

Abstract
Protein synthesis is a tightly controlled process and its deregulation plays an important role in tumorigenesis. Protein synthesis remains poorly understood with very few well identified validated targets for therapeutic purposes. In this study we employ nitric oxide (NO), which suppresses protein synthesis by inactivating initiation factor eIF2-αto examine the mechanism by which low and high oxidative stress inhibits protein synthesis. In breast cancer cells, low NO stress induced heme regulated inhibitor (HRI) activation, which facilitated gradual decline in short half-life proteins. High NO stress induced HRI and protein kinase R (PKR) activation, leading to a sharp decline in protein synthesis as accessed by a decline in short and long half-life proteins and dramatic morphological changes. In contrast, human mammary epithelial (HME) and Ras transfected untransformed human mammary epithelial (MCF-10A1 neo N) cells were less susceptible to NO-induced inhibition of protein synthesis and cytostasis. Our results suggests that NO-induced cytostasis in breast cancer cells was due to PKR activation and increased phosphorylation of eIF2-αwhile the reduced susceptibility of normal mammary epithelial cells to NO could be due to the inaccessibility of PKR, which is bound to inhibitor p58.
Keywords: NO, Translation, PKR, Cyclin D1, eIF2-α
Introduction
Protein synthesis is a tightly regulated process that plays a critical role in proliferation and differentiation (1, 2). Raf/MEK/ERK1/2 and PI-3 kinase/Akt are the prominent signaling cascades that deregulate mTOR, a master regulator of protein synthesis and its down-stream specific translation factors like eIF4E and p70 S6 kinase (3, 4). Deregulation of proliferation pathways and protein synthesis has been strongly implicated in the pathogenesis of cancer and metastasis (5–7). In spite of intense research, protein synthesis is poorly understood with little or no success to targeted therapeutic intervention. It is recently becoming evident that cap-dependent protein synthesis pathway is an integrator and amplifier of many essential oncogenic signals (8). Radiation therapy has been found to act on cancer cells largely due to generation of reactive oxygen species (ROS), which also induce normal tissue toxicity (9). One of the areas that remain unexplored is the sensitivity of initiation of protein synthesis in untransformed and cancer cells to various oxidative stresses. In this study we utilize nitric oxide (NO), which is a potent bioactive signaling molecule (10–13), to examine the effect of low and high oxidative stress on protein synthesis in breast cancer and normal mammary epithelial cells. NO has been found to inhibit the initiation steps of protein synthesis in a number of cell types by phosphorylating and inactivating eukaryotic initiation factor 2-alpha (eIF2-α) (14–15).
We have previously reported that NO (released by 1mM DETA-NONOate) treatment of malignant breast cancer cell line MDA-MB-231 reduces the cyclin D1/retinoblastoma (Rb) signaling to induce cytostasis (16). This NO induced cytostasis was due to inhibition of protein synthesis resulting in the decline of cyclin D1, a short half-life protein. In this study we took advantage of the availability of DETA NONOate because it is a soluble chemical compound that enabled us to quantitate high and low stress levels. The amount of high and low NO stress was kept within the range that activated macrophages produce in vivo (17). To simulate the range of NO (1–2 μM) produced by activated macrophages, breast cancer cells and normal mammary epithelial were treated with 1 or 2mM DETA-NONOate, a long acting NO donor with a half-life of 20h. DETA-NONOate at 1mM release 0.5μM NO, which was used to induce low stress, while 2mM of the donor was chosen to induce high NO stress to cells (16–17). We found that breast cancer cells with low NO stress induced a small upregulation of p-eIF2-α levels, which led to the decline in short half-life proteins; whereas high NO stress induced a prominent increase in p-eIF2-α, and induced decline of both short and long half-life proteins. In contrast, human mammary epithelial (HME) and Ras transfected human mammary epithelial (MCF-10A1 neo N) cells were less susceptible to NO-induced inhibition of protein synthesis and cytostasis.
We conclude that protein synthesis in breast cancer cells are more susceptible to NO induced stress, leading to PKR activation and inhibition of protein synthesis through phosphorylation of eIF2-α compared to normal mammary epithelial cells.
Materials and Methods
Materials
DETA-NONOate, SNAP, ODQ, and 2–4-carboxyphenyl)-4, 4, 5, 5-tetramethyl-1-oxyl-3-oxide (cPTIO) were purchased from Cayman Biochemicals (Ann Arbor, MI). Actinomycin D, hydrogen peroxide, cycloheximide, dithiothreitol (DTT), leupeptin, aprotinin, phenylmethylsulfonyl fluoride (PMSF), hemin, 2-aminopurine, ponceau S solution and protein A-Sepharose beads were purchased from Sigma (St. Louis, MO). All of the cell culture media were purchased from Mediatech, Inc. (Hendon, VA). Rabbit polyclonal anti–phosphorylated Akt (anti-pAkt; 559029) was from PharMingen, BD Biosciences (San Diego, CA). ERK1/2 MAPK (9102) antibody was from New England Biolabs (Beverly, MA). Cyclin D1, Akt, ODC, CDK4, Cyclin E, eIF2α, and ERK1/2 antibodies were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Phosphorylated mTOR, p-eIF4E, phosphorylated p70s6k (p-p70s6k), p-Akt, HRI, phosphorylated eIF2-α (p-eIF2-α), p58 and phosphorylated ERK1/2 (p-ERK1/2) were from Cell Signaling Technology (Beverly, MA). Rabbit anti-pPKR (pT451) was obtained from Invitrogen BioSource (Carlsbad, CA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was purchased from Chemicon (Temecula, CA). Tran 35S-label was from MP Biomedicals (Solon, OH). PKR antibody was purchased from Sigma (St. Louis, MO).
Cell Culture
Human breast cancer cell lines MDA-MB-231, MDA-MB-468 and MCF-7 were obtained from American Type Culture Collection. MDA-MB-231 and MCF-7 were cultured in DMEM containing sodium pyruvate, 10 mM non-essential amino acids, 2 mM L-glutamine, 1 μg/ml insulin, and 10% fetal bovine serum (FBS). MDA-MB-468 were cultured in DMEM containing 10 mM non-essential amino acids, 2 mM L-glutamine, 1 μg/ml insulin, and 10% fetal bovine serum (FBS) without sodium pyruvate. Human mammary epithelial (HME) was obtained from Cambrex Bio Sciences (Walkersville, MD). MCF-10A1 (R) cells, a spontaneously immortalized untransformed human mammary epithelial cell line (MCF-10A transformed with H-Ras oncogene) was obtained from Robert J. Pauley (Barbara Ann Karmanos Cancer Institute, Detroit, Michigan). HME and MCF-10A1 (R) cells were cultured in DMEM:Ham’s F-12 (1:1) supplemented with 5% equine serum, 10 mM HEPES, 10 μg/ml insulin, 20 ng/ml epidermal growth factor, 100 ng/ml cholera enterotoxin, and 0.5 μg/ml hydrocortisone. For experimental purposes, cells were grown in 5% FBS, allowed to seed overnight and treated with drugs for various time periods.
Cytostasis Assay
Cells (7.5 × 105 cells/well) were seeded in 6-well plates. After overnight seeding, the medium was changed and fresh drugs were added. The cells were collected from 0 to 24 h, and viability was determined by trypan blue exclusion method and counted on a hemocytometer.
Cell Cycle Analysis
Cells suspended in hypotonic DNA staining buffer (0.1% sodium citrate/0.3% Triton X-100/0.01% propidium iodide/0.005% ribonuclease A) were incubated for 15 min at 4°C and subjected to fluorescence-activated cell sorting (FACS) to analyze the percentage of cells in the different phases of the cell cycle (16).
Measurement of Total Protein Synthesis by Metabolic Labeling
MDA-MB-231 cells (2×106) were plated overnight in media containing DMEM and 5% FBS. Cells treated with or without DETA NONOate (1 or 2mM) for 6 or 16h were starved for 3h in methionine- cysteine-free media (GIBCO-BRL), after which they were labeled with 100μCi/ml of a mixture of [35 S] methionine for 3h. After removal of the radioactive media, the cells were collected, washed twice with PBS and lysed on ice for 30 min in 1ml of lysis buffer [10mM Tris/HCl buffer, pH 7.4/150mM NaCl/1mM EDTA, pH 8.0/0.1% (v/v) Triton X-100] containing 0.2mM PMSF, 1μM pepstatin A and 1μM leupeptin. Cell lysates were clarified by centrifugation at 2200g for 5min and the protein concentrations of the supernatant was determined using Protein Assay Dye Reagent Concentrate (Bio-Rad Laboratories, Inc). Equal amounts of lysates were spotted on Whatman 3MM and washed with TCA. Proteins with incorporated [35S] methionine were measured in a scintillation counter.
Western Analysis
Cells were lysed in cell lysis buffer containing (50 mM HEPES (pH 7.5); 1 mM DTT, 150 mM NaCl, 1 mM EDTA, 0.1% Tween 20, 10% glycerol, 10 mM β-glycerophosphate, 1 mM NaF, 0.1 mM orthovanadate, 10 μg/ml leupeptin, 10 μg/ml aprotinin and 0.1 mM PMSF) and incubated at 4° C for 30 min for analysis of cytosolic proteins. Protein concentration was measured using Bio-Rad protein assay reagent. For analysis of total cellular protein, the cells were lysed in RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 1% Nonidet P-40 and 0.5% sodium deoxycholate) containing 0.5% SDS and protein concentrations were measured using BCA dye concentrate (Pierce). For denaturing gels, lysates (30 μg) were boiled in Laemmli sample buffer containing beta-mercaptoethanol (β-ME) and resolved electrophoretically on 10 % SDS-polyacrylamide gel. For native gels, the lysates (30 μg) were treated with Laemmli sample buffer without β-ME and loaded on 4–20% gradient polyacrylamide gel without boiling. The gels were electrotransferred to a polyvinylidine difluoride membrane (Bio-Rad) using a tank blot procedure (Bio-Rad Mini Protean II). The membranes were incubated with various primary antibodies (1:1000 dilution) and respective horseradish peroxidase-linked to secondary antibody (1:1000 dilution) (Amersham Corp., Piscataway, NJ) for 1h. Immunoreactive bands were visualized by ECL detection system (Amersham).
Immunoprecipitation and Kinase Assay
Cells were lysed in lysis buffer containing 50mM Tris/HCl, pH8.0, 300mM NaCl, 10mM MgCl2, 0.5% Igepal Ca-630 (Sigma), 1mM EDTA and antiprotease cocktail (Roche). 500μg of supernatant were precleared by incubation for 4h at 4°C with Protein A/G–agarose (Santa Cruz Biotechnology) and with Protein A/G-agarose preincubated with pre-immune serum. The pre-cleared lysate was adjusted to 150mM NaCl and 0.25% Igepal Ca-630 and subjected to overnight immunoprecipitation on a rotating wheel at 4°C using rabbit antiserum or anti- HRI or PKR antibody. The mixtures were incubated further for 4h at 4°C with 50μl of Protein A/G–agarose. After 5 washes in 50mM Tris/HCl, pH8.0, 0.25% Igepal Ca-630, 1mM EDTA and 150mM NaCl, the immunoprecipitates were analyzed by kinase assay using myelin basic protein as the substrate.
Inhibition of PKR by small interfering RNA (si-RNA)
Synthetic si-RNA targeting human PKR (Gene accession number NM_002759, Gene ontology, 4694) in MDA-MB-231 cells and random oligo VIII as a control for nonspecific si-RNA effects were purchased from Dharmacon Research (Lafayette, CO). Amaxa nucleofecter was used for the transfection (~85% efficiency) for 24 hours following which the cells were treated with and without DETA-NONOate for another 24 hours. We also used another PKR si-RNA (Dharmacon, NM_002759, Gene ontology, 6915) in some of our experiments to confirm the specificity of PKR siRNA and rule out the possibility of off-target si-RNA effects in our experiments.
Cell Fractionation
The cell fractionation was done using nuclear and cytoplasmic extraction reagents from Pierce. The fractionation was done according to the manufacturer’s protocol.
Statistical Analysis
Each value represents the mean SE of at least three separate experiments in each group. Comparisons of means were made by using Student’s t test for unpaired values. When more than two means were compared, an ANOVA with repeated measurements was used. If a significant p value was found, Scheffé’s test for multiple comparisons was used to identify differences among groups. Values were considered significant when p was <0.05.
Results
Low and high NO stresses affect protein synthesis at different levels
It is well established that the phosphorylation of eIF2-α, a key player in protein synthesis, attenuates the rate of translation of capped cellular mRNAs (8, 18). NO has been found in a few cell types to suppress protein synthesis by eliciting the phosphorylation of eIF2-α (14). We initially assessed the effect of low and high NO stress on eIF2-α and total protein synthesis in breast cancer cell line MDA-MB-231. The level of Cyclin D1, with its short half-life, was used as an indicator of protein synthesis. Low NO stress as generated by 1mM DETA NONOate induced ~ 2.1 fold up regulation of p-eIF2-α levels and ~50% reduction of Cyclin D1 levels at 16h (Fig. 1A). However, a prominent ~5 fold increase of p-eIF2-α levels and a sharper decline of Cyclin D1 (~80%) were observed with high NO stress as generated by 2mM DETA-NONOate at 16h (Fig 1A). The levels of eIF2-α remained unaltered in all the above treatments (Fig 1A). We also found that pretreatment of cells with an NO quencher, cPTIO (50μM), attenuated the effect of low and high stress on of eIF2-α (data not shown).
Figure 1.
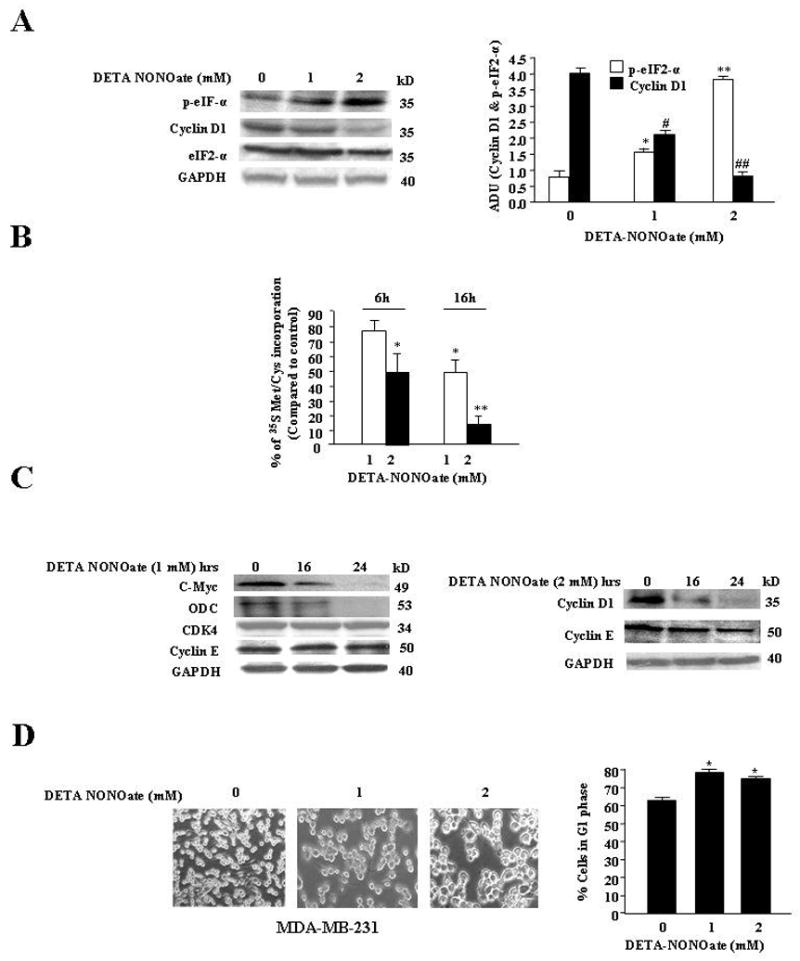
A, High and low NO stress induced differential increase of p-eIF2-α levels and decrease in Cyclin D1 levels while eIF2-α level remain unaltered. Left, MDA-MB-231 cells were treated with DETA-NONOate (1mM or 2mM) and prepared for immunoblot analysis at 24hrs. The membrane was probed with Cyclin D1, p-eIF2-αeIF2-αand GAPDH Abs. Results are representative of three different experiments. Right, Arbitrary densitometric units (ADU) of Figure 1A. Results are expressed as a mean of three different experiments ± SE. *, p≤ 0.02; and **, p≤ 0.001 compared to control untreated p-eIF2-α and #, p≤ 0.02; ##, p≤ 0.001 compared to control untreated Cyclin D1 group. B, High NO stress induced sharper decline in protein synthesis. MDA-MB-231 cells were treated with DETA-NONOate (1mM or 2mM) before exposure to [35S] methionine and [sS] cysteine. The cells were harvested at 6 and 16 hours as described in “Materials and Methods.” The labeled proteins were measured with a scintillation counter. Results are representative of three different experiments. *, p≤ 0.02; **, p≤ 0.005 compared to control untreated cells. C Left, Low NO stress induced decline in the levels of short half-life proteins but not long half-life proteins. MDA-MB-231 cells were treated with DETA-NONOate (1mM) at different time points and prepared for Western analysis as described in “Materials and Methods.” The membrane was probed with Cyclin D1, c-myc, ODC, Cyclin E, CDK4 and GAPDH Abs. Results are representative of three different experiments. Right Panel: High NO stress induced decline in the levels of short and long half-life proteins. MDA-MB-231 cells were treated with DETA-NONOate (2mM) at different time points and prepared for Western analysis. The membrane was probed with Cyclin D1, Cyclin E and GAPDH Abs. Results are representative of three different experiments. D, High NO stresses induced morphological changes in MDA-MB-231 cells. Left, MDA-MB-231 cells were treated with and without DETA-NONOate (1 and 2mM). Cell numbers for 1mM and 2mM NO treated cells were comparable. After 24hrs, cells were photographed with LEICA DM IRBE microscope at 10× magnification. Results are representative of three different experiments. D Right, High NO stress induced increase in G1 phage of cell cycle. FACS analysis of control and 1 mM and 2mM DETA-NONOate treated MDA-MB-231 cells at 24hrs. The cells were prepared for FACS analysis, as described in Materials and Methods, and analyzed in a FACScalibur flow cytometer. The number of cells in each phase of the cell cycle was obtained by MODFIT software. Results are expressed as a mean of three different experiments. *, p≤ 0.05 compared to untreated control without DETA NONOate treatment.
To determine if NO-induced stress inactivated the translational machinery to induce a global decline of protein synthesis, cells pretreated with DETA NONOate (1and 2 mM) were metabolically labeled with 35S methionine for 4h. Incorporation of the labeled methionine in newly synthesized protein was measured by liquid scintillation counter. Low and high NO stress led to a ~40% and 85% decline in labeled proteins respectively at 16h compared to untreated cells (Fig 1B). Since the above experiments indicated that NO induced a global decline in protein synthesis, we assessed the levels of Cyclin D1, other cyclins, CDKs and proteins with a similar half-life. In MDA-MB-231 cells, low NO stress induced a down regulation of short half-life proteins like ODC and C-Myc, while levels of longer half-life proteins (Cyclin E and CDK4) remained unaltered (Fig 1C, left). High NO stress (2mM DETA NONOate) however, led to a sharp decline in Cyclin D1, while Cyclin E, a long half-life protein was also reduced by 50% (1C, right).
We further examined the effect of NO stress on cell proliferation. We found that both low and high NO stress induced cytostasis as there was no increase in cell number, which was observed as early as 16h (data not shown). However, there was a distinct difference in the effect on the cellular morphology between the levels of NO stress. With low levels of NO stress the cells were cytostatic but retained the flattened morphology of control cell, while with high NO stress, the cytostatic cells changed their flattened morphology to become round (Fig 1D, left). In the cell cycle analysis, we observed that with low and high NO stress, there was a 20–22% increase in G1 cell cycle arrest (Fig 1D, right). Treatment of MCF-7 and MDA-MB-468 with low and high NO stress also increased p-eIF2-α levels and reduced protein synthesis (data not shown).
Low NO stress activated HRI while high NO stress also activated PKR in breast cancer cells
We further examined the kinases that are up stream of eIF2-α to understand the mechanism by which low and high NO stress differentially inactivates eIF2-α. Specific stresses such as oxidative stress, heme deprivation or viral infection in mammalian cells have been found to phosphorylate eIF2-α via four upstream kinases: double stranded RNA-dependent protein kinase R (PKR), heme regulated inhibitor (HRI), general control non-derepressible-2 (GCN2), and double-stranded RNA-activated protein kinase-like ER kinase (PERK) (19–20). Since NO has an affinity for heme, we initially examined whether HRI contributed to NO-induced up regulation of p-eIF2-α levels and cell cycle arrest in breast cancer cells. HRI is reported to be present in an inactive closed conformation under normal conditions. Stress or appropriate stimuli induces auto-phosphorylation of HRI and its maturation to an open active configuration (21–25). Low amounts of active HRI were detected in the cytosolic fraction of MDA-MB-231 cells and NO (low and high stress) increased these levels by 24hr (Fig 2A, left). Similarly, NO treatment also induced activation of HRI in MDA-MB-468 and MCF-7 cells (data not shown). To access the role of NO induced activation of HRI in up regulating p-eIF2-α, HRI was rendered inactive with carbon monoxide (CO) or hemin (data not shown), both of which have been shown to inhibit HRI activation (22, 23, and 26). Cells pretreated with CORM A1 (500uM), a CO-donor, attenuated the increase in p-eIF2-α and decline in Cyclin D1 by low NO stress (Fig 2A, right). Densitometric quantitation of increase in p-eIF2-α and decline in Cyclin D1 by low NO stress is shown in Fig 2 B. However, CORM-A1 or hemin treatment could not attenuate the high levels of p-eIF2-α seen with high NO stress (data not shown). This suggested an involvement of additional kinases in high NO stress induced stimulation of eIF2-α.
Figure 2.
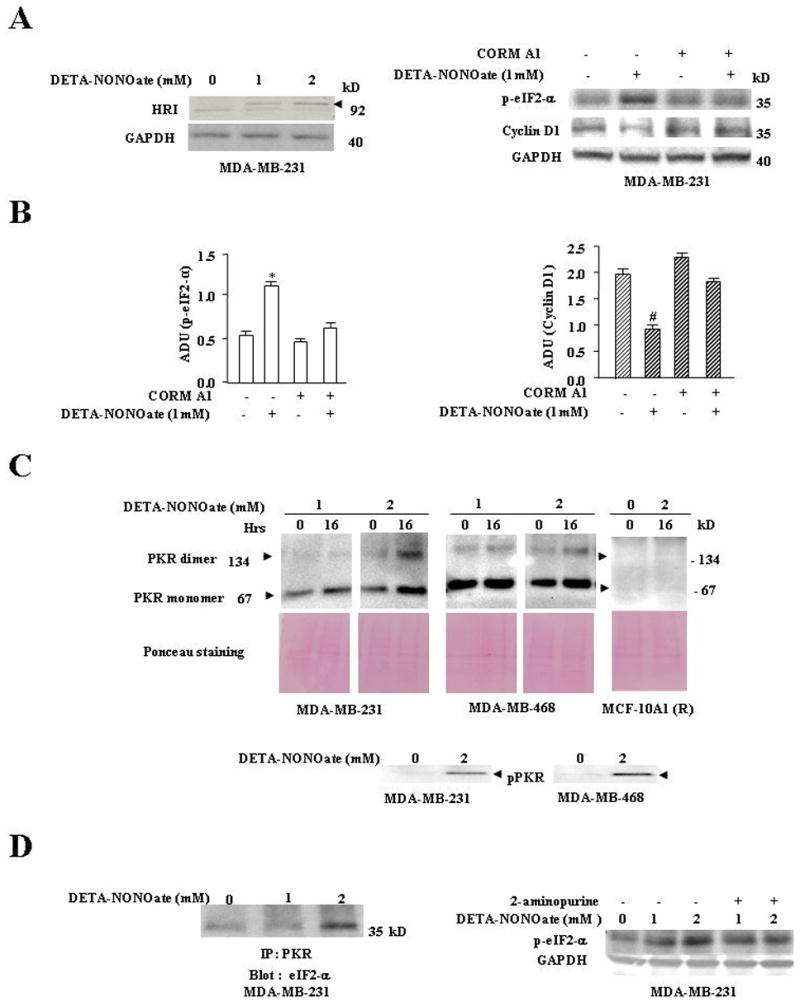
A Left, NO induced increase in the levels of HRI. MDA-MB-231 cells were treated with DETA-NONOate (1mM or 2mM) and harvested at 24hrs. The cells were prepared for Western analysis as described in “Materials and Methods.” The membrane was probed with HRI and GAPDH Abs. Results are representative of three different experiments. Right, CO attenuated NO-induced increase of p-eIF2-α levels and decrease of Cyclin D1 levels. MDA-MB-231 cells were treated as follows: Lane 1, untreated cells; Lane 2, cells treated with DETA-NONOate (1mM); Lane 3, cells treated with CORM A1 (500 μmol); Lane 4, cells pretreated CORM A1 (500 μmol) for 1hr before exposure to DETA-NONOate (1mM). The cells were harvested at 24hrs and prepared for western analysis. The membranes were probed with p-eIF2-αCyclin D1, and GAPDH Abs. Results are representative of three different experiments. B, Arbitrary densitometric units (ADU) of Fig. 2A, right. Results are expressed as a mean of three different experiments ± SE. *, p≤ 0.02 compared to control p-eIF2-α group without CORM A1and DETA NONOate treatment; and #, p ≤ 0.01 compared to control Cyclin D1 group without CORM A1and DETA NONOate treatment. C, High NO stress induced significant increase of dimeric and monomeric forms of PKR. MDA-MB-231, MDA-MB-468 and MCF-10A1® cells were treated as follows: 0 hrs, cells untreated; 16 hours, cells treated with DETA-NONOate (1mM or 2mM) for 16 hrs as indicated. The cells were harvested at their respective time points and prepared for western analysis as described in “Materials and Methods.” The membranes were probed with PKR Abs and Ponceau-S stained to confirm equal loading of sample in the gel. C, lower panel shows western blots probed with pPKR Ab. Results are representative of three different experiments. D, Left Panel, High NO stress sharply increased PKR association with eIF2-α. MDA-MB-231 cells were treated with 1mM and 2mM DETA-NONOate for 16hrs. The cells were prepared for immunoblot analysis as described in “Materials and Methods.” Immunoprecipitated PKR was analyzed for eIF2-α association. Results are representative of three different experiments. Right Panel, High NO stress mediated up-regulation of eIF2-α levels was attenuated with 2-aminopurine. Treatment of 2-aminopurine, a PKR inhibitor, did not affect the increase of p-eIF2-α levels with low NO stress (1mM) but did attenuate the increase of p-eIF2-α levels with high NO stress (2mM). MDA-MB-231 cells were treated as follows: Lane 1, untreated cells; Lane 2 and 3, cells treated with DETA-NONOate 1mM and 2mM respectively; Lane 4 and 5, cells pretreated with 2-aminopurine (2mM) for 45 minutes before exposure to DETA-NONOate 1mM and 2mM; The cells were harvested at 24hrs and prepared for western analysis as described in “Materials and Methods.” The membranes were probed with p- eIF2-α and GAPDH Abs. The results are representative of three different experiments.
PKR, a serine/threonine protein kinase phosphorylates eIF2-α and is over expressed in human breast cancer (27–28). We assessed the role of PKR in low and high NO-induced inhibition of cell proliferation. PKR was analyzed on native PAGE gels, where both the inactive monomeric and active dimeric forms of PKR could be detected in MDA-MB-231 and MDA-MB-468 cells after 16 hrs of treatment with high NO (2mM DETA NONOate) (Fig 2C top panel). This dimeric form of PKR at high NO was associated with phosphorylation of PKR in human breast cancer cells (Fig 2C bottom panel), whereas phosphorylated PKR was not detected in normal HME cells (data not shown). Low NO (1mM DETA NONOate) stress up regulated the cytosolic monomeric forms of PKR by ~2 fold in MDA-MB-231 and MDA-MB-468 without significant dimeric forms; whereas in normal mammary epithelial MCF-10A1 (R) cells, we were unable to detect either forms of PKR (Fig 2C top panel). We also found that these cell lines when repeatedly treated with SNAP, another NO donor, there was significant up regulation of dimeric forms of PKR (data not shown). We next assessed whether PKR was associated with eIF2-α in these breast cancer cells after NO treatment. We observed that NO treatment of both MDA-MB-231 and MDA-MB-468 cells increased association of PKR with eIF2-α as determined by immunoprecipitation (Fig 2D, left). To determine the role of PKR activation in the phosphorylation of eIF2-α during NO treatment, PKR was inactivated by 2-aminopurine, a pharmacological inhibitor of PKR (29). Pretreatment with 2-aminopurine attenuated high NO stress induced increase in p-eIF2-α levels by ~55% in MDA-MB-231 cells (Fig 2D, right). Similar results were obtained in MCF-7 and MDA-MB-468 cells, where 2-aminopurine pretreatment also attenuated the increase in p-eIF2-α (data not shown). In addition, we assessed whether high NO stress-induced increase of p-eIF2-α could be completely attenuated by inhibition of both PKR and HRI in MDA-MB-231 cells. Cells were pretreated with the 2-aminopurine (PKR inhibitor) and hemin (HRI inhibitor) before they were exposed to NO. We found that while high NO stress up regulated p-eIF2-α levels, pretreatment with 2-aminopurine and hemin attenuated the increase in p-eIF2-α levels to near control (data not shown).
These results demonstrated that HRI was activated by both low and high NO stress and PKR was activated only by high stress, leading to low and high levels of p-eIF2-α respectively.
Breast cancer cells were more susceptible to high NO stress induced inhibition of protein synthesis, compared to normal cells
To further validate the exclusive role of PKR in mediating the effects of high NO stress on protein synthesis we used PKR si-RNA to specifically reduce PKR levels in breast cancer cells. PKR si-RNA induced a ~70% decline in PKR levels after 48h, as accessed by immunoblot analysis (Fig 3A). PKR si-RNA treated cells when further exposed to high stress did not increase in p-eIF2α levels, which contrasts to ~2. fold increase found with random si-RNA treatment (Fig 3A). NO-induced a sharp decline in Cyclin D1 levels in random si-RNA treated cells, which was found attenuated in PKR si-RNA treated cells (Fig 3A). Further examination of NO-induced cytostasis in PKR and random si-RNA treated cells showed that NO induced only ~25% decline in proliferation of PKR si-RNA treated cells compared to ~50% decline in random si-RNA treated cells (Fig 3B, left). Morphological changes associated with high NO stress were also found attenuated with PKR si-RNA treatment (data not shown). We also examined the reversibility of NO induced cell cycle arrest in random and PKR si-RNA treated cells. Cells were treated with low and high NO stress for 24h before the media was changed and cells allowed to recover for another 24h. While recovery of random si-RNA treated cells from low NO stress was reversible, recovery from high NO stress was irreversible even till 48h (Fig 3B, right panel). In PKR si-RNA cells, however, recoveries from low and high NO stress was reversible (Fig 3B, right). These results suggest that NO stress induced PKR activation led to a stronger and less reversible effect on protein synthesis.
Figure 3.
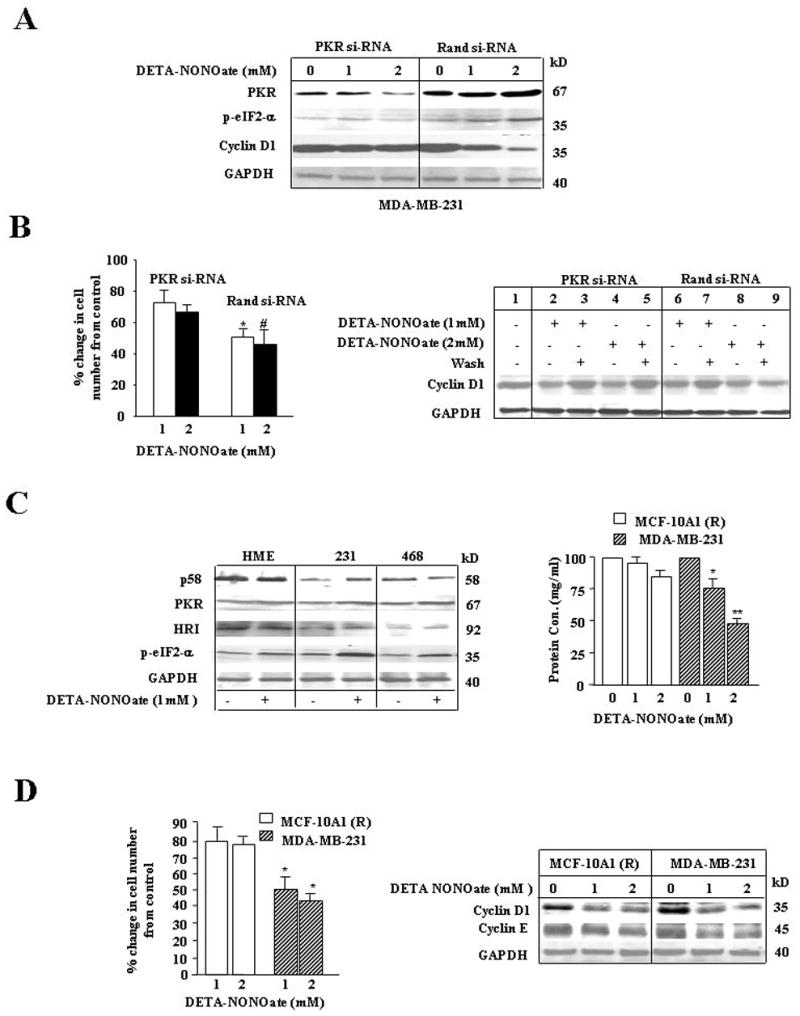
A, NO exposure to PKR si-RNA transfected cells compared to control had lower levels of p-eIF2-α. NO treated control random si-RNA transfected cells upregulated PKR and p-eIF2-α levels and downregulated Cyclin D1 levels. MDA-MB-231 cells were transfected with random si-RNA or PKR si-RNA using solution V from the AMAXA nucleofector kit. 24hrs after transfection, cells were treated with DETA-NONOate (1 or 2mM) and harvested after another 24hrs. The cells were prepared for western analysis. The membrane was probed with PKR, p-eIF2-αCyclin D1 and GAPDH Abs. Results are representative of three different experiments. B, Left, NO induced decline in proliferation was more dramatic in cells treated with random si-RNA compared to PKR si-RNA. MDA-MB-231 cells transfected with PKR si-RNA or random si-RNA were treated with DETA-NONOate (1mM or 2mM). The cells were harvested at 24hrs and counted using the trypan blue method. Results are expressed as a mean of three experiments ± SE. *, p≤ 0.02 compared to PKR si-RNA group treated with 1mM DETA NONOate; #, p≤ 0.05 compared to PKR si-RNA group treated with 2mM DETA NONO ate. B, Right, Cells with PKR si-RNA had complete recovery of cyclin D1 levels after removal of NO. MDA-MB-231 cells were treated as follows: Lane 1, untreated control cells; PKR si-RNA and random si-RNA transfected cells were treated with DETA-NONOate (1mM) for Lanes 2, 3, 6 and 7. PKR si-RNA and random si-RNA transfected cells were treated with DETA-NONOate (2mM) for Lanes 4, 5, 8 and 9; after 24hrs of DETA-NONOate treatment, cells were exposed to NO-free media for another 48hrs in Lanes 3, 5, 7, and 9. All cells were harvested and prepared for western analysis. The membrane was probed with Cyclin D1 and GAPDH Abs. Results are representative of three different experiments. C, Left, Human mammary epithelial (HME) cells had higher levels of p58, a PKR inhibitor, and low levels of p-eIF2-α compared to breast cancer cell lines. With NO stress, breast cancer cell lines had a higher increase in PKR, HRI and p58 levels compared to the levels of PKR, HRI and p58 in human mammary epithelial HME cells. HME, MDA-MB-231 (231) and MDA-MB-468 (468) cell lines were treated with DETA-NONOate (1mM) and prepared for western analysis at 24 hrs. The membranes were probed with p58, PKR, HRI, p-eIF2-α and GAPDH Abs. Results are representative of three different experiments. C, right, NO induced sharper decline in protein concentration in breast cancer cells compared to MCF-10A1 (R) cells. MCF-10A1 (R) and MDA-MB-231 cells were treated with or without DETA-NONOate (1 or 2 mM). After 24hrs, cells were harvested and lysed. Protein concentrations were measured using Bradford reagent. Results are expressed as a mean of three experiments ± SE. *, p≤ 0.05 and **, p≤ 0.001 compared to MDA-MB-231 control group without DETA NONOate treatment. D Left, NO stress induced sharper decline in MDA-MB-231 cell proliferation compared to MCF-10A1 (R). No significant change in cell number was found between high and low NO stress treated MCF-10A1 (R) or MDA- MB-231 cells. MCF-10A1 (R) and MDA-MB-231 cells were treated with DETA-NONOate (1 or 2mM) and viable cell numbers were counted using the trypan blue method. Results are expressed as a mean of three experiments ± SE. *, p≤ 0.02 compared to control MDA-MB-231 group without DETA NONOate treatment. D Right, High NO stress induced decline in Cyclin E levels in MDA-MB-231 cells but not MCF-10A1 (R) cells. MCF-10A1 (R) and MDA-MB-231 cells were treated with or without DETA-NONOate (1or 2mM) and prepared for western analysis. The membrane was probed with Cyclin D1, Cyclin E and GAPDH Abs. Results are representative of three different experiments.
It has been reported that p58 is an inhibitor of PKR and remains tightly associated with PKR in cells that over expresses this inhibitor. The levels of inhibitor p58 were assessed in human mammary epithelial (HME), as well as MDA-MB-231 and MDA-MB-468 cells. We found higher levels of inhibitor p58 in HME cells compared to MDA-MB-231 and MDA-MB-468 cells, while HRI levels were comparable in both cell types (Fig 3C, left). Although the levels of HRI was comparable, we found that high NO stress induced lower levels of p-eIF2-α (1–1.5 –fold) in HME cells, compared to the higher levels of p-eIF2-α (2–2.5-fold) observed in breast cancer cells (Fig 3C, left). Due to low free PKR and p-eIF2-α levels in HME and MCF-10A1 (R) (data not shown) cells, we examined the effect of low and high NO stress on the total cellular protein concentration and also cell proliferation. In mammary epithelial cells there was no significant decline of total cellular proteins when exposed to low and high NO stress (Fig 3C, right). This is in sharp contrast to breast cancer cell line MDA-MB-231, where the decline of total cellular protein was prominent with both low and high NO stress (Fig 3C, right). We also compared the effect of low and high NO stress on cell proliferation in mammary epithelial and breast cancer cells. In response to low and high NO stress, the decline in cell proliferation was more prominent in breast cancer than mammary epithelials (Fig 3D, left). We also investigated the effect of high and low NO stress on Cyclin D1 and E levels in MCF-10A1 (R) cells. We observed that while the decline in Cyclin D1 in MCF-10A1 (R) cells was comparable to cancer cells, there was no significant decline in Cyclin E (Fig 3D, right). These results suggested that activated PKR in breast cancer cells led to a prominent increase of p-eIF2-α and a strong inhibition of protein synthesis.
NO stress potentiated PKR activation after serum starvation or hydrogen peroxide treatment
Although PKR is over expressed in breast cancer cells, we observed that high NO stress was required for its activation. Since tumors are subjected to numerous stresses in vivo, we wanted to assess whether low NO stress in the presence of another cellular stress could potentiate PKR activation. To determine NO-induced potentiation of PKR activation in the presence of another stress, cells were exposed to 300μM hydrogen peroxide (H2O2), an oxidative stress, and lower NO stress (DETA-NONOate 300μM or 500μM). We found that while H2O2 or lower NO stress did not induce PKR activation alone, simultaneous exposure of the cells to lower NO and H2O2 dramatically increased PKR activation when analyzed by native gel electrophoresis (Fig 4A). Similar potentiation of PKR activation was observed in serum starved cells exposed to lower NO stress (Fig 4A). We further examined the effect of these stresses, alone or in combination, on eIF2-α activation. Increased levels of p-eIF2-α was observed in cells with a combination of NO and another stress (Fig 4B, left). We examined the effect of simultaneous exposure of NO and H2O2 on cell proliferation and observed that the two stresses led to dramatic decline in cell number compared to either stress alone (Fig 4B, right). We also assessed the effect of these stresses had on protein synthesis by monitoring the levels of total cellular protein, Cyclin D1 and Cyclin E in these cells. We observed that NO exposure to H2O2 or serum starved cells had a lower total protein concentration (data not shown) and lower levels of Cyclin D1 and Cyclin E than either stress alone. Cell cycle analysis was also performed in cells where NO potentiated PKR activation. The potentiation of PKR activation with NO treatment was also found in cells pre-treated with either tumor necrosis factor-α (TNF-α) or wortmannin (PI-3 kinase inhibitor) compared to stress alone (data not shown).
Figure 4.
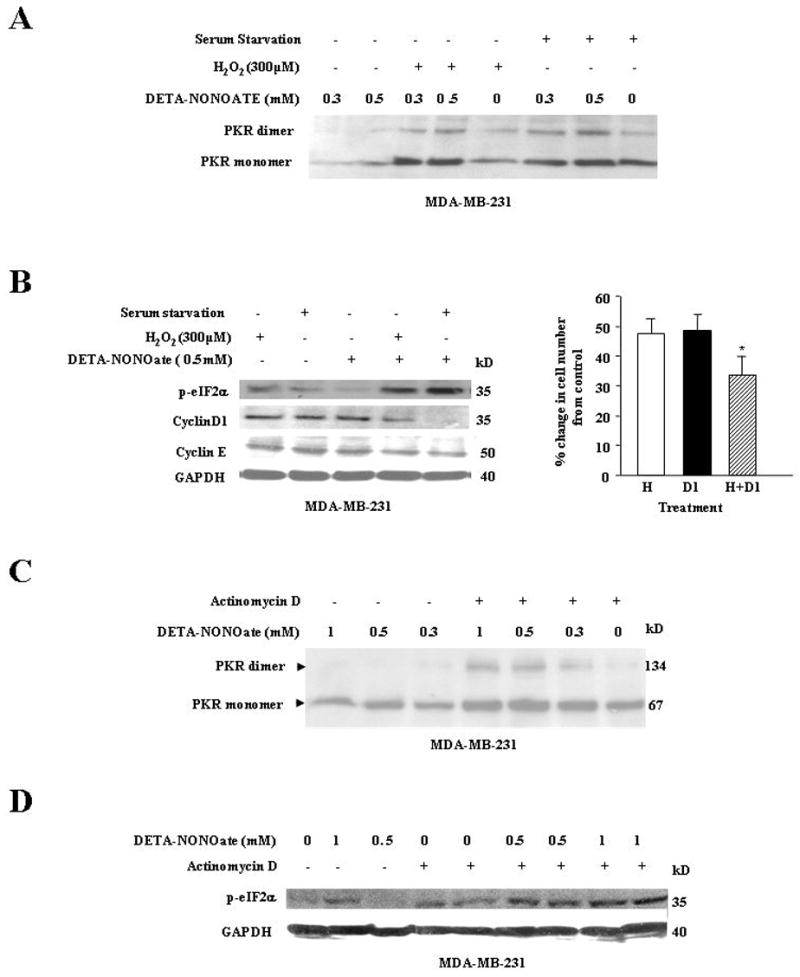
A, Lower NO stress (0.5 mM) with either hydrogen peroxide or serum starvation stress sharply increased PKR dimerization. MDA-MB-231 cells were treated at follows: Lane 1 and 2, cells treated with DETA-NONOate (0.3 or 0.5mM); Lane 3 and 4, cells co-exposed to hydrogen peroxide (50μM) and DETA-NONOate (0.3 or 0.5mM) exposure; Lane 5, cells treated with hydrogen peroxide (50μM); Lane 6 and 7, cells were serum starved for 24hr before DETA-NONOate (0.3 or 0.5mM) exposure; Lane 8, cells were serum starved. The cells were harvested at 24hrs and prepared for western analysis. The membrane was probed with PKR Ab and Ponceau-S stained to confirm equal loading of sample in the gel. Results are representative of three different experiments. B Left, Low NO (0.5 mM) stress with either hydrogen peroxide or serum starvation stress sharply increased p-eIF2-α levels and decreased Cyclin D1 and Cyclin E levels. MDA-MB-231 cells were treated as follows: Lane 1, cells treated with hydrogen peroxide (50μM); Lane 2, cells were serum starved; Lane 3, cells treated with DETA-NONOate (0.5mM); Lane 4, cells co-exposed to hydrogen peroxide (50μM) and NO (0.5mM); Lane 5, cells were serum starved for 24hrs before NO (0.5mM) exposure. The cells were harvested at 24hrs and prepared for western analysis. The membrane was probed with p-eIF2-αCyclin D1, Cyclin E and GAPDH Abs. Results are representative of three different experiments. B Right, Hydrogen peroxide with low NO stress (1mM) compared to either stress alone induced a greater decrease in cell proliferation. MDA-MB-231 cells were treated with hydrogen peroxide (50μM) or DETA-NONOate (1mM) alone or with both stresses. The cells were harvested and counted using the trypan blue method. Results are expressed as a mean of three different experiments ± SE. (H=H2O2 and D1=1mM DETA NONOate). *, p≤ 0.02 compared to control untreated cells. C, Low NO stress with actinomycin D induced a sharper increase of PKR dimerization compared to NO or actinomycin D stress alone. MDA-MB-231 cells were treated as follows: Lane 1, 2, and 3, cells were treated with 1mM, 0.5 mM and 0.3mM of DETA-NONOate respectively; Lane 4, 5, and 6, cells were pretreated with actinomycin D (10 μM) before exposure to 1mM, 0.5mM and 0.3mM of DETA-NONOate respectively; Lane 7, cells treated with actinomycin D (10 μM). The cells were harvested at 24h and analyzed on a native PAGE. The membrane was probed with anti-PKR Ab. Results are representative of three different experiments. D, Low NO stress with actinomycin D sharply up regulated p-eIF2-α levels compared to either stress alone. NO induced PKR dimerization and phosphorlyation of eIF2-α was dependent on NO concentration. MDA-MB-231 cells were treated at follows: Lane 1, cells untreated; Lane 2 and 3, cells treated with DETA-NONOate (1 and 0.5mM); Lane 4 and 5, cells treated with actinomycin D (10 μM), Lane 6, 7, 8, and 9, cells pretreated with actinomycin D (10 μM) before exposure to DETA-NONOate (0.5 or 1 mM). Cells were harvested at 24hrs and prepared for western analysis. The membrane was probed with anti-p-eIF2α; and anti-GAPDH Abs. Results are representative of three different experiments.
Most chemotherapeutic agents like paclitaxel and tamoxifen when administered in high concentrations, target cellular DNA replicating machinery to induce cell cycle arrest and induce apoptosis (30, 31). High concentrations of these agents could also induce toxicity to normal cells (30, 31). We investigated whether NO could potentiate PKR activation in the presence of actinomycin D, which targets cellular DNA. While actinomycin D or NO itself could up regulate the monomeric forms of PKR, simultaneous exposure of these stresses to cells dramatically increased PKR activation (Fig 4C). This increase was also reflected in the levels of p-eIF2-α, where increased phosphorylation was observed in cells exposed to NO and actinomycin D (Fig 4D). We, therefore, concluded that lower or low NO stress in the presence of another stress potentiated PKR activation in breast cancer cells.
NO stress induced inactivation of eIF2-α was independent of p- ERK, p-AKT or cGMP levels in breast cancer cells
In this study we found that in breast cancer cells, low NO stress increases monomeric cytosolic forms of PKR, while active forms of PKR was up regulated by high NO stress. To further assess the mechanism by which NO induced up regulation of PKR, we examined the possible involvement of pERK1/2 and pAkt, which have been implicated to be responsive to NO and other stimuli (32–34). We have reported in MDA-MB-231 cells that with NO treatment, there was decline in cyclinD1 levels, Rb was hypophosphorylated and cells underwent cytostasis (16). In the present study we observed that with low and high NO stress (data not shown); the levels of p-ERK1/2 remain elevated, while ERK remain unchanged (Fig 5A, top panel). To further access the role of pERK1/2 in NO induced PKR activation, cells were treated with PD 98059, which inhibits MEK1/2, the up stream kinase of ERK1/2. NO-induced PKR activation occurred even in the absence of ERK activation suggesting no role of ERK1/2 in NO induced PKR activation (data not shown). We also examined the levels of pAkt, which was found high with NO treatment while levels of Akt remain unaltered. We examined the effect of NO on the down stream targets of Akt such as mTOR, eIF-4E and p70s6k, which are critical for the translation of cap-dependent mRNA. We found that in NO treated MDA-MB-231 cells, key players mediating the translation of Cyclin D1 like p-mTOR, p-eIF-4E (data not shown) and p-70s6k remained highly activated, despite the decline in Cyclin D1 levels. (Fig 5A, top panel).
Figure 5.
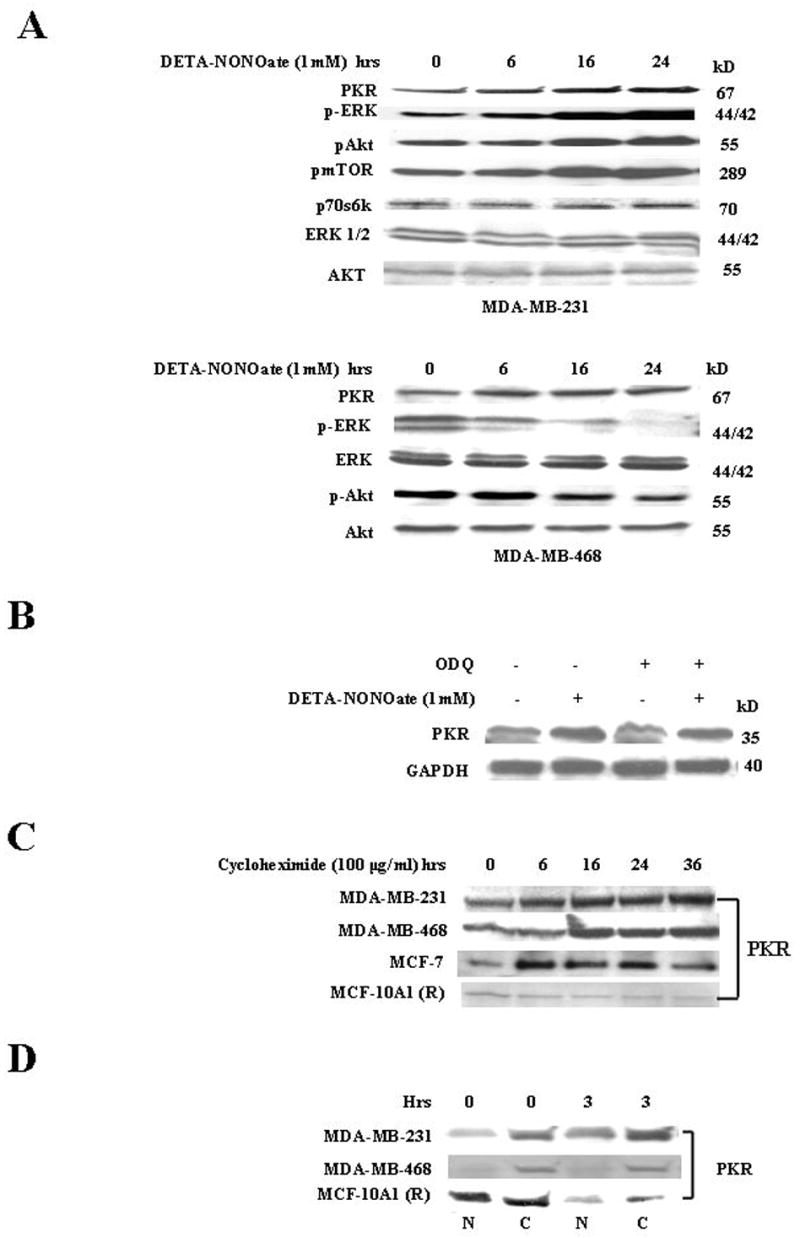
A, DETA-NONOate- induced up regulation of PKR is independent of ERK/AKT pathways Top Panel. MDA-MB-231 cells were treated with DETA-NONOate (1mM) at different time points and prepared for western analysis. The membrane was probed with anti-PKR, p-ERK, pAkt, p-mTOR, Akt, ERK1/2 and p70s6k Abs. Results are representative of three different experiments. Bottom Panel: MDA-MB-468 cells were treated with DETA-NONOate (1mM) at different time points and harvested at 24hrs. The cells were prepared for western analysis and the membrane was probed with anti-PKR, pERK, ERK, p-Akt, and Akt Abs. Results are representative of three different experiments. B, NO induced decline of PKR is independent of cGMP pathway. MDA-MB-231cells were used as follows: Lane 1, untreated cells; Lane 2, cells treated with DETA-NONOate (1mM); Lane 3, cells treated with ODQ (50μM); Lane 4, cells pretreated with ODQ (50μM) for 45 minutes before exposure to NO (1mM). Cells were harvested at 24 hrs and prepared for western analysis. The membrane was probed with anti-Cyclin D1 and anti-GAPDH Abs. Results are representative of three different experiments. C, Cycloheximide-mediated increase of PKR monomers in human breast cancer cells. The cells were treated with cycloheximide (200 μg/mL) and harvested at different time points. The cells were prepared for western analysis and the membrane probed with anti-PKR Ab. Results are representative of three different experiments. D, PKR monomers are equally distributed in cystolic and nuclear fractions of MCF-10A1 (R) cells compared to the predominantly cystolic PKR monomers of breast cancer cells. The cell fractionation was done according to the manufacturer’s protocol. The cells were treated with or without DETA-NONOate (1mM) and harvested at 0 and 3h. The cells were prepared for western analysis and the membranes probed with anti-PKR Ab. Results are representative of three different experiments. N, nuclear and C, cytoplasmic fractions.
We also examined another cell line MDA-MB-468 where we have reported that NO treatment induces decline in the levels of pERK1/2 due to the induction of MAP kinase phosphatase-1 (MKP-1) (32). NO treatment of MDA-MB-468 cells activated PKR even though p-ERK was down regulated; suggesting that activation of PKR was independent of p-ERK levels (Fig 5A, bottom panel). The levels of pAkt were also down regulated in this cell line while Akt remain unchanged. Thus we conclude that NO induced PKR activation is independent of the levels of pERK and pAkt in breast cancer cells. Since cGMP is a well characterized second messenger of NO-mediated signaling, we examined whether it had a role in NO- induced PKR activation. Treatment of cells with ODQ, an inhibitor of NO-mediated cGMP generation, did not significantly affect NO-induced up regulation of PKR (Fig 5B). These results demonstrated that NO-induced PKR activation and up regulation of p-eIF2-α was cGMP independent and did not involve p-ERK or p-AKT/mTOR/eIF-4E pathways.
We assessed PKR stability in a number of breast cancer cell lines MCF-7, MDA-MB-468 and MDA-MB-231 and also in MCF-10A1 (R) cells using cycloheximide. There was a sustained increase in cytosolic PKR, which was high even at 36h in the breast cancer cells (Fig 5C) with cycloheximide treatment. In MCF-10A1 (R) cells, cycloheximide treatment led to a slight decline in PKR levels as early as 3–6h. This PKR decline in MCF-10A1 (R) cells was a sharp contrast to the stability of PKR in breast cancer cells (Fig 5C). The cycloheximide-induced increase of PKR in MDA-MB-231 cells was also attenuated by MG-132 treatment (chemical inhibitor of the 26S proteosome), suggesting a possible role of the ubiquitin proteosomal machinery in PKR up regulation (data not shown).
Since there was a sharp contrast in PKR stability between breast cancer and normal mammary epithelial cells, we examined the nuclear and cytosolic localization of PKR in these cells. In cancer cell lines MDA-MB-231 and MDA-MB-468, PKR was predominant in the cytosolic fraction, whereas in MCF-10A1 (R) cells, PKR was equally distributed in the cytosolic and nuclear fractions (Fig 5D).
Since NO-induced increase in PKR activation was independent of the protein synthesis, total cellular levels of PKR in control and after DETA-NONOate treatment was examined. This was accomplished by lysis of the cells in RIPA buffer, which solubilizes all the cellular membranes, and analysis of PKR in SDS PAGE gels. We did not observe any change in PKR levels in control and DETA-NONOate treated cells suggesting that total cellular PKR levels remain unaltered (data not shown).
Discussion
In this study, we found that breast cancer cells over expressing PKR were more susceptible to NO-stress induced inhibition of protein synthesis than HME and MCF-10A1 (R) cells. PKR is a component of the signal transduction pathways mediating cell growth and responses to stress. It consists of two functionally distinct domains: an N- terminal double stranded RNA (dsRNA) binding regulatory domain, which consists of two ds RNA binding motifs and a C- terminal kinase catalytic domain. PKR exists in a latent monomeric state in which ds RNA binding domains auto inhibit the kinase domain. Binding of dsRNA ligand induces conformational changes in PKR to promote its dimerization and autophosphorylation (28, 35). PKR can also be activated by cytokines and stress signaling pathways that likely operate independent of dsRNA. The most well characterized role of PKR is phosphorylation of eIF2-α at Ser 51, which inhibits translation of all capped mRNA (27, 28). PKR has been found to be a potent negative regulator of cell growth and proliferation in yeast and mammalian cells (36).
Our model system using DETA-NONOate, in breast cancer cells enabled us to expose cells to different levels of stress. Adjustable concentrations of the NO donor and use of pharmacological inhibitors (2-aminopurine, CO and hemin) helped us elucidate the sequential steps in low and high NO stress induced inhibition of protein synthesis, where HRI activation preceded PKR activation (Fig. 6C). We found that NO stress in breast cancer cells up regulated the levels of cytosolic monomeric and dimeric PKR. This activation of PKR led to high levels of p-eIF2-α, sharp decline in protein synthesis, changed cellular morphology, and irreversible cytostasis. Reducing the levels of PKR in these cells by PKR siRNA treatment, however, led to only diminished effects of NO stress on protein synthesis and reversibility of the cytostatic process. HRI consists of two heme-binding domains: a stable heme-binding site and a reversible heme binding site. During activation, HRI is phosphorylated at multiple sites by autophosphorylation, and exists as an active dimer held by non-covalent interactions (21–25). It is reported that CO and NO compete for the stable heme-binding site on HRI (22–23). We found that treatment with a CO donor, CORM-A1, attenuated the activation of HRI by low NO stress. High and low NO stress activated HRI in breast cancer cells, inducing small up regulation of p-eIF2-α and inhibition of protein synthesis with a decline in only short half-life proteins (Fig. 6C).
Figure 6.
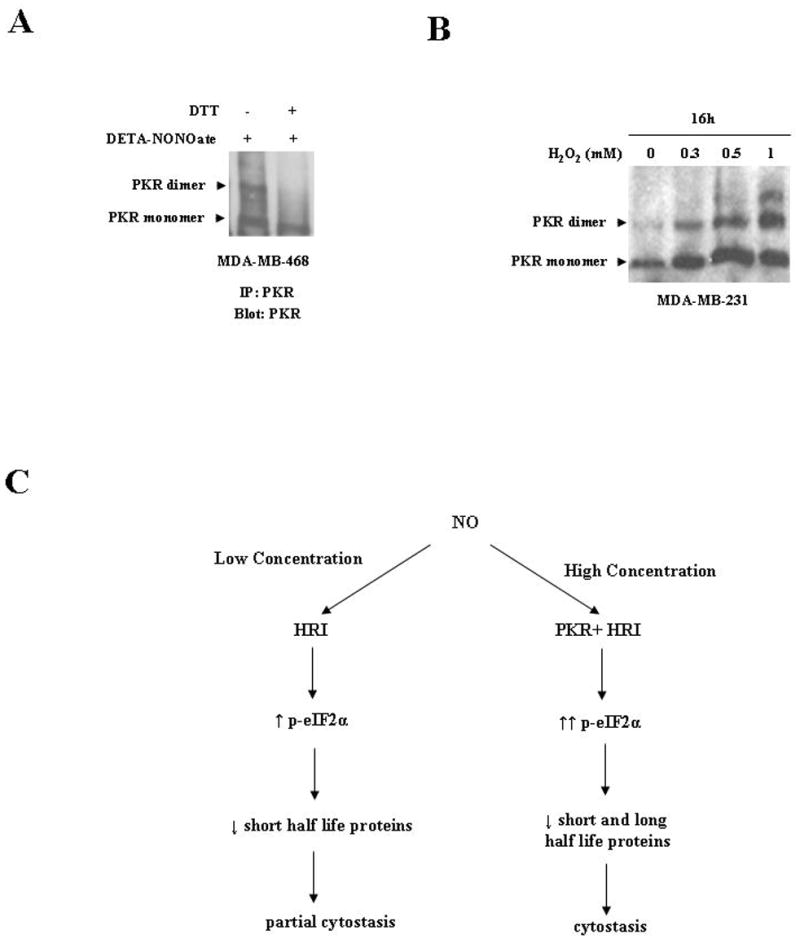
A, Co-incubation of DTT, a reducing agent, with NO immunprecipitated PKR monomers attenuated the increase in PKR dimeric forms. MDA-MB-468 cells were harvested and immunoprecipitated with anti-PKR antibody. The immunoprecipiated PKR was treated as follows: Lane 1, lysates treated with DETA-NONOate (2mM) only; Lane 2, lysates treated with DETA-NONOate (2mM) and DTT (1mM). Cells were incubated for 30 minutes at 37° C and run on native PAGE. The membrane was probed with anti-PKR Ab. Results are representative of three different experiments. B, Hydrogen peroxide treatment induced an increase in PKR dimerization in MDA-MB-231 cells in a concentration dependent manner. MDA-MB-231 cells were treated with various concentrations of hydrogen peroxide (0.3, 0.5, and 1mM) for 16hrs. The cells were harvested at respective time points and prepared for western analysis on native PAGE as described in “Materials and Methods.” The membrane was probed with anti-PKR Abs. C, Flow charts of differential NO stress leading to inhibition of protein synthesis and cytostasis. Low NO concentrations (1mM DETA NONOate) mediate HRI activation, leading to low levels of p-eIF2-α. Phosphorylated eIF2-α induced decline in shorter half life proteins and caused partial cytostasis. High NO concentration (2mM DETA NONOate) mediates the activation of both PKR and HRI, leading to high levels of p-eIF2-α. Elevated peIF2-α level induced sharper decline in long and short half-life proteins, leading to cytostasis.
The effect of “low” and “high” NO stress levels mediating HRI activity is likely the result of a direct interaction with its heme group. This is a first-order process involving direct ligation of NO to the heme moiety. However, the somewhat selective effect of “high” NO stress to mediated PKR activity is likely due to the generation of nitrogen oxide-mediated thiol nitrosation, leading to PKR activation. N2O3 is a predominant nitrosating species derived from NO. Since the generation of N2O3 exhibits higher order kinetics (2nd order in NO), this chemistry is relevant only at high NO levels. Moreover, since this process is dependent on NO concentration to the second power, even a seemingly minor increase in NO (2-fold in this study) can result in an exponential (and not linear) increase in the generation of N2O3. This likely explains the large differences in activity between “low” (approx. 1 μM) and “high” (approx. 2μM) even though there is only a 2-fold difference in the concentration of NO.
Over the past years, there have been efforts to develop methods of using ds RNA to activate PKR selectively in cancer cells to efficiently kill them. One of the strategies took advantage of the many chromosomal rearrangements, truncations and alternative splicing of pre-mRNA that produced mRNA species unique to the cancer cells. Upon hybridization with mRNA, ds RNA molecules were generated that caused selective death of cells through PKR phosphorylation (37, 38). In another study, this strategy has proved effective in inhibiting glioblastoma growth within mouse brain (38). Activation of PKR has been found to also play a role in esophageal cancer cell apoptosis induced by adenoviral vectors expressing TNF-α gene (39). Breast cancer cells over expressing PKR appear to be a strong candidate for the ds RNA therapeutic approach. Here, we report for the first time, a novel strategy for PKR activation. We demonstrate that high NO stress by itself or co exposure of low NO stress simultaneously with another stress (H2O2 or serum deprivation) can activate PKR selectively in breast cancer cells, while HME and MCF-10A1 (R) remained largely unaffected. This strategy also appears highly promising as a non toxic modality for breast cancer treatment, because PKR in normal mammary epithelials remains bound to p58 inhibitor. Although we assessed only the p58 inhibitor of PKR in normal mammary and cancer cells, there are other intracellular regulators, some of which are heat shock proteins, mRNA of p23/TCTP, ribosome, p67 and protein phosphatase 1 (40–44). These intracellular regulators could also contribute to the decreased susceptibility of normal mammary epithelials to NO. In ovarian cancer cells it has been reported that NO is capable of enhancing H2O2-mediated processes by inhibiting its catalytic degradation (45).
We also find that PKR is very stable in breast cancer cells, having a constant level even after 36h of cycloheximide treatment. In examining PKR cellular localization, we find that PKR is predominantly cytosolic in breast cancer cells; while in HME cells, they are equally distributed between nuclear and cytosolic fractions. Mammalian ribosomes have been proposed to be a reservoir of inactive PKR monomers in which PKR is prevented from binding to dsRNA. PKR must be displaced from the ribosomes by dsRNA in order to become activated (42). We think that NO induced a redistribution of compartmentalized PKR, leading to an increase of the cytosolic monomers, which facilitated the formation of dimers. Studies have shown that NO and its derivatives could influence cellular signal transduction and modify main classes of proteins through S-nitrosylation, the coupling of an NO moiety to a reactive cysteine thiol (46, 47). S-Nitrosylation could also promote or inhibit disulfide linkages within or between proteins depending on thiol proximity (47, 48). Cysteine thiols with acidic (Asp, Glu) or basic side chains (Arg, His, Lys) nearby could readily undergo S-nitrosylation through a concerted acid-base catalysis (47). Since the PKR protein has 2 arginine and cysteine residues in close proximity to each other (within 5 amino acids), we have preliminary data where NO binds directly to PKR via S-nitrosylation to induce PKR dimerization. In our study, we immunoprecipitated PKR from control cell lysates and treated it with DTT, a reducing agent which abolished DETA NONOate-induced dimerization (Fig. 6A). It has been reported that H2O2 induced dimerization via disulfide linkages in Yap1p (49). To assess whether H2O2 induced disulfide linkages in PKR, MDA-MB-231 cell lines were treated with H2O2 (0.3, 0.5, and 1mM). Upon western analysis we found H2O2 induced PKR dimerization (Fig. 6B). These results provide preliminary evidence that NO induces disulfide linkages between PKR monomers, leading to an increase in PKR oligomeric and dimeric forms. Further studies are underway to consolidate these observations. Although one study has tied HRI to NO induced cytostasis via phosphorylation of eIF2-α in neuroepithelial and myoblast cells (22), this is the first report of NO inducing cytostasis via PKR activation.
Most therapeutic strategies against cancer use cytotoxic drugs or gene therapies that are directed at the DNA (30, 31). However, an increasing body of data is emerging about the involvement of the translational processes in tumorigenesis, indicating that protein synthesis can be an additional target for anticancer strategies (50). Although our and other studies consistently demonstrate tumoricidal activity of NO in vitro, there have been controversies regarding its effect in vivo. This is due to the fact that macrophages, which produce NO, have been found infiltrating high grade breast tumors in vivo. Recently it is becoming clear that macrophages, which are associated with Th2 response, could be suppressing Th1 like response of NO. So, strategies that would stimulate Th1 responses in vivo may help manifest the tumoricidal effect of NO.
We have previously reported interactions between the protein synthesis machinery and the proliferative signal transduction pathways, where breast cancer cells are dependent on Cyclin D1 synthesis for proliferation (3). Dependence of cancer cells on Cyclin D1, a short half-life protein, for proliferation reinforces the emerging importance of the translational machinery to maintain oncogenesis. Our studies strongly suggest the importance of targeting the translational machinery for therapeutic purposes. We can exploit the over expression of PKR in breast cancer cells by activating it to induce suppression of protein synthesis, cytostasis and possibly apoptosis.
Acknowledgments
This work was supported in part by Palomba Weingarten, the Allegra Charach Cancer Research Fund, USPHS Grant CA-78357 (G.C), 1R03 HD053888-01A1 (S.P), and NIH/NIGMS 1S06-GM068510-03 (R.S). We thank Svetlana Arutyonova and Janis Cuevas for their technical assistance.
References
- 1.Bilanges B, Stokoe D. Mechanism of translational deregulation in human tumors and therapeutic intervention strategies. Oncogene. 2007;26:5973–90. doi: 10.1038/sj.onc.1210431. [DOI] [PubMed] [Google Scholar]
- 2.Holland EC, Sonenberg N, Pandolfi PP, Thomas G. Signaling control of mRNA translation in cancer pathogenesis. Oncogene. 2004;23:3138–44. doi: 10.1038/sj.onc.1207590. [DOI] [PubMed] [Google Scholar]
- 3.Mamane Y, Petroulakis E, Le Bacquer O, Sonenberg N. mTOR, translation initiation and cancer. Oncogene. 2006;25:6416–22. doi: 10.1038/sj.onc.1209888. [DOI] [PubMed] [Google Scholar]
- 4.Pervin S, Singh R, Hernandez E, Wu G, Chaudhuri G. Nitric oxide in physiological concentrations targets the translational machinery to increase the proliferation of human breast cancer cells: Involvement of mTOR/eIF-4E pathway. Cancer Res. 2007;67:289–99. doi: 10.1158/0008-5472.CAN-05-4623. [DOI] [PubMed] [Google Scholar]
- 5.Armengol G, Rojo F, Castellvi J, et al. 4E-binding protein 1: a key molecular “funnel factor” in human cancer with clinical implications. Cancer Res. 2007;67:7551–5. doi: 10.1158/0008-5472.CAN-07-0881. [DOI] [PubMed] [Google Scholar]
- 6.Graff JR, Konicek BW, Vincent TM, et al. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J Clin Invest. 2007;117:2385–8. doi: 10.1172/JCI32044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Larsson O, Li S, Issaenko OA, Avdulov S, Peterson M. Eukaryotic translation initiation factor 4E induced progression of primary human mammary epithelial cells along the cancer pathway is associated with targeted translational deregulation of oncogenic drivers and inhibitors. Cancer Res. 2007;67:6814–24. doi: 10.1158/0008-5472.CAN-07-0752. [DOI] [PubMed] [Google Scholar]
- 8.Polunovsky VA, Bitterman PB. The cap-dependent translation apparatus integrates and amplifies cancer pathways. RNA Biology. 2006;3:10–17. doi: 10.4161/rna.3.1.2718. [DOI] [PubMed] [Google Scholar]
- 9.Kuptsova N, Chang-Claude J, Kropp S, et al. Genetic predictors of long-term toxicities after radiation therapy for breast cancer. Int Cancer. 2007 doi: 10.1002/ijc.23138. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 10.Hibbs JB, Jr, Taintor RR, Vavrin Z. Iron depletion: possible cause of tumor cell cytotoxicity induced by activated macrophages. Biochem Biophys Res Commun. 1984;123:716–23. doi: 10.1016/0006-291x(84)90288-2. [DOI] [PubMed] [Google Scholar]
- 11.Lala PK, Orucevic A. Role of nitric oxide in tumor progression: lessons from experimental tumors. Cancer and Metastasis Reviews. 1998;17:91–106. doi: 10.1023/a:1005960822365. [DOI] [PubMed] [Google Scholar]
- 12.Stuehr DJ, Nathan CF. Nitric oxide a macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J Exp Med. 1989;169:1543–55. doi: 10.1084/jem.169.5.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lavnikova N, Burdelya L, Lakhotia A, Patel N, Prokhorova S, Laskin DL. Macrophage and interleukin-1 induced nitric oxide production and cytostasis in hamster tumor cells varying in malignant potential. J Leukoc Biol. 1997;61:452–8. doi: 10.1002/jlb.61.4.452. [DOI] [PubMed] [Google Scholar]
- 14.Kim YM, Son K, Hong SJ, et al. Inhibition of protein synthesis by nitric oxide correlates with cytostatic activity: nitric oxide induces phosphorylation of initiation factor eIF2-alpha. Mol Med. 1998;4:179–90. [PMC free article] [PubMed] [Google Scholar]
- 15.Clemens MJ, Bushell M, Jeffrey IW, Pain VM, Morley SJ. Translation initiation factor modifications and the regulation of protein synthesis in apoptotic cells. Cell Death and Differentiation. 2000;7:603–15. doi: 10.1038/sj.cdd.4400695. [DOI] [PubMed] [Google Scholar]
- 16.Pervin S, Singh R, Chaudhuri G. Nitric Oxide induced cytostasis and cell cycle arrest of a human breast cancer cell line (MDA-MB-231): potential role of cyclin D1. Proc Natl Acad Sci USA. 2001;98:3583–8. doi: 10.1073/pnas.041603998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Espey MG, Miranda KM, Pluta RM, Wink DA. Nitrosative capacity of macrophages is dependent on nitric-oxide synthase induction signals. J Biol Chem. 2000;275:11341–47. doi: 10.1074/jbc.275.15.11341. [DOI] [PubMed] [Google Scholar]
- 18.Grant CM, Miller PF, Hinnebusch AG. Requirement for intercistronic distance and level of eukaryotic initiation factor 2 activity in reinitiation on GCN4 mRNA vary with downstream cistron. Mol Cell Biol. 1994;14:2616–28. doi: 10.1128/mcb.14.4.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Society Transactions. 2006;34:7–11. doi: 10.1042/BST20060007. [DOI] [PubMed] [Google Scholar]
- 20.Hinnebusch AG. eIF2α kinases provide a new solution to the puzzle of substrate specificity. Nature Structural and Molecular Biology. 2005;12:835–38. doi: 10.1038/nsmb1005-835. [DOI] [PubMed] [Google Scholar]
- 21.Igarashi J, Sato A, Kitagawa T, et al. Activation of heme-regulated eukaryotic initiation factor 2α kinase by nitric oxide is induced by the formation of a five-coordinate NO-heme complex. J Biol Chem. 2004;279:15751–62. doi: 10.1074/jbc.M310273200. [DOI] [PubMed] [Google Scholar]
- 22.Uma S, Yun BG, Matts RL. The heme-regulated eukaryotic initiation factor 2α kinase. A potential regulatory target for control of protein synthesis by diffusible gases. J Biol Chem. 2001;276:14875–83. doi: 10.1074/jbc.M011476200. [DOI] [PubMed] [Google Scholar]
- 23.Yun BG, Matts JAB, Matts RL. Interdomain interactions regulate the activation of the heme-regulated eIF2α kinase. Biochim Biophy Acta. 2005;1725:174–81. doi: 10.1016/j.bbagen.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 24.Lu L, Han AP, Chen JJ. Translation initiation control by heme-regulated eukaryotic initiation factor 2α kinase in erythoid cells under cytoplasmic stresses. Mol and Cell Biol. 2001;21:7971–80. doi: 10.1128/MCB.21.23.7971-7980.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen JJ, London IM. Regulation of protein synthesis by heme-regulated eIF2α kinase. TIBS. 1995;20:105–8. doi: 10.1016/s0968-0004(00)88975-6. [DOI] [PubMed] [Google Scholar]
- 26.Yang JM, London IM, Chen JJ. Effects of hemin and porphyrin compounds on intersubuint disulfide formation of heme-regulated eIF2α kinase and the regulation of protein synthesis in reticulocyte lysates. J Biol Chem. 1992;267:20519–24. [PubMed] [Google Scholar]
- 27.Savinova O, Joshi B, Jagus R. Abnormal levels and minimal activity of the dsRNA-activated protein kinase, PKR, in breast cancer cells. Inter J Biochem Cell Bio. 1999;31:175–89. doi: 10.1016/s1357-2725(98)00140-x. [DOI] [PubMed] [Google Scholar]
- 28.Kim SH, Forman AP, Matthews MB, Gunnery S. Human breast cancer cells contain elevated levels and activity of the protein kinase, PKR. Oncogene. 2000;19:3086–94. doi: 10.1038/sj.onc.1203632. [DOI] [PubMed] [Google Scholar]
- 29.Jarrous N, Osman F, Kaempfer R. 2-Aminopurine selectively inhibits splicing of tumor necrosis factor alpha mRNA. Mol Cell Bio. 1996;16:2814–22. doi: 10.1128/mcb.16.6.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calaf GM. Susceptibility of human breast epithelial cells in vitro to hormones and drugs. Int J Oncol. 2006;28:285–95. [PubMed] [Google Scholar]
- 31.Dziadyk JM, Sui M, Zhu X, Fan W. Paclitaxel-induced apoptosis may occur without a prior G2/M-phase arrest. Anticancer Res. 2004;24:27–36. [PubMed] [Google Scholar]
- 32.Pervin S, Singh R, Chaudhuri G. Nitric oxide-induced Bax integration into the mitochondrial membrane commits MDA-MB-468 cells to apoptosis: essential role of Akt. Cancer Res. 2003;63:5470–79. [PubMed] [Google Scholar]
- 33.Raines KW, Cao GL, Porsuphatana S, Tsai P, Rosen GM, Shapiro P. Nitric oxide inhibition of ERK1/2 activity in cells expressing neuronal nitric-oxide synthase. J Biol Chem. 2004;279:3933–40. doi: 10.1074/jbc.M304813200. [DOI] [PubMed] [Google Scholar]
- 34.Shinohara M, Mikhailov AV, Aguirre-Ghiso JA, Rieder CL. Extracellular signal-regulated kinase 1/2 activity is not required in mammalian cells during late G2 for timely entry into or exit from mitosis. Mol Biol Cell. 2006;17:5227–40. doi: 10.1091/mbc.E06-04-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dey M, Cao C, Dar AC, et al. Mechanistic Link between PKR dimerization, autophosphorylation, and eIF2α substrate recognition. Cell. 2005;122:901–13. doi: 10.1016/j.cell.2005.06.041. [DOI] [PubMed] [Google Scholar]
- 36.Scheuner D, Patel R, Wang F, et al. Double-stranded RNA-dependent protein kinase phosphorylation of the alpha-subunit of eukaryotic translation initiation factor 2 mediates apoptosis. J Biol Chem. 2006;281:21458–68. doi: 10.1074/jbc.M603784200. [DOI] [PubMed] [Google Scholar]
- 37.Shir A, Friedrich I, Levitzki A. Tumor specific activation of PKR as a non-toxic modality of cancer treatment. Semin Cancer Biol. 2003;13:309–14. doi: 10.1016/s1044-579x(03)00045-2. [DOI] [PubMed] [Google Scholar]
- 38.Friedrich I, Eizenbach M, Sajman J, Ben-Bassat H, Levitzki A. A cellular screening assay to test the ability of PKR to induce cell death in mammalian cells. Molecular Therapy. 2005;12:969–75. doi: 10.1016/j.ymthe.2005.06.442. [DOI] [PubMed] [Google Scholar]
- 39.von Holzen U, Bocangel D, Pataer A, et al. Role for the double-stranded RNA-activated protein kinase PKR in Ad-TNF-alpha gene therapy in esophageal cancer. Surgery. 2005;138:261–8. doi: 10.1016/j.surg.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 40.Fremont M, Vaeyens F, Herst CV, De Meirleir KL, Englebienne P. Double-stranded RNA-dependent protein kinase (PKR) is a stress-responsive kinase that induces NFkappaB-mediated resistance against mercury cytotoxicity. Life Sci. 2006;78:1845–56. doi: 10.1016/j.lfs.2005.08.024. [DOI] [PubMed] [Google Scholar]
- 41.Bommer UA, Borovjagin AV, Greagg MA, et al. The mRNA of the translationally controlled tumor protein P23/TCTP is a highly structured RNA, which activates the dsRNA-dependent protein kinase PKR. RNA. 2002;8:478–96. doi: 10.1017/s1355838202022586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raine DA, Jeffrey IW, Clemens MJ. Inhibition of the double-stranded RNA-dependent protein kinase PKR by mammalian ribosomes. FEBS Letters. 1998;436:343–48. doi: 10.1016/s0014-5793(98)01163-6. [DOI] [PubMed] [Google Scholar]
- 43.Gil J, Esteban M, Roth D. In vivo regulation of the dsRNA-dependent protein kinase PKR by the cellular glycoprotein P67. Biochem. 2000;39:16016–25. doi: 10.1021/bi001754t. [DOI] [PubMed] [Google Scholar]
- 44.Tan SL, Tareen SU, Melville MW, Blakely CM, Katz MG. The direct binding of the catalytic subunit of protein phosphatase 1 to the PKR protein kinase is necessary but not sufficient for inactivation and disruption of enzyme dimer formation. J Bio Chem. 2002;277:36109–17. doi: 10.1074/jbc.M205109200. [DOI] [PubMed] [Google Scholar]
- 45.Farias-Eisner R, Chaudhuri G, Aeberhard E, Fukuto JM. The chemistry and tumoricidal activity of nitric oxide/hydrogen peroxide and the implications to cell resistance/susceptibility. J Biol Chem. 1996;271:6144–51. doi: 10.1074/jbc.271.11.6144. [DOI] [PubMed] [Google Scholar]
- 46.Pfeilschifter J, Eberhardt W, Huwiler A. Nitric oxide and Mechanisms of Redox Signaling. J Am Soc Nephrol. 2003;14:S237–S40. doi: 10.1097/01.asn.0000077409.55250.84. [DOI] [PubMed] [Google Scholar]
- 47.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: Purview and Parameters. Nature Rev Mol Cell Bio. 2005;6:150–66. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 48.Arnelle DR, Stamler JS. NO+, NO and NO− Donation by S-Nitrosothiols: Implications for Regulation of Physiological Functions by S-Nitrosylation and Acceleration of Disulfide Formation. Arch Biochm Biophy. 1995;318:279–85. doi: 10.1006/abbi.1995.1231. [DOI] [PubMed] [Google Scholar]
- 49.Kuge S, Arita M, Murayama A, Maeta K, Izawa S, Inoue Y, et al. Regulation of the yeast Yap1p nuclear export signal is mediated by redox signal-induced reversible disulfide bond formation. Mol Cell Biol. 2001;21:6139–50. doi: 10.1128/MCB.21.18.6139-6150.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holland EC, Sonenberg N, Pandolfi PP, Thomas G. Signaling control of mRNA translation in cancer pathogenesis. Oncogene. 2004;23:3138–44. doi: 10.1038/sj.onc.1207590. [DOI] [PubMed] [Google Scholar]