

Abstract
In health, long-lived plasma cells (LLPC) are essential for durable protective humoral immunity, and, conversely, in disease are a major source of pathogenic Abs in autoimmunity, graft rejection, and allergy. However, the molecular basis for their longevity is largely unknown. We have recently found that CD28 signaling in plasma cells (PC) is essential for sustaining Ab titers, by supporting the survival of LLPC, but not short-lived PC (SLPC). We now find that, unlike SLPC, CD28 activation in LLPC induces prosurvival downstream Vav signaling. Knockin mice with CD28 cytoplasmic tail mutations that abrogate Vav signaling (CD28-AYAA) had significantly fewer LLPC but unaffected SLPC numbers, whereas mice with mutations that abrogate PI3K signaling (CD28-Y170F) were indistinguishable from wild-type controls. This was consistent with the loss of CD28’s prosurvival effect in LLPC from CD28-AYAA, but not CD28-Y170F, mice. Furthermore, the CD28 Vav motif in the B lineage was essential for the long-term maintenance of Ag-specific LLPC populations and Ab titers in vivo. Signaling downstream of the CD28 Vav motif induced previously undescribed transcriptional regulation of B lymphocyte–induced maturation protein-1, a key mediator of PC differentiation and maintenance. These findings suggest CD28 signaling in LLPC modulates the central B lymphocyte–induced maturation protein-1 transcriptional nexus involved in long-term survival and function.
Introduction
Durable immunity against many pathogens is critically dependent on the long-term maintenance of protective Ab titers, and conversely, sustained Ab levels mediate the disease pathology in autoimmunity, allergy, and allograft rejection (1–3). Because the t1/2 of Ig molecules is on the order of days, long-term maintenance of Ab titers requires continuous Ig production by persistent populations of terminally differentiated B-lineage plasma cells (PC). In the current paradigm, maintenance of these populations is a function of two broadly and incompletely defined subsets of PC that are intrinsically distinct in terms of longevity and anatomic localization (1, 4, 5). The first subset are short-lived PC (SLPC), which survive for weeks/months and are the predominant PC population in secondary lymphoid organs such as the spleen. The SLPC populations are sustained by B cells activated by persistent or recurrent Ag exposure. This mechanism, however, does not explain how protective Ab titers can be maintained for years without re-exposure to the inciting Ag (6), and can persist in mice (7, 8) and humans (9) despite depletion of a replenishing B cell pool. These Ag/B cell–independent Ab titers are thought to be maintained by the second subset of PC that are long-lived (LLPC), surviving for up to the lifetime of the host and found predominantly in the bone marrow (BM) (6). Importantly, LLPC are not autonomously long-lived, but rather their survival is dependent upon their intrinsic capability to access and use a fixed number of specific prosurvival niches within the BM (1). However, what molecular mechanisms underlie this intrinsic capability and LLPC longevity remain largely uncharacterized.
Despite considerable indirect evidence supporting the existence of separate SLPC and LLPC subsets, there has been little direct proof they are in fact distinct cell populations. Anatomic localization has remained the “gold standard” distinguishing characteristic, although evidence suggests this distinction is not absolute (10–12). Only recently have cellular and molecular characteristics been identified that more clearly define LLPC and SLPC as being intrinsically distinct subsets (13–15). We have recently reported that one such characteristic is the requirement for CD28 function for the survival of LLPC, but not SLPC (14). CD28 has been best characterized as the prototypic T cell costimulatory receptor that augments TCR-mediated T cell proliferation, survival, metabolic fitness, and effector function (16). CD28 is also expressed on normal murine and human PC (14, 17, 18) and the malignant BM-resident PC of multiple myeloma (MM) (19, 20), but its function in B lineage has been largely uncharacterized. Previous studies have shown that the loss of CD28 activation significantly diminishes humoral responses, although this was attributed to defects in T cell help and germinal center formation (21–30). However, overexpression of CD28 on myeloma cells is a poor prognostic marker (18, 31), and, consistent with this, we have found that activation of CD28 on MM cells directly transduces a prosurvival signal (32). In normal PC, an intrinsic role for CD28 was implicated by the finding that its expression in normal B cells is specifically repressed by the B cell master regulator Pax-5, derepressed during B→PC differentiation, and that its loss in the B lineage diminished short-term primary Ab responses (33). Extending these observations, we found that B lineage loss of CD28 caused a significant reduction in in situ LLPC survival within the BM (from a t1/2 of 426 to 63 d), and manifested as a loss of the BM LLPC population and inability to sustain Ag-specific Ab titers long-term following vaccination (14). In contrast, loss of CD28 had no effect on splenic SLPC survival or population numbers. Although LLPC and SLPC express equivalent levels of CD28, only in the LLPC did CD28 activation lead to NF-κB signaling. Thus, these findings suggest it is this ability to signal in LLPC that allows these cells to receive a prosurvival signal from the CD80/CD86+ stromal cells within the long-term BM survival niches, and conversely, the inability to signal prevents the SLPC from doing so.
It is not known how CD28 signals in LLPC, or how these signals regulate survival and function. In T cells, specific CD28 cytoplasmic motifs initiate distinct downstream signaling and cellular responses (21, 23, 34). Phosphorylation of the Y170MNM motif leads to binding the p85 subunit of PI3K and activation of the PI3K→PDK1→Akt→NF-κB pathway (34). Phosphorylation of the P187YAP190 motif leads to binding by Lck and Grb2, with the latter activating downstream Vav→Rac1/Cdc42 →ras→AP-1 and Vav→phospholipase C (PLC)γ→NF-κB/NFAT pathways (34). We have previously reported in CD28 knockin mice that mutation of the Y170MNM motif (Y→F, Y170F) unexpectedly had little effect on T cell function, but mutation of the P187YAP190 motif to AYAA significantly impaired T cell activation and generation of OVA-specific Ab responses—although the humoral defect was attributed to the loss of T cell help, and PC were not examined (21, 23). Separate studies in PC have demonstrated that Vav signaling (from unidentified origins) is essential for the expression of the transcriptional modulator B lymphocyte–induced maturation protein 1 (BLIMP-1, prdm1) (35), a central regulator of PC differentiation required for maintenance of LLPC in the BM (36, 37). Altogether, these preceding studies led us to examine what signaling pathways are activated by CD28 in normal PC, and what molecular and cellular responses are regulated by these pathways.
Materials and Methods
Animals
Female and male C57BL/6J (wild-type [WT]), B6.129S2-Igh-6tm1Cgn/J (μMT), and B6.129S2-Cd28tm1Mak/J (CD28−/−) mice were purchased from The Jackson Laboratory at 5–6 wk of age. Female C57BL/6J retired breeders ∼9 mo old were purchased from The Jackson Laboratory. BLIMP-1gfp mice were a gift of S. Nutt (The Walter and Eliza Hall Institute of Medical Research, Parkville, VIC, Australia). Female and male breeding pairs of CD28-AYAA knockin (CD28-AYAA) and CD28-Y170F knockin (CD28-Y170F) were derived, as previously reported (23). Upon receipt, animals were housed and bred at the Department of Laboratory Animal Resources (Roswell Park Cancer Institute, Buffalo, NY) in a pathogen-free barrier facility. All animal experiments were approved by the Roswell Park Cancer Institute Institutional Animal Care and User Committee.
Abs and flow cytometry
BLIMP-1 (C14A4) was purchased from Cell Signaling. Cells were stained with anti-CD45.1 (clone A20), anti-CD45.2 (clone 104), anti–B220-PE/Cy7 (clone RA3-6B2), anti–I-A/I-E-PerCP/Cy5.5 (clone M5/114.15.2), anti-CD19 (clone 6D5), and anti–CD3-PE (clone 17A2) purchased from BioLegend; anti-CD28 (clone PV1), anti-hamster IgG (H + L)-FITC, and isotype control rat IgG2a-PE purchased from Beckman Coulter; anti–CD138-PE (clone 281-2) from BD Pharmingen; anti-mouse IL-6R (R&D Systems); and anti-goat IgG-FITC (United States Biochemical). Polyclonal control hamster-IgG was purchased from Genetex. Polyclonal control hamster IgG and anti-CD28 mAb were conjugated to Dynabeads goat anti-mouse IgG (Invitrogen) per manufacturer’s instructions, and were cultured with cells at 2:1 bead to cell ratio, respectively. Cells were incubated with staining reagents in staining media (PBS-1% FCS, 5 mM HEPES, 5 mM 10% sodium azide) for 30 min in 4°C. Analysis was performed by flow cytometry (LSR II and FACSCalibur). For the immunoprecipitation and Western blot experiments, Abs against PI3K p85, Vav1 (both from Cell Signaling), phospho-Vav (Santa Cruz Technology), and phosphotyrosine (clone 4G10; EMD Millipore) were used.
For intracellular staining for phospho-AKT, J558 cells or purified BM PC were treated with control hamster Ig or anti-CD28 mAb (PV1) for 1 h in serum-free media, fixed, and stained for phosphorylated Akt (threonine 308; BD Biosciences [558275]).
For intracellular staining of pPLCγ1, PC enrichment was performed on BM and spleens of WT C57/B6 mice using the MACS CD138+ PC isolation kit (Miltenyi Biotec). Cells were plated in serum-free media at 1 × 105/well in a 96-well plate and incubated at 37°C for 30 min prior to activation. For activation, cells were treated with 20 μg/well control hamster Ig or anti-CD28 mAb (PV1) for 15 min. Cells were immediately washed with cold PBS and stained with anti–B220-Pacific Blue (clone RA3-6B2) and anti–CD138-allophycocyanin (clone 281-2) from BioLegend in cold PBS. Cells were then fixed and permeabilized with the Foxp3 intracellular staining kit from eBioscience using an anti–phospho-PLCγ1 Ab from Cell Signaling (2821S) and a goat–anti-rabbit FITC from Santa Cruz (sc2012). For analysis, CD138+/B220− cells were gated, and the mean fluorescent intensity was assessed on the FITC channel. Flow analysis was done using FlowJo 9.4.5.
PC purification and cell culture
PC isolation was performed as described previously (14). Briefly, PC were isolated using the MACS CD138+ PC isolation kit (Miltenyi Biotec). Cells were labeled with non-PC depletion mixture and anti-biotin microbeads for non-PC depletion. Cells were then labeled with CD138 microbeads and run over the magnetic column twice to adhere CD138− cells. The purity of the CD138+ population was >83%. Serum starvation studies and survival assays were performed, as we have previously described (14, 32).
BM-derived dendritic cell
BM cells from WT C57/B6 mice were differentiated in culture with GM-CSF (20 ng/ml, derived from supernatant) for 7 d, as we have reported (14). PC ± BM-derived dendritic cells (BMDC) were cultured in a 48-well flat-bottom plate tissue culture plate (2 or 2.5 × 104 PC and 2 × 105 BMDC in 0.8 ml culture medium/well) at 37°C in 5% CO2 with 20 ng/ml GM-CSF.
Immunoprecipitation and Western blot analysis
The plasmacytoma cell line J558 or purified BM and splenic PC were cultured, as previously described (14). For Western blots, whole-cell lysates were prepared, run on 8% SDS-PAGE gels, and transferred to polyvinylidene difluoride membrane. The membrane was probed with anti–BLIMP-1 (Cell Signaling). Blots were then probed for rabbit polyclonal anti-actin (Sigma-Aldrich). Membranes were developed with ECL (Thermo Scientific), according to manufacturer’s instructions.
For the immunoprecipitations, 1 × 107 J558/conditions were activated with control hamster Ig (10 μg/ml) or anti-CD28 mAb (PV1.1, 2 μl/100 μl) for 15 min, then lysed in radioimmunoprecipitation assay buffer. A total of 1–10 μg primary immunoprecipitation Ab (CD28 or phosphotyrosine) was incubated with the protein lysate for 2–4 h, and immunocomplexes were captured by rProtein G agarose (Invitrogen) and analyzed by Western blot for the p85 subunit of PI3K or for Vav1, as detailed above.
BM reconstitution and immunizations
BM chimeras were generated as previously described (14); briefly, the chimeras were generated by retro-orbitally injecting 106 BM cells, depleted of T cells (Miltenyi Biotec), at a 40:60 ratio of μMT and WT, μMT and CD28−/−, μMT and CD28-AYAA, or μMT and CD28-Y170F BM into lethally irradiated WT mice. Mice were immunized s.c. with 1:1 ratio of 100 μg NIP-OVA (Biosearch Tech) in CFA (Thermo Scientific) on day 0 and boosted on day 7 with 1:1 ratio of 100 μg NIP-OVA in IFA (Thermo Scientific).
ELISAs
Murine IgG1 and NIP-IgG1 Ab titers were determined by ELISA per the manufacturer’s instructions (Bethyl) and as we have previously reported (14). Briefly, NUNC 96-well plates were precoated with capture Ab in coating buffer or NIP-BSA in PBS–0.2 M NaCl (15 μg/ml; Biosearch Technologies) overnight in 4°C. Murine IL-6 was assayed by ELISA per manufacturer’s instructions (R&D Systems).
ELISPOT assays
NIP-specific and total IgG Ab-secreting cells (ASC) were quantified by ELISPOT assay as per manufacturer’s instructions (Mabtech) and our previous report (14). Briefly, Millipore 96-well plates (MAIPSWU) were precoated with anti-IgG capture Ab (15 μg/ml) or NIP-BSA (20 μg/ml) in PBS.
Prdm1 quantitative PCR
Equal loading volumes of actin were used as an endogenous control target using a SD.2 quantitative PCR machine, with subsequent triplicate wells used for prdm1 expression. Primer sets used were as follows: prdm1 forward, 5′-CGTAGAAAAGGAGGGACCGC-3′; prdm1 reverse, 5′-TCTTTGCTGTTGTTGGCAGC-3′; actin forward, 5′-CCTAAGGCCAACCGTGAAAAG-3′; and actin reverse, 5′-GAGGCATACAGGGACAGCACA-3′.
Plasmids and dual luciferase assay
The 7000-bp BLIMP-1, 4500-bp BLIMP-1, and 1500-bp BLIMP-1 luciferase constructs were constructed in the PGL3 basic plasmid (Promega; also the source for the CMV-Renillia luciferase construct). A total of 2 × 106 J558 cells was transfected with 2 μg promoter construct/100 ng Renilla DNA per group with the Nucleofector system (Amaxa) as per protocol for kit V. The transfected J558 cells were cultured in 10% FCS media alone or with polyclonal hamster Ig or anti-CD28 mAb beads, and after 16 h the cells were harvested and lysed. Luciferase activity was assayed using Dual Luciferase Reporter Assay (Promega) per the manufacturer’s instructions. Briefly, cells were lysed in 1× passive lysis buffer, the passive lysis buffer lysate was then mixed with LARII buffer, and firefly luciferase activity was measured by the Monolight 3010 luminometer (BD Pharmingen). Relative luciferase activity was determined as follows: luciferase activity/Renilla activity.
Chromatin immunoprecipitation assay
J558 cells were treated for 30 min with anti-CD28 (PV1.1, 2 μg/ml) or isotype control (anti-Syrian hamster Ig, 2 μg/ml). After 30 min, DNA and DNA-bound molecules were cross-linked by formaldehyde fixation and sheared into 300- to 500-bp fragments by sonication. A total of 500 μg chromatin was immunoprecipitated by overnight incubation with 2 μg anti-c-Rel, or isotype control, followed by incubation with recombinant protein A agarose beads (Repligen, Waltham, MA). DNA/protein cross-links were subsequently reversed by overnight incubation at 65°C. DNA was isolated by phenyl/chloroform extraction and isopropanol precipitation. Relative prdm1-promoter enrichment/binding was determined by quantitative PCR for a region of the promoter putatively identified as containing c-Rel binding sites. Primers are as follows: −7316 bp, forward, 5′-CGGCCCCGACAAGATTAGTT-3′, and reverse, 5′-TCTCTCGTTTACTCTGCGGC-3′; −6923 bp, forward, 5′-AGTTTTCGCCTCCTGTTGCT-3′, and reverse, 5′-TTCTGAGGCGGATAAGGTGC-3′; −5821 bp, forward, 5′-AAGTCCAGGCTGATGGCTTC-3′, and reverse, 5′-CTTTCCCACCTGAGAGGCTG-3′; −5120 bp, forward, 5′-ACTACACCCCTTGGAGCAGA-3′, and reverse, 5′-CAGGTTAGCCTAGCATAGAGGC-3′; −4756 bp, forward, 5′-CCAGGCTCTCACTGATGTGG-3′, and reverse, 5′-GCAAACGGGGAAAACCACTG-3′.
Statistical analysis
Student t test was performed for statistical analysis using two-tailed, nonequal variances, and 95% confidence interval. Mann–Whitney rank sum test was used for equal variances. Analysis of mean fluorescent intensities was done by one-way ANOVA (and nonparametric) with Newman–Keuls posttest. The 4-hydroxy-5-indo-3 nitrophenyl conjugated to ovalbumin (NIP-OVA)–specific PC population t1/2 was calculated as follows: t1/2 = (change in time) × Log (2)/Log(beginning amount/end amount), where change in time is 63 d − 21 d for BM or 42 d, 21 d for spleen, then beginning amount = number of NIP-OVA–specific ASCs at day 21 and ending amount = number of ASCs at day 63 for BM and 42 for spleen.
Results
CD28 activation in PC induces downstream PI3K and Vav signaling
We first examined whether CD28 activation in PC resulted in downstream activation of the PI3K and Vav pathways, as has been reported in T cells (34) (Supplemental Fig. 1). Utilizing the murine CD28+ PC cell line J558 (which expresses high levels of CD28 comparable to PC [Fig. 1A]), we found in coimmunoprecipitation studies that direct activation with αCD28 mAb resulted in association of the p85 subunit of PI3K to the CD28 receptor by 15 min (Fig. 1B). Further downstream of PI3K, CD28 activation induced phosphorylation of Akt in both J558 (Fig. 1C, left) and purified primary murine BM PC (Fig. 1C, right). Similarly, CD28 activation triggered Vav signaling, as indicated by Vav1 phosphorylation (Fig. 1D). This Vav signaling would be predicted to result in downstream PLCγ1 activation, which was seen following CD28 activation in purified BM, but not in splenic PC (Fig. 2; PLCγ1 activation measured by its phosphorylation). It should be noted that, for our studies, we are using the conventional prevailing equivalence of BM PC = LLPC and splenic PC = SLPC, with its attendant caveats. Altogether these findings suggest that, similar to T cells, CD28 activation in PC leads to downstream signaling through the PI3K and Vav pathways.
FIGURE 1.

CD28 activation induces PI3K and Vav signaling in PC. J558 murine plasmacytoma cells were activated with control hamster Ig (con) or anti-CD28 mAb (αCD28, PV1.1) for 15 min and then analyzed by coimmunoprecipitation, followed by Western blot. (A) CD28 expression by J558, by flow cytometry. (B) PI3K. Left, Protein lysates from J558 activated as indicated were immunoprecipitated with αCD28 mAb, and the immune complexes were analyzed by Western blot for coimmunoprecipitation of the p85 subunit of PI3K. Total p85 and tubulin (as a loading control) were determined by Western blot from total cell lysates. Data representative of three independent experiments. Right, Relative densitometry combined from three experiments. (C) Downstream AKT activation. J558 cells (left) or purified BM PC (right) were treated with control Ig (filled histograms) or αCD28 mAb (open histograms) for 1 h in serum-free media, fixed, stained for phospho-Akt, and analyzed by flow cytometry. Data representative of two independent experiments. (D) Vav. Protein lysates from J558 activated as indicated were immunoprecipitated for phosphotyrosine, and the immunocomplexes were analyzed by Western blot for the presence of Vav1 (arrow). Data are representative of three independent experiments. Right, Relative densitometry combined from three experiments. The same results were seen when the Western blot was probed for phospho-Vav1 (data not shown). *p < 0.05, ***p < 0.001.
FIGURE 2.

Differential PLCγ signaling in BM versus splenic PC. BM and splenic PC were isolated and treated with αCD28 mAb or isotype control Ab for 15 min, fixed, gated on CD138+/B220− PC populations, and stained for phospho-PLCγ1. Top, Histograms representative of three independent experiments with the solid gray peak representing the isotype control, gray line the control Ig-treated group, and black the αCD28 mAb-treated PC. Bottom, Mean fluorescent intensity for pPLCγ1 is pooled for two wells for each experiment; data representative of three independent experiments. Statistical analysis done by one-way ANOVA (and nonparametric) with Newman–Keuls posttest. Data representative of two independent experiments. *p < 0.05.
The PYAP proline motif of CD28 is required for its prosurvival effect in BM PC
To more precisely define the roles of the pathways downstream of CD28, we next examined the CD28-Y170F and CD28-AYAA knockin mice, which are disabled in CD28’s ability to signal to PI3K or Vav, respectively (34). Although the CD28 PYAP motif also binds Lck in T cells (34), Lck has not been detected in either myeloma cells (38) or primary BM PC (39). The CD28 expression on BM (Supplemental Fig. 2A) and splenic (Supplemental Fig. 2B) CD138+ PC purified from CD28-Y170F and CD28-AYAA mice was equivalent to WT, as was the expression on T cells from the BM (Supplemental Fig. 2C) and spleen (Supplemental Fig. 2D). Consistent with the previous T cell studies, CD28 activation significantly induced AKT phosphorylation (as a measure of downstream PI3K signaling) in BM PC from WT and CD28-AYAA mice, which was lost in the CD28-Y170F BM PC (Fig. 3A). Similarly, CD28 activation significantly induced PLCγ1 phosphorylation (as a measure of Vav signaling) in BM PC from WT and CD28-Y170F mice, but not in CD28-AYAA BM PC (Fig. 3B). Interestingly, CD28-induced PLCγ1 phosphorylation was less in the CD28-Y170F compared with WT, suggesting that this CD28 motif or downstream signaling components may also contribute to Vav signaling. In naive mice, multiparametric flow cytometric analysis demonstrated that the total number of splenic PC was equivalent for all genotypes (Fig. 3A), in agreement with our previous findings (14). In contrast, whereas WT and CD28-Y170F mice had equivalent total numbers of BM PC (Fig. 3B), the CD28-AYAA mice had significantly fewer BM PC versus WT or CD28-Y170F mice, with the greatest reduction of BM PC in the CD28−/− global knockout mice (Fig. 3A, left). The consistent finding that the CD28−/− mice have fewer BM PC than the CD28-AYAA may reflect the contribution of other motifs in the cytoplasmic tail [as we have recently shown in T cells (40)]. An additional possibility is that even a signaling-incompetent CD28 receptor can still induce prosurvival factors from the niche, based on our previous observation that PC CD28 induces stromal DC production of the PC survival cytokine IL-6 via binding to DC CD80/CD86 (14, 18). Consistent with this, we found that BM PC from CD28-AYAA mice can still induce DC IL-6 production at levels similar to BM PC from WT and CD28-Y170F animals (Supplemental Fig. 3).
FIGURE 3.
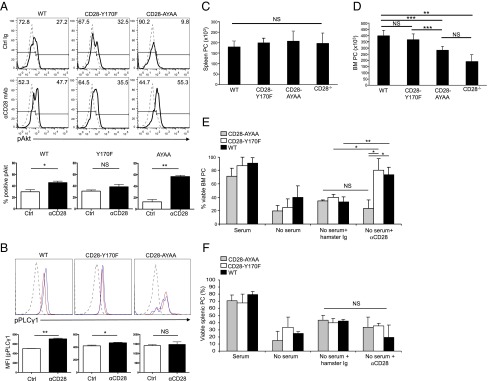
The prosurvival effect of the CD28 PYAP proline motif in BM PC. (A) PI3K signaling. BM PC were purified from WT, CD28-Y170F, and CD28-AYAA mice; treated with control hamster Ig or CD28-activating Ab for 1 h; and stained for intracellular pAKT; and percentage of cells positive for pAKT was determined (inset numbers). Gray histogram, control Ig; black histogram, CD28 mAb. All conditions done in duplicate, representative of two independent experiments. (B) Vav signaling. BM PC were purified from WT, CD28-Y170F, and CD28-AYAA mice; treated with control hamster Ig or CD28-activating Ab for 1 h; stained for intracellular pPLCγ1; and assessed for mean fluorescent intensity (MFI). Solid gray histogram, isotype control staining; red histogram, control Ig; blue histogram, CD28 mAb. All conditions done in duplicate; representative of two independent experiments. (C and D) Total numbers of PC in the (C) spleen or (D) BM of WT, CD28-Y170F, CD28-AYAA, and CD28−/− mice were determined from total mononuclear cells by multiparametric flow cytometry using CD138+B220− to identify PC. Mean ± SD of five mice. (E and F) Purified (E) BM or (F) splenic PC from WT, CD28-Y170FF, and CD28-AYAA mice were cultured ± FCS for 24 h ± control hamster or αCD28 mAb-coated beads, and viability was assessed by trypan blue exclusion. Mean ± SD of three independent experiments are shown. *p < 0.05, **p < 0.01, ***p < 0.001.
The diminished BM PC population in the CD28-AYAA mice may be due to a number of factors, including impaired PC generation, PC homing to the BM, or in situ survival once in the BM. Our previous studies have pointed to a prosurvival role for CD28, and we found that in serum starvation conditions in vitro the viability of BM PC purified from WT or CD28-Y170F mice was significantly increased by activating αCD28 mAb (Fig. 3C). In contrast, CD28 activation did not enhance the survival of BM PC from CD28-AYAA or CD28−/− mice, consistent with the in vivo findings. CD28 activation also did not rescue any of the splenic PC from the different strains (Fig. 3D). These findings indicate that CD28-mediated BM LLPC survival is primarily dependent on signals arising from its PYAP motif.
The CD28 PYAP motif in the B lineage is necessary for the long-term maintenance of Ag-specific BM LLPC populations and Ab titers
The CD28-AYAA mice have T cell defects in activation and function (21, 23) that could be indirectly inhibiting the generation of BM LLPC in vivo, although this seems less likely given the generation of Ig class-switched splenic SLPC is unaffected. To more clearly delineate the T versus B lineage effect of the CD28-AYAA mutation in the humoral response, we generated BM chimeric mice by cotransplanting lethally irradiated WT hosts with BM from μMT mice that lack B cells (33, 41) plus BM from WT, CD28-Y170F, CD28-AYAA, CD28−/− mice (Fig. 4A). Thus, in the experimental mice, the B lineage arises solely from the CD28 knockin/knockout BM, whereas the T cell compartment is chimeric for WT CD28 receptor from the μMT BM. Analysis of chimerization showed expected percentages of CD3+CD28+ T cells and CD138+ PC in both the spleen and BM (Supplemental Fig. 4). After BM reconstitution, the chimeras were primed/boosted with the T-dependent Ag NIP-OVA in CFA/IFA, and serum Ig and PC numbers were assessed over time. All four chimeras had equivalent total serum IgG1 levels out to 63 d (Fig. 4B), which was not unexpected given our previous findings that the SLPC populations are not dependent on CD28 for survival. In contrast, whereas all the chimeras had similar NIP-specific IgG1 titers at day 7, by day 21 the titers in the CD28-AYAA:μMT and CD28−/−:μMT chimeras were significantly lower compared with the WT:μMT or CD28-Y170F:μMT animals (Fig. 4C). This difference was even more pronounced by days 42 and 63. At day 21, there was no significant difference between the WT:μMT versus CD28-Y170F:μMT chimeras, but by day 42 through 63 the anti-NIP titers in the CD28-Y170F:μMT animals were lower by a small, but statistically significant amount. Similarly, anti-NIP Ab levels were equivalent between CD28-AYAA:μMT and CD28−/−:μMT chimeras on days 21 and 42, with a small, but statistically significant reduction in titers in the CD28−/−:μMT chimeras by day 63. To determine whether the inability to sustain anti-NIP Ab titers was due to accelerated loss of the LLPC population versus alterations in Ig production, the number of total and Ag-specific PC was assessed. The total number of PC in the spleen was equivalent between all four chimeras across all the time points (Fig. 4D), but was significantly lower in the BM of the CD28-AYAA:μMT and CD28−/−:μMT mice versus WT:μMT or CD28-Y170F:μMT chimeras from day 21 through 63 (Fig. 4E). Consistent with the anti-NIP Ab levels, the total number of BM PC was significantly reduced in the CD28-AYAA:μMT and CD28−/−:μMT chimeras, and largely maintained in the WT:μMT and CD28-Y170F:μMT mice. There are fewer BM PC in the CD28-Y170F:μMT compared with WT:μMT chimeras that is significant at day 42 and appreciable (but not significant) at day 63, which may account for the slightly lower titers in the knockin chimeras. The frequency of NIP-specific Ab-secreting cells (ASC) was also similar in the spleens of all four chimeras (Fig. 4F), and the loss of NIP-specific SLPC between day 21 and 42 was consistent with their short-lived nature. Whereas the numbers of NIP-specific ASC in the BM were comparable at day 21, by day 63 there were significantly fewer in the CD28-AYAA:μMT and CD28−/−:μMT mice compared with the WT:μMT or CD28-Y170F:μMT chimeras (Fig. 4G). In this assay, there was no statistical difference between the CD28-AYAA:μMT and CD28−/−:μMT chimeras, nor between the WT:μMT and CD28-Y170F:μMT animals. Altogether these data indicate that whereas the initial generation of humoral responses at day 7 appeared unaffected by defects in PC-intrinsic CD28 signaling, the ability to sustain NIP-specific Ab levels and BM LLPC populations long-term was dependent on signaling downstream of the CD28 PYAP motif.
FIGURE 4.

Requirement of the CD28 PYAP motif in the B lineage for long-term BM LLPC survival and humoral responses. (A) Experimental design. (B) Total serum IgG1 and (C) NIP-specific IgG1 levels were quantified by ELISA at the time points indicated. Each point represents one mouse. (D) Total PC numbers in spleen, and (E) BM was determined from total mononuclear cells by flow cytometry using CD138+B220− to identify PC, five mice per group. Spenic (F) and BM (G) NIP-IgG ASC numbers were determined by ELISPOT. Each point represents the tripicate mean of one mouse. Mean of the group indicated by black bars. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, *****p < 0.00001, ******p < 0.000001.
As noted above, the diminished BM PC population in the CD28-AYAA:μMT and CD28−/−:μMT chimeras could be due to impaired PC generation, PC homing to the BM, or in situ survival once there. However, the initial generation of NIP-OVA–specific splenic PC is similar across all genotypes (Fig. 4F), suggesting that intrinsic B→PC differentiation and extrinsic T cell help are not significantly impaired. Furthermore, because the loss of NIP-OVA–specific splenic PC in all the genotypes by day 42 indicates there is no sustained Ag-driven generation of new PC, alterations in PC homing to BM versus in situ survival can be distinguished by the rate of decrease (as measured by t1/2) of NIP-OVA–specific PC populations within the BM over time. Defects in homing would be manifested by lower starting BM PC numbers, but the same t1/2 compared with WT, whereas defects in in situ survival would be manifested by similar starting BM PC numbers (as suggested by Fig. 4C, 4G), but shortened t1/2. Consistent with the latter, the t1/2 of the WT NIP-OVA–specific BM PC population was 424 d, compared with 27 d for CD28−/− and 49 d for CD28-AYAA. The t1/2 of the CD28-Y170F NIP-OVA–specific BM PC population was 157 d, which reflects the intermediate decrease in NIP-OVA–specific IgG titers compared with the WT and CD28−/−/CD28-AYAA chimeras (Fig. 4C). In contrast, the t1/2 of the splenic NIP-OVA–specific PC were similar across all genotypes (18 d for WT, 11 d for CD28−/−, 9 d for CD28-AYAA, 10 d for CD28-Y170F). Altogether, these data indicate an essential role for the CD28 PYAP motif in supporting the in vivo survival of LLPC within the BM, and a possible additional role for the YMNM motif.
CD28 activation upregulates BLIMP-1 expression
CD28 signaling in T cells induces transcription of gene targets that modulate a diverse array of biological responses (34). We have reasoned that CD28 activation in LLPC may similarly regulate transcriptional hubs, in particular those modulated by LLPC residency in the BM niche. One candidate is BLIMP-1, which was initially characterized as a transcriptional repressor essential for PC differentiation and maintenance (36, 37). More recent studies have suggested BLIMP-1 acts to reinforce terminal PC differentiation and function (15), which is consistent with the observation that BLIMP-1 expression is most highly upregulated after the PC enter the BM niche (42). This further upregulation of BLIMP-1 may be triggered by the successful LLPC engagement of the niche, and results in responses necessary for long-term survival and function. To begin to assess whether CD28 may be involved in upregulating BLIMP-1 expression, we used PC from the BLIMP-1 (prdm1)gfp reporter knockin mice (42) and found that CD28 activation further upregulated prdm1 reporter expression in a subset of BM PC (Fig. 5A), but had no effect in splenic PC. Although the nature of the nonresponsive BM PC subset is not yet clear, we predict this is the residual CD28-independent BM PC population seen in the CD28−/− mice (Fig. 3E), possibly BM-resident SLPC. Evidence that CD28 upregulation of BLIMP-1 induces additional responses is suggested in Fig. 5B (top), in which the BM PC that upregulated BLIMP-1 expression also upregulated expression of a known BLIMP-1 target gene (43), the B cell maturation Ag [BCMA, one of the receptors for BAFF/APRIL (44)]. Both the BLIMP-1gfp/BCMA two-color staining and analysis of BCMA expression alone (Fig. 5B, bottom) demonstrate that, in addition to upregulating expression in BM PC that are already positive, CD28 activation can restore expression in a subpopulation of PC that have lost expression—possibly due to in vitro culture without a supportive microenvironment.
FIGURE 5.

CD28 activation increases BLIMP-1 and BMCA expression in BM PC. (A) BM and splenic PC were purified from BLIMP-1gfp reporter mice and cultured ± control Ig-coated beads or αCD28 mAb-coated beads for 16 h, and analyzed for BLIMP-1-GFP expression by flow cytometry. Results representative of three independent experiments. Quantification of three independent experiments shown on right and represents the aggregate mean ± SD of these experiments. (B) BM and splenic PC purified from BLIMP-1gfp mice were cultured with control Ig-coated beads or αCD28 mAb-coated beads as above, and stained for BCMA and analyzed by flow cytometry. Data representative of two independent experiments. **p < 0.01.
CD28 signaling downstream of the PYAP motif regulates the prdm1 promoter
Previous studies have shown that the Vav proteins are required for the induction of BLIMP-1 gene expression necessary for PC differentiation and function (35), which suggests that Vav signaling downstream of the CD28 PYAP motif may be regulating BLIMP-1 expression in BM-resident LLPC. Consistent with this, we found that CD28 activation in vitro induced a 1.4- to 1.7-fold increase in BLIMP-1 protein expression in BM PC purified from WT (Fig. 6A) and CD28-Y170F (Fig. 6B) mice, while having no effect in CD28-AYAA BM PC (Fig. 6C) or splenic PC from all genotypes (Fig. 6D–F). Quantitation by quantitative PCR also demonstrated CD28-induced induction of prdm1 mRNA expression in purified BM PC from WT and CD28-Y170F mice, but not from CD28-AYAA animals (Fig. 6G). The same pattern of expression was also seen for CD28-induced BCMA expression (data not shown).
FIGURE 6.

CD28 activation-induced BLIMP-1 expression is dependent on the CD PYAP motif. Purified BM (A) WT, (B) CD28-Y170FF, (C) CD28-AYAA PC, and purified splenic (D) WT, (E) CD28-Y170F, and (F) CD28-AYAA PC were cultured ± control Ig or αCD28 mAb-coated beads for 16 h. Lysates were made and BLIMP-1 expression was analyzed by Western blot analysis. Blots representative of two independent experiments; densitometry relative to actin are the aggregate mean ± range of two independent experiments. (G) Quantitative PCR. BM PC purified from WT, CD28-Y170F, and CD28-AYAA mice were incubated for 4 h in serum-free media with control Ig or activating CD28 mAb; prdm1 expression was assessed by quantitative PCR and shown as fold induction (relative quantity values) of prdm1 expression in αCD28-treated over control Ig-treated conditions. Data representative of three independent experiments. *p < 0.05, ***p = 0.0001, ****p < 0.0001.
We more closely examined CD28-mediated regulation of BLIMP-1 expression in the J558 PC cell line, in which αCD28 mAb treatment upregulated BLIMP-1 protein expression 1.7-fold (Fig. 7A). The finding in the BLIMP-1GFP mice that CD28 activation upregulated reporter expression suggests this is happening at the promoter level. Transfection of J558 with three prdm1 promoter constructs [7000, 4500, or 1500 bp from the PC-specific prdm1 start site in exon 1A (45)] demonstrated all three had significant and equivalent basal promoter activity (Fig. 7B). Following activation with αCD28 mAb, J558 transfected with the 7000-bp promoter construct had a significant induction of luciferase activity that was lost with truncation to 4500 bp. In silico analysis of this region did not identify any canonical CD28 RE/AP sites originally described in T cells (46). However, CD28 activation in T cells induces binding of the canonical NF-κB family member c-Rel to the IL-2 promoter (46), and we have shown that CD28 activation induces c-rel activation in MM cells (32). Furthermore, NF-κB binding to the prdm1 promoter in the region 5′ of exon 1A upregulates BLIMP-1 expression in PC (45). Our in silico analysis of the prdm1 promoter identified five putative NF-κB–binding motifs between −7000 and −4500 bp (Fig. 8A). CD28 activation of J558 induced significant c-Rel binding to three of the five sites (−6923, −5821, and −5120 bp) (Fig. 8B–F), consistent with CD28-mediated upregulation of prdm1 promoter activity. Altogether, these results support a model in BM PC in which CD28 (PYAP)→Vav→NF-κB activation regulates a central molecular nexus via upregulation of BLIMP-1, which then may induce broader transcriptional programming that allows for long-term LLPC function and survival.
FIGURE 7.

CD28 activation upregulates the prdm1 promoter activity. (A) J558 cells were cultured ± control Ig or αCD28 mAb-coated beads for 24 h. Lysates were made, and BLIMP-1 expression was analyzed by Western analysis. Densitometry relative to actin is the mean ± SD of three independent experiments. *p < 0.05. (B) J558 cells were transfected with 10 μg each reporter and Renilla constructs. Two hours later, cells were left unstimulated (black bars) or stimulated with control (white) or αCD28 mAb-coated beads (gray) for 16 h, and then assayed for luciferase activity relative to the Renilla transfection control. Results representative of three independent experiments. *p < 0.05, **p < 0.01.
FIGURE 8.

CD28 ligation induces c-Rel binding to the prdm1 promoter. (A) NF-κB binding sites in the prdm1 promoter. In silico analysis identified five putative NF-κB binding sites in the portion of the prdm1 promoter demonstrating CD28 responsiveness (−7500 to −4500 bp). (B–F) Chromatin immunoprecipitation. J558 were treated with αCD28 mAb or control Ig in serum-free media for 1 h, fixed, and sonicated, and chromatin was immunoprecipitated with αc-Rel or isotype control Ig. DNA was isolated and c-Rel binding to the prdm1 promoter was measured by quantitative PCR using primers flanking the indicated putative binding sites. Results of five independent experiments. NS versus control Ig, *p < 0.05 [Mann–Whitney rank sum test (B–D and F) or Student t test (E)].
Discussion
Although protection against reinfection was first recognized in 430 bc by Thucydides (5) and durable protection by vaccination demonstrated by Edward Jenner over 200 y ago, the specific molecular and cellular components of long-lived humoral immunity are only now being identified. Seminal studies have led to the current paradigm in which sustained Ab titers can be maintained by SLPC in a B cell/Ag-dependent fashion and/or by LLPC in a B cell/Ag-independent fashion (5, 6, 8, 9), in which SLPC play the primary role in protecting against frequent endemic infections and LLPC against infrequent epidemic infections. A key aspect of this paradigm is that the different PC subsets require specific niches for survival, and that LLPC longevity is dependent upon their ability to access prosurvival niches that SLPC cannot (1). Thus, the molecular components that allow LLPC to interact with the niche both define this PC subset and play a fundamental role in maintaining durable humoral responses. We have recently reported that signaling through CD28 is one such component, selectively supporting the survival of BM LLPC via interaction with CD80/CD86+ stromal cells in the BM niche, and resulting in long-term maintenance of Ab titers (14). This is consistent with previous studies demonstrating that CD28 is necessary for humoral responses to viral infection, vaccination, and in allergy models (21–25), although this was largely attributed to CD28’s role in Th cell activation [with the notable exception of (33)]. Nonetheless, a direct prosurvival role for CD28 in PC is supported by considerable evidence of such a role in malignant PC of MM (19, 20, 31, 32, 47).
Because CD28 signaling plays a significant role in conferring PC longevity, defining the components of this pathway may begin to unravel the mechanisms underlying long-term PC survival and function. We have found that, similar to T cells, CD28 activation in the BM PC induces both downstream PI3K→Akt and Vav→PLCγ1 signaling. Further delineation of the roles of these pathways in CD28 knockin mice demonstrated that disabling Vav signaling in the CD28-AYAA mice abrogated the prosurvival effect of CD28 activation in BM LLPC, comparable to the global loss of CD28 expression. In vivo, this was manifested by the markedly shorter t1/2 of Ag-specific CD28-AYAA PC in the BM and the inability to sustain Ag-specific Ab titers. That total IgG levels were normal in the CD28-AYAA mice points to the specificity of the defect in LLPC, and suggests that LLPC contribute only a small fraction of total IgG in the organism (alternatively, there could be a compensatory expansion of short-lived PC/B cells to restore total IgG levels to normal). In contrast, disabling PI3K signaling in the CD28-Y170F mice had only a modest effect on BM LLPC survival and maintenance of Ab titers. This stands in contrast to our findings in MM, in which pharmacological inhibitors of PI3K signaling completely abrogated the prosurvival effect of CD28 activation (48). However, one key difference between these studies is the strength of CD28 activation; the myeloma studies were done with agonistic Abs, whereas the studies reported in this work are primarily examining loss of endogenous CD28 signaling. Another difference is that Y170F mutation only blocks PI3K signaling arising from that motif, whereas the inhibitors block PI3K signaling from all sources—raising the possibility that the downstream Vav signaling is interacting with other pathways.
As downstream Vav signaling mediates CD28’s prosurvival effect in BM PC, and splenic PC do not activate PLCγ1 following CD28 stimulation, this suggests that a potential basis for differential signaling in SLPC versus LLPC is differential expression of the signaling components between CD28 and PLCγ1, namely Grb2, Vav, linker for activation of T cells, or SLP76. Our analysis of the publicly available gene expression database of human B cell→PC differentiation (http://amazonia.transcriptome.eu) (49, 50) found that, whereas there were no major differences in Grb2, Vav1-3, or linker for activation of T cells expression across the stages of differentiation, SLP76 expression was low in B cells, plasmablasts, and early PC—but is substantially elevated in BM PC (A. Utley, unpublished data). SLP76 is an adapter protein that has been shown to link CD28 costimulation to downstream Vav pathways during T cell activation (51, 52), but a role in PC biology has not been previously appreciated.
Further characterization of the CD28 (PYAP)→Vav pathway in PC uncovered a previously undescribed regulation of the central transcriptional regulator BLIMP-1, via NF-κB–binding elements in the promoter region proximal to exon 1A previously shown to be required for PC differentiation (45). This is consistent with other studies implicating Vav proteins as upstream regulators of BLIMP-1 expression in PC development (35). BLIMP-1 was initially characterized as a master regulator of PC differentiation (36, 53), and even in fully differentiated PC its continued expression is required for survival (37). More recent studies suggest that BLIMP-1 may be dispensable for key events early in PC differentiation [e.g., induction of the BCMA/Mc1-1 antiapoptotic axis (15)], but may be essential to maintain terminal differentiation and function—which is also suggested by the further upregulation of BLIMP-1 after PC enter the BM niche (42). Our data indicate that one mechanism by which high-level BLIMP-1 expression is induced in BM-resident LLPC is via CD28 activation upon engagement with CD80+/CD86+ cells in the BM niche.
These findings suggest a model in which PC-intrinsic CD28 activation in the context of the LLPC prosurvival niche reinforces BLIMP-1 expression, and BLIMP-1 expression reinforces long-term LLPC differentiation and function. However, it is not yet clear whether BLIMP-1 upregulation is directly playing a role in mediating CD28’s prosurvival effects. This question is further complicated by the observation that CD28 is not involved in regulating basal BLIMP-1 expression, but only the inducible response. It is unlikely that the CD28→BLIMP-1→BCMA upregulation is primarily responsible for PC survival, because CD28 activation protects BM PC from serum starvation in vitro, in which no exogenous growth factors are present. However, we have found in both normal PC (A. Utley, unpublished results) and MM (48) that CD28 regulates a number of metabolic, oxidative stress, antiapoptotic pathways, mitochondrial mass, and function as well as expression of other transcriptional regulators such as IFN regulatory factor 4. Whether these are mediated through BLIMP-1 is being actively investigated.
The obvious correlate of the expanding number of distinct PC subsets that occupy different niches that cut across the simple BM versus secondary lymphoid organ anatomic dichotomy is that subset-specific PC-niche interactions will dictate the survival/function of each particular subset. Whereas CD28 appears essential for the PC subset that maintains long-term IgG titers, it is not for IgG-secreting SLPC nor IgA-secreting PC (14). Even for CD28, recent studies have pointed to additional complexity. Njau et al. (54) have reported that loss of CD28 increased both T-independent and T-dependent Ab responses as well as the number of splenic and BM PC, although the T-dependent IgG responses were not long-lived. Aside from technical differences between this study and ours, another explanation for the observed differences is that, under certain conditions, CD28 activation may inhibit plasmablast proliferation and limit the initial expansion of the PC population. This is consistent with our previous observation that CD28 activation inhibits myeloma cell proliferation (32). Altogether, these studies point to an important but previously unexplored role of CD28 and its ligands directly in PC biology and generation/maintenance of humoral responses. Defining this role may provide insights into novel therapeutic strategies to enhance beneficial Ab responses, and conversely, approaches to eliminate pathogenic humoral responses in autoimmunity and allergy as well as the malignant PC in MM.
Supplementary Material
Acknowledgments
We thank Dr. Mark Brady, Department of Biostatistics and Bioinformatics, Roswell Park Cancer Institute, for very helpful discussions.
This work was supported by National Institutes of Health Grants CA121044 and AI100157 (to K.P.L.) and HL62683 (to J.M.G.) and the Multiple Myeloma Research Foundation.
The online version of this article contains supplemental material.
- ASC
- Ab-secreting cell
- BCMA
- B cell maturation Ag
- BLIMP-1
- B lymphocyte–induced maturation protein 1
- BM
- bone marrow
- BMDC
- BM-derived dendritic cell
- DC
- dendritic cell
- LLPC
- long-lived PC
- MM
- multiple myeloma
- NIP-OVA
- 4-hydroxy-5-indo-3 nitrophenyl conjugated to ovalbumin
- PC
- plasma cell
- PLC
- phospholipase C
- SLPC
- short-lived PC
- WT
- wild-type.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Radbruch A., Muehlinghaus G., Luger E. O., Inamine A., Smith K. G., Dörner T., Hiepe F. 2006. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat. Rev. Immunol. 6: 741–750. [DOI] [PubMed] [Google Scholar]
- 2.Arce S., Cassese G., Hauser A., Dörner T., Odendahl M., Manz R., Radbruch A., Hiepe F. 2002. The role of long-lived plasma cells in autoimmunity. Immunobiology 206: 558–562. [DOI] [PubMed] [Google Scholar]
- 3.Luger E. O., Wegmann M., Achatz G., Worm M., Renz H., Radbruch A. 2010. Allergy for a lifetime? Allergol. Int. 59: 1–8. [DOI] [PubMed] [Google Scholar]
- 4.Slifka M. K., Ahmed R. 1998. Long-lived plasma cells: a mechanism for maintaining persistent antibody production. Curr. Opin. Immunol. 10: 252–258. [DOI] [PubMed] [Google Scholar]
- 5.Tangye S. G. 2011. Staying alive: regulation of plasma cell survival. Trends Immunol. 32: 595–602. [DOI] [PubMed] [Google Scholar]
- 6.Amanna I. J., Carlson N. E., Slifka M. K. 2007. Duration of humoral immunity to common viral and vaccine antigens. N. Engl. J. Med. 357: 1903–1915. [DOI] [PubMed] [Google Scholar]
- 7.Ahuja A., Anderson S. M., Khalil A., Shlomchik M. J. 2008. Maintenance of the plasma cell pool is independent of memory B cells. Proc. Natl. Acad. Sci. USA 105: 4802–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slifka M. K., Antia R., Whitmire J. K., Ahmed R. 1998. Humoral immunity due to long-lived plasma cells. Immunity 8: 363–372. [DOI] [PubMed] [Google Scholar]
- 9.Looney R. J., Anolik J. H., Campbell D., Felgar R. E., Young F., Arend L. J., Sloand J. A., Rosenblatt J., Sanz I. 2004. B cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose-escalation trial of rituximab. Arthritis Rheum. 50: 2580–2589. [DOI] [PubMed] [Google Scholar]
- 10.Mahévas M., Patin P., Huetz F., Descatoire M., Cagnard N., Bole-Feysot C., Le Gallou S., Khellaf M., Fain O., Boutboul D., et al. 2013. B cell depletion in immune thrombocytopenia reveals splenic long-lived plasma cells. J. Clin. Invest. 123: 432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Racine R., McLaughlin M., Jones D. D., Wittmer S. T., MacNamara K. C., Woodland D. L., Winslow G. M. 2011. IgM production by bone marrow plasmablasts contributes to long-term protection against intracellular bacterial infection. J. Immunol. 186: 1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reynolds, A., M. Kuraoka, and G. Kelsoe. 2013. Natural IgM is produced by B1a cell-derived bone marrow plasma cells that do not share a survival niche with IgG AFC. J. Immunol. 190: 74. 16 (Abstr.). [DOI] [PMC free article] [PubMed]
- 13.Chu V. T., Fröhlich A., Steinhauser G., Scheel T., Roch T., Fillatreau S., Lee J. J., Löhning M., Berek C. 2011. Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat. Immunol. 12: 151–159. [DOI] [PubMed] [Google Scholar]
- 14.Rozanski C. H., Arens R., Carlson L. M., Nair J., Boise L. H., Chanan-Khan A. A., Schoenberger S. P., Lee K. P. 2011. Sustained antibody responses depend on CD28 function in bone marrow-resident plasma cells. J. Exp. Med. 208: 1435–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peperzak V., Vikström I., Walker J., Glaser S. P., LePage M., Coquery C. M., Erickson L. D., Fairfax K., Mackay F., Strasser A., et al. 2013. Mcl-1 is essential for the survival of plasma cells. Nat. Immunol. 14: 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharpe A. H., Freeman G. J. 2002. The B7-CD28 superfamily. Nat. Rev. Immunol. 2: 116–126. [DOI] [PubMed] [Google Scholar]
- 17.Kozbor D., Moretta A., Messner H. A., Moretta L., Croce C. M. 1987. Tp44 molecules involved in antigen-independent T cell activation are expressed on human plasma cells. J. Immunol. 138: 4128–4132. [PubMed] [Google Scholar]
- 18.Nair J. R., Carlson L. M., Koorella C., Rozanski C. H., Byrne G. E., Bergsagel P. L., Shaughnessy J. P., Jr., Boise L. H., Chanan-Khan A., Lee K. P. 2011. CD28 expressed on malignant plasma cells induces a prosurvival and immunosuppressive microenvironment. J. Immunol. 187: 1243–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pellat-Deceunynck C., Bataille R., Robillard N., Harousseau J. L., Rapp M. J., Juge-Morineau N., Wijdenes J., Amiot M. 1994. Expression of CD28 and CD40 in human myeloma cells: a comparative study with normal plasma cells. Blood 84: 2597–2603. [PubMed] [Google Scholar]
- 20.Zhang X. G., Olive D., Devos J., Rebouissou C., Ghiotto-Ragueneau M., Ferlin M., Klein B. 1998. Malignant plasma cell lines express a functional CD28 molecule. Leukemia 12: 610–618. [DOI] [PubMed] [Google Scholar]
- 21.Dodson L. F., Boomer J. S., Deppong C. M., Shah D. D., Sim J., Bricker T. L., Russell J. H., Green J. M. 2009. Targeted knock-in mice expressing mutations of CD28 reveal an essential pathway for costimulation. Mol. Cell. Biol. 29: 3710–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferguson S. E., Han S., Kelsoe G., Thompson C. B. 1996. CD28 is required for germinal center formation. J. Immunol. 156: 4576–4581. [PubMed] [Google Scholar]
- 23.Friend L. D., Shah D. D., Deppong C., Lin J., Bricker T. L., Juehne T. I., Rose C. M., Green J. M. 2006. A dose-dependent requirement for the proline motif of CD28 in cellular and humoral immunity revealed by a targeted knockin mutant. J. Exp. Med. 203: 2121–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shahinian A., Pfeffer K., Lee K. P., Kündig T. M., Kishihara K., Wakeham A., Kawai K., Ohashi P. S., Thompson C. B., Mak T. W. 1993. Differential T cell costimulatory requirements in CD28-deficient mice. Science 261: 609–612. [DOI] [PubMed] [Google Scholar]
- 25.van Wijk F., Nierkens S., de Jong W., Wehrens E. J., Boon L., van Kooten P., Knippels L. M., Pieters R. 2007. The CD28/CTLA-4-B7 signaling pathway is involved in both allergic sensitization and tolerance induction to orally administered peanut proteins. J. Immunol. 178: 6894–6900. [DOI] [PubMed] [Google Scholar]
- 26.Ribeiro, A. C., I. M. Laurindo, L. K. Guedes, C. G. Saad, J. C. Moraes, C. A. Silva, and E. Bonfa. 2013. Abatacept severely reduces the immune response to pandemic 2009 influenza A/H1N1 vaccination in patients with rheumatoid arthritis. Arthritis Care Res. 65: 476-480. [DOI] [PubMed]
- 27.Phelps C. J., Ball S. F., Vaught T. D., Vance A. M., Mendicino M., Monahan J. A., Walters A. H., Wells K. D., Dandro A. S., Ramsoondar J. J., et al. 2009. Production and characterization of transgenic pigs expressing porcine CTLA4-Ig. Xenotransplantation 16: 477–485. [DOI] [PubMed] [Google Scholar]
- 28.Horspool J. H., Perrin P. J., Woodcock J. B., Cox J. H., King C. L., June C. H., Harlan D. M., St. Louis D. C., Lee K. P. 1998. Nucleic acid vaccine-induced immune responses require CD28 costimulation and are regulated by CTLA4. J. Immunol. 160: 2706–2714. [PubMed] [Google Scholar]
- 29.Borriello F., Sethna M. P., Boyd S. D., Schweitzer A. N., Tivol E. A., Jacoby D., Strom T. B., Simpson E. M., Freeman G. J., Sharpe A. H. 1997. B7-1 and B7-2 have overlapping, critical roles in immunoglobulin class switching and germinal center formation. Immunity 6: 303–313. [DOI] [PubMed] [Google Scholar]
- 30.Akalin E., Chandraker A., Russell M. E., Turka L. A., Hancock W. W., Sayegh M. H. 1996. CD28-B7 T cell costimulatory blockade by CTLA4Ig in the rat renal allograft model: inhibition of cell-mediated and humoral immune responses in vivo. Transplantation 62: 1942–1945. [DOI] [PubMed] [Google Scholar]
- 31.Mateo G., Montalbán M. A., Vidriales M. B., Lahuerta J. J., Mateos M. V., Gutiérrez N., Rosiñol L., Montejano L., Bladé J., Martínez R., et al. PETHEMA Study Group; GEM Study Group 2008. Prognostic value of immunophenotyping in multiple myeloma: a study by the PETHEMA/GEM cooperative study groups on patients uniformly treated with high-dose therapy. J. Clin. Oncol. 26: 2737–2744. [DOI] [PubMed] [Google Scholar]
- 32.Bahlis N. J., King A. M., Kolonias D., Carlson L. M., Liu H. Y., Hussein M. A., Terebelo H. R., Byrne G. E., Jr., Levine B. L., Boise L. H., Lee K. P. 2007. CD28-mediated regulation of multiple myeloma cell proliferation and survival. Blood 109: 5002–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delogu A., Schebesta A., Sun Q., Aschenbrenner K., Perlot T., Busslinger M. 2006. Gene repression by Pax5 in B cells is essential for blood cell homeostasis and is reversed in plasma cells. Immunity 24: 269–281. [DOI] [PubMed] [Google Scholar]
- 34.Boomer J. S., Green J. M. 2010. An enigmatic tail of CD28 signaling. Cold Spring Harb. Perspect. Biol. 2: a002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stephenson L. M., Miletic A. V., Kloeppel T., Kusin S., Swat W. 2006. Vav proteins regulate the plasma cell program and secretory Ig production. J. Immunol. 177: 8620–8625. [DOI] [PubMed] [Google Scholar]
- 36.Shapiro-Shelef M., Lin K. I., McHeyzer-Williams L. J., Liao J., McHeyzer-Williams M. G., Calame K. 2003. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity 19: 607–620. [DOI] [PubMed] [Google Scholar]
- 37.Shapiro-Shelef M., Lin K. I., Savitsky D., Liao J., Calame K. 2005. Blimp-1 is required for maintenance of long-lived plasma cells in the bone marrow. J. Exp. Med. 202: 1471–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Semenov I., Akyuz C., Roginskaya V., Chauhan D., Corey S. J. 2002. Growth inhibition and apoptosis of myeloma cells by the CDK inhibitor flavopiridol. Leuk. Res. 26: 271–280. [DOI] [PubMed] [Google Scholar]
- 39.Zhan F., Tian E., Bumm K., Smith R., Barlogie B., Shaughnessy J., Jr. 2003. Gene expression profiling of human plasma cell differentiation and classification of multiple myeloma based on similarities to distinct stages of late-stage B-cell development. Blood 101: 1128–1140. [DOI] [PubMed] [Google Scholar]
- 40.Boomer J. S., Deppong C. M., Shah D. D., Bricker T. L., Green J. M. 2014. Cutting edge: a double-mutant knockin of the CD28 YMNM and PYAP motifs reveals a critical role for the YMNM motif in regulation of T cell proliferation and Bcl-xL expression. J. Immunol. 192: 3465–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tuaillon N. 2000. Repertoire analysis in human immunoglobulin heavy chain minilocus transgenic, muMT/muMT mice. Mol. Immunol. 37: 221–231. [DOI] [PubMed] [Google Scholar]
- 42.Kallies A., Hasbold J., Tarlinton D. M., Dietrich W., Corcoran L. M., Hodgkin P. D., Nutt S. L. 2004. Plasma cell ontogeny defined by quantitative changes in blimp-1 expression. J. Exp. Med. 200: 967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deng S., Yuan T., Cheng X., Jian R., Jiang J. 2010. B-lymphocyte-induced maturation protein1 up-regulates the expression of B-cell maturation antigen in mouse plasma cells. Mol. Biol. Rep. 37: 3747–3755. [DOI] [PubMed] [Google Scholar]
- 44.O’Connor B. P., Raman V. S., Erickson L. D., Cook W. J., Weaver L. K., Ahonen C., Lin L. L., Mantchev G. T., Bram R. J., Noelle R. J. 2004. BCMA is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med. 199: 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morgan M. A., Magnusdottir E., Kuo T. C., Tunyaplin C., Harper J., Arnold S. J., Calame K., Robertson E. J., Bikoff E. K. 2009. Blimp-1/Prdm1 alternative promoter usage during mouse development and plasma cell differentiation. Mol. Cell. Biol. 29: 5813–5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shapiro V. S., Truitt K. E., Imboden J. B., Weiss A. 1997. CD28 mediates transcriptional upregulation of the interleukin-2 (IL-2) promoter through a composite element containing the CD28RE and NF-IL-2B AP-1 sites. Mol. Cell. Biol. 17: 4051–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robillard N., Jego G., Pellat-Deceunynck C., Pineau D., Puthier D., Mellerin M. P., Barillé S., Rapp M. J., Harousseau J. L., Amiot M., Bataille R. 1998. CD28, a marker associated with tumoral expansion in multiple myeloma. Clin. Cancer Res. 4: 1521–1526. [PubMed] [Google Scholar]
- 48.Murray, M. E., C. M. Gavile, J. R. Nair, C. Koorella, L. M. Carlson, D. Buac, A. Utley, M. Chesi, P. L. Bergsagel, L. H. Boise, and K. P. Lee. 2014. CD28-mediated pro-survival signaling induces chemotherapeutic resistance in multiple myeloma. Blood 123: 3770–3779. [DOI] [PMC free article] [PubMed]
- 49.Jourdan M., Caraux A., De Vos J., Fiol G., Larroque M., Cognot C., Bret C., Duperray C., Hose D., Klein B. 2009. An in vitro model of differentiation of memory B cells into plasmablasts and plasma cells including detailed phenotypic and molecular characterization. Blood 114: 5173–5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jourdan M., Caraux A., Caron G., Robert N., Fiol G., Rème T., Bolloré K., Vendrell J. P., Le Gallou S., Mourcin F., et al. 2011. Characterization of a transitional preplasmablast population in the process of human B cell to plasma cell differentiation. J. Immunol. 187: 3931–3941. [DOI] [PubMed] [Google Scholar]
- 51.Koretzky G. A., Abtahian F., Silverman M. A. 2006. SLP76 and SLP65: complex regulation of signalling in lymphocytes and beyond. Nat. Rev. Immunol. 6: 67–78. [DOI] [PubMed] [Google Scholar]
- 52.Raab M., Pfister S., Rudd C. E. 2001. CD28 signaling via VAV/SLP-76 adaptors: regulation of cytokine transcription independent of TCR ligation. Immunity 15: 921–933. [DOI] [PubMed] [Google Scholar]
- 53.Shapiro-Shelef M., Calame K. 2005. Regulation of plasma-cell development. Nat. Rev. Immunol. 5: 230–242. [DOI] [PubMed] [Google Scholar]
- 54.Njau M. N., Kim J. H., Chappell C. P., Ravindran R., Thomas L., Pulendran B., Jacob J. 2012. CD28-B7 interaction modulates short- and long-lived plasma cell function. J. Immunol. 189: 2758–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.