

Background: A protein product of the NAALADL1 gene is a homolog of glutamate carboxypeptidase II, a metallopeptidase studied as a promising theranostic cancer agent.
Results: We solved the x-ray structure and analyzed the substrate specificity of the NAALADL1 gene product.
Conclusion: We demonstrated that the protein represents a novel human ileal aminopeptidase.
Significance: This study describes a novel enzyme involved in protein/peptide digestion in the small intestine and clarifies controversial previous reports.
Keywords: Aminopeptidase, Intestinal Metabolism, Metalloprotease, Molecular Evolution, Protein Degradation, X-ray Crystallography, DPP IV Activity, PICS, Human Ileal Aminopeptidase
Abstract
N-acetylated α-linked acidic dipeptidase-like protein (NAALADase L), encoded by the NAALADL1 gene, is a close homolog of glutamate carboxypeptidase II, a metallopeptidase that has been intensively studied as a target for imaging and therapy of solid malignancies and neuropathologies. However, neither the physiological functions nor structural features of NAALADase L are known at present. Here, we report a thorough characterization of the protein product of the human NAALADL1 gene, including heterologous overexpression and purification, structural and biochemical characterization, and analysis of its expression profile. By solving the NAALADase L x-ray structure, we provide the first experimental evidence that it is a zinc-dependent metallopeptidase with a catalytic mechanism similar to that of glutamate carboxypeptidase II yet distinct substrate specificity. A proteome-based assay revealed that the NAALADL1 gene product possesses previously unrecognized aminopeptidase activity but no carboxy- or endopeptidase activity. These findings were corroborated by site-directed mutagenesis and identification of bestatin as a potent inhibitor of the enzyme. Analysis of NAALADL1 gene expression at both the mRNA and protein levels revealed the small intestine as the major site of protein expression and points toward extensive alternative splicing of the NAALADL1 gene transcript. Taken together, our data imply that the NAALADL1 gene product's primary physiological function is associated with the final stages of protein/peptide digestion and absorption in the human digestive system. Based on these results, we suggest a new name for this enzyme: human ileal aminopeptidase (HILAP).
Introduction
Human N-acetylated α-linked acidic dipeptidase-like protein (NAALADase L)3 is encoded by the NAALADL1 gene localized at chromosome 11q12. The gene spans more than 14 kbp and contains 18 exons (1). Translation of the mRNA transcript yields a 740-amino acid type II transmembrane protein. According to the MEROPS database, NAALADase L belongs to the M28B metallopeptidase subfamily (MEROPS ID: M28.011). Glutamate carboxypeptidase II (GCPII), which shares 37% sequence identity and 50% similarity with NAALADase L, is another member of this subfamily.
In 1997, Shneider et al. (2) identified a NAALADase L protein (designated as I100) in rat ileum. They analyzed mRNA expression in rat and human tissues, raised polyclonal antibody against I100, and detected DPP IV activity in an immunoprecipitate from rat ileum. The only other report to date describing the NAALADL1 gene product was published by Pangalos et al. in 1999 (1). These researchers performed a comparative analysis of the human NAALADL1 gene product and its two close paralogs NAALADase I and NAALADase II, which are now known as GCPII and GCPIII, respectively. They gave the protein its current name, NAALADase L, based on the close homology of these enzymes. They cloned NAALADase L cDNA, identified its position in the human genome, analyzed its alternative splicing, and detected DPP IV activity in NAALADase L-transfected cell lysate. Additionally, they detected DPP IV activity in GCPII- and GCPIII-transfected cell lysates. However, this activity was subsequently not confirmed for GCPII and GCPIII in experiments with purified recombinant protein (3, 4). In the present study, we performed a thorough structural and biochemical characterization of the human NAALADL1 gene product using a purified recombinant protein preparation to elucidate the physiological role of the protein.
EXPERIMENTAL PROCEDURES
Cloning NAALADase L cDNA and Preparation of Expression Plasmids
Two plasmids, A (ID LIFESEQ95147340) and B (ID LIFESEQ4181072), containing cDNA coding for NAALADase L were purchased from Open Biosystems (now GE Healthcare). The extracellular portion of NAALADase L (aa 28–740) was amplified from plasmid A, and BclI and XhoI restriction sites were introduced by PCR using primers FNAL28BclI (aaatgatcaatccccaaaaaagccaactcactggc) and RNAL740XhoI (tttctcgagtcatcagaggtcagccacaggcc). The PCR product was then ligated via BglII and XhoI restriction sites into pMT/BiP/V5-HisA (Life Technologies, Inc.), resulting in pMT/BiP/rhNaalL28–740. Sequencing identified two mutations in pMT/BiP/rhNaalL28–740 (resulting in aa mutations L364P and L393P). Therefore, the mutated part of DNA was replaced with the corresponding DNA from plasmid B utilizing NcoI and KpnI cleavage sites to insert leucine residues at the proper positions in the plasmid DNA. Additional sequencing confirmed that the mutations were repaired.
For preparation of N-terminal His-tagged NAALADase L, the extracellular portion of the protein (aa 28–740) was amplified from pMT/BiP/rhNaalL28–740 using primers FNAL28NdeI (aaacatatgatccccaaaaaagccaactcactggc) and RNAL740XhoI. The PCR product was then ligated into pET28b via NdeI and XhoI restriction sites, yielding pET28b-HisNaalL.
For preparation of N-terminal Avi-tagged NAALADase L, the extracellular portion of the protein (aa 28–740) was amplified from pMT/BiP/rhNaalL28–740 using primers FNAL28BclI and RNAL740XhoI. The PCR product was then ligated into pMT/BiP/AviTEV/rhGCPII (5) via BglII and XhoI restriction sites, yielding pMT/BiP/AviTEV/rhNaalL. The final protein construct contains an N-terminal AviTagTM (Avidity) sequence and TEV cleavage site.
The E416A mutation was introduced into pMT/BiP/AviTEV/rhNaalL using the QuikChange site-directed mutagenesis kit (Stratagene). Primers RNAL-E416A (gccaatgagcccaaactccgcagccccccagctcgc) and FNAL-E416A (gcgagctggggggctgcggagtttgggctcattggc) were used for the PCR, which was performed according to the manufacturer's protocol, yielding pMT/BiP/AviTEV/rhNaalL(E416A).
The plasmid pET24a_HIVprD25N, which encodes HIV-1(D25N) protease, was created from pET24a_HIVprWT (6), which encodes wild-type HIV-1 protease, using the QuikChange site-directed mutagenesis kit (Stratagene) with primers RHIV-D25N (gaagctctattaaatacaggagcagatgata) and FHIV-D25N (tatcatctgctcctgtatttaatagagcttc) according to the manufacturer's protocol. The sequences of all constructed plasmids were verified by DNA sequencing.
Preparation of His-NaalL Protein for Immunization
The plasmid pET28b-HisNaalL was used for large scale expression of the extracellular portion (aa 28–740) of NAALADase L with an N-terminal His tag (His-NaalL) in Escherichia coli BL21-CodonPlus-RIL cells using 1 mm isopropyl 1-thio-β-d-galactopyranoside for induction. His-NaalL was purified from inclusion bodies under denaturing conditions. First, the cell pellet was dissolved in running buffer (8 m urea, 0.5 m NaCl, 50 mm Na2HPO4, pH 8.0) and loaded onto a HisTrap HP (GE Healthcare) column using an ÄKTAexplorer LC system (GE Healthcare). The column was washed with running buffer containing 10 mm imidazole, and protein was eluted with 1 m imidazole. The elution fractions were pooled and used for mouse immunization.
Preparation of Monoclonal Antibody against NAALADase L
Monoclonal antibodies were prepared according to an established protocol (7). Briefly, BALB/c mice were intraperitoneally immunized with purified His-NaalL in 8 m urea (40 μg in 100 μl) mixed with an equal volume of incomplete Freund's adjuvant at 3-week intervals. Hybridoma cells, prepared by fusion of immune spleen cells and Sp/0 myeloma cells, were cloned by limited dilution and assayed for production of NAALADase L-specific antibodies by enzyme-linked immunosorbent assay. Positive selection was performed against His-NaalL, and negative selections were performed against rhGCPII and rhGCPIII prepared in our laboratory (3, 4). Four positive hybridoma clones (1C11, 3C11, 3D10, and 4E9) were expanded, and conditioned medium from the most sensitive hybridoma clone (1C11) was used for immunoblots. Additionally, we tested the ability of all four hybridoma supernatants to immunoprecipitate native Avi-NaalL. The results indicated that these monoclonal antibodies bind specifically to the denatured form of NAALADase L (data not shown).
Expression and Purification of Avi-NaalL and Avi-NaalL(E416A)
Preparation and purification of Avi-NaalL was performed as described previously (5). Briefly, Drosophila S2 cells expressing E. coli biotin-ligase (BirA) in the endoplasmic reticulum were transfected with pMT/BiP/AviTEV/rhNaalL (encoding the extracellular portion (aa 28–740) of NAALADase L with an N-terminal Avi tag and TEV protease cleavage site. Large scale expression of Avi-NaalL was performed, and the conditioned medium was concentrated 10-fold. The recombinant protein was purified on Streptavidin Mutein MatrixTM (Roche Applied Science), followed by size exclusion chromatography on SuperdexTM 200 10/300 GL (GE Healthcare) in 10 mm Bistris propane, 150 mm NaCl, pH 7.4. The concentration of the final protein preparation was determined by amino acid analysis using a Biochrom30 amino acid analyzer (Biochrom). The preparation of Avi-NaalL(E416A) mutant was performed analogously.
Expression and Purification of HIV-1 Protease and HIV-1(D25N) Protease
HIV-1 protease was prepared as described previously (6).
HIV-1(D25N) protein was overexpressed in E. coli BL21(DE3)RIL (Novagen) transformed with pET24a_HIVprD25N expression plasmid. Insoluble recombinant protein, accumulated in the form of inclusion bodies, was isolated and solubilized in 50% (v/v) acetic acid. Solubilized protein was purified by gel chromatography in 50% (v/v) acetic acid, using a Superdex G75 16/60 gel filtration column (GE Healthcare). Fractions containing mutant HIV-1 protease were pooled, and the protein was subsequently refolded by dropwise dilution into a 19-fold excess of water and additional dialysis against water for 3 h at 4 °C, followed by subsequent overnight dialysis against 50 mm MES, 10% (v/v) glycerol, 1 mm EDTA, 0.05% (v/v) 2-mercaptoethanol, pH 5.8. The purified protein was stored at −70 °C.
Crystallization and Data Collection
Avi-NaalL stock solution (5 mg/ml) was mixed in a 1:1 ratio with reservoir solution (0.16 m magnesium formate and 16% (w/v) PEG 3350). Crystals were grown using the hanging drop vapor diffusion method at 293 K. Diffraction quality crystals (0.3 × 0.2 × 0.2 mm3) were obtained by seeding. For diffraction experiments, crystals were cryogenized in liquid nitrogen after transferring to cryoprotectant containing 0.16 m magnesium formate, 16% (w/v) PEG 3350, and 14% (v/v) glycerol. The diffraction data were collected at 100 K using synchrotron radiation at the Southeast Regional Collaborative Access Team sector 22 (BM beamline) of the Advanced Photon Source (Argonne, IL) with the x-ray wavelength tuned to 1.0000 Å. The complete data set was collected from a single crystal and recorded on a MarMosaic 225-mm CCD detector. Data processing was performed with the HKL2000 software package (8).
Structure Determination and Refinement
The Avi-NaalL structure was solved by molecular replacement using the program Phaser (9) and coordinates of GCPII (PDB entry 2PVW) as a search model. Subsequent structural refinement was carried with the program Refmac 5.1 (10). During the refinement process, 1% of randomly selected reflections were kept aside for cross-validation (Rfree). Refinement was interspersed with manual corrections aided by the program Coot (11). The stereochemical quality of the final model was evaluated using the Web server MolProbity (12). The final model and experimental amplitudes were deposited in the RCSB Protein Data Bank (entry 4TWE).
Quantitative PCR (qPCR)
Amounts of NAALADL1 gene transcript were quantified using two distinct sets of primers. The first set, NaalL_A, contained forward and reverse primers (sequences cactgggctgccactacag and ttgtagacgctcacattcacct) and fluorescent hydrolysis Universal ProbeLibrary Probe 3 (Roche Applied Science). This set was designed to amplify nucleotides 945–1032 in NAALADL1 transcript NM_005468, which covers the region containing exons 6 and 7 and corresponds to aa 310–339 in the longest ORF. The expected size of the amplified product was 88 bases; the size of the possibly amplified genomic DNA was 758 bases. The second set, NaalL_B, consisted of forward and reverse primers (sequences atctcctccatggacattgc and gctgtgtggtaggtggggta) and fluorescent hydrolysis Universal ProbeLibrary Probe 41 (Roche Applied Science). This set was designed to amplify nucleotides 1580–1656 in NAALADL1 transcript NM_005468, which covers the region containing exons 13 and 14 and corresponds to aa 522–547 in the longest ORF. The expected size of the amplified product was 77 bases, whereas the size of the possibly amplified genomic DNA was 1024 bases.
All qPCRs (10 μl) were carried out in triplicate in sealed 96-well plates (FrameStar® 480, 4titude) using a LightCycler 480 II instrument (Roche Applied Science). Each reaction consisted of LightCycler 480 Probe Master (Roche Applied Science) diluted according to the manufacturer's protocol, forward and reverse primers (final concentration 1 μm each), fluorescent probe (final concentration of probe 3 was 100 nm; final concentration of probe 41 was 50 nm), and template DNA. Reaction mixtures without template DNA were prepared as controls. Moreover, interplate calibrators were included in each plate. Initial denaturation for 3 min at 95 °C was followed by 45 cycles of 10 s at 95 °C, 30 s at 66 °C, and 30 s at 72 °C. The threshold cycle numbers (Cq) were then determined from fluorescence intensities acquired during the qPCR runs by the second derivative maximum method using LightCycler 480 software (Roche Applied Science). The presence and size of PCR products for all qPCRs was analyzed by agarose gel electrophoresis.
A set of serial dilutions, ranging from 102 to 107 copies/reaction, of the pMT/BiP/AviTEV/rhNaalL plasmid was used for absolute quantification. The initial concentration of plasmid DNA prior to dilution was determined spectrophotometrically based on absorption at 260 nm (Nanodrop ND-1000, Thermo Scientific). The amount of NAALADL1 transcript was then measured in 0.1-μl aliquots of tissue cDNA libraries obtained from Clontech (Human MTC Panels I and II).
To assess the selectivity of qPCR amplification, pcDNA4 plasmids with subcloned protein coding sequences of homologous human transcripts (ORF from NM_004476 coding aa 1–750 of GCPII, ORF from NM_005467 coding aa 1–740 of GCPIII) at concentrations of 106 copies/reaction were amplified in the same qPCR setup with both primer sets. Possible products were inspected by agarose gel electrophoresis. Amplification of isolated human genomic DNA was tested in the same manner.
Human Tissue Lysate Preparation
Human tissue lysates were prepared as described previously (13). Briefly, human samples were obtained during autopsy performed 4–8 h post-mortem. The samples were homogenized in 50 mm Tris-HCl, 150 mm NaCl, pH 7.5, supplemented with EDTA-free protease inhibitors (Roche Applied Science). The samples were then sonicated using Soniprep 150 (Sanyo) three times for 10 s on ice, and 1% (v/v) Triton X-100 was added. After a 30-min incubation on ice, the suspension was centrifuged at 16,000 × g for 10 min, and the supernatants were stored at −80 °C for further experiments.
SDS-PAGE and Western Blotting
Protein samples were resolved by reducing SDS-PAGE. Gels were silver-stained for analysis of protein purification or stained with colloidal Coomassie G-250 for mass spectrometry analysis or electroblotted (100 V for 1 h) onto a nitrocellulose membrane using a Mini Trans-Blot® Cell wet blot system (Bio-Rad).
After blotting, membranes were blocked with casein (SDT), incubated overnight with hybridoma supernatant 1C11, washed three times with 0.05% (w/w) Tween 20 in PBS (PBST), incubated for 1 h with HRP-conjugated goat anti-mouse antibody (1 mg/ml; Thermo Scientific) diluted in casein in a 1:25,000 ratio, and washed three times with PBST. Blots were developed using SuperSignal West Dura chemiluminiscence substrate (Thermo Scientific) according to the manufacturer's protocol. Reactive bands were visualized on a ChemiDoc-ItTM600 imaging system (UVP; 20-min exposition, 1 × 1 binning).
MS Analysis
The chosen gel sections were excised, destained, reduced with DTT, alkylated with iodoacetamide, and digested with trypsin for 10 h. Peptides were extracted and dissolved in 0.1% formic acid (FA). Samples were analyzed with an UltiMate 3000 RSLCnano system (Thermo Scientific) coupled to a TripleTOF 5600 mass spectrometer with a NanoSpray III source (AB Sciex). Peptides were separated on an Acclaim PepMap100 analytical column (3 μm, 250 mm × 75-μm inner diameter; Thermo Scientific). The concentration of buffer B (0.1% FA in acetonitrile) was gradually increased from 5 to 30% in buffer A (0.1% aqueous FA) over 40 min. The MS mass range was set to 350–1250 m/z; in MS/MS mode, the instrument acquired fragmentation spectra within 100–1600 m/z. The program Protein Pilot 4.5 (AB Sciex) was used for protein identification against the UniProt Homo sapiens database (reviewed December 20, 2013).
Samples for proteomic identification of cleavage site (PICS) were analyzed in the same manner but without gel extraction and by applying a 95-min instead of a 40-min gradient. PICS results were searched against the UniProt H. sapiens database containing typical MS contaminants (reviewed June 23, 2014).
LNCaP Lysate Preparation
LNCaP cells were grown in RPMI 1640 medium (Sigma-Aldrich) with the addition of 10% (v/v) FBS. After reaching confluence, the cells were harvested and sonicated four times for 15 s in lysis buffer (50 mm Tris-HCl, 150 mm NaCl, 1% (w/v) C12E8 (Affymetrix), 50 mm NaF, 1 mm Na3VO4, 1 mm β-glycerol phosphate, 2.5 mm Na4O7P2, pH 7.4). Cell debris was removed by centrifugation (45,000 × g for 60 min at 4 °C), and protein concentration was determined by Bradford assay (14).
DPP IV Activity Assay
Protein samples, either purified recombinant Avi-tagged protein or LNCaP cell lysate, were incubated in 25 mm Bistris propane, 150 mm NaCl, pH 8.5, with 100 μm Gly-Pro-aminomethylcoumarin (Sigma) for 15 h at 37 °C. The reactions were performed in triplicate in white 96-well polypropylene plates with V-bottoms. The fluorescence was measured on an Infinite® M1000 PRO (TECAN) with excitation and emission wavelengths/bandwidths of 335/5 nm and 450/10 nm, respectively.
Synthesis of Dipeptides and Dipeptide Libraries
Substrate mixtures of the general formula Ac-Aaa-Xxx-OH (where Aaa is one of 19 proteinogenic amino acids (all except cysteine), and Xxx is an equimolar mixture of all 19 amino acids (all except cysteine)) were prepared by solid phase peptide synthesis on 2-chlorotritylchloride resin (for C-terminal amino acids Asp, His, Pro, and Trp) or on Wang resin (for all other C-terminal amino acids) using the Fmoc (N-(9-fluorenyl)methoxycarbonyl)/t-butyl protection and N,N′-diisopropylcarbodiimide/1-hydroxybenzotriazole coupling strategy described by Barlos et al. (15).
N-terminal acetylation was accomplished with acetic anhydride in the presence of N,N-diisopropylethylamine. The completeness of all couplings was controlled by a ninhydrin test (16). A mixture of 94% trifluoroacetic acid, 2% triisopropylsilane, 2% ethanedithiol, and 2% water was used for final cleavage from the resin. The peptides were freeze-dried from 10% acetic acid in water.
To confirm the presence and quantify the amount of all N-acetylated dipeptides in the mixtures, samples were analyzed by amino acid analysis using a Biochrom30 amino acid analyzer (Biochrom) following the manufacturer's protocol.
Carboxypeptidase Activity Assay
Reactions were performed in a round-bottom 96-well PP plate. The enzyme (final concentration 0.25 μm) and 5 μl of N-acetylated dipeptide library (the final concentration of each individual dipeptide in the library was ∼25 μm) were diluted with reaction buffer (25 mm Bistris propane, 150 mm NaCl, 0.001% (w/v) C12E8, pH 7.4) to a final volume of 50 μl. The reaction was then incubated at 37 °C for 15 h and subsequently analyzed by HPLC. The cleaved free amino acids in the reaction mixtures were analyzed on an Agilent 1260 HPLC system (Agilent Technologies) equipped with an AccQ-Tag Ultra RP 130 Å column (1.7 μm, 100 mm × 2.1-mm inner diameter; Waters) following a previously published protocol for amino acid analysis (17).
Enzymatic Assay with Peptides from PICS
Reactions were performed in 1.5-ml capped plastic vials in a final volume of 40 μl. The final enzyme concentration was 1 μm for 15-h incubations and 0.1 μm for time-dependent cleavage analysis. The final concentration of substrate (peptide 52 or peptide 71) was 100 μm. Reactions were buffered with 25 mm Bistris propane, 150 mm NaCl, 0.001% (w/v) C12E8, pH 7.4. After stopping reactions by the addition of 40 μl of 1 m glycine-HCl, pH 2.4, mixtures were analyzed on an Agilent 6230 TOF LC/MS (Agilent Technologies) equipped with a Dual AJS electrospray ionization source operated in the positive ion mode (acquisition 4 GHz, HiRes).
Separation of the peptides and amino acids was conducted on the Acquity UPLC BEH-C18 130 Å column (1.7 μm, 100 mm × 2.1-mm inner diameter; Waters) using a 10-min gradient from 2 to 100% mobile phase B (A, 0.1% aqueous FA; B, 0.1% FA in acetonitrile). The column temperature was 25 °C, and the flow rate was 0.3 ml/min.
HPLC Analysis of Aminopeptidase Activity
Reactions were performed in round-bottom 96-well PP plates. For determination of kinetic parameters of IIDPNG peptide cleavage by Avi-NaalL, the reactions with various concentrations of the peptide ranging from 6 to 1600 μm were performed. The Avi-NaalL was first diluted in reaction buffer (25 mm Bistris propane, 150 mm NaCl, 0.001% (w/v) C12E8, pH 7.4) in a final volume of 40 μl to ensure 10–20% conversion. The enzyme was then preincubated for 5 min at 37 °C, and then 10 μl of various concentrations of peptide substrate (IIDPNG) was added to start the reaction. After 15 min, the reactions were stopped by the addition of 50 μl of stopping buffer (1 m glycine-HCl, pH 2.4).
For determination of the inhibition constant of bestatin, 90 ng of Avi-NaalL, ensuring ∼10% conversion, and various concentrations ranging from 0.1 to 500 μm of bestatin hydrochloride (AppliChem) were mixed in reaction buffer in a final volume of 40 μl. The reactions were preincubated for 5 min at 37 °C, and then 10 μl of peptide substrate (IIDPNG; final concentration 50 μm) was added to start the reaction. After 15 min, the reactions were stopped by the addition of 50 μl of stopping buffer. Each reaction was performed in duplicate.
The reactions were subsequently analyzed on an Agilent 1260 HPLC system (Agilent Technologies) equipped with the Acquity UPLC BEH-C18 130 Å column (1.7 μm, 100 mm × 2.1-mm inner diameter; Waters) using a 6-min gradient from 2 to 100% mobile phase B (A, 0.1% aqueous FA; B, 0.1% FA in acetonitrile). The column temperature was 25 °C, and the flow rate was 0.3 ml/min. The obtained data were then processed using GraFit version 5.0.11 (Erithacus Software Ltd.).
The catalytic efficiency of Avi-NaalL(E416A) mutant was determined in reaction buffer by incubation with Avi-NaalL(E416A) (final concentration 10 μm) and peptide IIDPNG (final concentration 4 mm) at 37 °C. 2 μl of reaction mixture was withdrawn from the reaction after 60, 120, and 240 min, added to 100 μl of stopping buffer, and subsequently analyzed on HPLC.
PICS Assay
PICS was performed according to a previously published protocol with several modifications (18). Non-transfected HEK293T cells were grown in DMEM to 90% confluence and harvested by washing into PBS buffer. Cells were transferred into hypotonic lysis buffer containing protease inhibitors and lysed by sonication, followed by cysteine reduction and alkylation. Proteins were precipitated using TCA and resolubilized into 200 mm HEPES, pH 7.5. Following resolubilization, the proteome-derived peptide library was prepared by cleaving isolated denatured proteins into peptides with trypsin (Sigma-Aldrich). After abolishing trypsin activity using 1 mm PMSF, a second round of sulfhydryl reduction and alkylation was performed. Primary amines on peptide N termini and lysine side chains were blocked by formaldehyde-cyanoborohydride reductive dimethylation. Excess modification reagents were removed by gel filtration, and the peptide library was purified and transferred to HPLC-grade water using a C-18 solid phase extraction cartridge Sep-Pak (Waters) according to the manufacturer's protocol. The peptide concentration was adjusted to 2 mg/ml. The integrity of the peptide library was confirmed by LC-MS/MS analysis, and aliquots were stored at −80 °C until further use.
For the endopeptidase assay, the peptide library (final concentration 1 mg/ml) was incubated in 200 μl of 25 mm Bistris propane, 150 mm NaCl, pH 7.4, and 0.2 μm Avi-NaalL. The reaction was incubated for 12 h at 37 °C. Following incubation, the reaction was heat-inactivated for 30 min at 70 °C and titrated with 2 m HEPES, pH 8.5, to final pH 8.0.
Subsequently, newly formed free peptide N termini (products of proteolytic cleavage) were in vitro biotinylated by the addition of 350 μm sulfo-NHS-SS-biotin (Thermo Scientific) for 4 h at room temperature. Biotinylated products were then immobilized on streptavidin-agarose (Solulink) by a 2-h incubation with mild agitation at room temperature, followed by washing. Additional washing steps (2 m urea followed by 20% isopropyl alcohol, 5% DMSO, and 5% acetonitrile, all in washing buffer (50 mm HEPES, 150 mm NaCl, pH 7.5)) were added to the original protocol, followed by 10 washes with washing buffer. Immobilized peptides were eluted with 20 mm DTT, desalted using Pepclean C-18 reverse phase cartridges (Thermo Scientific) according to the manufacturer's protocol, and analyzed by mass spectrometry.
As negative controls, Avi-NaalL(E416A) and buffer alone (blank) were used. As positive controls, the HIV-1 protease cleavage profile in 100 mm sodium acetate, 300 mm NaCl, pH 4.7, was tested with wild-type and HIV-1(D25N) protease. The protease library ratio was 1:200, and the final enzyme concentration was 0.2 μm.
PICS Data Analysis
Data were analyzed using a series of predesigned queries in Microsoft Access database software. First, lists of identified peptides from each MS run were loaded into the database and filtered for peptides containing products of N-terminal modification by biotinylation. Second, peptides with confidence over 80% were picked for enzymatic reactions (Avi-NaalL or HIV-1 protease), whereas peptides with confidence over 10% were picked for control reactions (reactions with catalytically ineffective forms of the enzymes and blanks). To properly subtract the background signal, the list of peptides found in each enzymatic reaction was screened for peptides present in the blank reaction and the reaction with catalytically ineffective enzyme. Such peptides were then removed from processing. Finally, the enzymatic reactions were screened for peptides identified in the original unprocessed peptide library. Such peptides were also removed from the analysis.
The final cleared list of identified peptides was then mapped against the FASTA database used for proteomics database search. By identified peptide alignment with the database, the N-terminal portions of cleaved peptides (preceding the cleavage site) were determined. If there was more than one computationally identified amino acid for a given P position, the position was omitted from the processing, whereas the identified peptide sequence remained listed for downstream analysis. The final list of substrate peptides containing sequences of P′ amino acids identified in the MS experiment and four P amino acids identified computationally is shown in Table 2. The frequency of each amino acid in each position was then calculated and plotted, yielding substrate specificity matrix heat maps.
TABLE 2.
List of Avi-NaalL peptidic substrate sequences determined by PICS
Sequences are shown in one-letter amino acid coding from N to C terminus. A dash marks the putative cleavage site.
RESULTS
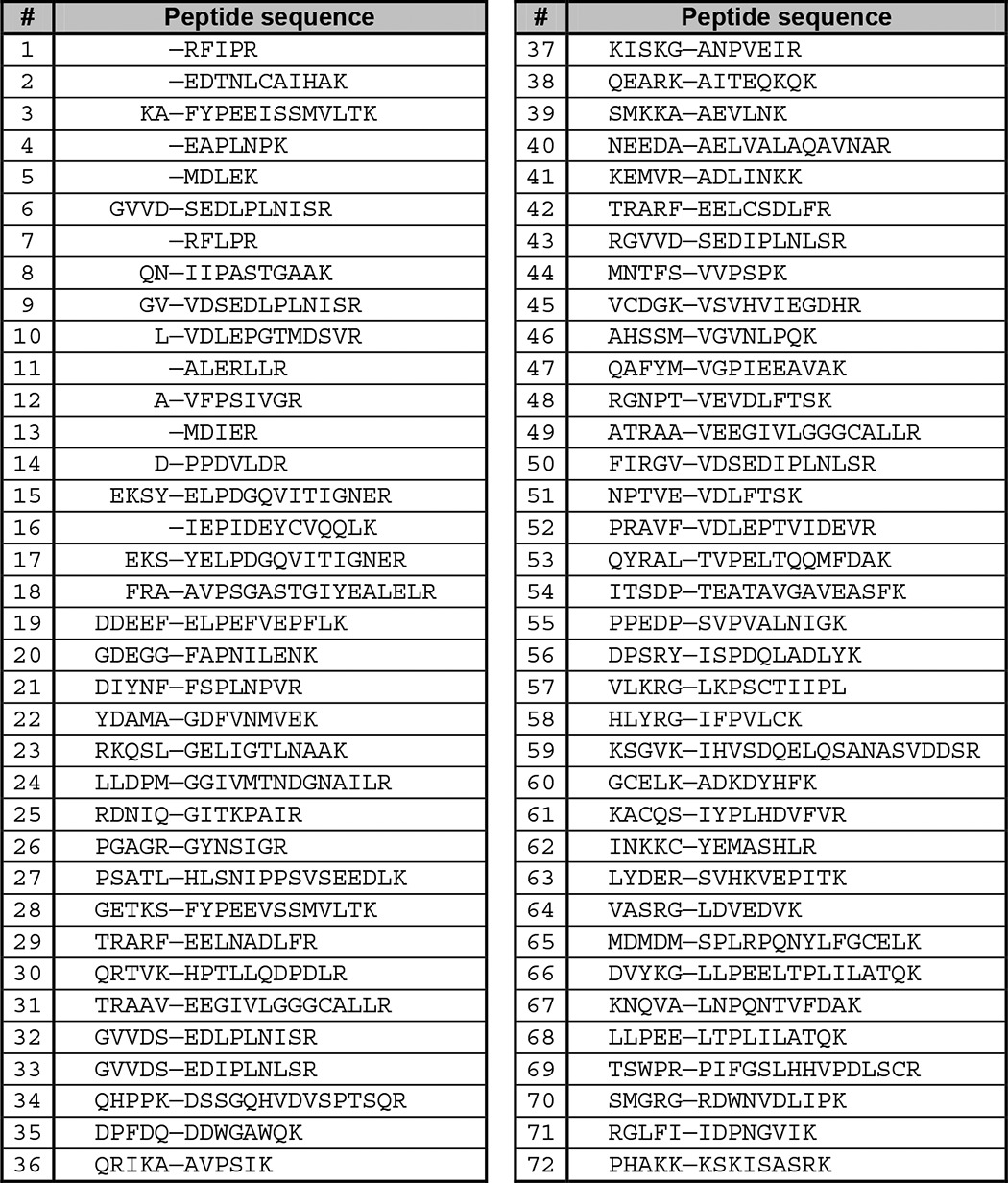
We used an expression and purification system established in our laboratory to produce the extracellular portion (aa 28–740) of human NAALADase L, designated Avi-NaalL, and its putative catalytically ineffective mutant Avi-NaalL(E416A) (5). These recombinant proteins, with a final purity greater than 95%, shown in Fig. 1, were used in all subsequent experiments.
FIGURE 1.
SDS-PAGE and size exclusion chromatography analysis of Avi-NaalL(E416A) affinity purification. Concentrated conditioned medium from S2 cells mixed with equilibration buffer (fraction L) was mixed with Streptavidin Mutein Matrix overnight. The medium was separated from the resin (fraction FT), and the resin was washed with washing buffer (fraction W1). The resin was then transferred to elution buffer (fraction W2). After a 1-h incubation, Avi-NaalL(E416A) was eluted with an excess of d-biotin (fractions E1–E4). Samples were loaded on the gel and silver-stained: fractions L and FT, 1 μl; fractions W1 and W2, 15 μl; fraction E1, 1 μl; fraction E2, 10 μl; fractions E3 and E4, 15 μl. The quaternary fold of Avi-NaalL(E416A) was subsequently analyzed using an FPLC system and SuperdexTM 200 10/300 column. Prior the analysis, the column was calibrated by low and high molecular weight gel filtration calibration kits (GE Healthcare) following the manufacturer's protocol. mAU, milliabsorbance units.
X-ray Structure of Avi-NaalL
We prepared diffraction quality crystals of human Avi-NaalL in hanging drops using a microseeding approach. We solved the structure by molecular replacement using the program Phaser and refined the final model to 1.75 Å resolution (PDB code 4TWE; data collection and refinement statistics are shown in Table 1). As shown in Fig. 2A, the overall fold of Avi-NaalL is very similar to that of GCPII (PDB code 3BXM) with a root mean square deviation of 1.41 Å for the 646 equivalent Cα atoms (19). Guided by the well described structural features of GCPII (20, 21), we annotated the Avi-NaalL structure accordingly.
TABLE 1.
Data collection and refinement statistics for Avi-NaalL
The data in parentheses refer to the highest resolution shell. Amino acids of the AviTEV-tag and first seven N-terminal amino acids of Avi-NaalL are disordered and not modeled in the structure. RMSD, root mean square deviation; AU, asymmetric unit.
| PDB code | 4TWE |
| Data collection statistics | |
| Space group | I222 |
| Temperature (K) | 100 |
| Cell parameters: a; b; c (Å) | 98.5; 174.8; 208.0 |
| No. of molecules in AU | 2 |
| Wavelength (Å) | 1.000 |
| Resolution limits (Å) | 30.0-1.75 |
| Highest resolution shell (Å) | 1.78-1.75 |
| No. of unique reflections | 177,979 (8822) |
| Multiplicity | 7.6 (7.5) |
| Completeness (%) | 99.7 (99.4) |
| Rpim (%)a | 2.9 (20.0) |
| Average I/σ(I) | 22.1 (4.2) |
| Wilson B (Å2)b | 31.7 |
| Refinement statistics | |
| Resolution range (Å) | 29.14-1.75 |
| Highest resolution shell (Å) | 1.80-1.75 |
| No. of reflections in working set | 172,688 (12,359) |
| No. of reflections in test set | 1757 (112) |
| R value (%)c | 0.167 (0.202) |
| Rfree value (%)d | 0.187 (0.231) |
| RMSD bond length (Å) | 0.020 |
| RMSD angle (degrees) | 1.74 |
| No. of atoms in AU | 13,236 |
| No. of protein atoms in AU | 11,535 |
| No. of ion atoms in AU | 6 |
| No. of solvent molecules in AU | 1695 |
| Mean B value (Å2) | 26.6 |
| Mean B value of protein atoms (Å2) | 25.0 |
| Mean B of solvent molecules (Å2) | 38.1 |
| Ramachandran plot statisticse | |
| Residues in favored regions (%) | 97.2 |
| Residues in allowed regions (%) | 99.8 |
| Outliers | Val373A/B, Ala450A |
a Rpim = Σhkl (1/(n − 1))∧0.5 Σnj = 1|Ihkl,j − <Ihkl>|/ΣhklΣjIhkl,j (30).
b Estimated by SFcheck.
c R value = |Fo| − |Fc‖/|Fo|, where Fo and Fc are the observed and calculated structure factors, respectively.
d Rfree is equivalent to the R value but is calculated for 1% of the reflections selected at random and omitted from the refinement process.
e As determined by MolProbity (12).
FIGURE 2.

X-ray structure of Avi-NaalL and its structural and sequential comparison to GCPII. A, alignment of Avi-NaalL (PDB code 4TWE) and GCPII (PDB code 3BXM) structures. Both are shown in ribbon representation. GCPII is shown in gray. Avi-NaalL is color-coded based on its domain organization: protease-like domain (red), apical domain (yellow), and C-terminal domain (orange). The zinc ions are shown as magenta spheres, and calcium ion is shown as a blue sphere. B, overall structure of Avi-NaalL dimer shown in ribbon representation. One monomer is shown in wheat, and the other is colored based on its domain organization. The zinc ions are shown as magenta spheres, and calcium ions are shown as blue spheres. The carbohydrate moieties are depicted as cyan sticks with oxygen atoms in red and nitrogen in blue. The disulfide bond between Cys-296 and Cys-313 is shown as a green stick. The top view of the dimer illustrates the extensive dimerization interface. C, sequential alignment of GCPII and NAALADase L. Conserved amino acids are highlighted in yellow, and similar amino acids are highlighted in cyan. The domain organization of NAALADase L is indicated by arrows above the sequence colored as follows: intracellular domain (black), transmembrane domain (gray), protease-like domain (red), apical domain (yellow), and C-terminal domain (orange). The catalytic acid/base Glu is marked with a blue star. Residues coordinating the zinc ions are black-boxed. N-Glycosylation sites are shown as blue circles, shaded if electron density for corresponding N-acetylglucoseamide was detected. Amino acids coordinating the calcium ion are shown as red circles, and Cys residues forming the disulfide bond are shown as green circles. Amino acids participating in N-acetyl-l-aspartyl-l-glutamate binding in GCPII are marked with magenta circles.
The Avi-NaalL monomer folds into three distinct domains: protease-like domain (residues 35–108 and 340–587), apical domain (residues 109–339), and C-terminal domain (residues 588–740). Two Avi-NaalL molecules in the asymmetric unit form a dimer with a buried surface area of more than 2300 Å2. The dimerization interface mainly involves interactions between the C-terminal domain of the first monomer and the protease-like/apical domains of the second monomer (Fig. 2B). Size exclusion chromatography data also indicated that the protein is dimeric with an approximate relative molecular mass of 250 kDa (Fig. 1), suggesting that this is probably its physiological quaternary structure. This quaternary arrangement is reminiscent of the dimerization pattern of both human transferrin receptor and GCPII (22).
Unlike GCPII, Avi-NaalL contains one intramolecular disulfide bridge, linking Cys-296 with Cys-313. The Avi-NaalL sequence also contains seven potential N-glycosylation motifs. We found clear electron density for at least one N-acetylglucoseamide moiety for all putative glycosylation sites except Asn-274. A sequential alignment of NAALADase L and GCPII is shown in Fig. 2C with several key functional amino acids highlighted.
A strong positive density peak representing a metal ion was observed in the Fo − Fc map of the Avi-NaalL structure. The metal is heptacoordinated by the γ-carboxylates of Glu-425 and Glu-428, the main chain carbonyls of Leu-261 and Thr-258, and the side chain hydroxyl group of Thr-258 (all at distances between 2.4 and 2.5 Å). The coordination sphere is completed by a water molecule (2.5 Å; Fig. 3A). In the final model, the Ca2+ ion was modeled into this position. Our assumption of modeling calcium ion was guided by several lines of indirect evidence, including the favored pentagonal bipyramidal geometry of the coordination sphere, agreement with the electron density, “optimal” interatomic distances, and matching atomic displacement parameters (B-factors) of the metal and coordinating residues (23). Additionally, the identity of the Ca2+ ion, occupying the equivalent site in closely related human GCPII, was confirmed experimentally before (20), thus strengthening our argument for calcium as a metal of choice. The exact structural and/or functional role of this cation, however, is not known at present and warrants further studies.
FIGURE 3.

Detailed features of the Avi-NaalL structure compared with the GCPII structure. Avi-NaalL residues are shown in green, and GCPII residues are shown in gray. Oxygens are colored red, and nitrogens are colored blue. A, calcium ion heptacoordination shell in Avi-NaalL with an electron density map. The calcium ion is coordinated by the Glu-425 and Glu-428 side chain carboxyl groups, the Thr-258 hydroxyl group, the carbonyl oxygens of Leu-261 and Thr-258, and a water molecule. Interactions are depicted as dashed lines with distances shown. The calcium ion is shown as a blue sphere, and water is shown as a red sphere. The 2Fo − Fc map (contoured to 1σ) is shown in blue, whereas the Fo − Fc map (contoured to 3σ) is colored green for positive and red for negative electron density peaks. B, superposition of the zinc ion coordination shells in Avi-NaalL and GCPII (PDB code 2PVW). The coordination shell of the active-site zincs is identical for both proteins, utilizing the side chains of His-368, Asp-378, Glu-417, Asp-445, and His-545 in Avi-NaalL. Additionally, the position of the Glu-416 side chain is identical to the position of the catalytic glutamate (Glu-424) in GCPII. Interactions are depicted as dashed lines with distances shown. C, electron density map around the Avi-NaalL active site. The 2Fo − Fc map (contoured to 1σ) is shown in blue, whereas the Fo − Fc map (contoured to 3σ) is colored green for positive and red for negative electron density peaks. Zinc ions are shown as magenta spheres, and waters are shown as red spheres. D, superposition of amino acids contributing to substrate binding in GCPII with the Avi-NaalL structure. The structure of GCPII E424A mutant (PDB code 3BXM) with bound N-acetyl-l-aspartyl-l-glutamate (NAAG), an endogenous GCPII substrate, is shown. Zinc ions in the GCPII structure are shown as gray spheres, and zinc ions in Avi-NaalL are shown as magenta spheres. Interactions of N-acetyl-l-aspartyl-l-glutamate with GCPII are depicted as dashed lines. E, superposition of the arginine patch in the GCPII (PDB code 3BXM) structure with Avi-NaalL. GCPII water molecule and chloride ion are depicted as gray spheres, and Avi-NaalL waters are shown as green spheres. Interactions of both GCPII and Avi-NaalL are depicted as dashed lines with distances shown.
Avi-NaalL binds two Zn2+ ions, which are coordinated by the side chains of His-368, Asp-378, Glu-417, Asp-445, and His-545 (Fig. 3B) at distances between 1.9 and 2.0 Å. These zinc-coordinating residues adopt the same conformation as in GCPII, forming the core of the Avi-NaalL active site. Glu-416, analogous to Glu-424 in GCPII (24), probably serves as a catalytic acid/base residue for peptide cleavage. Additionally, the presence of zinc ions was experimentally confirmed by fluorescent scanning of the Avi-NaalL protein crystal (data not shown). Similar to GCPII, Avi-NaalL also contains a cis peptide bond between Asp-378 and Pro-379, which is a hallmark of bimetallic peptidases.
Although Avi-NaalL was crystallized in the absence of a ligand, we detected an extensive positive electron density peak in the vicinity of the zinc ions (Fig. 3C). Unfortunately, we were unable to reliably model any molecule into this electron density. Moreover, this electron density peak spans over the expected position of the catalytic hydroxyl ion, which therefore could not be modeled in the Avi-NaalL structure. In GCPII, this hydroxyl ion completes the tetrahedral coordination of zinc ions and acts as a nucleophilic agent during peptidic substrate cleavage (24).
The comparison of amino acids that mediate ligand binding in GCPII with their Avi-NaalL counterparts is illustrated in Fig. 3D; the structure of inactive GCPII in complex with its endogenous substrate N-acetyl-l-aspartyl-l-glutamate was used for comparison. We found two conserved residues that probably mediate substrate binding to Avi-NaalL, Arg-198 and Tyr-544. Their GCPII counterparts, Arg-210 and Tyr-552, interact with the C-terminal α carboxylic group and the carbonyl oxygen of the cleaved peptide bond of the GCPII substrate. The residues Arg-534 and Arg-536, which form the so-called “arginine patch” in GCPII, thus providing the GCPII substrate binding cleft with a positive charge and contributing to its preference for negatively charged peptide substrates, are not conserved in Avi-NaalL (Fig. 3E). Consequently, the chloride ion, which stabilizes the energetically unfavorable “all-gauche” conformation of Arg-534 in GCPII, is not present in the Avi-NaalL structure.
Expression Pattern of the Human NAALADL1 Gene Product
We determined the amount of NAALADL1 gene transcript in commercially available cDNA libraries normalized to set of control genes from several human tissues by qPCR. We used two primer sets, each consisting of a primer pair for amplification and a fluorescent probe for detection. The first set of amplification primers targeted the region spanning exons 6 and 7 (NaalL_A), whereas the second targeted exons 13 and 14 (NaalL_B). The absolute amounts of NAALADL1 transcripts detected are shown in Fig. 4A. We identified NAALADL1 gene transcripts in several tissues, including the testis, colon, small intestine, ovary, spleen, and thymus.
FIGURE 4.

NAALADL1 gene product expression in human tissues. A, qPCR determination of NAALADL1 mRNA. Two sets of primers (NaalL_A and NaalL_B) were used to independently assess the level of NAALADL1 transcripts in different human tissues. No. of transcripts, number of transcripts determined in 0.1 μl of normalized cDNA library, with values showing the mean of triplicate measurements. Error bars, S.D. (for more experimental details, see “Experimental Procedures”). B, Western blot analysis of human tissue samples. Tissue lysates (50 μg of total protein) were loaded onto the gel with recombinant Avi-NaalL (5 ng) as a control. Samples were incubated with 1C11 hybridoma supernatant overnight, and the antibody-antigen complex was visualized using HRP-conjugated anti-mouse secondary antibody. The dashed squares indicate false positive signals, whereas the solid box marks NAALADase L (as determined by MS; data not shown).
On the other hand, using our newly developed mAb 1C11, we found that NAALADase L expression is primarily restricted to the small intestine (Fig. 4B). We interrogated the specificity of Western blots by mass spectrometry by analyzing several areas corresponding to positive Western blot signals (boxed in Fig. 4B). The small intestine tissue sample (solid box) was the only one in which we identified the NAALADL1 gene product (UniProt accession number Q9UQQ1). We found 33 distinct peptides covering 41% of the sequence entry in the database. On the other hand, gel sections from ovary tissue (dashed box) did not contain NAALADase L, suggesting that 1C11 has nonspecific cross-reactivity.
Enzymatic Activities of NAALADase L
Putative DPP IV Activity
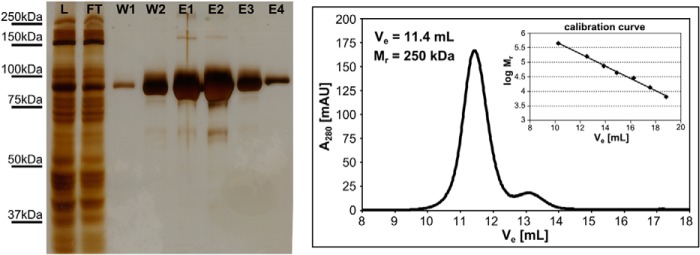
Because both human and rat NAALADase L were previously reported to possess DPP IV activity (1, 2), we attempted to confirm these findings using our purified protein preparations. As illustrated in Fig. 5, we were unable to detect any DPP IV activity for Avi-NaalL and Avi-GCPII, whereas the use of LNCaP lysate as a positive control resulted in a clear increase in fluorescence intensity.
FIGURE 5.
Determination of Avi-NaalL DPP IV activity. DPP IV activity was assayed using the fluorogenic substrate Gly-Pro-aminomethylcoumarin (excitation/emission, 335 nm/450 nm). Potential DPP IV activity of recombinant Avi-NaalL (1 μg) was assessed. Avi-GCPII (1 μg) and substrate alone (blank) were used as negative controls, whereas LNCaP lysate (10 μg of total protein) was used as a positive control. The reactions were incubated for 15 h at 37 °C. Each reaction was performed in triplicate and is shown as a mean with S.D. (error bar). RFU, relative fluorescence units.
Putative Carboxypeptidase Activity
Guided by our structural analysis of Avi-NaalL, we set out to investigate its putative carboxypeptidase activity. We synthesized and screened a dipeptide library encompassing 19 proteinogenic amino acids (all but cysteine). Our library consisted of 19 individual N-acetylated dipeptide mixtures, each mixture comprising 19 different dipeptides with defined N-terminal and variable C-terminal amino acids (e.g. Ac-Ala-Xxx-OH and Ac-Met-Xxx-OH). After incubating each mixture with Avi-NaalL, released C-terminal amino acids were detected using HPLC analysis. We used a catalytically ineffective mutant, Avi-NaalL(E416A), as a control. Additionally, we tested an active recombinant GCPII preparation (Avi-GCPII) and its catalytically ineffective E424A mutant as a corresponding control pair (5, 25).
The results of this carboxypeptidase activity assay presented as differential heat maps of active and inactive protein preparation are illustrated in Fig. 6. The control experiment with Avi-GCPII indicates that the assay is reliable because it confirms a clear preference for acidic residues (Glu/Asp) at both the C- and N termini of the dipeptides (3). On the other hand, Avi-NaalL shows negligible activity and no preference for any amino acid at either the C or N terminus. These results demonstrate that Avi-NaalL does not possess detectable carboxypeptidase activity against an N-acetylated peptide as a substrate.
FIGURE 6.

Heat maps reflecting qualitative determination of carboxypeptidase activity. Recombinant Avi-NaalL and Avi-GCPII were incubated with 19 different N-acetylated dipeptide libraries of the general formula Ac-Aaa-Xxx-OH (where Aaa represents a constant amino acid, and Xxx is a mixture of 19 proteinogenic amino acids). After 15 h of incubation at 37 °C, the free amine groups of cleaved C-terminal amino acids were modified, and the amino acids were detected by HPLC. Avi-NaalL(E416A) and Avi-GCPII(E424A), catalytically ineffective mutants of both enzymes, were used for background subtraction. The color coding corresponds to the percentage of conversion of the particular aa in the reaction mixture; the overall amount of each amino acid in dipeptide libraries was determined by amino acid analysis prior to the experiment.
Putative Endopeptidase Activity
We performed a PICS experiment to assess Avi-NaalL endopeptidase activity. This technique enables simultaneous characterization of both the S and S′ cleavage sites of an enzyme (18). We used a peptide library derived from the HEK293T cell proteome as substrate and included HIV-1 protease as a positive control to assess the functionality of the assay. Additionally, Avi-NaalL(E416A) and HIV-1(D25N), the corresponding catalytically ineffective mutants, were included to ensure reliable background subtraction.
The specificity matrices, presented in the form of heat maps, obtained from PICS are shown in Fig. 7. They summarize the sequences of peptide substrates for Avi-NaalL (72 individual sequences identified; a list of the peptides is shown in Table 2) and HIV-1 protease (over 160 individual sequences identified). For better clarity, we depicted matrices characterizing the percentage of individual amino acids at a given position in the identified cleaved peptides in Fig. 7A. Fig. 7B illustrates the ratios of individual amino acid occurrence at a given position compared with the overall occurrence of that amino acid in the human genome. Our data for HIV-1 protease showed high similarity to those found in the MEROPS database, demonstrating that our PICS experimental set-up provides a reliable account of protease substrate specificity. The specificity matrix for Avi-NaalL revealed substantial enrichment of acidic residues (Glu/Asp) in the P2′ position and Pro residue in the P3′ position.
FIGURE 7.

Heat maps of PICS results. PICS experiments were performed as described under “Experimental Procedures” with Avi-NaalL and its catalytically ineffective mutant Avi-NaalL(E416A) and with HIV-1 protease and its catalytically ineffective mutant HIV-1(D25N) protease (positive control). Briefly, the proteins were incubated overnight with a peptide library derived from the HEK293T proteome. Cleaved peptides were subsequently isolated from the reaction mixture via biotin modification of newly formed primary amino groups. The peptides were identified by LC-MS/MS analysis, and their corresponding prime parts were determined by sequence alignment with a human proteome database. The MEROPS substrate profile of HIV-1 protease is presented for comparison (see the MEROPS Web site). A, heat maps illustrate in grayscale the percentage of individual amino acids at a particular position relative to the scissile peptide bond. Four positions (P4′–P4) are shown. B, heat maps illustrate ratios of individual amino acid occurrence at a given position compared with the overall occurrence of that amino acid in the human genome. Ratios above 1 (i.e. preferred by the protease analyzed) are shown in shades of green, whereas ratios below 1 (i.e. rejected by the protease analyzed) are shown in shades of red.
Novel Aminopeptidase Activity
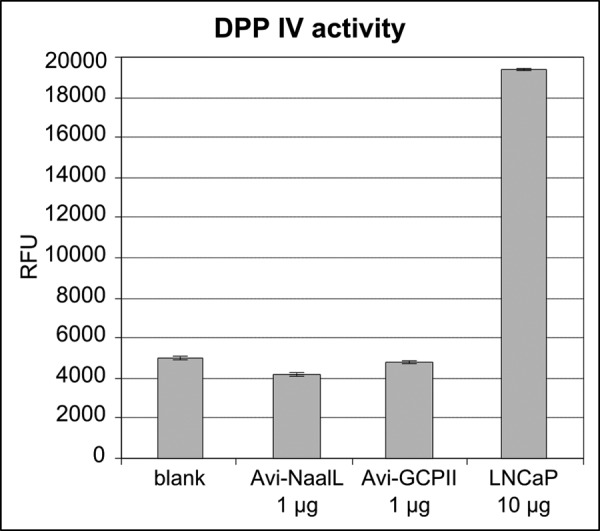
To elaborate on the PICS results, we synthesized two of the identified peptides, peptide 52 (AVF↓VDLEPT) and peptide 71 (GLFI↓IDPNG), and assayed their processing by Avi-NaalL and Avi-NaalL(E416A).
During overnight incubation, both peptides were completely processed by wild-type Avi-NaalL, whereas its E416A mutant showed minimal activity (Fig. 8). Interestingly, we could identify only N-terminally truncated parts of the assayed peptides. Moreover, we detected free phenylalanine in both reactions, which most likely originated from the N-terminal portion of the peptides. These data suggest that Avi-NaalL possesses aminopeptidase rather than endopeptidase activity. To confirm this assumption we performed a time-dependent analysis of hydrolysis of peptide 52 and found that Avi-NaalL indeed processes its peptide substrate sequentially from the N terminus (Fig. 9A).
FIGURE 8.
Avi-NaalL-catalyzed hydrolysis of two peptide substrates identified by PICS. Recombinant Avi-NaalL was mixed with peptide (peptide 52 or 71) in final concentrations of 1 and 100 μm, respectively. The specificity of cleavage was investigated using the catalytically ineffective mutant Avi-NaalL(E416A). Reaction mixtures were incubated for 15 h at 37 °C and analyzed using LC/MS. Peptide sequences or individual amino acids were identified based on their molecular mass and are shown in the key. The amounts of peptide or amino acid were determined using absorption at 210 nm. Each reaction was done in duplicate, and the result is shown as a mean with S.E (error bar).
FIGURE 9.

Quantitative analysis of Avi-NaalL aminopeptidase activity. A, time-dependent analysis of the peptide 52 cleavage by Avi-NaalL. Recombinant Avi-NaalL was mixed with peptide 52 in final concentrations of 0.1 and 100 μm, respectively. Reactions were performed at 37 °C and were stopped at times ranging from 5 to 90 min and analyzed by LC/MS. The relative amount of individual peptides or amino acids is plotted against reaction time in A1–A5. B, determination of kinetic parameters of model peptide substrate (I↓IDPNG) cleavage by Avi-NaalL. The enzyme was incubated with various concentrations of substrate, ranging from 6.3 to 1600 μm, for 15 min at 37 °C. The reaction mixtures were subsequently analyzed using HPLC. Each reaction was done in duplicate, and the result is shown as a mean with S.E. (error bar). The kinetic parameters were obtained by non-linear fit of the data using GraFit version 5.0.11 (Erithacus Software Ltd.). The reciprocal linear plot is also illustrated for comparison. C, determination of inhibition constant of aminopeptidase specific inhibitor bestatin toward Avi-NaalL. Various concentrations of bestatin, ranging from 0.1 to 500 μm, were incubated with Avi-NaalL and a model substrate (I↓IDPNG) for 15 min at 37 °C. The reaction mixtures were subsequently analyzed using HPLC. Each reaction was done in duplicate, and the result is shown as a mean with S.E. (error bar). The IC50 value was obtained by non-linear fit of the data using GraFit version 5.0.11 (Erithacus Software Ltd.), and the Ki value was then calculated using the Cheng-Prussoff equation for competitive mode of inhibition (31).
To obtain more quantitative data on Avi-NaalL enzymatic activity we designed model peptide substrate derived from peptide 71 (I↓IDPNG) from which only one Ile should be cleaved off by Avi-NaalL. Using this model substrate, we determined kinetic parameters of Avi-NaalL cleavage, Km = 210 μm and kcat = 2.16 s−1 (Fig. 9B), and also determined the inhibition constant of a common aminopeptidase inhibitor bestatin toward Avi-NaalL, Ki = 10.7 μm (Fig. 9C). Additionally, using high substrate concentration, thus modeling saturation state of the enzyme, we were able to determine the catalytic efficiency of the Avi-NaalL(E416A) mutant, kcat = 0.0011 s−1. This result confirmed our prediction that the Glu-416 indeed serves as a proton shuttle residue, which is crucial for proper catalytic efficiency of the enzyme.
Finally, to further corroborate the Avi-NaalL substrate specificity, we designed a random peptide (RIKLNWFS) and assayed its degradation by Avi-NaalL. As shown in Fig. 10, we identified that the peptide was efficiently processed to single amino acids by Avi-NaalL during overnight incubation, a finding that is in line with the expected cleavage pattern because the peptide contained neither an acidic residue nor Pro that could block the peptide processing. Moreover, this experiment also revealed the fact that Avi-NaalL is able to efficiently process longer oligopeptides as well as smaller tri- or dipeptides.
FIGURE 10.
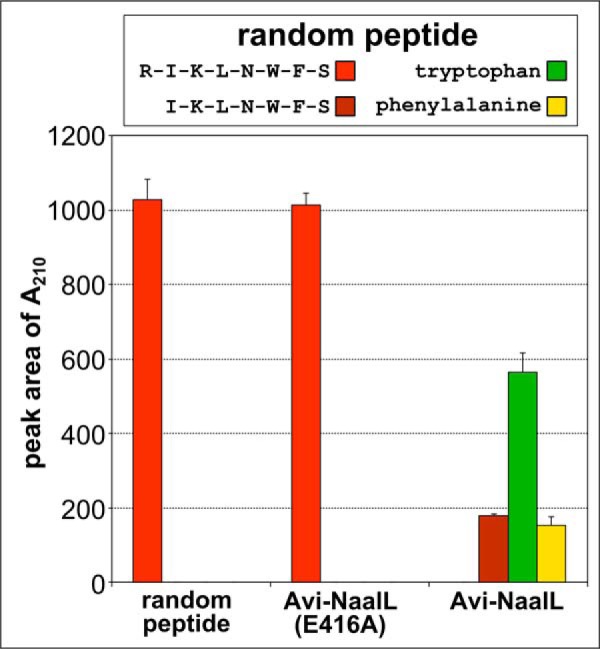
Processing of a random peptide by Avi-NaalL. Recombinant Avi-NaalL was mixed with the peptide RIKLNWFS in final concentrations of 1 and 100 μm, respectively. The specificity of cleavage was investigated using the catalytically ineffective mutant Avi-NaalL(E416A). Reaction mixtures were incubated for 15 h at 37 °C and analyzed using LC/MS. The peptide sequences or individual amino acids were identified based on their molecular mass and are shown in the key. The amounts of peptide or amino acids were determined by absorption at 210 nm. Each reaction was done in duplicate, and the result is shown as a mean with S.E. (error bar).
All of these results demonstrate that Avi-NaalL is an aminopeptidase with wide substrate specificity. Additionally, the results strongly suggest that the apparent Avi-NaalL endopeptidase-like substrate specificity determined by PICS (acidic residues in P2′ and Pro in P3′ position) is actually a motif preventing further N-terminal processing of peptides by Avi-NaalL.
DISCUSSION
Structural Features of the NAALADL1 Gene Product
This is the first study to experimentally describe the structure of the NAALADL1 gene product, which is highly homologous to that of GCPII. The amino acids coordinating two active site zinc ions and the catalytic acid/base residue (Glu-416; Fig. 3B) in Avi-NaalL are identical to those in GCPII. On the other hand, the GCPII amino acids participating in substrate recognition and formation of the arginine patch, which provides positive charge to the GCPII active site, are not conserved in Avi-NaalL. This structural comparison suggested that Avi-NaalL might bind the C terminus of its potential peptide substrate and position it toward the catalytic acid/base residue to enable proteolysis. However, it also indicated that Avi-NaalL substrate specificity is probably different from that of GCPII.
Both GCPII and NAALADase L belong to the MH clan (family M28) of metallopeptidases. The primary feature of these peptidases is a di-zinc active site center with conserved zinc-coordinating amino acids His, Asp, Glu, Asp/Glu, and His with the first Asp coordinating both metal ions. Additionally, for all peptidase from this clan, an additional residue (Asp or Glu) has been identified that is important for catalysis and is placed adjacent to coordinating amino acids in the motif His-Xxx-Asp or Glu-Glu. Based on the sequence and structural alignment of Avi-NaalL with other members of the M28 family, such as aminopeptidase S from Streptomyces griseus (SGAP) or GCPII, Glu-416 most probably plays the role of an acid/base catalytic residue in Avi-NaalL. This assumption is additionally supported by the enzymological data with the E416A mutant presented in this study.
Similarly to other families from the MH clan (e.g. the M20 family), the M28 family of peptidases contains both carboxypeptidases (GCPII) and aminopeptidases (SGAP). In the M20 family, the peptidase enzymatic activity is determined by the sequence, more specifically by the presence of either Glu or Asp residue at the ambiguous position in the zinc coordination center. If a Glu residue is present, the peptidase embodies carboxypeptidase activity (e.g. carboxypeptidase G2 from Pseudomonas sp.), whereas in the presence of an Asp residue, the peptidase acts as an aminopeptidase (e.g. aminopeptidase PepV from Lactobacillus delbrueckii) (26). Interestingly, this relationship is not observed in the M28 family, where an Asp coordinating residue is present in both GCPII and SGAP.
Considering tertiary and quaternary structure organization, Avi-NaalL adopts a structure identical to that of GCPII, containing three domains and folding into the dimer, whereas aminopeptidases from the M28 family, such as SGAP, are single-domain monomeric proteins. On the other hand, our enzymological data indicate that Avi-NaalL, like SGAP, possesses aminopeptidase activity. Therefore, we hypothesize that Avi-NaalL represents an evolutionary intermediate between GCPII and SGAP, sharing the same structural features with the former and the same enzymatic activity with the latter. The elucidation of an Avi-NaalL reaction mechanism and substrate binding may hopefully lead to the identification of structure-activity relationship similar to that described for the M20 family. However, additional crystallographic studies, targeting complexes of the wild-type enzyme with a potent inhibitor (e.g. bestatin) or complexes of a catalytically ineffective mutant (e.g. Avi-NaalL(E416A)) with a substrate, will be needed to address these questions in detail.
Distribution of the NAALADL1 Gene Product in Human Tissues
Although prior studies have described NAALADL1 gene product expression in human tissues at the mRNA level, this is the first study that analyzes its expression profile at a protein level. Interestingly, the restricted expression of NAALADase L, which we detected only in the small intestine, did not correspond well with the results from our qPCR analysis, which showed wide tissue distribution (Fig. 4). False positive results from the qPCR assay or alternative splicing of the NAALADL1 transcript are two possible explanations for these differences.
Amplification of plasmid NAALADL1 transcript in our qPCR assay conditions was ∼90% effective and showed linear dependence between the Cq values and the logarithm of the plasmid concentration over a complete range of standard concentrations for both primer sets. Moreover, we confirmed that both primer sets do not amplify transcripts of close NAALADase L homologs human GCPII/III and genomic DNA to a degree that would interfere with our qPCR assay (data not shown). These additional experiments and the use of commercial cDNA libraries, which are normalized to several different housekeeping genes and pooled from numerous individuals, help to ensure the reliability of our qPCR data.
Previously reported data on NAALADL1 transcript expression by Pangalos et al. (1), amplifying the region between exons 14 and 16, showed wide tissue distribution. On the other hand, a study by Shneider et al. (2), using a large probe spanning exons 10–15, detected NAALADL1 transcript only in the small intestine. The discrepancies between these two studies could be explained by alternative splicing of the NAALADL1 transcript; short amplicons are more prone to recognize several splice variants, whereas long probes are more specific.
Data from the AceView database also support a high degree of alternative splicing of the NAALADL1 transcript, describing more than 15 different splice variants (27). Among these variants, deletions of exon 11 (68 bp) or exon 13 (91 bp) are most common. The NaalL_A primer set, targeting exons 6 and 7, can detect both of these potential splice variants, whereas the NaalL_B primer set, targeting exons 13 and 14, cannot detect a variant lacking exon 13. This observation may be responsible for the different results in lung, ovary, and prostate tissues, in which NaalL_A but not NaalL_B primer sets amplified the NAALADL1 transcript (Fig. 4A).
Taken together, both the presented and previously published data support the hypothesis that the NAALADL1 transcript undergoes extensive alternative splicing. This processing may function as a regulatory mechanism of NAALADase L expression.
Putative Activities of NAALADase L
We were unable to detect the previously reported DPP IV activity in our enzyme preparation. The previous reports used either polyclonal antibody immunoprecipitate (2) or crude cell lysate (1, 2). Due to the ubiquitous presence of DPP IV activity in mammalian cells (28), we suggest that their results may have been false positive. Furthermore, one of these studies also reported DPP IV activity for GCPII and GCPIII, which was subsequently disproved by experiments with purified recombinant protein (3, 4).
The results from our carboxypeptidase assay, which suggest slow but specific Avi-NaalL cleavage of N-acetylated dipeptides with identical C and N termini, can be explained by sequential N-terminal degradation of the tested dipeptides. The free N-terminal amino acid, present in the reaction mixture 19 times more frequently than other amino acids, would be detected preferentially, leading to a false positive signal. The release of free N-terminal amino acid could be explained either by the ability of Avi-NaalL to cleave the N-acetyl moiety from the dipeptide or by incomplete N-acetylation during library synthesis.
Aminopeptidase Activity of NAALADase L
After ruling out the potential carboxypeptidase activity of Avi-NaalL, we used PICS, a more general method, to probe potential Avi-NaalL proteolytic activity. PICS enables not only analysis of endopeptidase activity but, in theory, also determination of aminopeptidase activity. Because the method is based on MS analysis of P′ regions of the cleaved peptides (i.e. the C-terminal fragments of putative peptide substrates), which are assigned by the peptide spectrum matching against the proteomic database, the cleavage of even one amino acid from the N terminus of putative peptide substrate can be detected. However, because the downstream sequences of different peptides are often identical, the determination of the “P sequence” can be ambiguous. We identified ∼15% of peptides in our assay with more than one possible P sequence. We omitted these P sequences from the analysis (see Table 2 and “Experimental Procedures” for more details about data analysis).
The PICS data suggested that Avi-NaalL prefers an acidic residue at P2′ and a Pro residue at P3′. Interestingly, individual analysis of 72 peptide sequences revealed that Asp/Glu in the P2′ position (31 peptides identified) and Pro in the P3′ position (25 peptides identified) are almost mutually exclusive. Of those 56 peptides, Glu/Asp and Pro were present simultaneously only in two cases. This observation led us to assume that these amino acids do not form a conserved substrate recognition motif for Avi-NaalL. Indeed, subsequent enzymatic analyses revealed that the identified cleavage pattern acts as a stopping point for N-terminal peptide degradation rather than as a substrate recognition motif. Nevertheless, these results confirmed the potential of PICS for characterization of aminopeptidase substrate specificity.
It should be noted that the peptides used for our PICS experiments were dimethylated at their N termini. This suggests that Avi-NaalL is capable of cleaving off N-terminal amino acids with blocked primary amine groups. However, this assumption conflicts somewhat with the results from the carboxypeptidase assay, in which N-acetylated dipeptides were inefficiently processed by Avi-NaalL. A likely explanation for this phenomenon is incomplete dimethylation of the peptide library, which would leave some peptides accessible to Avi-NaalL degradation.
Subsequent analysis of two individual peptides identified by PICS confirmed the aminopeptidase activity of Avi-NaalL. We were able to detect only the P′ portion of peptide substrates. Additionally, we identified individual amino acids that were present in the N-terminal portions of the analyzed peptides (Phe in peptides 52 and 71 or Phe and Trp in random peptide). Moreover, reactions with prolonged incubation times revealed that either Asp/Glu at P2′ or Pro at P3′ is sufficient to significantly decrease the cleavage efficacy, but only the combination of both amino acids completely prevents peptide degradation (Fig. 8, peptide 52). The inhibition experiment with bestatin together with detection of complete degradation of random peptide showed that Avi-NaalL probably belongs to the group of aminopeptidases that are able to cleave a variety of amino acids from the peptide N terminus, such as bovine lens aminopeptidase and SGAP (29). Although we perform quantitative analysis of Avi-NaalL cleavage of a model substrate, an additional analysis of Avi-NaalL substrate specificity and cleavage efficacy will be necessary for proper classification of this novel aminopeptidase.
Nevertheless, our data clearly demonstrate that the NAALADL1 gene product possesses different enzymological activity than GCPII. Because the protein product (“NAALADase L”) currently bears the same EC number as GCPII (EC 3.4.17.21), a new EC number for this enzyme should be appointed. Additionally, considering that the name NAALADase L (N-acetylated α-linked acidic dipeptidase-like) does not in fact describe the enzyme's features and is rather misleading, we suggest renaming the enzyme human ileal aminopeptidase (HILAP), to reflect its actual enzymatic activity and expression profile.
To conclude, we solved the x-ray structure of the extracellular portion of the human NAALADL1 gene product and showed that it is very similar to the GCPII structure, including complete conservation of all amino acids necessary for peptidase activity. Analysis of NAALADL1 gene product expression showed high levels of corresponding mRNA in several tissues. However, the protein was identified predominantly in small intestine, implying that the corresponding mRNA undergoes extensive alternative splicing. We disproved the previously reported DPP IV-like activity of the enzyme and demonstrated that a recombinant protein derived from the NAALADL1 gene possesses aminopeptidase activity. The enzyme degrades peptide substrates from their N termini until it reaches an acidic residue at P2′ or proline residue at P3′. This activity is inhibited by the specific aminopeptidase inhibitor bestatin.
Given its restricted expression profile and wide substrate specificity, we suggest that the physiological functions of the NAALADL1 gene product are probably associated with the final stages of protein digestion in the human body and suggest a new name for this enzyme: human ileal aminopeptidase (HILAP).
Acknowledgments
We acknowledge Jana Starková and Karolína Šrámková for excellent technical support, Helena Jindrová for performing size exclusion chromatography column calibration, Mirka Blechová for peptide synthesis, and Pavla Angelisová (Institute of Molecular Genetics, Academy of Sciences of the Czech Republic) for preparation of mAbs.
This work was supported by Grant Agency of the Czech Republic (Grant P304–12-0847) and InterBioMed Project LO 1302 from the Ministry of Education of the Czech Republic. This work was also supported in part by the National Institutes of Health, NCI, Center for Cancer Research, Intramural Research Program (to J. L.) and also in part by project “BIOCEV” (CZ.1.05/1.1.00/02.0109) from the European Regional Development Fund (to C. B.). The use of the Advanced Photon Source was supported by United States Department of Energy Contract W-31-109-Eng38.
The atomic coordinates and structure factors (code 4TWE) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- NAALADase I/II/L
- N-acetylated α-linked acidic dipeptidase I/II/-like
- aa
- amino acid(s)
- Avi-GCPII
- extracellular portion of GCPII with N-terminal AviTEV tag
- Avi-NaalL
- extracellular portion of NAALADase L with N-terminal AviTEV tag
- DPP IV
- dipeptidyl peptidase IV
- FA
- formic acid
- GCPII/III
- glutamate carboxypeptidase II/III
- HILAP
- human ileal aminopeptidase
- His-NaalL
- extracellular portion of NAALADase L with N-terminal His tag
- His tag
- hexahistidine tag
- I100
- 100-kDa brush border membrane protein
- PDB
- Protein Data Bank
- PICS
- proteomic identification of cleavage site
- PP
- polypropylene
- qPCR
- quantitative polymerase chain reaction
- rhGCPII/III
- extracellular portion of recombinant human GCPII/III
- SGAP
- aminopeptidase from S. griseus
- TEV
- tobacco etch virus
- Bistris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane.
REFERENCES
- 1. Pangalos M. N., Neefs J. M., Somers M., Verhasselt P., Bekkers M., van der Helm L., Fraiponts E., Ashton D., Gordon R. D. (1999) Isolation and expression of novel human glutamate carboxypeptidases with N-acetylated α-linked acidic dipeptidase and dipeptidyl peptidase IV activity. J. Biol. Chem. 274, 8470–8483 [DOI] [PubMed] [Google Scholar]
- 2. Shneider B. L., Thevananther S., Moyer M. S., Walters H. C., Rinaldo P., Devarajan P., Sun A. Q., Dawson P. A., Ananthanarayanan M. (1997) Cloning and characterization of a novel peptidase from rat and human ileum. J. Biol. Chem. 272, 31006–31015 [DOI] [PubMed] [Google Scholar]
- 3. Barinka C., Rinnová M., Sácha P., Rojas C., Majer P., Slusher B. S., Konvalinka J. (2002) Substrate specificity, inhibition and enzymological analysis of recombinant human glutamate carboxypeptidase II. J. Neurochem. 80, 477–487 [DOI] [PubMed] [Google Scholar]
- 4. Hlouchová K., Barinka C., Klusák V., Sácha P., Mlcochová P., Majer P., Rulísek L., Konvalinka J. (2007) Biochemical characterization of human glutamate carboxypeptidase III. J. Neurochem. 101, 682–696 [DOI] [PubMed] [Google Scholar]
- 5. Tykvart J., Sácha P., Bařinka C., Knedlík T., Starková J., Lubkowski J., Konvalinka J. (2012) Efficient and versatile one-step affinity purification of in vivo biotinylated proteins: expression, characterization and structure analysis of recombinant human glutamate carboxypeptidase II. Protein Expr. Purif. 82, 106–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kozísek M., Sasková K. G., Rezácová P., Brynda J., van Maarseveen N. M., De Jong D., Boucher C. A., Kagan R. M., Nijhuis M., Konvalinka J. (2008) Ninety-nine is not enough: molecular characterization of inhibitor-resistant human immunodeficiency virus type 1 protease mutants with insertions in the flap region. J. Virol. 82, 5869–5878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Langone J. J., Van Vunakis H. (1986) Immunological techniques. Part I. Hybridoma technology and monoclonal antibodies. Methods Enzymol. 121, 1–947 [PubMed] [Google Scholar]
- 8. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 9. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 11. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 12. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rovenská M., Hlouchová K., Sácha P., Mlcochová P., Horák V., Zámecník J., Barinka C., Konvalinka J. (2008) Tissue expression and enzymologic characterization of human prostate specific membrane antigen and its rat and pig orthologs. Prostate 68, 171–182 [DOI] [PubMed] [Google Scholar]
- 14. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 15. Barlos K., Chatzi O., Gatos D., Stavropoulos G. (1991) 2-Chlorotrityl chloride resin: studies on anchoring of Fmoc-amino acids and peptide cleavage. Int. J. Pept. Protein Res. 37, 513–520 [PubMed] [Google Scholar]
- 16. Kaiser E., Colescott R. L., Bossinger C. D., Cook P. I. (1970) Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal. Biochem. 34, 595–598 [DOI] [PubMed] [Google Scholar]
- 17. Woodward C., Henderson J. K., Wielgos T. (2007) High-speed Amino Acid Analysis (AAA) on 1.8 μm Reversed-phase (RP) Columns. Agilent Technologies Application Note 5989-6297EN, Agilent Technologies, Inc., Wilmington, DE [Google Scholar]
- 18. Schilling O., Huesgen P. F., Barré O., Auf dem Keller U., Overall C. M. (2011) Characterization of the prime and non-prime active site specificities of proteases by proteome-derived peptide libraries and tandem mass spectrometry. Nat. Protoc. 6, 111–120 [DOI] [PubMed] [Google Scholar]
- 19. Krissinel E. (2012) Enhanced fold recognition using efficient short fragment clustering. J. Mol. Biochem. 1, 76–85 [PMC free article] [PubMed] [Google Scholar]
- 20. Mesters J. R., Barinka C., Li W., Tsukamoto T., Majer P., Slusher B. S., Konvalinka J., Hilgenfeld R. (2006) Structure of glutamate carboxypeptidase II, a drug target in neuronal damage and prostate cancer. EMBO J. 25, 1375–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pavlícek J., Ptácek J., Barinka C. (2012) Glutamate carboxypeptidase II: an overview of structural studies and their importance for structure-based drug design and deciphering the reaction mechanism of the enzyme. Curr. Med. Chem. 19, 1300–1309 [DOI] [PubMed] [Google Scholar]
- 22. Lawrence C. M., Ray S., Babyonyshev M., Galluser R., Borhani D. W., Harrison S. C. (1999) Crystal structure of the ectodomain of human transferrin receptor. Science 286, 779–782 [DOI] [PubMed] [Google Scholar]
- 23. Zheng H., Chruszcz M., Lasota P., Lebioda L., Minor W. (2008) Data mining of metal ion environments present in protein structures. J. Inorg. Biochem. 102, 1765–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Klusák V., Barinka C., Plechanovová A., Mlcochová P., Konvalinka J., Rulísek L., Lubkowski J. (2009) Reaction mechanism of glutamate carboxypeptidase II revealed by mutagenesis, x-ray crystallography, and computational methods. Biochemistry 48, 4126–4138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Navrátil M., Ptáček J., Šácha P., Starková J., Lubkowski J., Bařinka C., Konvalinka J. (2014) Structural and biochemical characterization of the folyl-poly-γ-l-glutamate hydrolyzing activity of human glutamate carboxypeptidase II. FEBS J. 281, 3228–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lindner H. A., Lunin V. V., Alary A., Hecker R., Cygler M., Ménard R. (2003) Essential roles of zinc ligation and enzyme dimerization for catalysis in the aminoacylase-1/M20 family. J. Biol. Chem. 278, 44496–44504 [DOI] [PubMed] [Google Scholar]
- 27. Thierry-Mieg D., Thierry-Mieg J. (2006) AceView: a comprehensive cDNA-supported gene and transcripts annotation. Genome Biol. 7, S12.1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sedo A., Malík R. (2001) Dipeptidyl peptidase IV-like molecules: homologous proteins or homologous activities? Biochim. Biophys. Acta 1550, 107–116 [DOI] [PubMed] [Google Scholar]
- 29. Taylor A. (1996) in The Aminopeptidases (Taylor A., ed) pp. 1–20, Landes Bioscience Publishers, Austin, TX [Google Scholar]
- 30. Weiss M. S. (2001) Global indicators of x-ray data quality. J. Appl. Crystallogr. 34, 130–135 [Google Scholar]
- 31. Cheng Y., Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108 [DOI] [PubMed] [Google Scholar]