

Abstract
The Rho GTPase Cdc42 coordinates regulation of the actin and the microtubule cytoskeleton by binding and activating the Wiskott–Aldrich syndrome protein. We sought to define the role of intrinsic expression of Cdc42 by mature B cells in their activation and function. Mice with inducible deletion of Cdc42 in mature B cells formed smaller germinal centers and had a reduced Ab response, mostly of low affinity to T cell–dependent Ag, compared with wild-type (WT) controls. Spreading formation of long protrusions that contain F-actin, microtubules, and Cdc42-interacting protein 4, and assumption of a dendritic cell morphology in response to anti-CD40 plus IL-4 were impaired in Cdc42-deficient B cells compared with WT B cells. Cdc42-deficient B cells had an intact migratory response to chemokine in vitro, but their homing to the B cell follicles in the spleen in vivo was significantly impaired. Cdc42-deficient B cells induced a skewed cytokine response in CD4+ T cells, compared with WT B cells. Our results demonstrate a critical role for Cdc42 in the motility of mature B cells, their cognate interaction with T cells, and their differentiation into Ab-producing cells.
Introduction
Motility and adhesion are critical functions of B cells that depend on an intact actin cytoskeleton. Newly differentiated B lymphocytes migrate from the bone marrow to secondary lymphoid organs. Within the lymphoid organs, B cells can migrate between follicles and marginal zones (MZs) of the spleen (1), and repeatedly circle between the dark and light zones of the germinal centers (GCs) (2). Adhesion molecules, such as LFA-1 and VLA-4, as well as their respective ligands ICAM-1 and VCAM-1, are necessary for the localization of the B cells in the MZs and the GCs (3, 4). Chemokines and their receptors, as well as sphingosine 1-phosphate and its receptors, are involved in the positioning and migration of B cells to the MZ and the GC (1, 5, 6).
The Wiskott–Aldrich syndrome protein (WASp) and its relative the neuronal-WASp (N-WASp) link cell surface receptors to the actin cytoskeleton. WASp-deficient B cells have reduced ability to form long protrusions and microvilli in cell-to-cell contacts (7), but undergo normal Ig class switching in vitro. Mice lacking WASp exhibit a deficiency in mature B cell subpopulations and mount a decreased Ab response (8–10). Likewise, mice with B cells lacking both WASp and N-WASp show severe defects in such responses (11). In two recent studies, B cell–intrinsic WASp deficiency was shown to result in B cell hyperactivity and autoimmunity in vivo (12, 13).
The small GTPase Cdc42 activates actin polymerization via the activation of WASp and N-WASp (14, 15), thereby regulating cell adhesion, migration, proliferation, and survival (16). Cdc42 is also linked to microtubules by binding to the Cdc42-interacting protein (CIP4) that regulates microtubule assembly and induces membrane deformation (17). Thus, Cdc42 can mediate the interaction between actin and microtubules (18, 19) and regulate membrane protrusions. Dominant-negative Cdc42 mutants interfere with B cell morphology and function (7, 20). By the use of conditional gene targeting, it has been shown that Cdc42 is essential for B lymphocyte development, as well as for Ag- and mitogen-driven B cell activation (21, 22). However, the exact role of Cdc42 in the function of mature B cells remains unknown, because in these studies deletion of Cdc42 early in B cell development resulted in severe reduction in the numbers of mature B cells and may have had nonspecific effects on the function of the residual mature B cells. In this work, we specifically deleted Cdc42 in mature B cells to investigate its role in the in vitro and in vivo immune response of mature B cells independent of its role in B cell development. We demonstrate that Cdc42 plays a critical role in the motility, adhesion, and Ab response of mature B cells.
Materials and Methods
Mice and immunizations
Cdc42flox mice have been described previously (23). OT-II mice were purchased from The Jackson Laboratory. The Mb1-cre-ERT2 mouse strain was a gift of M. Reth (University of Freiburg). It was made by Cre-ERT2 inserted into the Cd79a locus that encodes Igα (24). The CD23-cre mice were a gift of M. Busslinger (Vienna Biocenter) (25). CIP4−/− mice have been described previously (26). All strains were on a C57BL/6 background. Breedings were set up so that wild-type (WT), heterozygotes (HZ), and knockouts (KO) could be obtained in the same breeding. Mice were bred in specific pathogen-free conditions. To achieve Cdc42 deletion, mice were given tamoxifen (5 mg in 50 μl) by gavage for consecutive 5 d. Nonimmunized mice were sacrificed on day 3 or 4 after the final tamoxifen treatment. Mice were immunized 4 d after the final tamoxifen treatment. Trinitrophenyl (TNP) was conjugated to SRBC, as described (27). SRBC, TNP-SRBC (10% mixture in PBS, 0.2 ml/mouse), and 4-hydroxy-3-nitrophenylacetyl coupled to keyhole limpet hemocyanin (NP-KLH) (100 μg/mouse in alum or 20–70 μg/mouse in PBS or alum for recall) were injected i.p. Mice were bled from the tail and sacrificed at 6 wk to 1 y of age. The concentrations of anti-NP Abs were measured by ELISA, using 4-hydroxy-3-nitrophenylacetyl coupled to BSA, with different hapten/carrier ratios, to enable affinity measurements. All animal experiments were approved by Stockholm North Animal Ethics Committee, or in accordance with the Animal Care and Use Committee of the Children’s Hospital Boston.
Cell culture
B and CD4+ T cells were purified from spleens by negative selection (Stem Cell Technologies or Miltenyi Biotec), sometimes followed by separation in a Percoll gradient (GE Healthcare). For analysis of Ig class switching, spleen B cells were enriched, as described previously (7). Monoclonal rat anti-mouse CD40 (1C10) was purified, as previously described (28), and was used at 10–20 μg/ml. IL-4 was used at 8–10 ng/ml (PeproTech). LPS (Sigma-Aldrich) was used at 10 μg/ml. Cell-spreading assays were performed on glass coverslips coated with anti-CD44 Abs (BD Biosciences).
Chemokine response
In vitro migration of B cells to CXCL12 was assessed, as previously described (8). In brief, 105 splenic B cells were added to the upper chamber of Transwell cultures, and 25 ng CXCL12 was added to the lower chamber. Migration of B cells to the lower chamber was analyzed by flow cytometry after 2 h of incubation at 37°C and expressed as percentage of total B cells. Each condition was set up in triplicates.
Homing of B lymphocytes in vivo
Single-cell suspensions of spleen B cells were prepared from mice immunized 6 d previously with SRBC and 10 d postinduction with tamoxifen. Cells were labeled with CFSE (Molecular Probes), and 107 labeled cells were injected i.v. into recipient WT mice immunized with SRBC 6 d previously. Spleens were removed after 16 h and frozen in OCT medium (Dalab).
T–B cell coculture assay
Cdc42-deficient or -sufficient splenic B cells were activated with anti-CD40 plus IL-4 for 2 d, thereafter harvested, washed, and incubated first with affinity-purified biotinylated F(ab′)2 goat anti-mouse IgM at 0.25 (high), 0.05 (medium), or 0.01 (low) μg/ml in BSS plus 1% FCS for 20 min. B cells were then washed and incubated with diluted OVA Ag delivery agent (Miltenyi Biotec) for 20 min on ice. B cells were thereafter washed and cultured at 2 × 105 cells/culture (2:1 ratio, as compared with T cell numbers) in a total volume of 200 μl. CD4+ T cells purified from OT-II mice were added to the cultures at 105/culture. Cultures were incubated for 3 d. Fours hours before harvest, PMA (Sigma-Aldrich; 0,5 μg/ml), ionomycin (Sigma-Aldrich; 0.5 μg/ml), and GolgiPlug (BD Biosciences; diluted 715 times) were added. Cells were harvested, washed, and stained with B220 and Abs to CD4, IL-2, and IFN-γ and analyzed by FACS.
Ag uptake
B cells were activated with anti-CD40 plus IL-4 for 2 d, thereafter harvested, and washed. They were incubated with 0.25 μg/ml affinity-purified biotinylated F(ab′)2 goat anti-mouse IgM for 20 min on ice. Cells were washed, resuspended in medium plus 10% FCS, and incubated for 0–120 min at 37°C. Thereafter, cells were placed on ice, washed, and treated with streptavidin-FITC (Dako) for 20 min on ice, washed again, and analyzed by FACS.
Western blot
Fifteen to twenty μg total cell extracts were separated by SDS-PAGE and analyzed by Western blot, using monoclonal rabbit anti–α-actin (Sigma-Aldrich) or polyclonal rabbit anti-Cdc42 (Abcam) Abs and HRP-conjugated swine anti-rabbit Ig (Dako). Monoclonal mouse anti-CIP4 (BD Biosciences) was used in combination with HRP-conjugated goat anti-mouse IgG1 (H chain specific; Jackson ImmunoResearch). Visualization was done using a Bio-Rad system with Image Lab software. Quantification of the bands was performed using ImageJ software (NIH), and relative density was obtained by dividing intensities of Cdc42 to intensities of α-actin. The result was normalized to the relative density of WT.
Measurement of Rac activity
Active GTP-bound Rac1/2 was quantified using the G-LISA Rac1,2,3 Activation Assay Biochem kit (Cytoskeleton), as described in the manufacturer’s protocol.
Flow cytometry
Single-cell suspensions were labeled with fluorescently conjugated anti-mouse Abs, including B220, CD4, CD21, CD23, CD93, CD95 (Fas), CD138, IFN-γ, IgM, IgD, IgG1, and IL-2 (BD Biosciences, eBioscience, BioLegend). A Live/Dead kit (Molecular Probes) was used to identify viable cells. Fluorescence minus one controls were included in all flow cytometry experiments. Data were acquired on FACSCalibur, FACSVerse, or LSRFortessa (BD Biosciences) and analyzed using FlowJo software, version 9.4.
Immunohistochemistry
Spleen sections were fixed in ice-cold acetone and labeled with fluorescently conjugated or biotinylated anti-mouse Abs, including CD1d, CD169 (MOMA), B220, IgM, IgG1, peanut agglutinin, TCR-β, and fluorescently conjugated streptavidin (BioLegend, Molecular Probes, and Serotec). Images of GCs, MZ B cells, and plasma cells were collected with a Leica DM IRBE confocal laser-scanning microscope (Leica Microsystems) equipped with one Argon and two HeNe lasers, using a HC PL APO lens at 10×/0.40 CS or 20×/0.70 IMM CORR oil and 90% glycerol (MP Biomedicals). The system was controlled by the LCS Lite software.
Stained slides from homing experiments were observed using Zeiss LSM 780 confocal microscope, equipped with a diode laser with wavelength 405 nm, an Argon laser with wavelengths 458/488/514 nm DPSS 561 nm, and a HeNe 633 nm laser. The following lenses were used: Plan-Apo 10×/0.45 and Plan-Apo 20×/0.8. The system was controlled by the ZEN Black 2011 software.
All images were processed with Adobe Photoshop CS4 Version 11.0.2 (Adobe Systems).
Live cell imaging
Cells were analyzed using Zeiss Axiovert 200M Cell Observer and SlideBook 5.0 Software. A HAL100 halogen lamp was used for bright field imaging. Images were taken with digital camera AxioCam MRc. The following lenses were used: EC-Plan-Neofluar 10×/0.3 Ph1 and EC-Plan-Neofluar 40×/0.75 Ph2. Alternatively, cells were analyzed using Nikon Eclipse Ti microscope with NIS-Elements BR 3.2 Software. A mercury lamp was used for bright field imaging. Images were taken with digital camera CoolSNAP HQ2. EC-Plan-Neofluar 10×/0.3 Ph1 lens was used. Experiments were performed at 37°C, and 10 mM HEPES buffer was added to the medium to keep constant pH. ImageJ (NIH) was used for different quantifications.
Immunofluorescence microscopy
B cells were activated for 20 h with anti-CD40 plus IL-4, transferred to anti-CD44 glass coverslips, and incubated for additional 22 h. They were thereafter fixed, washed, permeabilized, treated with anti-mouse CD16/CD32, and then stained with fluorescently conjugated phalloidin (Sigma-Aldrich), Hoechst 33258 (Sigma-Aldrich), rat anti-mouse α-tubulin (Abcam), mouse anti-human CIP4 (BD Biosciences), fluorescently conjugated goat anti-mouse IgG1 (H chain specific; Jackson ImmunoResearch), and/or donkey anti-rat Ig (which had minimal cross-reactivity with mouse Ig; Jackson ImmunoResearch). Spread cells were defined as cells with at least one protrusion longer than one cell diameter. Cells were observed using Zeiss LSM 780 confocal microscope, as described above, with a Plan-Apo 63×/1.4 oil-immersion lens. Images were processed with Adobe Photoshop CS4 Version 11.0.2 (Adobe Systems).
Synapse formation in vitro
Anti-CD40 plus IL-4–activated B cells were mixed with OTII CD4+ T cells in ratio 1:5 and incubated for 2 h at 37°C. Thereafter, cells were placed on 50 μg/ml fibronectin (Life Technologies)-precoated eight-well slides and incubated at 37°C for 1 h. Cells were fixed, washed, permeabilized, and washed again. Cells were stained with α-tubulin primary Abs and later with fluorescently conjugated phalloidin-FITC (Sigma-Aldrich) and donkey anti-rat Ig Cy3 (Jackson ImmunoResearch), B220-PB (BioLegend), and anti–TCR-β allophycocyanin (BD Biosciences) together with anti-CD4 allophycocyanin (BD Biosciences). Cells were observed using Zeiss LSM 780 confocal microscope, as described above, with a Plan-Apo 63×/1.4 oil-immersion lens. Images were processed with ZEN Black 2011 software (Zeiss).
Scanning electron microscopy
Cells grown on glass were fixed by immersion in 2.5% glutaraldehyde in 0.1 M phosphate buffer. Specimens were briefly rinsed in distilled water and placed first in ethanol of different percentages in cold and thereafter into acetone. They were dried using a critical point dryer (Balzer; CPD 010) using carbon dioxide. After drying, specimens were mounted on an aluminum stub and coated with platinum (Bal-Tec SCD 005). The specimens were analyzed in an Ultra 55 field emission-scanning electron microscope (Zeiss) at 3 kV.
Statistical analysis
The GraphPad Prism software was used to make graphs and analyze data (GraphPad Software). Data are expressed as medians or means ± SD, as indicated. Statistical significance between groups was assessed by Student t test. Differences were considered significant when p ≤ 0.05: *p ≤ 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Results
Generation of mice with Cdc42 deletion in mature B cells
B cells devoid of the Rho-type GTPases, Rac1, and Rac2 do not enter into the white pulp of the spleen (29). To avoid the possibility of potential effect of Cdc42 deficiency on altered positioning of Cdc42-deficient B cells in the white pulp during their development, we constructed mice with inducible deletion of Cdc42. To this purpose, we crossed Cdc42flox/flox mice with mb1-cre-ERT2 mice. In the mb1-cre-ERT2 mouse strain, expression of Cre-gene is regulated by the mb1 promoter. Its fusion product Cre-ERT2 is localized in the cytoplasm but undergoes nuclear translocation following administration of tamoxifen. Crossing mb1-cre-ERT2 mice with Cdc42flox/flox mice gave the expected Mendelian genotypes in the offspring. To simplify the nomenclature, the Cdc42flox/flox/mb1cre-ERT2/+, Cdc42flox/wt/mb1cre-ERT2/+, and mb1+/+ mice are designed KO, HZ, and WT, respectively. To induce Cdc42 deletion, we fed the mice tamoxifen for consecutive 5 d. Analysis of purified splenic B cells 3 d later revealed that the relevant region of cdc42 was efficiently deleted from the genome and that Cdc42 protein levels were reduced by ∼90% in Cdc42flox/flox/mb1cre-ERT2/+ mice (Fig. 1A, 1B). In the weeks after tamoxifen induction of Cdc42 excision, the Cdc42-deficient B cells were gradually replaced by newly differentiating Cdc42-sufficient B cells. Genomic analysis revealed that B cells that lack Cdc42 remained dominant for 2 wk postinduction with tamoxifen, following which Cdc42-sufficient B cells gradually dominated with very few Cdc42-deficient B cells being detected after 6 wk (Supplemental Fig. 1A, 1B). There were lower numbers of total leukocytes per spleen on day 3 postinduction with tamoxifen, which corresponded to reduced numbers of B220+ B cells in Cdc42flox/flox/mb1cre-ERT2/+ mice (Fig. 1C). Comparing the numbers of B cells in the spleen, KO mice had 41% of the numbers in WT. Comparing the B cell/spleen ratios, KO mice had 75% of WT levels. In agreement with previous data obtained from crossing Cdc42flox/flox mice with mice expressing Cre controlled by the B cell–specific CD19 promoter (21) or by the mb1 promoter (22), the numbers of transitional (T)1, T2-MZ precursors, and follicular B cells were all reduced (Fig. 1D). Remarkably, MZ B cells were almost absent in Cdc42flox/flox/mb1cre-ERT2/+ mice, both by fluorescent microscopy of spleen sections and by flow cytometry (B220+, CD21high, CD23low; Fig. 1E [left panel], 1F).
FIGURE 1.

Cdc42 deletion in B cells leads to lower numbers of follicular splenic B cells and impairs the formation of MZs and GCs. (A) PCR analysis of the Cdc42 gene locus in the DNA of purified splenic B cells in Cdc42flox/wt/mb1+/+ (WT) Cdc42flox/wt/mb1cre-ERT2/+ (HZ) and Cdc42flox/flox/mb1cre-ERT2/+ (KO) mice 3 d after tamoxifen induction. (B) Immunoblot analysis of Cdc42 in lysates from purified splenic B cells of WT, HZ, and KO mice, showing expression of Cdc42 or α-actin (left panel) and density of the Cdc42 bands relative to the corresponding ones of α-actin and normalized to WT (right panel). Columns and bars represent mean and SD of four experiments. (C–E) Flow cytometry of different subpopulations of B cells. (C) Total numbers of cells per spleen (left panel), numbers of B220+ cells per spleen (middle panel), and fraction of splenic cells that are B cells (right panel). (D) Numbers of T1 cells, defined as B220+, IgM+, CD21−/low, and CD23− (left panel); numbers of T2-MZ precursor cells, defined as B220+, IgM+/high, CD21+/high, and CD23+ (middle panel); and numbers of follicular B cells, defined as B220+, IgM+/low, CD21+/low, and CD23+ (right panel) per spleen. (E) Numbers of MZ B cells defined as B220+, IgM+, CD21high, and CD23low (MZ B cells) (left panel), and numbers of GC B cells defined as B220+, GL7+, and CD95+ (right panel) per spleen. Controls (Ctrl) in (C)–(E) refer to mice not given tamoxifen, WT (mb1+/+), HZ (Cdc42flox/+/mb1cre-ERT2/+), and KO (Cdc42flox/flox/mb1cre-ERT2/+) were all given tamoxifen. Ag: mice given Ag. Each symbol represents one mouse; 5–10 mice per group were tested. (F and G) Immunohistochemistry of spleen sections from TNP-SRBC–immunized mice on day 6 after Ag injection (10 d postinduction with tamoxifen). (F) For the visualization of MZs, B220 (blue) was used to stain B cells, MOMA (green) to stain MZ metallophilic macrophages, and anti-CD1d (red) to stain MZ B cells. The lower panels are blow-ups of the upper ones, as indicated. (G) For the analysis of GCs, peanut agglutinin (red) was used to stain GCs, anti–TCR-β (green) to stain T cells, and B220 (blue) to stain B cells. Peanut agglutinin also stains the outer regions of the MZ due to binding to endothelial cells. (F and G) Representative images of three mice per group are shown; scale bars, 300 μm. *p ≤ 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
As an alternative strategy to delete Cdc42 in mature B cells, we crossed CD23-cre mice with Cdc42flox/flox mice. In Cdc42flox/flox/CD23cre/+ mice, Cdc42 will be deleted in B cells just after entering the spleen, as they transit from the T1 to the T2 stage (Supplemental Fig. 2A). As shown in Supplemental Fig. 1C, the level of Cdc42 in purified B cells from Cdc42flox/flox/CD23cre/+ mice was ∼50% of WT. Cdc42 was more efficiently deleted in B cells from these mice following stimulation for 2 d with anti-CD40 plus IL-4 in vitro (Supplemental Fig. 1D). This stimulation upregulates CD23 expression and consequently cre expression in B cells from Cdc42flox/flox/CD23cre/+ mice, resulting in more efficient cre-mediated deletion of Cdc42.
Cdc42 deletion in mature B cells results in a decreased GC formation in response to immunization with a T cell–dependent particulate Ag
T cell–dependent (TD) and T cell–independent Ab responses to soluble Ags are reduced in mice with B cell–specific deletion of Cdc42 (21). We investigated in detail the T cell–dependent immune response to particulate Ag in mice with inducible Cdc42 deletion in mature B cells. To this end, we immunized mice with TNP-SRBC, 3 d postinduction with tamoxifen, and analyzed GC in the spleen 6 d later by peanut agglutinin staining of spleen section and by FACS analysis of B220+, GL7+, and CD95+ GC B cells. On the day of analysis, the majority of B cells in the spleen lacked Cdc42 (Supplemental Fig. 1E). GCs were significantly smaller, and the numbers of B220+, GL7+, and CD95+ GC B cells were lower in Cdc42flox/flox/mb1cre-ERT2/+ mice compared with controls (Fig. 1E [right panel], 1G).
Despite smaller GCs, the Cdc42flox/flox/mb1cre-ERT2/+ mice mounted a normal humoral immune response to the particulate Ag TNP-SRBC (Fig. 2A). Furthermore, the numbers of IgM- and IgG1-expressing plasma cells as well as the percentage of plasmablasts and plasma cells were similar in mice with Cdc42-sufficient and -deficient B cells (Supplemental Fig. 2B, 2C). Interestingly, very few IgG1+ cells were found in the B cell areas in the Cdc42flox/flox/mb1cre-ERT2/+ KO mice, whereas such cells were easily detected in WT and HZ mice (Fig. 2B, 2C). This suggests that IgG1-producing plasma cells are mainly localized to extrafollicular regions in the Cdc42flox/flox/mb1cre-ERT2/+ KO mice. Curiously, the IgG1 response was significantly higher in HZ mice, compared with WT controls (Supplemental Fig. 2C), suggesting an advantage for haploinsufficiency of Cdc42 in B cells. Switching in vitro was not reduced in Cdc42-deficient B cells. In fact, switching to IgG2b was slightly enhanced (Supplemental Fig. 3A). These findings suggest that Cdc42 is not important for isotype switching and plasma cell differentiation in response to TNP-SRBC.
FIGURE 2.

Deletion of Cdc42 in B cells leads to impaired humoral immune response. (A) TNP-specific serum Abs in mice immunized with TNP-SRBC. Arrows indicate the time of Ag injection. IgM anti-TNP (left panel) and IgG anti-TNP (right panel); each time point represents mean and SD of three to six mice per group. (B) Immunohistochemistry of spleen sections from unimmunized mice (upper panels) or mice immunized with TNP-SRBC 6 d previously (lower panels) and stained with MOMA (green), anti-IgM (blue), and anti-IgG1 (red). Arrows point to the follicular areas. Representative images from three mice per group are shown. Scale bars, 150 μm. (C) Ratio of the level of IgG1 staining in the follicular regions to the staining in extrafollicular regions, four images per group; vertical bars represent median. (D) Anti-NP response in mice immunized with NP-KLH at weeks 0 and 21. Arrows indicate the times of immunization. Each time point represents mean and SD of five to seven mice per group. The amount of serum Abs was determined in ELISA using 4-hydroxy-3-nitrophenylacetyl coupled to BSA with either 8 (left panel) or 25 (middle panel) NP molecules per molecule of BSA. The ratio of the two responses is a measure of the affinity of Abs and is shown in the right panel. The response in KO mice was significantly different from those in WT and HZ (p < 0.0001). For the experiments shown in (A)–(C), Cdc42 was deleted by crossing Cdc42-floxed mice with mb1-cre-ERT mice; for the experiments shown in (D), Cdc42-floxed mice were crossed with mice expressing CD23-cre. *p ≤ 0.05.
Deletion of Cdc42 in mature B cells results in an Ab response of reduced magnitude and lower affinity to soluble TD Ag
To determine whether the decreased GC response in mice with deletion in mature B cells affects the affinity maturation of B cells, we immunized mice with NP-KLH, against which Ab responses of low and high affinity can be measured, and examined their secondary response. We used the Cdc42flox/flox/CD23cre mouse strain in these experiments, because induction of Cdc42 deletion in Cdc42flox/flox/mb1cre-ERT2/+ mice is transient and thus is not suitable for the long period needed for the experiment. The NP-specific Ab titer was severely reduced in Cdc42flox/flox/CD23cre/+ KO mice immunized with NP-KLH. The levels of both low and high anti-NP Abs were significantly reduced in Cdc42flox/flox/CD23cre/+ KO mice, compared with Cdc42flox/wt/CD23cre/+ HZ or CD23+/+ WT controls. In addition, the secondary response following boosting with NP-KLH was significantly lower in Cdc42flox/flox/CD23cre/+ KO mice compared with controls (Fig. 2D). Follicular dendritic cells, which express CD23, were present in normal numbers in the spleens of Cdc42flox/flox CD23cre/+ KO mice, as determined by CD35 staining (Supplemental Fig. 3C). We also performed a similar immunization experiment using mice from the mb1-cre-ERT2 crossing with Cdc42-floxed mice (Supplemental Fig. 3B). In this experiment, the Cdc42flox/flox/mb1cre/ERT2/+ KO mice produced mainly low-affinity Abs. Thus, Cdc42 expression in B cells is crucial for the magnitude and affinity of the Ab response to soluble TD Ag.
Deletion of Cdc42 in mature B cells alters their ability to activate T cells
The reduced humoral immune response to NP-KLH raised the question whether deletion of Cdc42 in mature B cells alters their ability to present Ag to T cells. We used the OVA-Ag delivery agent system (Miltenyi Biotec) to test the ability of anti-CD40 plus IL-4–activated B cells, loaded with OVA via the BCR, to present Ag in vitro to CD4+ T cells from OVA-TCR–transgenic OT-II mice. The numbers of CD4+ T cells were not significantly different, except at high Ag concentration, in which T cell numbers in cultures containing Cdc42-deficient B cells were lower than in cultures containing control B cells (Fig. 3A). Ag presentation by Cdc42-deficient B cells resulted in a lower percentage of T cells producing IL-2 and a higher percentage of T cells producing IFN-γ compared with Ag presentation by WT B cells (Fig. 3B, 3C). The experiment was also performed, using different T:B cell ratios, varying from 1:2 to 1:0.5, with similar results (data not shown). The difference in T cell activation was not due to a difference in the ability of B cells to take up Ag or to form an immunological synapse with the T cells (Supplemental Fig. 3D–F). We did not observe an IL-4 response, either at the mRNA or protein level. The experiment in Fig. 3 was performed using mice expressing cre from the CD23 locus. Similar results were obtained when using mice expressing cre from the Mb1 locus (Mb1cre-ERT2; Supplemental Fig. 3G). We conclude that Cdc42 expression in B cells is essential for efficient IL-2 secretion of CD4+ T cells in cognate T–B cell interaction.
FIGURE 3.
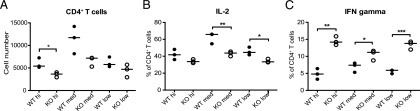
Impaired T cell activation by Ag-pulsed Cdc42-deficient B cells. Effect of Ag concentration on CD4+ OT-II T cell numbers (A), IL-2 expression (B), or IFN-γ expression (C). OT-II T cells were cultured with OVA-loaded preactivated B cells from Cdc42flox/flox/CD23cre/+ (KO) or CD23+/+ controls (WT). Anti-CD40 + IL-4–activated B cells were pretreated with different concentration of Ag (high, 0.25 μg/ml; medium, 0.05 μg/ml; low, 0.01 μg/ml), followed by OVA delivery agent, and cultured with T cells at a T:B cell ratio of 1:2. Each symbol represents one mouse; horizontal lines correspond to median values. *p ≤ 0.05, **p < 0.01, ***p < 0.001.
Deletion of Cdc42 in mature B cells reduces their homing to B cell follicles
In agreement with previous observations on B cells that lack Cdc42 (21), we found that deletion of Cdc42 in mature B cells had no effect on their migratory response toward the chemokine CXCL12 in vitro (Fig. 4A). We investigated whether deletion of Cdc42 in mature B altered their ability to home to the B cell areas in the spleen. To this end, mice in which Cdc42 was deleted in B cells by tamoxifen treatment were immunized with SRBC on day 4 postinduction with tamoxifen and sacrificed 6 d later. B cells were purified and labeled with CFSE. They were subsequently injected i.v. into SRBC-preimmunized WT mice. The injected host mice were sacrificed on the following day, and spleen sections were analyzed. As shown in Fig. 4B and 4C, WT and HZ B cells homed to the B cell areas of the spleen, whereas significantly fewer Cdc42flox/flox/mb1cre-ERT2/+ KO B cells had entered the B cell follicles. Thus, although the chemokine response in vitro is normal, homing in vivo is markedly reduced for B cells with inducible deletion of Cdc42 expression.
FIGURE 4.

Reduced homing of Cdc42-deficient B cells to spleen follicles, but normal chemokine response. (A) Migratory response of B cells to the chemokine CXCL12 in vitro. The percentage of cells migrating to the lower chamber in relation to total numbers of cells per culture was determined. Background refers to cultures without chemokine. Columns and bars represent mean and SD of two mice per group. (B) In vivo homing of B cells to spleen follicles. Ten million CFSE-labeled purified B cells from immunized mice were transferred to preimmunized WT mice, and spleen sections were analyzed the next day. The T cell area was stained with anti–TCR-β (blue) and the follicular area with B220 (red). CFSE+ cells are green. Scale bars, 100 μm. (C) Quantitative analysis of the numbers of CFSE-labeled cells per B cell area or total spleen area. Columns and bars represent mean and SD of three mice per group. (A–C) WT (mb1+/+), HZ (Cdc42flox/wt/mb1cre-ERT2/+), KO (Cdc42flox/flox/mb1cre-ERT2/+). *p ≤ 0.05.
B cells that lack Cdc42 fail to form long protrusions on Ab-coated surfaces
To determine whether deletion of Cdc42 in mature B cells affects the actin and microtubule cytoskeleton in B cells, we examined the ability of the B cells to spread on adhesive surfaces after being exposed to anti-CD40 plus IL-4. B cells from Cdc42flox/wt/mb1cre-ERT2/+ HZ and mb1+/+ WT mice spread and formed long, thin protrusions, when cultured on anti-CD44–coated surfaces. However, B cells from Cdc42flox/flox/mb1cre-ERT2/+ KO mice showed a severely reduced spreading response (Fig. 5A, 5B). Whereas WT B cells formed finely branched protrusions that stained for polymerized actin, B cells lacking Cdc42 had brushlike nonbranched protrusions (Fig. 6A, 6B). Many cells lacking Cdc42 expressed ruffles (Fig. 6A), possibly as a compensatory mechanism. When staining for microtubules, both cell types formed distinct microtubule-organizing centers. In WT cells, actin and microtubule staining was evident along the entire length of the long protrusions, whereas tubulin was not detectable in the short protrusions of B cells lacking Cdc42 (Fig. 6C–F). Next, we stained for CIP4, which can mediate interaction among Cdc42, WASp, and microtubules (18, 19). Interestingly, CIP4 was present along the entire length of the long extensions in WT B cells. It was also abundant in the nucleus, forming a speckled pattern, in both WT and Cdc42-deficient B cells. However, in Cdc42-deficient cells, CIP4 was not detectable in extensions, but found mostly in the body of the cytoplasm and in the nucleus (Fig. 6C, 6D, 6G, 6H). In addition, immunoblots revealed that the level of CIP4 was increased more than twice in Cdc42-deficient B cells, as compared with WT B cells (Fig. 5C). The different localization of CIP4 in Cdc42-deficient B cells prompted the question whether CIP4 expression in B cells would be necessary for spreading. B cells from CIP4 KO mice, which totally lacked CIP4 expression, spread normally (Supplemental Fig. 4A, 4B).
FIGURE 5.

Reduced spreading and upregulation of CIP4 in Cdc42-deficient B cells. (A) Spreading visualized using bright field microscopy (left panel) and quantification of the percentage of spread cells (right panel). Columns and bars represent mean and SD of at least 500 cells per group. The experiment was repeated four times with similar results. (B) Mean length of the longest protrusion per cell determined using ImageJ program. Boxes indicate the values for the majority of the cells; vertical bars indicate the range from highest to lowest values of 200 cells per group. The experiment was repeated four times with similar results. (C) CIP4 expression in B cells activated for 2 d with anti-CD40 + IL-4. Upper panel, Representative immunoblot of CIP4 and α-actin; lower panel, quantitative analysis of CIP4 expression relative to that of α-actin and normalized to WT. Columns and bars represent mean and SD of three mice per group. (D) Selected still pictures from Supplemental Video 3 of WT B cells (upper 4 images) and Supplemental Video 4 of KO B cells (lower 4 images). A single cell each of WT and KO is followed and is marked with an arrow. Numbers represent minutes after start of videos. (A and D) Scale bars, 10 μm. (A–D) WT (mb1+/+), HZ (Cdc42flox/wt/mb1cre/+), and KO (Cdc42flox/flox/mb1cre/+). **p < 0.01, ****p < 0.0001.
FIGURE 6.

Altered morphology and different localization of α-tubulin and CIP4 in Cdc42-deficient B cells. (A) Scanning electron microscopy. Pseudo-coloring was used to highlight the cells. (B) Confocal images of spread cells, stained for F-actin (green), α-tubulin (red), and Hoechst 33258 (blue). (C, E, and G) Confocal images of cells stained for F-actin (red), CIP4 (green), and Hoechst 33258 (blue). (E and G) Blow-ups of selected areas of (C). (D, F, and H) Confocal images of cells stained for α-tubulin (red), CIP4 (green), and Hoechst 33258 (blue). (F and H) Blow-ups of (D). (A–D) Scale bars, 10 μm. The experiment was performed with WT (mb1+/+) and KO (Cdc42flox/flox/mb1cre/+) B cells activated for 2 d with anti-CD40 + IL-4 and incubated on anti-CD44–coated cultures and was repeated three times with similar results.
Anti-CD40 plus IL-4–stimulated WT B cells formed large, round, evenly shaped cell aggregates. Similar types of aggregates were formed by B cells with inducible deletion of Cdc42 (Supplemental Fig. 4C). As shown in Supplemental Fig. 4D, at later time points, the diameter of the aggregates was slightly smaller when B cells lacked Cdc42. There was no significant difference at earlier times (data not shown). When aggregates were dissociated by pipetting, the B cells reassociated with similar kinetics, independent of Cdc42 expression (Supplemental Fig. 4E).
Time-lapse photography of B cells activated with anti-CD40 plus IL-4 in suspension showed that some WT cells formed long, trailing uropods, resembling the spread cells (Supplemental Video 1), whereas Cdc42-deficient B cells were much rounder and only formed short protrusions (Supplemental Video 2). Cdc42-sufficient B cells contacted several cells at the same time using the long extensions, and the morphology of individual cells was variable and very plastic. The contact to the underlying surface appeared very firm (Supplemental Video 3, Fig. 5D, upper panel). In contrast, Cdc42-deficient B cells (Supplemental Video 4, Fig. 5D, lower panel) seemed mainly to make contacts of short duration and did not firmly attach to the surface. Thus, Cdc42 is necessary for the cellular shape and formation of contacts in B cells. The presence of ruffles in B cells lacking Cdc42 prompted us to examine whether there was a compensatory increase in Rac activity. As shown in Supplemental Fig. 4F, there was no difference in Rac activation in activated Cdc42-negative B cells, as compared with that in WT B cells.
Discussion
Cdc42 is a key coordinator of the crosstalk between the actin and the microtubule cytoskeleton; this led us to determine the specific features of Cdc42 deficiency in mature B cells. We found that absence of Cdc42 in B cells led to decreased numbers of total, follicular, and MZ B cells; a reduced humoral immune response; reduced GCs; and impaired production of Abs to TD Ags, which were mostly of low-affinity Abs. The ability of B cells that lack Cdc42 to activate T cells was impaired. This correlated with reduced spreading of B cells and altered morphology of migrating B cells, involving an inability to form long extensions containing tubulin.
In the absence of Cdc42, T1, T2-MZ precursors, MZ, and follicular B cells were all reduced in number, in agreement with previous results (21, 22). In addition, we found that GCs were reduced in size, and there were fewer GC B cells in the spleen after immunization with a TD Ag. Recent results have shown that both MZ and GC B cells migrate extensively, the former between the MZ and follicular area (1), the latter intrazonally (2). Motility is regulated by actin polymerization, and thus, it is logical that these responses depend on Cdc42. The anti-TNP Ab response was normal in Cdc42flox/flox/mb1cre-ERT2/+ mice immunized with TNP-SRBC, but was significantly reduced and of lower affinity in response to immunization with TNP-KLH. This is not surprising in view of the fact that TNP-SRBC is a very potent Ag. Thus, expression of Cdc42 in B cells is essential for an efficient humoral response to a soluble TD Ag.
We presented evidence that Ag-specific T cell production of IL-2 is reduced in response to Ag presented by Cdc42-deficient B cells. In contrast, IFN-γ production was significantly higher when presented by Cdc42-deficient B cells, compared with control B cells. Impaired stability of cognate interactions between SAP-deficient T and B cells is also associated with increased IFN-γ production (30, 31), suggesting that instability of T–B cell interaction may contribute to the increased IFN-γ secretion by T cells when Ag is presented by Cdc42-deficient B cells.
Spreading in vitro is a phenomenon that occurs only in B cells activated by a T cell–dependent stimulus, such as CD40 ligation with or without IL-4, and LPS plus IL-4 but not LPS alone (32). The spreading depends on actin polymerization and presence of microtubules (32, 33). We show in this study that Cdc42-deficient B cells failed to form long protrusions and that the shorter protrusions they formed lacked microtubules. CIP4 is an effector molecule for Cdc42 that connects WASp activity to the microtubule cytoskeleton (19). Interestingly, CIP4 was expressed along the complete length of the long dendrites formed by WT cells, but not in the shorter dendrites that predominated in Cdc42-deficient B cells. However, B cell spreading occurred in the absence of CIP4. This indicates that either there is a redundancy with another F-bar protein, such as transducer of Cdc42-dependent actin assembly-1 (Toca-1) (34) or that this class of proteins is not important for spreading in B cells. Curiously, CIP4 was upregulated in B cells lacking Cdc42, as compared with WT B cells, indicating that signaling pathways of these two molecules are connected. Moreover, CIP4 was also abundantly expressed in a speckled pattern in the nucleus of both Cdc42-sufficient and -deficient B cells, as evidenced by confocal microscopy, ruling out the possibility that the staining in fact would be cytoplasmic. To our knowledge, nuclear staining of CIP4 has not been demonstrated previously. B cells from CIP4 KO mice proliferate and class switch normally in vitro in response to LPS, anti-CD40, and anti-IgM Abs. However, GC formation and the humoral immune response are reduced in these mice, which also exhibit a T cell migration deficiency (26). Additional experiments are needed to determine the function of CIP4 and other F-bar proteins in B cells.
An intriguing observation is that WASp-deficient B cells exhibit reduced chemokine responses (8), whereas B cells that lack Cdc42 are normal in this respect. In addition, homotypic adhesion is reduced more in WASp-deficient B cells than in Cdc42-deficient B cells (7). Further experiments are needed to elucidate the differences in signaling pathways involving Cdc42 and WASp.
Our experiments indicate that the spreading response was associated with the formation of long trailing uropods during the migration of activated B cells in vitro. These uropods are able to maintain a connection with one cell, as the cell moves forward to connect to another cell. It is tempting to speculate that there is a correlation between this in vitro response and the ability of B cells to home to the B cell follicles and to form GCs and MZs. Similar patterns of dendritic B cell morphology have been observed in vivo, in GCs and MZs (1, 35). It is possible that the dendritic morphology allows more efficient interactions between T and B cells or between two B cells and that this is crucial for formation of the GCs and MZs.
While preparing this article for resubmission, Burbage et al. (22) reported that Cdc42flox/Mb1-cre mice in which Cdc42 is deleted early in B cell development have very few mature B cells in the spleen, absence of plasma cells, and virtually no serum Ig. Similar findings were reported by Guo et al. (21), who studied Cdc42flox/CD19-cre mice to delete Cdc42 early in B cell development. In neither of these studies is it possible to distinguish the contribution of the developmental effects of Cdc42 deletion in early B cells and of the severe reduction in peripheral mature B cells to the impairment of the Ab response. Comparing the results presented in this paper with the previous ones (21, 22), we report at least 2-fold higher numbers of KO B cells in the spleen. If newly differentiating B cells cannot enter into the secondary lymphoid organs, it will affect the outcome of an immune response, without necessarily meaning that there is a deficiency at later stages in B cell differentiation. Our results clearly show that Cdc42 plays a critical role also in the function of mature B cells. Indeed, mature B cells were significantly depleted shortly after tamoxifen induction of Cdc42 deletion, indicating a role of Cdc42 for their survival. Furthermore, Cdc42 was necessary for the migration of the remaining B cells, their ability to engage T cells in cognate interactions, and their capacity to form GCs and mount an Ab response to TD Ag. Our functional results and the previous demonstrations that signaling via the BCR and CD40 activates Cdc42 and is impaired in its absence (21, 22) suggest that Cdc42 activation by the BCR and CD40 is essential for mature B cell function. The guanosine nucleotide exchange factors that activate Cdc42 following BCR and CD40 ligation in B cells will be the subject of future investigations.
Note added in proof.
The Mb1-cre-ERT2 mouse strain is described in an article by Hobeika et al. (36).
Supplementary Material
Acknowledgments
We thank Dr. Kjell Hultenby, Karolinska Institutet, for help with electron microscopy. We also thank Dr. Zheng, University of Cincinnati; Dr. Reth, University of Freiburg; and Dr. Busslinger, Vienna Biocenter, for the gifts of Cdc42flox, Mb1-cre-ERT2, and CD23-cre mice, respectively. We thank the imaging facility at Stockholm University for being able to use their microscopes. Dr. Tybulewicz (Medical Research Council, National Institute for Medical Research, London, U.K.) is thanked for helpful advice. Dr. Narayanaswamy Ramesh and Dr. Marton Keszei are thanked for experimental guidance and help, respectively.
This work was supported by the Swedish Research Council (to E.S. and L.S.W.), the Department of Molecular Biosciences, Stockholm University (to E.S.), the Karolinska Institutet, the European Commission 7th Framework Programme, the Ragnar Söderberg Foundation, the Groschinsky Foundation, the Åke Wiberg Foundation, the Bergvall Foundation, the Swedish Society of Medicine (to L.S.W.), and U.S. Public Health Service Grant HL-059561 (to R.S.G.). M.A.P.B. was supported by a fellowship from Fundação para a Ciência e a Tecnologia (SFRH/BD/47926/2008), and C.I.M.D. was supported by a fellowship from the Karolinska Institutet.
The online version of this article contains supplemental material.
- CIP4
- Cdc42-interacting protein
- GC
- germinal center
- HZ
- heterozygote
- KLH
- keyhole limpet hemocyanin
- KO
- knockout
- MZ
- marginal zone
- NP
- 4-hydroxy-3-nitrophenylacetyl
- N-WASp
- neuronal-WASp
- T
- transitional
- TD
- T cell–dependent
- TNP
- trinitrophenyl
- WASp
- Wiskott–Aldrich syndrome protein
- WT
- wild-type.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Arnon T. I., Horton R. M., Grigorova I. L., Cyster J. G. 2013. Visualization of splenic marginal zone B-cell shuttling and follicular B-cell egress. Nature 493: 684–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Victora G. D., Schwickert T. A., Fooksman D. R., Kamphorst A. O., Meyer-Hermann M., Dustin M. L., Nussenzweig M. C. 2010. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 143: 592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koopman G., Parmentier H. K., Schuurman H. J., Newman W., Meijer C. J., Pals S. T. 1991. Adhesion of human B cells to follicular dendritic cells involves both the lymphocyte function-associated antigen 1/intercellular adhesion molecule 1 and very late antigen 4/vascular cell adhesion molecule 1 pathways. J. Exp. Med. 173: 1297–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu T. T., Cyster J. G. 2002. Integrin-mediated long-term B cell retention in the splenic marginal zone. Science 297: 409–412. [DOI] [PubMed] [Google Scholar]
- 5.Bannard O., Horton R. M., Allen C. D. C., An J., Nagasawa T., Cyster J. G. 2013. Germinal center centroblasts transition to a centrocyte phenotype according to a timed program and depend on the dark zone for effective selection. Immunity 39: 912–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Green J. A., Suzuki K., Cho B., Willison L. D., Palmer D., Allen C. D. C., Schmidt T. H., Xu Y., Proia R. L., Coughlin S. R., Cyster J. G. 2011. The sphingosine 1-phosphate receptor S1P₂ maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat. Immunol. 12: 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Westerberg L., Greicius G., Snapper S. B., Aspenström P., Severinson E. 2001. Cdc42, Rac1, and the Wiskott-Aldrich syndrome protein are involved in the cytoskeletal regulation of B lymphocytes. Blood 98: 1086–1094. [DOI] [PubMed] [Google Scholar]
- 8.Westerberg L., Larsson M., Hardy S. J., Fernández C., Thrasher A. J., Severinson E. 2005. Wiskott-Aldrich syndrome protein deficiency leads to reduced B-cell adhesion, migration, and homing, and a delayed humoral immune response. Blood 105: 1144–1152. [DOI] [PubMed] [Google Scholar]
- 9.Meyer-Bahlburg A., Becker-Herman S., Humblet-Baron S., Khim S., Weber M., Bouma G., Thrasher A. J., Batista F. D., Rawlings D. J. 2008. Wiskott-Aldrich syndrome protein deficiency in B cells results in impaired peripheral homeostasis. Blood 112: 4158–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Westerberg L. S., de la Fuente M. A., Wermeling F., Ochs H. D., Karlsson M. C., Snapper S. B., Notarangelo L. D. 2008. WASP confers selective advantage for specific hematopoietic cell populations and serves a unique role in marginal zone B-cell homeostasis and function. Blood 112: 4139–4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Westerberg L. S., Dahlberg C., Baptista M., Moran C. J., Detre C., Keszei M., Eston M. A., Alt F. W., Terhorst C., Notarangelo L. D., Snapper S. B. 2012. Wiskott-Aldrich syndrome protein (WASP) and N-WASP are critical for peripheral B-cell development and function. Blood 119: 3966–3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Becker-Herman S., Meyer-Bahlburg A., Schwartz M. A., Jackson S. W., Hudkins K. L., Liu C., Sather B. D., Khim S., Liggitt D., Song W., et al. 2011. WASp-deficient B cells play a critical, cell-intrinsic role in triggering autoimmunity. J. Exp. Med. 208: 2033–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Recher M., Burns S. O., de la Fuente M. A., Volpi S., Dahlberg C., Walter J. E., Moffitt K., Mathew D., Honke N., Lang P. A., et al. 2012. B cell-intrinsic deficiency of the Wiskott-Aldrich syndrome protein (WASp) causes severe abnormalities of the peripheral B-cell compartment in mice. Blood 119: 2819–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aspenström P., Lindberg U., Hall A. 1996. Two GTPases, Cdc42 and Rac, bind directly to a protein implicated in the immunodeficiency disorder Wiskott-Aldrich syndrome. Curr. Biol. 6: 70–75. [DOI] [PubMed] [Google Scholar]
- 15.Rohatgi R., Ma L., Miki H., Lopez M., Kirchhausen T., Takenawa T., Kirschner M. W. 1999. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell 97: 221–231. [DOI] [PubMed] [Google Scholar]
- 16.Hall A. 2005. Rho GTPases and the control of cell behaviour. Biochem. Soc. Trans. 33: 891–895. [DOI] [PubMed] [Google Scholar]
- 17.Aspenström P. 2009. Roles of F-BAR/PCH proteins in the regulation of membrane dynamics and actin reorganization. Int. Rev. Cell Mol. Biol. 272: 1–31. [DOI] [PubMed] [Google Scholar]
- 18.Aspenström P. 1997. A Cdc42 target protein with homology to the non-kinase domain of FER has a potential role in regulating the actin cytoskeleton. Curr. Biol. 7: 479–487. [DOI] [PubMed] [Google Scholar]
- 19.Tian L., Nelson D. L., Stewart D. M. 2000. Cdc42-interacting protein 4 mediates binding of the Wiskott-Aldrich syndrome protein to microtubules. J. Biol. Chem. 275: 7854–7861. [DOI] [PubMed] [Google Scholar]
- 20.Yoshida H., Tomiyama Y., Ishikawa J., Oritani K., Matsumura I., Shiraga M., Yokota T., Okajima Y., Ogawa M., Miyagawa Ji., et al. 2000. Integrin-associated protein/CD47 regulates motile activity in human B-cell lines through CDC42. Blood 96: 234–241. [PubMed] [Google Scholar]
- 21.Guo F., Velu C. S., Grimes H. L., Zheng Y. 2009. Rho GTPase Cdc42 is essential for B-lymphocyte development and activation. Blood 114: 2909–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burbage M., Keppler S. J., Gasparrini F., Martínez-Martín N., Gaya M., Feest C., Domart M.-C., Brakebusch C., Collinson L., Bruckbauer A., Batista F. D. 2015. Cdc42 is a key regulator of B cell differentiation and is required for antiviral humoral immunity. J. Exp. Med. 212: 53–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang L., Wang L., Zheng Y. 2006. Gene targeting of Cdc42 and Cdc42GAP affirms the critical involvement of Cdc42 in filopodia induction, directed migration, and proliferation in primary mouse embryonic fibroblasts. Mol. Biol. Cell 17: 4675–4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schweighoffer E., Vanes L., Nys J., Cantrell D., McCleary S., Smithers N., Tybulewicz V. L. J. 2013. The BAFF receptor transduces survival signals by co-opting the B cell receptor signaling pathway. Immunity 38: 475–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwon K., Hutter C., Sun Q., Bilic I., Cobaleda C., Malin S., Busslinger M. 2008. Instructive role of the transcription factor E2A in early B lymphopoiesis and germinal center B cell development. Immunity 28: 751–762. [DOI] [PubMed] [Google Scholar]
- 26.Koduru S., Kumar L., Massaad M. J., Ramesh N., Le Bras S., Ozcan E., Oyoshi M. K., Kaku M., Fujiwara Y., Kremer L., et al. 2010. Cdc42 interacting protein 4 (CIP4) is essential for integrin-dependent T-cell trafficking. Proc. Natl. Acad. Sci. USA 107: 16252–16256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karlsson M. C. I., Wernersson S., Diaz de Ståhl T., Gustavsson S., Heyman B. 1999. Efficient IgG-mediated suppression of primary antibody responses in Fcgamma receptor-deficient mice. Proc. Natl. Acad. Sci. USA 96: 2244–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ström L., Laurencikiené J., Miskiniené A., Severinson E. 1999. Characterization of CD40-dependent immunoglobulin class switching. Scand. J. Immunol. 49: 523–532. [DOI] [PubMed] [Google Scholar]
- 29.Henderson R. B., Grys K., Vehlow A., de Bettignies C., Zachacz A., Henley T., Turner M., Batista F., Tybulewicz V. L. 2010. A novel Rac-dependent checkpoint in B cell development controls entry into the splenic white pulp and cell survival. J. Exp. Med. 207: 837–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cannons J. L., Yu L. J., Jankovic D., Crotty S., Horai R., Kirby M., Anderson S., Cheever A. W., Sher A., Schwartzberg P. L. 2006. SAP regulates T cell-mediated help for humoral immunity by a mechanism distinct from cytokine regulation. J. Exp. Med. 203: 1551–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qi H., Cannons J. L., Klauschen F., Schwartzberg P. L., Germain R. N. 2008. SAP-controlled T-B cell interactions underlie germinal centre formation. Nature 455: 764–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davey E. J., Thyberg J., Conrad D. H., Severinson E. 1998. Regulation of cell morphology in B lymphocytes by IL-4: evidence for induced cytoskeletal changes. J. Immunol. 160: 5366–5373. [PubMed] [Google Scholar]
- 33.Sumoza-Toledo A., Santos-Argumedo L. 2004. The spreading of B lymphocytes induced by CD44 cross-linking requires actin, tubulin, and vimentin rearrangements. J. Leukoc. Biol. 75: 233–239. [DOI] [PubMed] [Google Scholar]
- 34.Ho H. Y., Rohatgi R., Lebensohn A. M., Le Ma J., Li J., Gygi S. P., Kirschner M. W. 2004. Toca-1 mediates Cdc42-dependent actin nucleation by activating the N-WASP-WIP complex. Cell 118: 203–216. [DOI] [PubMed] [Google Scholar]
- 35.Hauser A. E., Junt T., Mempel T. R., Sneddon M. W., Kleinstein S. H., Henrickson S. E., von Andrian U. H., Shlomchik M. J., Haberman A. M. 2007. Definition of germinal-center B cell migration in vivo reveals predominant intrazonal circulation patterns. Immunity 26: 655–667. [DOI] [PubMed] [Google Scholar]
- 36.Hobeika E., Levit-Zerdoun E., Anastasopoulou V., Pohlmeyer R., Altmeier S., Alsadeq A., Dobenecker M. W., Pelanda R., Reth M. CD19 and BAFF-R can signal to promote B-cell survival in the absence of Syk. EMBO J. 34: 925–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.