

Abstract
Discovery-based proteomics has found its place in nearly every facet of biological research. A key objective of this approach is to maximize sequence coverage for proteins across a wide concentration range. Fractionating samples at the protein level is one of the most common ways to circumvent challenges due to sample complexity and improve proteome coverage. Of the available methods, one-dimensional sodium dodecyl sulfate-polyacrylamide gel electrophoresis followed by liquid chromatography-tandem mass spectrometry (GeLC-MS/MS) is a robust and reproducible method for qualitative and quantitative proteomic analysis. Here we describe a general GeLC-MS/MS protocol and include technical advice and outline caveats to increase the probability of a successful analysis.
Keywords: GeLC-MS/MS, SDS-PAGE, Protein fractionation, Protein digestion, Liquid chromatography, Mass spectrometry, Protein identification
1 Introduction
MS-based proteomic methods are currently unparalleled in their ability to rapidly characterize protein mixtures from biological samples. MS-based methods, however, often fail to reach the sensitivity of immunohistochemical methods that benefit from massive amplification of signal. Identification of low-abundance proteins to obtain as global of an analysis as possible is an ongoing challenge in the field [1]. While there is a substantial effort to improve proteome coverage during data acquisition steps, some of the most straightforward and effective methods are at the sample preparation phase of analysis. Broadly speaking, there are four levels of preparative fractionation methods: (1) cellular and (2) subcellular fractionation, followed by (3) protein- and (4) peptide-level fractionation. Popular examples of each include cell sorting, differential centrifugation, one-dimensional sodium dodecyl sulfate- polyacrylamide gel electrophoresis (1DSDS-PAGE), and orthogonal chromatography of digested peptides, respectively. The first two methods are highly dependent on the sample type, are compatible with the protein- and peptide-level fractionation methods, and have been reviewed elsewhere [2]. Peptide-level fractionation has been a popular approach for improved sequence and proteome coverage and has gained widespread popularity with the advent of the online tandem SCX-RP mud-pit approach [3] as discussed in Chapter 3. While these methods have proven successful on many levels, there are some distinct advantages to fractionating at the protein level. For example, if samples contain a wide range of protein abundances (wide dynamic range), peptides from the abundant proteins tend to dominate the MS acquisition time and hence impede the identification of lower level proteins. Protein-level separation allows for isolation of abundant proteins for improved low-level protein coverage. In addition, there is the potential to obtain isoform information based on the physical principle of the separation and gel migration by PAGE, in this example, to resolve isoforms that differ by molecular weight.
Considerations for selecting a protein separation method depend on many factors including resolving power, concentration constraints, reagent compatibility, and post-separation sample compatibility to name a few. The 1D SDS-PAGE method is a logical choice for a wide range of protein samples due to high resolving power, protein capacities in excess of sample loads for modern nLC-MS/MS instruments, compatibility with many detergents and chaotropes used in sample extraction, cost, and availability. For many years 2D-PAGE was the method of choice for protein-level fractionation prior to MS analysis. The approach has the capacity for high resolution based on two orthogonal physical properties of a protein and the ability to focus identification efforts (albeit only for more abundant proteins). However, the method is poorly suited for more global protein identification. While it is always possible to increase proteome overage by performing higher dimensional fractionation, there is a trade-off in sample loss and instrument time required for sample analysis. Many proteomic projects are constrained by limited sample quantities and instrument availability. The combination of protein-level separation by 1D-SDS-PAGE followed by RP LC-MS/MS analysis of digests from all bands, referred to as GeLC-MS/MS, offers a powerful analytical approach that balances real-world constraints with obtaining optimal proteome coverage.
The protocol described here is based on the original in-gel digestion approach presented by Rosenfeld et al. with subsequent modifications [4, 5] (see Fig. 1). Proteins are separated by SDS-PAGE, and the entire gel lanes are excised and subdivided into bands. The proteins in these gel sections are subsequently digested in-gel, and the extracted peptides are subjected to nano-flow reversed-phase LC-MS/MS analysis to obtain peptide sequence identifications that can be mapped to proteins in a sequence database [6, 7]. The database search results can be queried by band to yield information regarding intact MW and search for evidence of protein isoforms or combined to yield the total sample identifications and relative quantification per sample. Here we present the steps for performing GeLC-MS/MS analysis and give suggestions to optimize peptide identification results using this method.
Fig. 1.
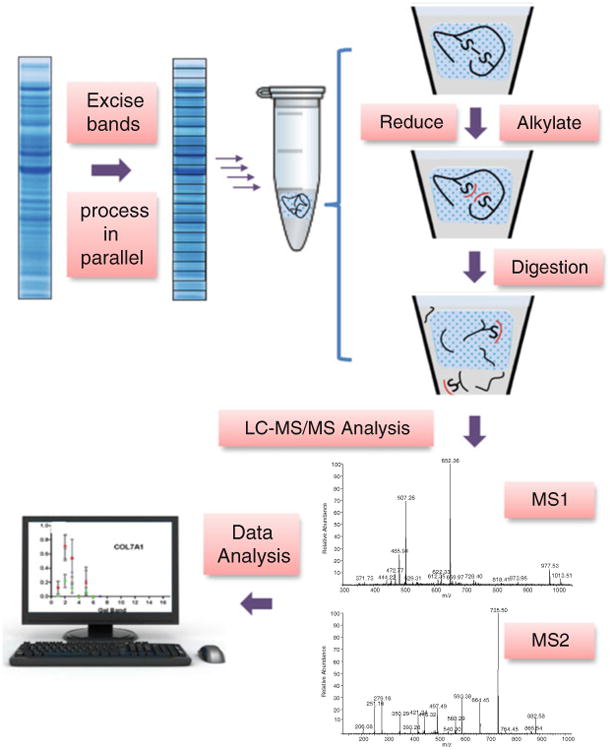
A schematic overview of the in-gel digestion procedure. Protein sample is separated by 1D SDS-PAGE, and the lane is cut into bands for parallel processing. Reduction and alkylation are performed prior to enzymatic digestion of proteins. Peptides are extracted for LC-MS/MS analysis followed by protein sequence database searches
2 Materials
2.1 1D SDS-Polyacrylamide Gel Electrophoresis
Pre-stained protein ladder.
Sample loading buffer (4×): 10 % Glycerol, 141 mM Tris base, 106 mM Tris-HCl, 2 % LDS, 0.51 mM EDTA, 0.22 mM SERVA® Blue G250, 0.175 mM phenol red, pH 8.5.
MES SDS running buffer (20×): 50 mM MES, 50 mM Tris base, 0.1 % SDS, 1 mM EDTA, pH 7.3.
Polyacrylamide gel: 40 % Acrylamide, 1 % bisacrylamide, 1.5 M Tris–HCl (pH 8.7), 10 % ammonium persulfate, TEMED, 10 % SDS (see Note 1).
Reducing agent (10×): 500 mM Dithiothreitol or 2.5 % β-mercaptoethanol.
Antioxidant: 10 % N,N-dimethylformamide, 15 % sodium bisulfite.
Coomassie Brilliant Blue-based stain: 0.1 % Coomassie Brilliant Blue G250, 10 % glacial acetic acid, 40 % methanol.
Destain solution: 25 % methanol, 7.5 % glacial acetic acid.
Electrophoresis gel box (e.g., XCell SureLock or other small-format gel box).
2.2 In-Gel Trypsin Digestion
Clean glass plate (large enough to place the entire gel on and room for a working area, 8″ × 8″).
Gel-cutting devices: Two steel razor blades, surgical scalpel, or the MEG-1.5 Gel Cutter (The Gel Company).
Low-protein-binding microcentrifuge tubes (0.65 or 1.5 mL).
Gel-loading pipette tips.
Autosampler vials with perforated caps.
SpeedVac concentrator.
25 mM ammonium bicarbonate: Dissolve 100 mg ammonium bicarbonate in 50 mL double-distilled water.
Destain solution: 25 mM ammonium bicarbonate/50 % acetonitrile (ACN). Weigh 100 mg ammonium bicarbonate, and prepare a 50 mL solution.
Extraction solution: 1 % formic acid.
Tris[2-carboxyethyl]phosphine (TCEP)-HCl stock solution: 5 mM TCEP in 25 mM ammonium bicarbonate.
Iodoacetamide (IAM) stock solution: 20 mM in 25 mM ammonium bicarbonate (always prepare fresh, light sensitive).
10 ng/μl Trypsin, sequencing grade (use 25 mM ice-cold ammonium bicarbonate to dilute stock trypsin solution, immediately before adding to gel pieces).
2.3 Liquid Chromatography-Tandem MS Analysis
Formic acid (LC/MS grade).
Solvent A: Double-distilled water from an all glass still, with 0.1 % formic acid. Add acid using a glass syringe.
Solvent B: ACN (LC/MS grade) with 0.1 % formic acid. Add acid using a glass syringe.
Trap column: ZORBAX 300SB-C18, dimensions 5 × 0.3 mm, 5 μm (Agilent Technologies).
Analytical column: Self-packed, KaSil fritted 100 μm i.d. × 150 mm fused silica capillary packed with Synergi C18 resin (Phenomenex; Torrance, CA), 15 μm tip ID (New Objective).
High-performance LC system, capable of nanoliter flow rates, with a chilled autosampler (e.g., Agilent 1260 nano system).
Mass spectrometer with tandem MS capabilities (e.g., LTQ Orbitrap Velos, Thermo Fisher Scientific).
Peak list extraction software (e.g., manufacture-supplied extractor, Mascot distiller, ProteoWizard [8])
MS/MS data analysis software (e.g., ProteinProspector or Mascot).
Optional: Software for differential analysis (spreadsheet program with statistical analysis package or dedicated programs for this purpose, e.g., Scaffold, Progenesis LC-MS)
3 Methods
3.1 1D SDS-PAGE
SDS-PAGE offers size-based separation and provides a convenient method for sample cleanup prior to trypsin digestion. Efficient protein extraction and isolation prior to GeLC-MS/MS are critical for obtaining an accurate representation of the proteome under study. Proteins can be prepared from in vivo (tissue or bodily fluids) and in vitro (cell culture or immunoprecipitations) sources. While preparation methods are sample specific, protocols commonly call for one or more of the following steps: (1) mechanical lysis of tissues or cells, or the use of alternative homogenization techniques, (2) solubilization in buffer (some classes of protein require strong detergents or chaotropes for solubilization), (3) subcellular fractionation approaches [9], and (4) protein complex isolation. The most basic form of protein isolation is by direct solubilization in SDS-PAGE sample buffer with heat [10]. However, if this is the method of choice, it is important to remove salts and incompatible detergents and chaotropes (such as guanidine hydrochloride) in order to avoid streaking and aberrant protein migration. This can readily be performed by precipitation of proteins prior to solubilization in sample buffer [11].
Add LDS sample buffer (4×) to the protein sample (with or without reducing agent), and heat at 70 °C for 10 min. Centrifuge the heated samples at 2,400 × g for 30 s to bring down insoluble material (see Notes 2 – 4).
Obtain 1.5 mm thick precast Bis-Tris 4-12 % gradient gel (see Notes 5 and 6).
Remove the Bis-Tris 4–12 % gradient gel from the storage pouch, and pull the plastic strip from the bottom of the cassette.
Place gel into mini gel box electrophoresis system with the gel comb opening facing towards the inside of the buffer tank.
Dilute MES SDS running buffer (10×) to a 1× solution using ddH2O. Make up to 1 L (see Note 7).
Fill the inner tank with the 1× running buffer. The buffer level must exceed the level of the wells.
Add 500 μL of antioxidant in the inner tank.
Gently pull the comb out of the cassette, and rinse the sample wells thoroughly with 1× running buffer to remove air bubbles and displace any storage buffer.
Apply protein sample up to 40 μg protein concentration per gel (see Notes 8 – 10).
Load 5–8 μL of the marker into a sample well of the gel to be run.
Attach the electrophoresis gel box lid, and connect the electrode cords to the power supply [red to (+) jack, black to (−) jack].
Set power supply for 180 V constant voltage and 35 min run time. Start the run. The bromophenol blue tracking dye elutes from the gel at approximately 30–35 min.
After electrophoresis is complete, shut off the power, disconnect electrodes, and remove gel(s) from electrophoresis gel box.
Insert the gel knife into the gap between the cassette's two plates. Twist to open gel cassette (see Note 15).
Carefully remove and discard the top plate, allowing the gel to remain on the bottom plate.
Let the gel fall off into 100 mL of ddH2O in a clean staining container.
Rinse the gel three times for 10 min with ddH2O (see Note 11).
Discard water from gel, and add 50 mL of Coomassie stain solution or sufficient volume to completely cover the gel (see Note 12).
Stain for 1 h at room temperature with gentle shaking. Bands will begin to develop within minutes. After staining is complete, discard the stain solution.
Rinse gel with 100 mL ddH2O to remove residual stain from container. Add 100 mL of destaining solution and microwave gel on high power for an additional 30 s. Gently shake the gel at room temperature for at least 1 h. Gel will have a clear background after 1 h in destain solution. The gel can be stored at 4 °C until processing for in-gel digestion (see Notes 13 and 14).
3.2 Band Excision
Place the gel on a clean glass plate. Cover the gel with just enough ddH2O water to prevent dehydration during the slicing process (see Note 16).
Cut the gel lane using (new, if possible) scalpel, razor blade, or OneTouch GridCutter.
Cut each of the excised bands into 1–2 mm cubes, and transfer these cubes to a 0.65 mL siliconized microcentrifuge tube (see Note 17).
3.3 In-Gel Digestion
4. Add ∼100 μL (or enough to cover) of 25 mM ammonium bicarbonate/50 % ACN and vortex for 10 min. Using gel-loading pipet tip, extract the supernatant and discard. The procedure should be repeated until the stain is completely removed. Two additional washes should be sufficient for moderately intense bands.
5. Add 100 μL of 5 mM TCEP, and incubate for 30 min at 56 °C. Spin. Discard all the liquid afterwards (see Note 18).
6. Allow samples to cool to room temperature.
7. Add 100 μL of 20 mM IAM, and incubate the gel pieces in the dark for 45 min at room temperature. Spin. Discard the liquid afterwards (see Note 19).
8. Wash the gel pieces with 100 μL of 25 mM ammonium bicarbonate, vortex for 10 min, and spin. Discard the liquid afterwards.
9. Wash the gel pieces with ∼100 μL (or enough to cover) of 25 mM ammonium bicarbonate in 50 % ACN, vortex for 10 min, and spin. Discard the liquid (see Note 20).
10. Dehydrate the gel pieces in 100 % ACN for 10 min, spin, and discard the liquid afterwards.
11. Dry the sample in a SpeedVac for 10 min. The gel pieces are now ready for tryptic digestion.
12. Just before use, dilute or reconstitute trypsin with 50 mM ice-cold ammonium bicarbonate to give final concentration of the 10 ng/μL (see Note 21).
13. Add trypsin solution to just cover the gel pieces (see Note 22).
14. Rehydrate dried gel pieces on ice or at 4 °C for 30–45 min [12].
15. Check that the gel pieces are covered with trypsin solution.
16. Add 25 mM ammonium bicarbonate as needed to cover the gel pieces.
17. Spin briefly, and incubate at 37 °C for 4 h–overnight (see Note 23).
18. Stop digestion by adding 20 μL of 5 % formic acid.
19. Vortex for 15–20 min, spin, and transfer the digest solution (aqueous extraction) into a clean autosampler vial appropriate for LC/MS-MS (see Note 24).
20. To the gel pieces, add 30 μL (enough to cover) of 1 % formic acid, vortex for 15–20 min, spin, and transfer solution to the tube used above. Repeat this step once (see Note 25).
21. Concentrate peptide extracts using a SpeedVac concentrator to a volume that is slightly larger than will be used for injection during LC-MS/MS analysis (see Notes 26 and 27).
22. Store the vial with the extracted peptides at −20 °C if the samples will not be run on the same day.
3.4 Mass Spectrometry Analysis
A high-pressure liquid chromatography system running at nano-flow rates should be used for peptide fractionation prior to mass spectrometry analysis (see Note 28). Reverse-phase liquid chromatography columns (15 cm × 100 μm ID) packed in-house with C18 resins (4 μm, 100 Å beads, Phenomenex, Torrance, CA) or commercial columns can be used. An LTQ Orbitrap Velos™ mass spectrometer (Thermo Fisher Scientific) or equivalent rapid scanning mass spectrometer may be used. Specific values in the methods below will be laboratory dependent and are provided simply as a benchmark.
Prior to analysis, calibrate the instrument using a standard calibration mixture according to a validated SOP or the manufacturer's instructions.
Perform a QC run to evaluate instrument performance with LC-MS/MS analyses of 1–20 fmol of a protein digest standard. Ideally the standard protein will not be present in the sample of interest. We commonly use yeast alcohol dehydrogenase digests for this purpose.
Load 1–10 μL of each peptide sample using the autosampler. Optional: Desalt on a trapping column with a flow rate of 10 μL/min for 3 min (see Note 29).
Program the switching valve to place the trapping column online with the analytical column.
Separate the peptides by reverse-phase LC with a 45- or a 75-min linear gradient (60–90-min total run time) from 5 % ACN to 35 % ACN with 0.1 % FA at 300 nL/min fl ow rate (see Note 30).
Acquire MS/MS in a data-dependent mode, in which MS/MS fragmentation is performed on the most intense ions of every full MS scan (10–20 is typical). Perform full MS scan (m/z 400– 2,000) in the Orbitrap with 60,000 resolution (see Note 31).
Run standard protein digest at routine intervals and at the end of the analysis to evaluate instrument performance.
3.5 Data Analysis
Several computational algorithms have been developed to match peptide fragmentation spectra to peptides for protein identification, as the complexity of tandem MS/MS data fi les precludes comprehensive manual interpretation of all spectra. Software packages and associated algorithms, such as Mascot [13], X!Tandem [14], and ProteinProspector [15 – 17], may be used to search a given sequence database for peptides with theoretical fragmentation spectra best matching the observed spectra and subsequent assignment of the matched peptides to the corresponding protein/s. An important component of the data analysis process is evaluation of the false discovery rate for peptide- and protein-level identifications; this is performed by including appropriate decoy sequences in the queried protein database. Results from individual gel bands can be combined and a statistical comparative analysis performed across different samples. Additional information regarding protein isoforms that resolve by gel band can be obtained by comparing identification results across samples by gel band.
4 Notes
Adjusting the ratio of acrylamide to cross-linker (bisacrylamide) allows for tailoring the gel to the specifi c sample under consideration. In general, low-percent acrylamide gels will separate high-molecule proteins more efficiently and high- percent acrylamide gels will separate low molecular proteins more efficiently. For GeLC-MS/MS applications we suggest the use of commercially available gels as they typically yield more reproducible results.
Heating the sample at 100 °C in SDS containing buffer can result in proteolysis. It is recommend to heat samples for 5–10 min for denaturing electrophoresis at 70 °C for reduced and 90 °C for non-reduced samples for optimal results [18].
SDS precipitates at 4 °C. Therefore, LDS sample buffer needs to be warmed prior to use if stored at 4 °C for increased shelf life.
Reducing agents such as TCEP and DTT are used to reduce disulfide bonds permitting more complete protein unfolding/denaturing prior to electrophoresis.
Free acrylamide may react with primary amines and free thiols on proteins during polyacrylamide gel electrophoresis [19]. We recommend using commercial precast gels for GeLC-MS/MS experiments due to our experiences with reproducibility of hand-cast gels. If hand-cast gels will be used we recommend using fresh reagents and allowing overnight polymerization. Precast gels are ready to use and offer greater convenience, more stringent quality control, and higher reproducibility than hand-cast gels in general.
Use gradient gels to separate samples containing a broad range of molecular weights. Gradient gels allow resolution of both high- and low-molecular-weight bands on the same gel.
Other buffer systems such us MOPS may also produce comparable results.
Overloading will result in poor to no resolution of protein bands.
High salt concentrations result in increased conductivity that affects protein migration and can result in gel artifacts in adjacent lanes containing samples with lower salt concentrations. Precipitate protein, and bring the pellet up in LDS sample loading buffer or perform dialysis (micro) using a lower salt buffer prior to electrophoresis.
Samples solubilized in guanidine-HCl have high ionic strength and produce increased conductivity similar to high salt concentrations. In addition, guanidine precipitates in the presence of SDS leading to various types of gel artifacts. If possible, change the solubilization agent by dialysis prior to electrophoresis or precipitate with cold ethanol (1:9 ratio: sample to ethanol) prior to electrophoresis [20].
The washing step is necessary to remove residual SDS, which interferes with dye binding.
A colloidal Coomassie (G250)-based stain is recommended for visualizing proteins. In GeLC-MS/MS experiments staining usually serves two purposes: location of intense protein bands to direct band cutting and to provide a global overview of gross differences by visual comparison of samples (usually this is of limited utility). If silver staining is used make sure that the protocol is compatible with mass spectrometry analysis. Glutaraldehyde, used in some protocols as a sensitizer, crosslinks proteins which results in decreased trypsin digestion and protein extraction from the gel.
Microwaving the gels speeds up both the staining and destaining process and can be circumvented with longer incubation times [21]. Commercially available stains are available that do not require the addition of heat or destain solution to visualize gel bands.
Gel migration is a very crude method for estimating molecular weight of a protein. Proteins migrate at different rates based on amino acid composition and presence of modifications. Acidic proteins repel SDS and thus will migrate slower towards the anode, whereas basic proteins bind a higher concentration of SDS and tend to migrate faster than most proteins that are used as molecular weight markers [22].
Reduce exposure of the gel, gel equipment, and supplies to primary sources of keratins, such as skin, hair, and clothing. Reduce the amount of exposure to dust and particulates, both of which can be a rich source of keratins. Perform as much work as possible in an area cleaned of dust, ideally in laminar flow hood. Wear powder-free gloves and sleeve protectors or a lab coat; wipe down ALL surfaces with 50 % ethanol solution, moistened lint-free cloth, or tissue, including glass plate used for cutting, the SpeedVac and centrifuge, racks, and tubes.
Always use siliconized or low-binding polypropylene tubes and low-retention tips (important once peptides have been generated) to minimize sample loss due to adsorption to tube or tip surfaces.
Dithiothreitol (DTT) is a commonly used alternative for reduction of disulfide bonds. TCEP offers several advantages including greater resistance to oxidation; it is a stronger reductant and is not prone to side reactions with peptide functional groups. Check the pH of your TCEP stock solution as it may be acidic when brought up in solution depending on buffering conditions and needs to be brought to neutral pH prior to addition to the protein sample.
The most commonly used agents for alkylation of cysteine thiols in protein samples prior to digestion are IAM followed by iodoacetic acid (IAA).
Sufficient washing of the gel pieces after alkylation is a critical step of in-gel digestion processing. Insufficient washing leaves residual alkylating agents in the gel. If present during digestion, the N-terminus of peptides is commonly alkylated which has two negative effects: (1) It splits the population of your peptides which decreases the probability of identification and (2) if not accounted for in database searches increases the odds of false-positive identifications [23].
Modified trypsin is preferred for protein digestion as it is less susceptible to autolysis.
Although trypsin is the most common enzyme of choice for proteomic methods, additional enzymes such as Asp-N, chymotrypsin, or Glu-C can be used to improve sequence coverage [24, 25].
37 °C has been suggested as the optimal temperature and is the temperature most commonly used for overnight in-gel-and in-solution-based tryptic digestion.
This is a potential point of contamination; all plastic vials have the potential to contain polymer. Confirm that the batch of autosampler vials has been used without incident prior to usage here.
A potential disadvantage of the GeLC-MS/MS approach when compared to tandem LC/LC-MS/MS approaches is the loss of peptides due to extraction efficiency from the gel. Speicher et al. have explored this issue using isotopically labeled protein and scintillation counting to determine sample losses and extraction efficiencies [26]. Their findings indicate that approximately 15–20 % of sample is lost during the entire in-gel digestion procedure. Sample is lost during destaining steps (∼4 %) and to the digestion tube (∼6 %), pipette tip (∼1 %), and the gel (7 %). Presumably in-solution digestion methods would be subject to losses due to the digestion tube and pipette tip/s. The overall recovery of peptides was approximately 78 % in the first round of extraction and an additional 6 and 1 % in subsequent rounds.
Generate an autosampler vial that contains a known amount of liquid for reference. The outer tube should be removed if it is difficult to visualize liquid level.
If possible do not let the peptide extract dry completely; resolubilization is a source of potential sample loss. If sample volume needs to be added use buffer A from the LC system.
Many investigators will need to utilize a core facility or work with a collaborator to have peptide digests analyzed by mass spectrometry. Important aspects of this analysis to consider and discuss with a facility or a collaborator include the following: (1) wait time between digestion and analysis: ideally digests will be run within the first 48 h of digestion; (2) complexity of sample and objectives of the analysis; these parameters will dictate the length of LC runs, the optimal mass spectrometer, and acquisition parameters to use.
For differential analysis we prefer to run band digests in groups of five from one lane (sample A, bands 1–5) followed by a blank run and the same five bands from the sample/s to be compared (sample B, C, etc.) before moving to the next five bands (sample A, 6–10). This serves to minimize artifacts due to variations in instrument performance over time while keeping blank runs to a minimum. The blank runs should be analyzed for QC purposes.
Longer columns improve chromatographic separation and generally increase peptide identifications; further improvements are observed with longer gradients [27].
The type of fragmentation used will depend on experimental considerations. Combinations of ETD and HCD have proven useful for improving protein coverage, and ETD has some advantages for mapping posttranslational modifications [28].
References
- 1.Zubarev RA. The challenge of the proteome dynamic range and its implications for in-depth proteomics. Proteomics. 2013;13:723–726. doi: 10.1002/pmic.201200451. [DOI] [PubMed] [Google Scholar]
- 2.Michelsen U, von Hagen J. Isolation of subcellular organelles and structures. Methods Enzymol. 2009;463:305–328. doi: 10.1016/S0076-6879(09)63019-6. [DOI] [PubMed] [Google Scholar]
- 3.Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 4.Rosenfeld J, Capdevielle J, Guillemot JC, et al. In-gel digestion of proteins for internal sequence analysis after one- or two- dimensional gel electrophoresis. Anal Biochem. 1992;203:173–179. doi: 10.1016/0003-2697(92)90061-b. [DOI] [PubMed] [Google Scholar]
- 5.Hellman U, Wernstedt C, Góñez J, et al. Improvement of an “In-Gel” digestion procedure for the micropreparation of internal protein fragments for amino acid sequencing. Anal Biochem. 1995;224:451–455. doi: 10.1006/abio.1995.1070. [DOI] [PubMed] [Google Scholar]
- 6.Baldwin MA. Protein identification by mass spectrometry. Mol Cell Proteomics. 2004;3:1–9. doi: 10.1074/mcp.R300012-MCP200. [DOI] [PubMed] [Google Scholar]
- 7.Chalkley RJ, Hansen KC, Baldwin MA. Bioinformatic methods to exploit mass spectrometric data for proteomic applications. Methods Enzymol. 2005;402:289–312. doi: 10.1016/S0076-6879(05)02009-4. [DOI] [PubMed] [Google Scholar]
- 8.Kessner D, Chambers M, Burke R, et al. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics (Oxford, England) 2008;24:2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Fonslow BR, Shan B, et al. Protein analysis by shotgun/bottom-up proteomics. Chem Rev. 2013;113:2343–2394. doi: 10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ericsson C, Nistér M. Protein extraction from solid tissue. Methods Mol Biol (Clifton, NJ) 2011;675:307–312. doi: 10.1007/978-1-59745-423-0_17. [DOI] [PubMed] [Google Scholar]
- 11.Jiang L, He L, Fountoulakis M. Comparison of protein precipitation methods for sample preparation prior to proteomic analysis. J Chromatogr A. 2004;1023:317–320. doi: 10.1016/j.chroma.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 12.Havlis J, Thomas H, Sebela M, et al. Fast-response proteomics by accelerated in-gel digestion of proteins. Anal Chem. 2003;75:1300–1306. doi: 10.1021/ac026136s. [DOI] [PubMed] [Google Scholar]
- 13.Perkins DN, Pappin DJ, Creasy DM, et al. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 14.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics (Oxford, England) 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 15.Clauser KR, Baker P, Burlingame AL. Role of accurate mass measurement (+/− 10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal Chem. 1999;71:2871–2882. doi: 10.1021/ac9810516. [DOI] [PubMed] [Google Scholar]
- 16.Chalkley RJ, Baker PR, Hansen KC, et al. Comprehensive analysis of a multidimensional liquid chromatography mass spectrometry dataset acquired on a quadrupole selecting, quadrupole collision cell, time-of- flight mass spectrometer: I. How much of the data is theoretically interpretable by search engines? Mol Cell Proteomics MCP. 2005;4:1189–1193. doi: 10.1074/mcp.D500001-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Chalkley RJ, Baker PR, Huang L, et al. Comprehensive analysis of a multidimensional liquid chromatography mass spectrometry dataset acquired on a quadrupole selecting, quadrupole collision cell, time-of-flight mass spectrometer: II. New developments in protein prospector allow for reliable and comprehensive automatic analysis of large datasets. Mol Cell Proteomics MCP. 2005;4:1194–1204. doi: 10.1074/mcp.D500002-MCP200. [DOI] [PubMed] [Google Scholar]
- 18.Kubo K. Effect of incubation of solutions of proteins containing dodecyl sulfate on the cleavage of peptide bonds by boiling. Anal Biochem. 1995;225:351–353. doi: 10.1006/abio.1995.1167. [DOI] [PubMed] [Google Scholar]
- 19.Chiari M, Righetti PG, Negri A, et al. Preincubation with cysteine prevents modifi cation of sulfhydryl groups in proteins by unreacted acrylamide in a gel. Electrophoresis. 1992;13:882–884. doi: 10.1002/elps.11501301193. [DOI] [PubMed] [Google Scholar]
- 20.Pepinsky RB. Selective precipitation of proteins from guanidine hydrochloride-containing solutions with ethanol. Anal biochem. 1991;195:177–181. doi: 10.1016/0003-2697(91)90315-k. [DOI] [PubMed] [Google Scholar]
- 21.Lill JR, Nesatyy VJ. Microwave-assisted protein staining, destaining, and in-gel/in-solution digestion of proteins. Methods Mol Biol (Clifton, NJ) 2012;869:521–532. doi: 10.1007/978-1-61779-821-4_46. [DOI] [PubMed] [Google Scholar]
- 22.Shi Y, Mower y RA, Ashley J, et al. Abnormal SDS-PAGE migration of cytosolic proteins can identify domains and mechanisms that control surfactant binding. Protein Sci. 2012;21:1197–1209. doi: 10.1002/pro.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woods AG, Sokolowska I, Darie CC. Identification of consistent alkylation of cysteine- less peptides in a proteomics experiment. Biochem Biophys Res Commun. 2012;419:305–308. doi: 10.1016/j.bbrc.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 24.Choudhar y G, Wu SL, Shieh P, et al. Multiple enzymatic digestion for enhanced sequence coverage of proteins in complex proteomic mixtures using capillary LC with Ion trap MS/MS. J Proteome Res. 2003;2:59–67. doi: 10.1021/pr025557n. [DOI] [PubMed] [Google Scholar]
- 25.Swaney DL, Wenger CD, Coon JJ. Value of using multiple proteases for large-scale mass spectrometry-based proteomics. J Proteome Res. 2010;9:1323–1329. doi: 10.1021/pr900863u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Speicher K, Kolbas O, Harper S, et al. Systematic analysis of peptide recoveries from in-gel digestions for protein identifications in proteome studies. J Biomol Tech JBT. 2000;11:74–86. [PMC free article] [PubMed] [Google Scholar]
- 27.Hsieh EJ, Bereman MS, Durand S, et al. Effects of column and gradient lengths on peak capacity and peptide identification in nanoflow LC-MS/MS of complex proteomic samples. J Am Soc Mass Spectrom. 2013;24:148–153. doi: 10.1007/s13361-012-0508-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swaney DL, McAlister GC, Coon JJ. Decision tree-driven tandem mass spectrometry for shotgun proteomics. Nat Methods. 2008;5:959–964. doi: 10.1038/nmeth.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]