the initial depolarization required to initiate persistent spiking, since in the absence of cholinergic stimulation brief depolarization failed to evoke persistent spiking (Fig. 11C). Furthermore, during persistent spiking evoked by cholinergic stimulation, brief hyperpolarization of the membrane inhibited persistent spiking, only for spiking to resume after the hyperpolarizing pulse (Fig. 11D). Presumably, an additional depolarizing conductance is activated (or hyperpolarizing conductance deactivated) by nAChR activation or by another receptor coactivated with nAChRs and this additional conductance remains active for several seconds, long after the decay of the nAChR PSP. The spike waveform changed little during persistent spiking (Fig. 12), suggesting that this additional current is unlikely to arise from one of the sodium or potassium conductances that shape the spike waveform.
Fig. 12.
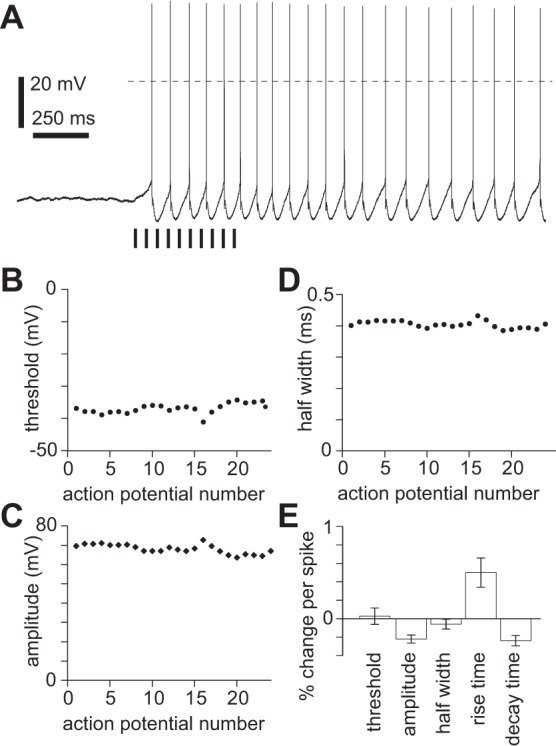
Persistent spiking and intrinsic conductances. A: example of persistent spiking in atropine, NBQX, CPP, gabazine, and CGP 52432; 400-pA constant current injection. Baseline membrane potential −47 mV; 10 × 5-ms illumination at 20 Hz. Dashed horizontal line denotes 0 mV. Virus-injected mouse. B–D: spike threshold (B), amplitude (threshold to peak; C), and half-width (width at half-amplitude; D) during persistent spiking for example in A. Each point represents 1 spike. E: summary of changes in spike threshold, amplitude, half-width, 10–90% rise time, and 10–90% decay time, expressed as mean ± SE % change per spike throughout persistent spiking for neuron in A.
Persistent spiking was eliminated by 10 mM intracellular BAPTA. With or without intracellular BAPTA, nAChR PSPs evoked 1 or more spikes (Fig. 11E), but in BAPTA spiking did not continue after the decay of the underlying PSP. Without BAPTA spiking continued until the holding current was removed, with the last spike 2,155 ± 261 ms after the start of a single 2- to 5-ms illumination and 3,357 ± 169 ms after a burst of stimuli (10 × 5 ms at 20 Hz; 13 neurons). With BAPTA, spiking ended 185 ± 38 ms after a single illumination and 445 ± 115 ms after a burst (3 neurons; single illumination and burst each P < 0.05, unpaired 2-tailed t-test). As a result, cholinergic stimuli evoked fewer spikes with BAPTA (single 5-ms illumination, 1.8 ± 0.1 spikes, 2 neurons; burst 3.3 ± 1.7 spikes, 3 neurons) than without BAPTA (single 5-ms illumination, 6.6 ± 0.8 spikes, 7 neurons; burst 21.7 ± 0.6 spikes, 7 neurons; single illumination and burst each P < 0.05, unpaired 2-tailed t-test). Hence activation of cholinergic axons evokes persistent spiking that requires activation of nAChRs and a calcium-activated current that does not affect the spike waveform.
Our results indicate that ACh has both brief, additive and prolonged, nonlinear effects on the spiking of layer 5B pyramidal neurons, with neurons being particularly sensitive to cholinergic activity when their membrane potentials are within ∼10 mV of spike threshold, such that a nAChR-mediated increase in spiking can be short-lived or can be more dramatic, evoking spiking that persists for many seconds.
Cholinergic responses by projection target.
In primary sensory neocortices, nAChRs are expressed by selected subpopulations of presynaptic terminals. For example, ACh can enhance the transmission of sensory information to neocortex via the activation of nAChRs on thalamocortical terminals in primary somatosensory and visual cortices (Disney et al. 2007; Gil et al. 1997; Metherate 2004). Neuromodulators can also act selectively on different projection pathways out of neocortex (Avesar and Gulledge 2012; Beique et al. 2007; Gaspar et al. 1995; Gee et al. 2012; Seong and Carter 2012; Sheets et al. 2011). For example, in medial prefrontal cortex mAChR activation by ACh has a greater effect on the excitability of layer 5 pyramidal neurons that project to the pons than on neurons that project to contralateral cortex (Dembrow et al. 2010) and nAChR activation evokes larger-amplitude currents from corticothalamic layer 6 pyramidal neurons than from layer 6 pyramidal neurons that do not project to thalamus (Kassam et al. 2008). Might ACh differentially modulate the output of motor cortex via expression of nACh receptors in pyramidal neurons that project to some subcortical targets but not pyramidal neurons that project to other target tissues?
In motor cortex descending axons of layer 5 pyramidal neurons project into the pyramidal tract or to the contralateral striatum, and these two pathways are mutually exclusive (Shepherd 2013). Within layer 6 the primary subcortical output is to thalamus, and layer 6 pyramidal neurons may therefore be divided into corticothalamic and noncorticothalamic, or intracortical, neurons. We compared the incidence of nAChR- and mAChR-mediated potentials in each of these subpopulations in motor cortex after retrograde labeling of pyramidal neurons by injection of fluorescent beads into spinal cord, contralateral striatum, or ipsilateral thalamus (Fig. 13A). In slices, we identified neurons with different projection targets by the somatic accumulation of fluorescent beads (Fig. 13B).
Fig. 13.
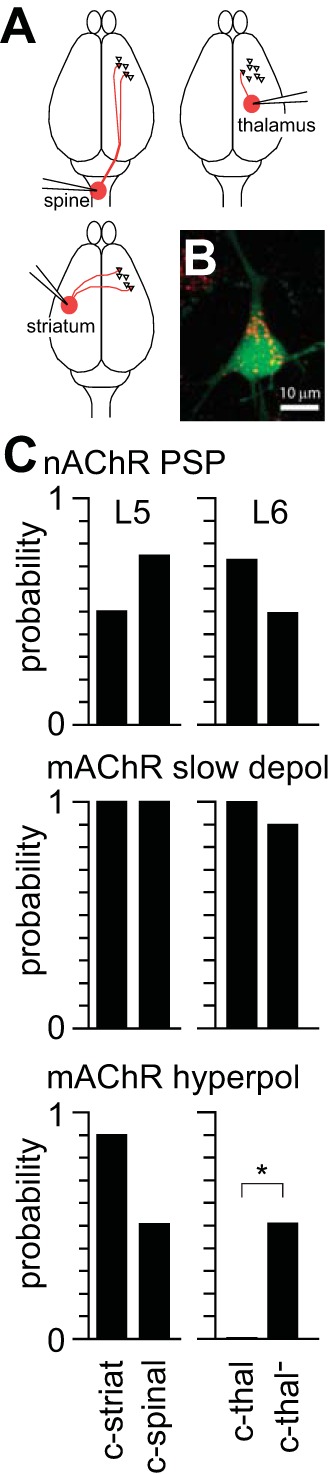
nAChR-mediated PSPs by projection target. A: schematic illustrating stereotaxic injections of fluorescent beads into subcortical projection targets. Red lines indicate approximate axonal projections from M1. B: 2-photon maximum intensity projection acquired during recording from a L5B pyramidal neuron from a ChAT-ChR2(Ai32) mouse, 5 days after bead injection into contralateral striatum. Neuron was filled with Alexa Fluor 488 (green) during whole cell recording. Numerous beads (red) are located in the soma. C: relative incidence of nAChR PSPs and mAChR-mediated slow depolarization and hyperpolarization in pyramidal neuron subpopulations identified based on somatic accumulation of fluorescent beads. c-striat, corticostriatal, 10 neurons; c-spinal, corticospinal, 12 neurons; c-thal, corticothalamic, 11 neurons; c-thal−, noncorticothalamic, 12 neurons. *Significant difference (P < 0.05, Fisher's exact test). Probability of recording nAChR PSPs was determined at rest; probability of observing mAChR responses was determined near threshold.
Synaptically released ACh frequently evoked nAChR PSPs in all four subpopulations of deep-layer M1 pyramidal neurons: corticospinal layer 5 pyramidal neurons, corticostriatal layer 5 pyramidal neurons, corticothalamic layer 6 pyramidal neurons, and noncorticothalamic (intracortical) layer 6 pyramidal neurons (Fig. 13C). The incidence of nAChR PSPs was not different between the two populations of layer 5 neurons or between the two populations of layer 6 neurons (nAChR PSPs in 5 of 10 corticospinal layer 5 pyramidal neurons, 8 of 11 corticostriatal layer 5 pyramidal neurons, P > 0.05, Fisher's exact test; nAChR PSPs in 8 of 11 corticothalamic layer 6 pyramidal neurons, 6 of 12 noncorticothalamic layer 6 pyramidal neurons, P > 0.05, Fisher's exact test). Hence our experiments revealed no evidence for different probabilities of nAChR PSPs in subpopulations of deep-layer pyramidal neurons with different projection targets. However, the amplitudes of nAChR PSPs were greater in layer 6 pyramidal neurons that projected to thalamus (9.5 ± 2.6 mV, 7 neurons) than in layer 6 neurons that did not project to thalamus (6.0 ± 2.3 mV, 6 neurons; P < 0.05, paired 2-tailed t-test). nAChR-mediated currents are larger in corticothalamic than noncorticothalamic pyramidal neurons in PFC (Kassam et al. 2008), and our results suggest that this enhancement of nAChR responses in corticothalamic layer 6 pyramidal neurons extends to M1.
The mAChR-mediated slow depolarization was also common in neurons from all four projection-based populations of deep-layer pyramidal neurons (Fig. 13C; no difference in incidence of slow depolarization between layer 5 or layer 6 subpopulations, P > 0.05, Fisher's exact test). In contrast, the hyperpolarization displayed differential expression by projection target (Fig. 13C), occurring often in both layer 5 projection-based populations and in noncorticothalamic layer 6 pyramidal neurons (no difference in incidence of hyperpolarization between layer 5 subpopulations, P > 0.05, Fisher's exact test) but being completely absent from corticothalamic layer 6 pyramidal neurons (different incidence of slow depolarization between layer 6 subpopulations, P < 0.05, Fisher's exact test).
nAChR PSP responses across layers and cortical areas.
In M1, nAChRs are expressed by pyramidal neurons throughout the layers of neocortex (Duffy et al. 2009; Nakayama et al. 1995; van der Zee et al. 1992) and cholinergic axons ramify through all layers (Lysakowski et al. 1989; Wainer and Mesulam 1990). Hence nAChR PSPs might be expected in pyramidal neurons in all layers. To test this hypothesis, we determined the frequency with which synaptically released ACh evoked nAChR PSPs in pyramidal neurons in layers 2/3, 5, and 6 of M1, PFC, and V1.
In M1 slices with abundant ChR2-labeled axons in all neocortical layers (Fig. 14A) and nAChR PSPs in layer 5B pyramidal neurons, cholinergic stimuli (10 × 5 ms at 20 Hz) rarely evoked nAChR PSPs in layer 2/3 pyramidal neurons (4 of 21 layer 2/3 neurons; Fig. 14B; lower probability in layer 2/3 than in layer 5A, layer 5B, or layer 6, P < 0.05 for each, Fisher's exact test). In layers 5A and 5B nAChR PSPs were common (6 of 8 layer 5A neurons, 82 of 90 layer 5B neurons; Fig. 14B; no difference in probability between layers 5A and 5B, Fisher's exact test), and in layer 6 nAChR PSPs were evoked in approximately half of neurons (16 of 32 neurons; Fig. 14B; greater probability in layer 6 than in layer 2/3 and lower than in layer 5B, P < 0.05 for each, Fisher's exact test). Hence in M1, nAChR PSPs occur almost exclusively in deep-layer pyramidal neurons.
Fig. 14.
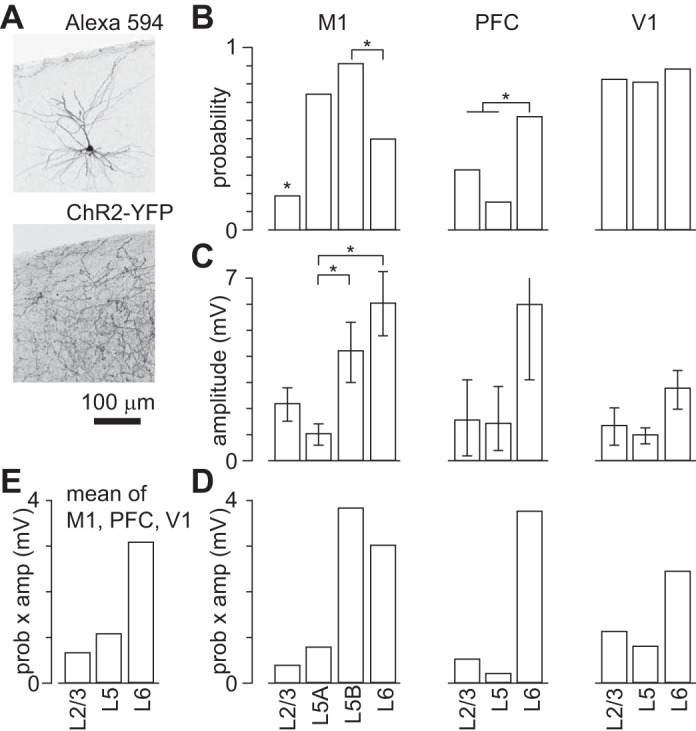
nAChR-mediated PSPs in M1, prefrontal cortex (PFC), and primary visual cortex (V1). A: images of layer 2/3 pyramidal neuron from a virus-injected mouse filled with biocytin during whole cell recording and visualized with Alexa Fluor 594-linked streptavidin and ChR2-YFP axons in the same field of view. Maximum-intensity projections from 2-photon stacks. B: probability of recording a nAChR PSP at rest in pyramidal neurons from each layer of M1, PFC, and V1. *Significant differences (P < 0.05, Fisher's exact test: M1 L2/3 different from L5A, from L5B, and from L6; M1 L5B different from L6; PFC L6 different from sum of L2/3 and L5). C: mean amplitudes of nAChR PSPs in pyramidal neurons in M1, PFC, and V1. Each bar denotes mean ± SE. *Significant differences (M1: P < 0.05, Kruskal-Wallis test; M1 L5A different from L5B, M1 L5B different from L6, each P < 0.05, Mann-Whitney test). D: plot to illustrate the net effect of nAChR activation (product of probability and amplitude of nAChR PSPs) on each layer of M1, PFC, and V1. Results in B–D are derived from 21, 8, 90, and 32 neurons from layers 2/3, 5A, 5B, and 6 of M1, respectively; 6, 13, and 8 neurons from layers 2/3, 5, and 6 of PFC, respectively; and 6, 11, and 9 neurons from layers 2/3, 5, and 6 of V1, respectively. E: summary of the strength of nAChR modulation of pyramidal neurons by cortical layer. Each bar shows the mean across M1, PFC, and V1 (equally weighted) of the product of probability and amplitude of the nAChR PSP.
In PFC and V1, the laminar pattern of nAChR PSPs was different from that in M1. In PFC stimuli (10 × 5 ms at 20 Hz) evoked nAChR PSPs in 2 of 6 layer 2/3 pyramidal neurons, 2 of 13 layer 5 pyramidal neurons, and 5 of 8 layer 6 pyramidal neurons (Fig. 14B). Hence nAChR PSPs were less common in all three layers of PFC than in layer 5B neurons in M1 (P < 0.05 for each layer, Fisher's exact test). Within PFC, nAChR PSPs were more common in layer 6 than in more superficial layers (P < 0.05, Fisher's exact test). As expected from previous studies (Bailey et al. 2010; Kassam et al. 2008; Poorthuis et al. 2013a), nAChR PSPs in layer 6 of prefrontal cortex arose from activation of non-α7 nAChRs, being unaffected by MLA and eliminated by DHβE (4 of 4 neurons). In V1, cholinergic stimuli (10 × 5 ms at 20 Hz) commonly evoked nAChR PSPs in pyramidal neurons in all cortical layers (5 of 6 layer 2/3 neurons, 9 of 11 layer 5 pyramidal neurons, 8 of 9 layer 6 pyramidal neurons; Fig. 14B; no differences in probability, Fisher's exact test).
The amplitudes of nAChR PSPs also differed across cortical layers and areas (Fig. 14C). The largest responses were observed in layer 5B of M1 (maximum 18.3 mV), but the mean nAChR PSP amplitude was greatest in layer 6 pyramidal neurons (M1 6.06 ± 1.21 mV, maximum 16.2 mV; PFC 5.99 ± 2.86 mV, maximum 15.9 mV; V1 2.76 ± 0.74 mV, maximum 5.8 mV; for M1, P < 0.05, Kruskal-Wallis test; layer 5A different from layer 5B, layer 5B different from layer 6, each P < 0.05, Mann-Whitney test). In all areas, mean peak amplitudes in layers 2–5 were between 1 and 2 mV, with the exception of M1 (Fig. 14C), where responses in layer 5B were larger than in layer 5A (P < 0.05, Mann-Whitney test) and larger than in layer 5 of PFC or V1 (layer 5B of M1 14.21 ± 1.14 mV; amplitude larger in M1 layer 5B than PFC layer 5 and V1 layer 5; P < 0.05, Kruskal-Wallis test).
To compare the effects of nAChR activation on pyramidal neurons in different layers and areas, we multiplied the probability and amplitude of nAChR PSPs for each layer (Fig. 14D). This analysis provides a measure of the overall effect of nAChR PSPs on laminar excitability and reveals that, in all three cortical areas, the effects of nAChR activation are greater in deep layers than in superficial layers. To summarize the average effect of ACh via nAChRs across cortical areas, we plot the mean effect by layer by averaging the effects in M1, PFC, and V1 (Fig. 14E). Hence in these cortical areas there is a general pattern of increased effectiveness of nAChR activation in deep layers, on which is superimposed area-specific variations in laminar sensitivity to ACh.
Hence our results indicate that ACh, acting via nAChRs, can directly excite pyramidal neurons in many cortical areas and layers. Our experiments reveal differences in the nAChR-mediated responsiveness of pyramidal neurons between cortical areas and neocortical layers. However, these local variations appear to operate within a more general framework that is common to neocortical areas, in which ACh exerts greater nAChR-mediated effects on deep-layer pyramidal neurons.
DISCUSSION
nAChRs on neocortical pyramidal neurons have proven difficult to activate in brain slice preparations, limiting the study of nAChRs. We have overcome this barrier by expressing channelrhodopsin in cholinergic axons and evoking ACh release in neocortical slices. Our results indicate that ACh activates nAChRs on pyramidal neurons in multiple layers and three cortical regions, probably via synapses between cholinergic axons and pyramidal neurons. Hence cholinergic activation of pyramidal neurons via nAChRs is common across neocortex.
Effects of ACh on pyramidal neurons.
Several authors have reported nAChR-mediated responses from pyramidal neurons (Chu et al. 2000; Guillem et al. 2011; Kassam et al. 2008; Poorthuis et al. 2013a; Roerig et al. 1997; Zolles et al. 2009), but in other studies no such responses were observed (Gil et al. 1997; Porter et al. 1999; Vidal and Changeux 1993) and in many the actions of ACh were mediated by mAChRs (Giessel and Sabatini 2010; Gulledge et al. 2007; Gulledge and Stuart 2005; Haj-Dahmane and Andrade 1996; Krnjevic 1971; McCormick 1992; McCormick and Prince 1986; Schwindt et al. 1988). The absence of nAChR responses is puzzling, as nAChRs are expressed in the dendrites of pyramidal neurons (Duffy et al. 2009; Levy and Aoki 2002; Lubin et al. 1999; Nakayama et al. 1995; van der Zee et al. 1992).
In most studies, ACh was applied by bulk perfusion or from a nearby pipette, which results in a relatively slow, widespread increase in concentration. nAChR desensitization during ACh application might be significant, reducing nAChR-mediated currents. Furthermore, many mAChRs are located extrasynaptically (Mrzljak et al. 1998; Yamasaki et al. 2010) and might be more strongly activated by applied ACh than by ACh released from cholinergic axons. Our results suggest that ACh application favors mAChR- over nAChR-mediated currents in pyramidal neurons, and this weighting may account for the paucity of nAChR-mediated responses in the literature.
ACh can act on interneurons in layer 1 of sensorimotor cortex via α7 nAChR-mediated synaptic and non-α7 nAChR-mediated diffuse mechanisms (Bennett et al. 2012), and there is a wider debate on whether ACh acts in neocortex primarily via synaptic contacts or volume transmission (Arroyo et al. 2014; Sarter et al. 2009). The short latency of nAChR-mediated responses in our experiments suggests that ACh forms cholinergic synapses with pyramidal neurons, but via non-α7 nAChRs. We found no evidence for an α7 nAChR-mediated effect or nAChR-mediated actions via volume transmission, but our results do not exclude such responses in pyramidal neurons. Although our recordings were from somata, there are nAChRs throughout the dendritic trees of pyramidal neurons (van der Zee et al. 1992), and it therefore seems likely that there are additional effects of ACh on the dendrites of pyramidal neurons.
Persistent spiking evoked by nAChRs.
Our results indicate that nAChR activation can evoke persistent spiking when paired with additional depolarization. Persistent spiking was prevented by intracellular BAPTA, suggesting that a rise in intracellular calcium concentration is also required. Calcium might enter through nAChRs or arise from a secondary source, such as voltage-activated calcium channels or a transmitter coreleased by cholinergic axons. Cholinergic axons may corelease glutamate (Allen et al. 2006; Gritti et al. 2006; Henny and Jones 2008; Manns et al. 2001), but in our experiments persistent spiking was unaffected by GluR antagonists. Other potential cotransmitters in the basal forebrain include neurotensin, somatostatin, neuropeptide Y, and galanin (Koliatsos et al. 1990).
nAChR-dependent persistent spiking has been reported in dopaminergic neurons in the substantia nigra pars compacta and subthalamic nucleus (Yamashita and Isa 2003a, 2003b) but not cortical neurons. In entorhinal, perirhinal, cingulate, and somatosensory cortices, persistent spiking can be evoked in excitatory neurons, but via mAChRs and a rise in intracellular calcium concentration (Egorov et al. 2002; Navaroli et al. 2011; Rahman and Berger 2011; Zhang and Séguéla 2010). Hence nAChR-dependent persistent spiking shares common mechanistic elements with mAChR-dependent persistent spiking but is initiated by activation of nAChRs, not mAChRs.
In spiking neurons, nAChR PSPs evoked a brief and modest increase in spike rate. Why was the effect during ongoing spiking not more prolonged, and why was the resulting spike rate typically lower than during nAChR-evoked persistent spiking? Presumably ongoing spiking suppresses the current that underlies persistent spiking or the resulting depolarization, perhaps by shunting the membrane.
Previous studies have revealed other mechanisms of prolonged spiking in pyramidal neurons, particularly deep-layer pyramidal neurons. For example, subthreshold DC current injection into the trunk of the apical dendrite can facilitate propagation of a dendritic spike from the distal apical dendrite to the soma, resulting in a burst of spikes (Larkum et al. 2001). Similarly, activation of GluRs in the basal dendrites can evoke a burst of spikes (Milojkovic et al. 2004, 2007). Presumably nAChR-persistent spiking and other mechanisms that can evoke prolonged spiking share some common mechanistic elements and differ important ways. Further experiments will be required to investigate these similarities and differences, but our results add nAChR activation to the collection of identified mechanisms that can evoke prolonged spiking from cortical pyramidal neurons.
In summary, nAChR activation increases the excitability of pyramidal neurons. The increase in spiking can be modest and transient or profound and persistent, depending on the membrane potential of the neuron. Hence ongoing synaptic drive to the pyramidal neuron determines the strength and duration of the increase in spiking evoked by ACh.
Laminar and regional variation in nAChR PSPs.
α3, α4, α7, and β2 nAChR subunits are located on neocortical pyramidal neuron somata, dendrites, and spines (Disney et al. 2007; Duffy et al. 2009; Levy and Aoki 2002; Lubin et al. 1999; Nakayama et al. 1995; Wevers et al. 1994), and several studies describe responses mediated by α4β2-, α7- and α5-containing nAChRs, the latter probably in heteromeric assembly with α4 and β2 subunits (Kassam et al. 2008; Poorthuis et al. 2013b; Zolles et al. 2009).
Our results are generally consistent with these previous studies, but there are contrasts. Layer 6 contains high expression of non-α7 nAChRs (Tribollet et al. 2004), including α5 (Proulx et al. 2013; Wada et al. 1990) and α4 (Lein et al. 2007) subunits, consistent with the high incidence of nAChR-mediated responses in layer 6 of PFC in previous studies (Bailey et al. 2010; Kassam et al. 2008; Poorthuis et al. 2013a). We found that nAChR PSPs in layer 6 are common and of large amplitude in all three areas studied, suggesting that the presence of α4/α5-mediated PSPs is a feature of layer 6 pyramidal neurons across cortical regions.
In layer 5, we found that nAChR PSPs are common in M1 and V1 and rare in PFC. In M1, nAChR PSPs were mediated by non-α7 nAChRs. In contrast, Poorthuis et al. (2012a) observed α7 nAChR-mediated responses in layer 5 pyramidal neurons in PFC. nAChR PSPs that we observed originated from nAChRs in the proximal dendrites. Hence one explanation for the lack of α7-mediated PSPs in our results might be that α7 nAChRs are located primarily in the distal dendrites and that α7-mediated depolarization of the distal dendrite failed to evoke depolarization at the soma in our experiments.
M1 is unusual in that the pyramidal neurons in layer 5B of M1 commonly display large-amplitude nAChR PSPs, unlike layer 5 pyramidal neurons in PFC and V1. The α5 subunit is not present in layer 5 (Wada et al. 1990). There is evidence for greater expression of α4 subunits in deep than in superficial layers (Lein et al. 2007; but see also Tribollet et al. 2004), but this expression pattern is not unique to M1. Hence it is unclear which nAChR subunit(s) underlies the unusually large nAChR PSPs in layer 5 of M1, but α5 subunits are unlikely to be involved.
In superficial layers, we found that pyramidal neurons in M1 and PFC rarely displayed nAChR PSPs, consistent with results from PFC (Poorthuis et al. 2013a), but that nAChR PSPs are common in superficial pyramidal neurons in V1. Layer 2/3 contains dense non-α7 labeling (Tribollet et al. 2004). Our results suggest that this dense labeling is from interneurons and perhaps in the dendrites of deep-layer pyramidal neurons.
Our results extend our knowledge of pyramidal neuron nAChRs from PFC into M1 and V1, revealing variation in laminar responsiveness to ACh between cortical regions; presumably the responsiveness of pyramidal neurons is tuned to the unique demands of each area. However, our results also reveal a general tendency for ACh to exert stronger effects in deep than superficial layers, suggesting that preferential modulation of deep-layer pyramidal neurons via nAChRs is a general property of the actions of ACh in neocortex.
nAChR-mediated modulation of neocortical circuits.
How might the layer-selective effect of ACh influence the flow of excitation through neocortex? The principal ascending excitatory drive to cortex is thalamocortical axons, which contact pyramidal neurons primarily in layers 5B and 4 [layers 5B and 3 in M1, which lacks layer 4 (Hooks et al. 2013; Shepherd 2009)]. From layer 4, information is passed by excitatory connections through layer 2/3 to layer 5 and to the subcortical projection targets of neocortex. Hence there are two primary excitatory pathways through neocortex: a short loop that connects thalamus with target structures through layer 5 and a longer loop that includes layers 4 and 2/3 (Armstrong-James et al. 1992; Constantinople and Bruno 2013; de Kock et al. 2007; Petreanu et al. 2009).
ACh enhances activation of neocortical pyramidal neurons by ascending thalamic drive. This enhancement arises from mAChR-mediated depolarization of pyramidal neurons and enhanced glutamate release from thalamocortical terminals in layer 4 (Metherate 2004; Disney et al. 2007; Gil et al. 1997). In addition, ACh activates non-fast spiking, non-parvalbumin-expressing interneurons in layers 1 and 2/3 via non-α7 nAChRs (Porter et al. 1999; Gulledge et al. 2007; Letzkus et al. 2011; Arroyo et al. 2012; Brombas et al. 2014). These interneurons inhibit parvalbumin- and somatostatin-expressing interneurons that target the somata and dendrites of pyramidal neurons. Hence nAChR-mediated inhibition of superficial interneurons reduces inhibition of superficial pyramidal neurons (Letzkus et al. 2011; Brombas et al. 2014). Our results indicate that ACh, again acting at nAChRs, directly promotes the spiking of deep-layer pyramidal neurons. Hence ACh modulates cortical output by at least three different nAChR-dependent mechanisms that enhance the responsiveness of neocortex to incoming sensory drive: by increasing the release of glutamate in layer 4, by indirectly enhancing the excitability of superficial pyramidal neurons, and by directly enhancing the excitability of deep-layer pyramidal neurons.
Why employ several mechanisms to modulate the excitability of pyramidal neurons in cortex? Multiple mechanisms may offer circuit specificity and semi-independent control. Multiple mechanisms of network modulation might allow ACh to independently modulate the excitability of deep- and superficial-layer pyramidal neurons, thereby gating the balance of sensory information processing in different cortical layers and controlling the balance of information flowing through the long and short loops from thalamus to the output structures of neocortex.
GRANTS
This work was supported by the National Institute of Mental Health (Grants 5T32 MH-067564 and 5R21 MH-085117), the National Institute of Neurological Disorders and Stroke (Grants 1R01 NS-078067), and the Brain Research Foundation (BRF SG 2010-13).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: T.H. and J.W. conception and design of research; T.H. and J.W. performed experiments; T.H. and J.W. analyzed data; T.H. and J.W. interpreted results of experiments; T.H. and J.W. prepared figures; T.H. and J.W. drafted manuscript; T.H. and J.W. edited and revised manuscript; T.H. and J.W. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Becky Imhoff and Lauren Sybert for technical assistance.
REFERENCES
- Allen TG, Abogadie FC, Brown DA. Simultaneous release of glutamate and acetylcholine from single magnocellular “cholinergic” basal forebrain neurons. J Neurosci 26: 1588–1595, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong-James M, Fox K, Das-Gupta A. Flow of excitation within rat barrel cortex on striking a single vibrissa. J Neurophysiol 68: 1345–1358, 1992. [DOI] [PubMed] [Google Scholar]
- Arroyo S, Bennett C, Aziz D, Brown SP, Hestrin S. Prolonged disynaptic inhibition in the cortex mediated by slow, non-α7 nicotinic excitation of a specific subset of cortical interneurons. J Neurosci 32: 3859–3864, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo S, Bennett C, Hestrin S. Nicotinic modulation of cortical circuits. Front Neural Circuits 8: 1–6, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avesar D, Gulledge AT. Selective serotonergic excitation of callosal projection neurons. Front Neural Circuits 6: 1–11, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CD, De Biasi M, Fletcher PJ, Lambe EK. The nicotinic acetylcholine receptor α5 subunit plays a key role in attention circuitry and accuracy. J Neurosci 30: 9241–9252, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beique JC, Imad M, Mladenovic L, Gingrich JA, Andrade R. Mechanism of the 5-hydroxytryptamine 2A receptor-mediated facilitation of synaptic activity in prefrontal cortex. Proc Natl Acad Sci USA 104: 9870–9875, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell KS, Shim H, Chen CK, McQuiston AR. Nicotinic excitatory postsynaptic potentials in hippocampal CA1 interneurons are predominantly mediated by nicotinic receptors that contain α4 and β2 subunits. Neuropharmacology 61: 1379–1388, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett C, Arroyo S, Berns D, Hestrin S. Mechanisms generating dual-component nicotinic EPSCs in cortical interneurons. J Neurosci 32: 17287–17296, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg RW, Friedman B, Schroeder LF, Kleinfeld D. Activation of nucleus basalis facilitates cortical control of a brain stem motor program. J Neurophysiol 94: 699–711, 2005. [DOI] [PubMed] [Google Scholar]
- Brombas A, Fletcher LN, Williams SR. Activity-dependent modulation of layer 1 inhibitory neocortical circuits by acetylcholine. J Neurosci 34: 1932–1941, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DB, Surmeier DJ. M1 muscarinic receptor modulation of Kir2 channels enhances temporal summation of excitatory synaptic potentials in prefrontal cortex pyramidal neurons. J Neurophysiol 97: 3432–3438, 2007. [DOI] [PubMed] [Google Scholar]
- Chu ZG, Zhou FM, Hablitz JJ. Nicotinic acetylcholine receptor-mediated synaptic potentials in rat neocortex. Brain Res 887: 399–405, 2000. [DOI] [PubMed] [Google Scholar]
- Conner JM, Culberson A, Packowski C, Chiba AA, Tuszynski MH. Lesions of the basal forebrain cholinergic system impair task acquisition and abolish cortical plasticity associated with motor skill learning. Neuron 38: 819–829, 2003. [DOI] [PubMed] [Google Scholar]
- Constantinople CM, Bruno RM. Deep cortical layers are activated directly by thalamus. Science 340: 1591–1594, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kock CP, Bruno RM, Spors H, Sakmann B. Layer- and cell-type-specific suprathreshold stimulus representation in rat primary somatosensory cortex. J Physiol 581: 139–154, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas P, Brown DA. Pathways modulating neural KCNQ/M(Kv7) potassium channels. Nat Rev Neurosci 6: 850–862, 2005. [DOI] [PubMed] [Google Scholar]
- Dembrow NC, Chitwood RA, Johnston D. Projection-specific neuromodulation of medial prefrontal cortex neurons. J Neurosci 30: 16922–16937, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilsaver S. Cholinergic mechanisms in depression. Brain Res 396: 285–316, 1986. [DOI] [PubMed] [Google Scholar]
- Disney AA, Aoki C, Hawken MJ. Gain modulation by nicotine in macaque V1. Neuron 56: 701–713, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy AM, Zhou P, Milner TA, Pickel VM. Spatial and intracellular relationships between the α7 nicotinic acetylcholine receptor and the vesicular acetylcholine transporter in the prefrontal cortex of rat and mouse. Neuroscience 161: 1091–1103, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorov AV, Hamam BN, Fransen E, Hasselmo ME, Alonso AA. Graded persistent activity in entorhinal cortex neurons. Nature 420: 173–178, 2002. [DOI] [PubMed] [Google Scholar]
- Fedorov N, Benson L, Graef JD, Hyman J, Sollenberger J, Perrefsson F, Lippiello PM, Bencherif M. A method for bidirectional solution exchange—“liquid bullet” applications of acetylcholine to alpha7 nicotinic receptors. J Neurosci Methods 206: 23–33, 2012. [DOI] [PubMed] [Google Scholar]
- Frazier CJ, Buhler AV, Weiner JL, Dunwiddie TV. Synaptic potentials mediated via alpha-bungarotoxin-sensitive nicotinic acetylcholine receptors in rat hippocampal interneurons. J Neurosci 18: 8228–8235, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar P, Bloch B, Le Moine C. D1 and D2 receptor gene expression in the rat frontal cortex: cellular localization in different classes of efferent neurons. Eur J Neurosci 7: 1050–1063, 1995. [DOI] [PubMed] [Google Scholar]
- Gee S, Ellwood I, Patel T, Luongo F, Deisseroth K, Sohal VS. Synaptic activity unmasks dopamine D2 receptor modulation of a specific class of layer V pyramidal neurons in prefrontal cortex. J Neurosci 32: 4959–4971, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharbawie OA, Whishaw IQ. Cholinergic and serotonergic neocortical projection lesions given singly or in combination cause only mild impairments on tests of skilled movement in rats: evaluation of a model of dementia. Brain Res 970: 97–109, 2003. [DOI] [PubMed] [Google Scholar]
- Giessel AJ, Sabatini BL. M1 muscarinic receptors boost synaptic potentials and calcium influx in dendritic spines by inhibiting postsynaptic SK channels. Neuron 68: 936–947, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil Z, Connors BW, Amitai Y. Differential regulation of neocortical synapses by neuromodulators and activity. Neuron 19: 679–686, 1997. [DOI] [PubMed] [Google Scholar]
- Gritti I, Henny P, Galloni F, Mainville L, Mariotti M, Jones BE. Stereological estimates of the basal forebrain cell population in the rat, including neurons containing choline acetyltransferase, glutamic acid decarboxylase or phosphate-activated glutaminase and colocalizing vesicular glutamate transporters. Neuroscience 143: 1051–1064, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillem K, Bloem B, Poorthuis RB, Loos M, Smit AB, Maskos U, Spijker S, Mansvelder HD. Nicotinic acetylcholine receptor β2 subunits in the medial prefrontal cortex control attention. Science 333: 888–891, 2011. [DOI] [PubMed] [Google Scholar]
- Gulledge AT, Park SB, Kawaguchi Y, Stuart GJ. Heterogeneity of phasic cholinergic signaling in neocortical neurons. J Neurophysiol 97: 2215–2229, 2007. [DOI] [PubMed] [Google Scholar]
- Gulledge AT, Stuart GJ. Cholinergic inhibition of neocortical pyramidal neurons. J Neurosci 25: 10308–10320, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haj-Dahmane S, Andrade R. Muscarinic activation of a voltage-dependent cation nonselective current in rat association cortex. J Neurosci 16: 3848–3861, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henny P, Jones BE. Projections from basal forebrain to prefrontal cortex comprise cholinergic, GABAergic and glutamatergic inputs to pyramidal cells or interneurons. Eur J Neurosci 27: 654–670, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero JL, Roberts MJ, Delicato LS, Gieselmann MA, Dayan P, Thiele A. Acetylcholine contributes through muscarinic receptors to attentional modulation in V1. Nature 454: 1110–1114, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooks BM, Mao T, Gutnisky DA, Yamawaki N, Svoboda K, Shepherd GM. Organization of cortical and thalamic input to pyramidal neurons in mouse motor cortex. J Neurosci 33: 748–760, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BE. Modulation of cortical activation and behavioral arousal by cholinergic and orexinergic systems. Ann NY Acad Sci 1129: 26–34, 2008. [DOI] [PubMed] [Google Scholar]
- Kalmbach A, Hedrick T, Waters J. Selective optogenetic stimulation of cholinergic axons in neocortex. J Neurophysiol 107: 2008–2019, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassam SM, Herman PM, Goodfellow NM, Alves NC, Lambe EK. Developmental excitation of corticothalamic neurons by nicotinic acetylcholine receptors. J Neurosci 28: 8756–8764, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilgard M. Cholinergic modulation of skill learning and plasticity. Neuron 38: 678–680, 2003. [DOI] [PubMed] [Google Scholar]
- Koliatsos VE, Martin LJ, Price DL. Efferent organization of the mammalian basal forebrain. In: Brain Cholinergic Systems, edited by Steriade M, Biesold D. Oxford: Oxford Univ. Press, 1990. [Google Scholar]
- Krnjevic K. The mechanism of excitation by acetylcholine in the cerebral cortex. J Physiol 215: 247–268, 1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ, Sakmann B. Dendritic mechanisms underlying the coupling of the dendritic with the axonal action potential initiation zone of adult rat layer 5 pyramidal neurons. J Physiol 533.2: 447–466, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Hassani OK, Alonso A, Jones BE. Cholinergic basal forebrain neurons burst with theta during waking and paradoxical sleep. J Neurosci 25: 4365–4369, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz JM, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, Desaki AL, Desta T, Diep E, Dolbeare TA, Donelan MJ, Dong HW, Dougherty JG, Duncan BJ, Ebbert AJ, Eichele G, Estin LK, Faber C, Facer BA, Fields R, Fischer SR, Fliss TP, Frensley C, Gates SN, Glattfelder KJ, Halverson KR, Hart MR, Hohmann JG, Howell MP, Jeung DP, Johnson RA, Karr PT, Kawal R, Kidney JM, Knapik RH, Kuan CL, Lake JH, Laramee AR, Larsen KD, Lau C, Lemon TA, Liang AJ, Liu Y, Luong LT, Michaels J, Morgan JJ, Morgan RJ, Mortrud MT, Mosqueda NF, Ng LL, Ng R, Orta GJ, Overly CC, Pak TH, Parry SE, Pathak SD, Pearson OC, Puchalski RB, Riley ZL, Rockett HR, Rowland SA, Royall JJ, Ruiz MJ, Sarno NR, Schaffnit K, Shapovalova NV, Sivisay T, Slaughterbeck CR, Smith SC, Smith KA, Smith BI, Sodt AJ, Stewart NN, Stumpf KR, Sunkin SM, Sutram M, Tam A, Teemer CD, Thaller C, Thompson CL, Varnam LR, Visel A, Whitlock RM, Wohnoutka PE, Wolkey CK, Wong VY, Wood M, Yaylaoglu MB, Young RC, Youngstrom BL, Yuan XF, Zhang B, Zwingman TA, Jones AR. Genome-wide atlas of gene expression in the adult mouse brain. Nature 445: 168–176, 2007. [DOI] [PubMed] [Google Scholar]
- Letzkus JL, Wolff SB, Meyer EM, Tovote P, Courtin J, Herry C, Luthi A. A disinhibitory microcircuit for associative fear learning in the auditory cortex. Nature 480: 331–335, 2011. [DOI] [PubMed] [Google Scholar]
- Levy RB, Aoki C. Alpha7 nicotinic acetylcholine receptors occur at postsynaptic densities of AMPA receptor-positive and -negative excitatory synapses in rat sensory cortex. J Neurosci 22: 5001–5015, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JY, Lin MZ, Steinbach P, Tsien RY. Characterization of engineered channelrhodopsin variants with improved properties and kinetics. Biophys J 96: 1803–1814, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubin M, Erisir A, Aoki C. Ultrastructural immunolocalization of the alpha7 nAChR subunit in guinea pig medial prefrontal cortex. Ann NY Acad Sci 868: 628–632, 1999. [DOI] [PubMed] [Google Scholar]
- Lysakowski A, Wainer BH, Bruce G, Hersh LB. An atlas of the regional and laminar distribution of choline acetyltransferase immunoreactivity in rat cerebral cortex. Neuroscience 28: 291–336, 1989. [DOI] [PubMed] [Google Scholar]
- Madisen L, Mao T, Koch H, Zhuo JM, Berenyi A, Fujisawa S, Hsu YW, Garcia AL 3rd, Gu X, Zanella S, Kidney J, Gu H, Mao Y, Hooks BM, Boyden ES, Buzsaki G, Ramirez JM, Jones AR, Svoboda K, Han X, Turner EE, Zeng H. A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat Neurosci 15: 793–802, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manns ID, Alonso A, Jones BE. Discharge properties of juxtacellularly labeled and immunohistochemically identified cholinergic basal forebrain neurons recorded in association with the electroencephalogram in anesthetized rats. J Neurosci 20: 1505–1518, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manns ID, Mainville L, Jones BE. Evidence for glutamate, in addition to acetylcholine and GABA, neurotransmitter synthesis in basal forebrain neurons projecting to the entorhinal cortex. Neuroscience 107: 249–263, 2001. [DOI] [PubMed] [Google Scholar]
- McCormick DA. Neurotransmitter actions in the thalamus and cerebral cortex. J Clin Neurophysiol 9: 212–223, 1992. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Prince DA. Two types of muscarinic response to acetylcholine in mammalian cortical neurons. Proc Natl Acad Sci USA 82: 6344–6348, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Prince DA. Mechanisms of action of acetylcholine in the guinea-pig cerebral cortex in vitro. J Physiol 375: 169–194, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Williamson A. Convergence and divergence of neurotransmitter action in human cerebral cortex. Proc Natl Acad Sci USA 86: 8098–8102, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metherate R. Nicotinic acetylcholine receptors in sensory cortex. Learn Mem 11: 50–59, 2004. [DOI] [PubMed] [Google Scholar]
- Milojkovic BA, Radojicic MS, Antic SD. A strict correlation between dendritic and somatic plateau depolarizations in the rat prefrontal cortex pyramidal neurons. J Neurosci 25: 3940–3951, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milojkovic BA, Zhou WL, Antic SD. Voltage and calcium transients in basal dendrites of the rat prefrontal cortex. J Physiol 585.2: 447–468, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrzljak L, Levey AI, Belcher S, Goldman-Rakic PS. Localization of the m2 muscarinic acetylcholine receptor protein and mRNA in cortical neurons of the normal and cholinergically deafferented rhesus monkey. J Comp Neurol 390: 112–132, 1998. [PubMed] [Google Scholar]
- Mrzljak L, Levey AI, Goldman-Rakic PS. Association of m1 and m2 muscarinic receptor proteins with asymmetric synapses in the primate cerebral cortex: morphological evidence for cholinergic modulation of excitatory neurotransmission. Proc Natl Acad Sci USA 90: 5194–5198, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, Ollig D, Hegemann P, Bamberg E. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci USA 100: 13940–13945, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama H, Shioda S, Okuda H, Nakashima T, Nakai Y. Immunocytochemical localization of nicotinic acetylcholine receptor in rat cerebral cortex. Mol Brain Res 32: 321–328, 1995. [DOI] [PubMed] [Google Scholar]
- Navaroli VL, Zhao Y, Boguszewski P, Brown TH. Muscarinic receptor activation enables persistent firing in pyramidal neurons from superficial layers of dorsal perirhinal cortex. Hippocampus 22: 1392–1404, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivas ND, Quintanar-Zilinskas V, Nenadic Z, Xu X. Laminar circuit organization and response modulation in mouse visual cortex. Front Neural Circuits 6: 70, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Sarter M. Cholinergic mediation of attention: contributions of phasic and tonic increases in prefrontal cholinergic activity. Ann NY Acad Sci 1129: 225–235, 2008. [DOI] [PubMed] [Google Scholar]
- Petreanu L, Mao T, Sternson SM, Svoboda K. The subcellular organization of neocortical excitatory connections. Nature 457: 1142–1146, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrof I, Viaene AN, Sherman SM. Two populations of corticothalamic and interareal corticocortical cells in the subgranular layers of the mouse primary sensory cortices. J Comp Neurol 520: 1678–1686, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorthuis RB, Bloem B, Schak B, Wester J, de Kock CP, Mansvelder HD. Layer-specific modulation of the prefrontal cortex by nicotinic acetylcholine receptors. Cereb Cortex 23: 148–161, 2013a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorthuis RB, Bloem B, Verhoog MB, Mansvelder HD. Layer-specific interference with cholinergic signaling in the prefrontal cortex by smoking concentrations of nicotine. J Neurosci 33: 4843–4853, 2013b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JT, Cauli B, Tsuzuki K, Lambolez B, Rossier J, Audinat E. Selective excitation of subtypes of neocortical interneurons by nicotinic receptors. J Neurosci 19: 5228–5235, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proulx E, Piva M, Tian MK, Bailey CD, Lambe EK. Nicotinic acetylcholine receptors in attention circuitry: the role of layer VI neurons of prefrontal cortex. Cell Mol Life Sci 71: 1225–1244, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raedler TJ, Tandon R. Cholinergic mechanisms in schizophrenia: current concepts. Curr Psychos Ther Rep 1: 20–26, 2006. [Google Scholar]
- Rahman J, Berger T. Persistent activity in layer 5 pyramidal neurons following cholinergic activation of mouse primary cortices. Eur J Neurosci 34: 22–30, 2011. [DOI] [PubMed] [Google Scholar]
- Ramanathan D, Tuszynski MH, Conner JM. The basal forebrain cholinergic system is required specifically for behaviorally mediated cortical map plasticity. J Neurosci 29: 5992–6000, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roerig B, Nelson DA, Katz LC. Fast synaptic signaling by nicotinic acetylcholine and serotonin 5-HT3 receptors in developing visual cortex. J Neurosci 17: 8353–8362, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Parikh V, Howe WM. Phasic acetylcholine release and the volume transmission hypothesis: time to move on. Nat Rev Neurosci 10: 383–390, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwindt PC, Spain WJ, Foehring RC, Chubb MC, Crill WE. Slow conductances in neurons from cat sensorimotor cortex in vitro and their role in slow excitability changes. J Neurophysiol 59: 450–467, 1988. [DOI] [PubMed] [Google Scholar]
- Seong HJ, Carter AG. D1 receptor modulation of action potential firing in a subpopulation of layer 5 pyramidal neurons in the prefrontal cortex. J Neurosci 32: 10516–10521, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets PL, Suter BA, Kiritani T, Chan CS, Surmeier DJ, Shepherd GM. Corticospinal-specific HCN expression in mouse motor cortex: Ih-dependent synaptic integration as a candidate microcircuit mechanism involved in motor control. J Neurophysiol 106: 2216–2231, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GM. Intracortical cartography in an agranular area. Front Neurosci 3: 337–343, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GM. Corticostriatal connectivity and its role in disease. Nat Rev Neurosci 14: 278–291, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M. Aceylcholine systems and rhythmic activities during the waking-sleep cycle. Prog Brain Res 145: 179–196, 2004. [DOI] [PubMed] [Google Scholar]
- Tribollet E, Bertrand D, Marguerat A, Raggenbass M. Comparative distribution of nicotinic receptor subtypes during development, adulthood and aging: an autoradiographic study in the rat brain. Neuroscience 12: 405–420, 2004. [DOI] [PubMed] [Google Scholar]
- van der Zee EA, Streefland C, Strosberg AD, Schröder H, Luiten PG. Visualization of cholinoceptive neurons in the rat neocortex: colocalization of muscarinic and nicotinic acetylcholine receptors. Mol Brain Res 14: 326–336, 1992. [DOI] [PubMed] [Google Scholar]
- Vidal C, Changeux JP. Nicotinic and muscarinic modulations of excitatory synaptic transmission in the rat prefrontal cortex in vitro. Neuroscience 56: 23–32, 1993. [DOI] [PubMed] [Google Scholar]
- Wada E, McKinnon D, Heinemann S, Patrick J, Swanson LW. The distribution of mRNA encoded by a new member of the neuronal nicotinic acetylcholine receptor gene family (α5) in the rat central nervous system. Brain Res 526: 45–53, 1990. [DOI] [PubMed] [Google Scholar]
- Wainer BH, Mesulam MM. Ascending cholinergic pathways in the rat brain. In: Brain Cholinergic Systems, edited by Steriade M, Biesold D. Oxford, UK: Oxford Univ. Press, 1990. [Google Scholar]
- Wevers A, Jeske A, Lobron C, Birtsch C, Heinemann S, Maelicke A, Schroder R, Schroder H. Cellular distribution of nicotinic acetylcholine receptor subunit mRNAs in the human cerebral cortex as revealed by non-isotopic in situ hybridization. Mol Brain Res 25: 122–128, 1994. [DOI] [PubMed] [Google Scholar]
- Whitehouse PJ, Hedreen JC, White CL, Price DL. Basal forebrain neurons in the dementia of Parkinson disease. Ann Neurol 13: 243–248, 1983. [DOI] [PubMed] [Google Scholar]
- Whitehouse PJ, Price D, Struble RG, Clarke AW, Coyle JT, Delong MR. Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science 215: 1237–1239, 1982. [DOI] [PubMed] [Google Scholar]
- Winkler J, Suhr ST, Gage FH, Thal LJ, Fisher LJ. Essential role of neocortical acetylcholine in spatial memory. Nature 375: 484–487, 1995. [DOI] [PubMed] [Google Scholar]
- Yamasaki M, Matsui M, Watanabe M. Preferential localization of muscarinic M1 receptor on dendritic shaft and spine of cortical pyramidal cells and its anatomical evidence for volume transmission. J Neurosci 30: 4408–4418, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Isa T. Flufenamic acid sensitive, Ca2+-dependent inward current induced by nicotinic acetylcholine receptors in dopamine neurons. Neurosci Res 46: 463–473, 2003a. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Isa T. Ca2+-dependent inward current induced by nicotinic receptor activation depends on Ca2+/calmodulin-CaMKII pathway in dopamine neurons. Neurosci Res 47: 225–232, 2003b. [DOI] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Davidson TJ, Mogri M, Deisseroth K. Optogenetics in neural systems. Neuron 71: 9–34, 2011. [DOI] [PubMed] [Google Scholar]
- Zhang YP, Oertner TG. Optical induction of synaptic plasticity using a light-sensitive channel. Nat Methods 4: 139–141, 2007. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Séguéla P. Metabotropic induction of persistent activity in layers II/III of anterior cingulate cortex. Cereb Cortex 20: 2948–2957, 2010. [DOI] [PubMed] [Google Scholar]
- Zolles G, Wagner E, Lampert A, Sutor B. Functional expression of nicotinic acetylcholine receptors in rat neocortical layer 5 pyramidal cells. Cereb Cortex 19: 1079–1091, 2009. [DOI] [PubMed] [Google Scholar]