

Background: Deletion of the mouse Brpf1 gene causes embryonic lethality, but the resulting defects await characterization.
Results: The vasculature, neural tube, and cell proliferation are abnormal in the mutant.
Conclusion: Brpf1 is important for embryo development and cell cycle control.
Significance: This study identifies a critical role of a multivalent chromatin regulator in embryogenesis and cell proliferation.
Keywords: Chromatin Modification, Development, Embryo, Histone Acetylase, Mouse Genetics
Abstract
With hundreds of chromatin regulators identified in mammals, an emerging issue is how they modulate biological and pathological processes. BRPF1 (bromodomain- and PHD finger-containing protein 1) is a unique chromatin regulator possessing two PHD fingers, one bromodomain and a PWWP domain for recognizing multiple histone modifications. In addition, it binds to the acetyltransferases MOZ, MORF, and HBO1 (also known as KAT6A, KAT6B, and KAT7, respectively) to promote complex formation, restrict substrate specificity, and enhance enzymatic activity. We have recently showed that ablation of the mouse Brpf1 gene causes embryonic lethality at E9.5. Here we present systematic analyses of the mutant animals and demonstrate that the ablation leads to vascular defects in the placenta, yolk sac, and embryo proper, as well as abnormal neural tube closure. At the cellular level, Brpf1 loss inhibits proliferation of embryonic fibroblasts and hematopoietic progenitors. Molecularly, the loss reduces transcription of a ribosomal protein L10 (Rpl10)-like gene and the cell cycle inhibitor p27, and increases expression of the cell-cycle inhibitor p16 and a novel protein homologous to Scp3, a synaptonemal complex protein critical for chromosome association and embryo survival. These results uncover a crucial role of Brpf1 in controlling mouse embryo development and regulating cellular and gene expression programs.
Introduction
Combinatorial modifications of histones form unique signaling platforms important for governing chromatin structure and function (1–4). Such modifications are frequently recognized by readers containing various structural modules, including the bromodomain, chromodomain, PHD2 (plant homeodomain-linked) zinc finger, and PWWP (Pro-Trp-Trp-Pro tetrapeptide-containing) domain (5–7). Some readers possess multiple domains to recognize different modifications in a combinatorial fashion (5). BRPF1 (or BR140, for bromodomain protein of 140 kDa) is one such reader. Like the paralogs BRPF2 and BRPF3, BRPF1 possesses an N-terminal PHD-zinc knuckle-PHD (PZP) module, a central bromodomain, and a C-terminal PWWP domain (8). The first PHD finger of BRPF1 or BRPF2 recognizes the unmodified N terminus of histone H3 (9, 10). The bromodomain possesses acetyllysine binding ability (11), and the PWWP domain has an affinity for methylated histone H3 (12, 13).
Moreover, BRPF1 possesses two motifs similar to Drosophila enhancer of polycomb (EPC) (14, 15). These two motifs flank the PZP module. The N-terminal EPC-like motif interacts with MOZ (monocytic leukemia zinc finger protein; also known as KAT6A and MYST3) and MORF (MOZ-related factor; also known as KAT6B and MYST4), whereas the C-terminal motif associates with ING5 (inhibitor of growth 5) and EAF6 (homolog of yeast Esa1-associated factor 6) (14, 16). MOZ and MORF do not interact with ING5 and EAF6 directly, so BRPF1 serves as a scaffold to assemble tetrameric complexes containing MOZ (or MORF), ING5, and EAF6 (16). The acetyltransferase domain of MOZ/MORF is sufficient for interaction with BRPF1, which enhances the acetyltransferase activity of MOZ and MORF toward nucleosomal histone H3 (16). Recent studies have uncovered HBO1 complexes containing BRPF1/2, ING5, and EAF6 (10, 17). Association with BRPF1 regulates the substrate specificity of HBO1 (histone acetyltransferase bound to ORC1) (10). Thus, BRPF1 is crucial for assembling multisubunit acetyltransferase complexes to control their enzymatic activity and substrate specificity (18). An interesting issue is how BRPF1 interacts with MOZ, MORF, and HBO1 under different biological and pathological contexts in vivo.
Of the pathological relevance, the MOZ gene was initially identified as a fusion partner in a chromosomal translocation causing monocytic leukemia (19). It is fused to four different partners in leukemia-associated chromosomal rearrangements, and similar rearrangements have been reported for the MORF gene (8, 20). Moreover, the MOZ gene is mutated in esophageal adenocarcinoma (21), whereas the MORF gene is disrupted in leiomyomata (22, 23), mutated in breast cancer (24), and altered in castration-resistant prostate cancer (25). A recent pan-cancer analysis of copy number variations has identified both genes as top-ranking targets amplified in different cancers (26). Related to this, the mouse Moz gene is required for optimal lymphoma development induced by Myc (27). In addition to cancer, the MOZ and MORF genes are mutated in multiple developmental disorders with the common characteristic of intellectual disability (28–35). Thus, both MOZ and MORF are important in cancer and other diseases.
As a key partner of MOZ and MORF, BRPF1 may modulate related pathogenesis. Moreover, the BRPF1 gene itself is recurrently mutated in pediatric cancers (36) and adult medulloblastoma (37). To understand these pathological processes, it is important to know the normal biological functions of BRPF1. However, little is known in this regard. To address this, we recently deleted the mouse Brpf1 gene and found that it is required for embryonic survival (38). Here, we present systematic analyses of the resulting developmental, cellular, and molecular defects. These results reveal that Brpf1 regulates different developmental programs during embryogenesis and that it is important for growth and proliferation of embryonic fibroblasts and hematopoietic progenitors. While two recent reports are on the crucial function of Brpf1 in mouse forebrain development (39, 40), this study identifies an essential role in regulating developmental programs just before mid-gestation. These new findings are unexpected from published genetic studies of mouse Moz, Morf, and Hbo1 (41–44).
MATERIALS AND METHODS
Animals
Mice were maintained in an animal facility at McGill University, and all procedures involved in the use of mice were performed according to guidelines and protocols approved by the McGill Animal Use Committee. Brpf1l/+ mice were obtained from the European Conditional Mouse Mutagenesis Program as described (38). A promoterless LacZ cassette is located between two FRT sites, whereas two loxP sites flank exons 4–6 of the Brpf1 gene (38). Crossing of Brpf1l/+ mice with EIIa-Cre mice (The Jackson Laboratory) resulted in the heterozygote Brpf1l/+;EIIa-Cre (or Brpf1+/Δ) (38), and subsequent intercrosses yielded the homozygote Brpf1l/l;EIIa-Cre (or Brpf1Δ/Δ). Brpf1+/− mice were obtained after consecutively mating Brpf1l/+ mice with PGK1-FLPo and EIIa-Cre strains (The Jackson Laboratory), and further intercrosses generated Brpf1−/− mice (38). Genotyping was carried out as described (38). Brpf1f/+;ER-Cre mice were generated after consecutive mating of Brpf1l/+ mice with PGK1-FLPo and UBC-Cre/ERT2 strains (The Jackson Laboratory).
Histology and Immunohistochemistry
Paraffin sections of embryonic and extra-embryonic tissues were stained with hematoxylin and eosin for histological examination. Benzidine staining was performed to detect hemoglobin in yolk sac erythrocytes as described (45), with minor modifications. Briefly, dissected embryos with intact yolk sacs were fixed for 15 min in 12% glacial acetic acid containing 0.4% benzidine dihydrochloride (Sigma, B3383). The staining reaction was initiated by the addition of hydrogen peroxide to a final concentration of 0.3%. Incubation was ∼20 min at room temperature; color development was carefully monitored. Stained embryos were immediately documented under a dissecting microscope (SteREO Lumar.V12, Zeiss) linked to a digital camera.
Periodic acid Schiff staining on placental paraffin sections was performed according to the manufacturer's protocol (Sigma, 395B). Whole-mount CD31 immunohistochemistry was performed as described (46), except that primary and secondary antibodies were incubated overnight at 4°C. The reagents used were as follows: purified rat anti-mouse CD31 antibody (BD Biosciences, 550274, 1:100), biotin-SP-conjugated AffiniPure donkey anti-rat IgG (H+L) antibody (Jackson ImmunoResearch, 712-065-153, 1:50), ABC kit (Vector Laboratories, PK-4001), and DAB substrate kit (Vector Laboratories, SK-4100). Images were taken with a digital camera (AxioCam MRc, Zeiss) linked to the dissecting microscope.
β-Gal Staining of Placental Sections
The staining procedure was the same as described (38, 39, 47). Briefly, pregnant mice were euthanized, and placentae were dissected out and fixed in 1% paraformaldehyde at 4 °C for 1–2 h. After washing with PBS, placentae were further cryoprotected in 30% sucrose in PBS overnight at 4 °C, embedded in Tissue-Tek OCT compound (Sakura Finetek, 4583) on dry ice, and stored at −80 °C. Cryosections were cut at 15 μm on a Cryotome (Thermo Electron, 77200187).
Clonogenic Hematopoietic Progenitor Assays
Four pairs of control and mutant yolk sacs were isolated from E9.5 embryos. Yolk sac single-cell suspension was prepared by passing through a cell strainer (StemCell Technologies, 27305, 40 μm), and 3 × 104 cells were seeded in complete methylcellulose-based medium (Stem Cell Technologies, M3434). Colonies were counted after 8 days of culture at 37 °C in a CO2 incubator.
MEF Isolation and Culture
Primary MEFs derived from embryos at E9.5 from Brpf1+/Δ intercross were cultured in MEF medium (47) and monitored for cell growth by IncuCyte (Essen Bioscience). Brpf1Δ/Δ MEFs failed to grow, so the UBC-Cre/ERT2 strain was employed to generate Brpf1f/f;ER-Cre MEFs. The Brpf1 gene was then inactivated in vitro after treatment with 4-hydroxytamoxifen in vitro. The MEFs were obtained from E15.5 embryos from crosses between Brpf1f/f and Brpf1f/+; ER-Cre mice. Brpf1f/f;ER-Cre MEFs were isolated from three independent litters and used at passage 2 or 3. MEF isolation was carried out as described (47). The cells were cultured in MEF medium (47), treated with vehicle (ethanol) or 400 nm of 4-hydroxytamoxifen (Sigma, H7904, dissolved in ethanol) for 4 days, and then changed to fresh MEF medium (47). For cell cycle analysis, MEFs with or without 4-hydroxytamoxifen treatment were collected and stained with propidium iodide solution (48) for subsequent data acquisition on a BD LSRII flow cytometer and further analysis by use of FlowJo software (Treestar). For analysis of daily cell growth, MEFs were seeded at low density for live-cell monitoring by IncuCyte for 1 week. For RT-qPCR and Western blotting, MEFs grown to ∼80% confluency were used.
Immunofluorescence Microscopy
For immunostaining of whole conceptuses, paraffin sections were used. Genomic DNA was isolated from each dewaxed embryo tissue, and genotyped. Immunofluorescence microscopy was performed as described (39, 49). Primary antibodies used were rabbit anti-cleaved caspase 3 (Cell Signaling, 9661, 1:200), mouse anti-neuronal class III β-tubulin (Tuj1) (Covance, MMS-435P, 1:500), mouse anti-histone H3 phosphorylated at Ser-10 (USBiological Life Sciences, H5110-13K, 1:100), goat anti-human SOX2 (R&D Systems, AF2018, 1:200), goat anti-HBO1 (Santa Cruz Biotechnology, sc-13284, 1:50), and rabbit anti-γH2AX (phospho-S139) (Abcam, ab2893, 1:500). Secondary antibodies were Alexa Fluor 568-labeled goat anti-rabbit IgG (Invitrogen, A11011, 1:500), Alexa Fluor 568-labeled goat anti-mouse IgG (Invitrogen, A11031, 1:500), and Cy3-conjugated anti-goat IgG (Molecular Probes, 1:500).
Frozen sections were used for placental immunostaining as described except that the sodium citrate-based antigen retrieval step was omitted (39). Primary antibodies were mouse monoclonal Gcm1 (glial cell missing 1; Santa Cruz Biotechnology, sc-101173, 1:200), rat anti-mouse CD31 (BD Biosciences, 550274, 1:200), goat placental lactogen I (Santa Cruz Biotechnology, sc-34713, 1:200), or rabbit anti-Esx1 antibody (extraembryonic-spermatogenesis-homebox protein 1; Santa Cruz Biotechnology, sc-133566, 1:100). Secondary antibodies were Alexa Fluor 568-labeled goat anti-rabbit IgG (Invitrogen, A11011, 1:500), Alexa Fluor 568-labeled goat anti-mouse IgG (Invitrogen, A11031, 1:500), Cy3-conjugated anti-goat IgG (Molecular Probes, 1:500), and CFL 488-linked goat anti-rat IgG (Santa Cruz, sc-362263, 1:500).
Microarray-based Gene Expression Profiling
Three pairs of control and mutant E8.75 embryos were used to prepare total RNA for microarray-based gene expression analysis as described (39). The resulting gene list is included in supplemental Table S1.
RT-qPCR
The procedure was carried out as described (39) using primers listed in Table 1.
TABLE 1.
RT-qPCR primers and validation of selected genes
RT-qPCR results are shown as the mean ± S.D. ns, not statistically significant.
| Target genes | Primer sequence |
-Fold change |
Primer position | GenBankTM accession no. | ||
|---|---|---|---|---|---|---|
| 5′ | 3′ | E8.75 embryos | E15.5 MEFs | |||
| Brpf1-ex | CAGTAAGATCACCAACCGCC | GAGGAAAGGGGTCAGCTGCA | 0.19 ± 0.05a | 0.03 ± 0.01a | 1711–2050 | NM_030178.1 |
| Brpf1-N | CAGCCCCTCTGAAGTCTCAC | CTAGTGCATTGGGGTCACCT | 1.14 ± 0.26 ns | 0.24 ± 0.05b | 379–956 | NM_030178.1 |
| Brpf2 | AACACTGACCTACGCACAAGC | GCCTCTCGCTGTTCTCCTTATT | 0.84 ± 0.15 ns | 1.24 ± 0.29 ns | 87–247 | NM_001033274 |
| Brpf3 | ACAAGCTCAAGATGCTAGAAGGC | TAGCTGGAAGTGACAAAGGCA | 0.84 ± 0.22 ns | 1.42 ± 0.27 ns | 3485–3608 | NM_001081315 |
| Moz | ATGGTAAAACTCGCTAACCCG | CGTCCCGTCTTTGACGCTC | 0.90 ± 0.10 ns | 0.91 ± 0.16 ns | 1–177 | NM_001081149 |
| Morf | AGAAGAAAAGGGGTCGTAAACG | GTGGGAATGCTTTCCTCAGAA | 0.86 ± 0.09 ns | 1.02 ± 0.11 ns | 2474–2668 | NM_017479 |
| Hbo1 | ATGCCGCGAAGGAAGAGAAAT | TCTTGGGAACTCTGGCTTAGC | 0.99 ± 0.07 ns | 1.16 ± 0.12 ns | 1–164 | NP_808287 |
| Mof | CCGGATAGCACCTGGCATTC | CATACTTCACTTTGGTGATC | 1.14 ± 0.12 ns | 0.87 ± 0.53 ns | 211–550 | NM_026370 |
| Hoxa9 | CCCCGACTTCAGTCCTTGC | GATGCACGTAGGGGTGGTG | 0.93 ± 0.06 ns | 0.84 ± 0.26 ns | 144–277 | NM_010456 |
| Rpls14 | TGCCACATCTTTGCATCCTTC | ACTCATCTCGGTCAGCCTTCA | 0.76 ± 0.07 ns | 1.10 ± 0.04 ns | 91–205 | NM_020600 |
| Rps19 | CAGCAGGAGTTCGTCAGAGC | CACCCATTCGGGGACTTTCA | 0.75 ± 0.17 ns | 31–102 | NM_023133 | |
| Rpl10l | ACCCAAAGTCCCGTTTCTGC | CTCGTCCGACACCATGTGG | 0.21 ± 0.03c | 1.03 ± 0.49 ns | 50–168 | NM_001162933 |
| Rpl26 | ACTTCTGACCGAAGCAAGAAC | CCGAATGGGCATAGACCGAA | 0.72 ± 0.17 ns | 22–150 | NM_009080 | |
| Rpl41 | CATCTTCCTTGAGACTCCTGC | CATCCCTCACTTCTGCTCC | 0.78 ± 0.20 ns | 16–167 | NM_018860 | |
| Rps29 | GTCTGATCCGCAAATACGGG | AGCCTATGTCCTTCGCGTACT | 0.79 ± 0.20 ns | 86–154 | NM_009093 | |
| p16 | GGGTTTCGCCCAACGCCCCGA | TGCAGCACCACCAGCGTGTCC | 0.65 ± 0.55 ns | 2.16 ± 0.14b | 190 | NM_001040654 |
| p19 | GTTTTCTTGGTGAAGTTCGTGC | TCATCACCTGGTCCAGGATTC | 0.55 ± 0.57 ns | 0.93 ± 0.15 ns | 134 | NM_009877 |
| p15 | CCCTGCCACCCTTACCAGA | CAGATACCTCGCAATGTCACG | 1.36 ± 1.01 ns | 1.45 ± 0.20 ns | 204–372 | NM_007670 |
| p27 | TGTCAGCGGGAGCCGCCAGG | ATATCTTCCTTGCTTCATAA | 0.65 ± 0.08c | 843–1182 | NM_009875.4 | |
| p57 | CCAAGCTGGACAGGACAAGC | AGTCCCAGCGGTTCTGGTCC | 1.27 ± 0.41 ns | 141–400 | NM_001161624 | |
| p21 | CACAGCGACCATGTCCAATC | GCGGGGCTCCCGTGGGCACT | 1.26 ± 0.53 ns | 201–680 | U09507.1 | |
| Bax | TGAAGACAGGGGCCTTTTTG | AATTCGCCGGAGACACTCG | 1.23 ± 0.04b | 59–198 | NM_007527 | |
| Cxcl15 | CAAGGCTGGTCCATGCTCC | TGCTATCACTTCCTTTCTGTTGC | 0.79 ± 0.03 ns | 1.12 ± 0.28 ns | 10–192 | NM_011339 |
| Fgf2 | GCGACCCACACGTCAAACTA | TCCCTTGATAGACACAACTCCTC | 0.89 ± 0.36 ns | 164–225 | NM_008006 | |
| Thpo | GGCCATGCTTCTTGCAGTG | AGTCGGCTGTGAAGGAGGT | 0.73 ± 0.28 ns | 27–140 | NP_033405 | |
| Thsd7a | AGGTGCCCACCCTCTATCTG | TGTATGTAACGTAGTCCAGCCT | 0.51 ± 0.22 ns | 128–258 | NM_001164805 | |
| Tailless | GGGAAGCACTACGGGGTCTA | GTGTCTTGTCTACGGGGCAT | 0.33 ± 0.14 ns | 73–196 | NM_152229 | |
| Fbxo40 | TGCCTCAACTCTGAGTATGGC | GAATGTTTCTGAGTCCACGTTTG | 1.02 ± 0.24 ns | 193–330 | NM_001037321 | |
| unknown | AACCACCTGGGAAGACAGGA | TCCCTTCACACACACAGCCT | 1.08 ± 0.65 ns | 735–846 | NR045476 | |
| Scp3l | AGTGCCTTTTGTCAGCAACAG | AGCGTGTCATTTGCCTGCTT | 11.51 ± 6.38c | 3.20 ± 0.71c | 418–509 | NM_001033423 |
| Pax9 | CGCACGCAGTGAATGGATTG | GCTGGTGTAGGGTAAGGAGC | 0.97 ± 0.18 ns | 716–845 | NM_011041 | |
| Pcdhb22 | TGTGATGGAGGAAACACCCAG | GCTTCCCAGTCTGTAGATCCAG | 1.32 ± 0.02 ns | 96–116 | NM_053147 | |
| Fabp4 | AAGGTGAAGAGCATCATAACCCT | TCACGCCTTTCATAACACATTCC | 1.55 ± 0.70 ns | 238–370 | NM_024406 | |
| Tnfrsf13c | TCTGGTGAGAAACTGCGTGTC | GTCAGCGCCAGTATCAGTCC | 1.19 ± 0.52 ns | 90–260 | NM_028075 | |
| Pou2af1 | CACCAAGGCCATACCAGGG | GAAGCAGAAACCTCCATGTCA | 0.92 ± 0.36 ns | 44–224 | NM_011136 | |
| Hemgn | GGAGGCAGACATCACAATGG | CCTTTTGCTCCACGTTCCCTT | 1.45 ± 1.82 ns | 146–256 | NM_053149 | |
| Mecom | AAGTAATGAGTGTGCCTATGGC | AGTTGACTCTCGAAGCTCAAAC | 0.73 ± 0.21 ns | 36–264 | NP_067417 | |
| CD31 | ACGCTGGTGCTCTATGCAAG | TCAGTTGCTGCCCATTCATCA | 1.12 ± 0.74 ns | 22–130 | NP_032842 | |
| Inpp4a | ACTCCATCGCTAGATCGAAAACC | AGGCAATGCTGCTTAGAAAGAT | 0.83 ± 0.51 ns | 169–307 | NP_084542 | |
| Nek10 | ATTACGTTCGTGATATTGGTGCT | TGACCACTACGCTTTCTAACCT | 0.78 ± 0.26 ns | 1418–1625 | NM_001195229 | |
| Il22ra1 | ATGAAGACACTACTGACCATCCT | CAGCCACTTTCTCTCTCCGT | 0.62 ± 0.43 ns | 1–198 | NM_178257 | |
| Gapdh | TGATGACATCAAGAAGGTGGTGAA | TCTTACTCCTTGGAGGCCATGT | as reference | 814–1059 | XM_001476707.3 | |
a p < 0.001.
b p < 0.01.
c p < 0.05.
Western Blotting
The procedure was the same as described (16, 47). Primary antibodies were rabbit anti-histone H3 (Abcam, ab1791, 1:5000), anti-histone H3 (acetyl Lys-14) (Millipore, 07-353, 1:4000), anti-histone H3 (acetyl Lys-9) (Abcam, ab10812, 1:4000), anti-acetyl-histone H3 (Millipore, 06-599, 1:4000), and goat anti-HBO1 (Santa Cruz Biotechnology, sc-13284, 1:200).
RESULTS
Brpf1 Loss Leads to Arrested Vasculogenesis in the Yolk Sac and Embryo Proper
To determine the biological function of Brpf1, we have recently characterized a mutant Brpf1l allele containing a promoterless LacZ knockin cassette as an efficient reporter (38). Using this reporter, we found that Brpf1 is highly expressed in the placenta, yolk sac, and neural tube (38), suggesting potentially important roles in related developmental processes. We also generated two mutant alleles, Brpf1Δ and Brpf1−, both of which are inactive (38). No viable embryos homologous for these alleles could be recovered after E9.5, indicating that loss of Brpf1 causes embryonic lethality (38).
While dissecting the embryos, we noticed that from E9.5 onward the mutant yolk sac contained no visible blood vessels (Fig. 1, A and B). Even in less severe cases, the mutant yolk sac contained a nonhierarchial vasculature, as opposed to the large vessels and small branches found in the control (Fig. 1C). Moreover, the blood vasculature was disorganized in the mutant embryo (Fig. 1D). Hemorrhage occurred in the cephalic region of the mutant embryos (Fig. 1D, right). Arrested vasculogenesis was also found in the mutant chorionic plate at E10.5 (Fig. 1E). H&E staining was used to compare control and mutant yolk sac sections (Fig. 1F). Whereas nucleated red blood cells were found in both control and mutant yolk sacs at E8.5 (Fig. 1F, top two panels), this situation changed dramatically at E9.5. In those severely affected mutant yolk sacs, only a few nucleated erythrocytes could be found in serial sections (Fig. 1F, middle two panels). Similarly, at E10.5, there were no nucleated erythrocytes in the mutant yolk sac (Fig. 1F, bottom two panels). These results suggest that embryonic lethality in Brpf1-deficient embryos is at least partially due to global vascular defects and massive hemorrhage.
FIGURE 1.
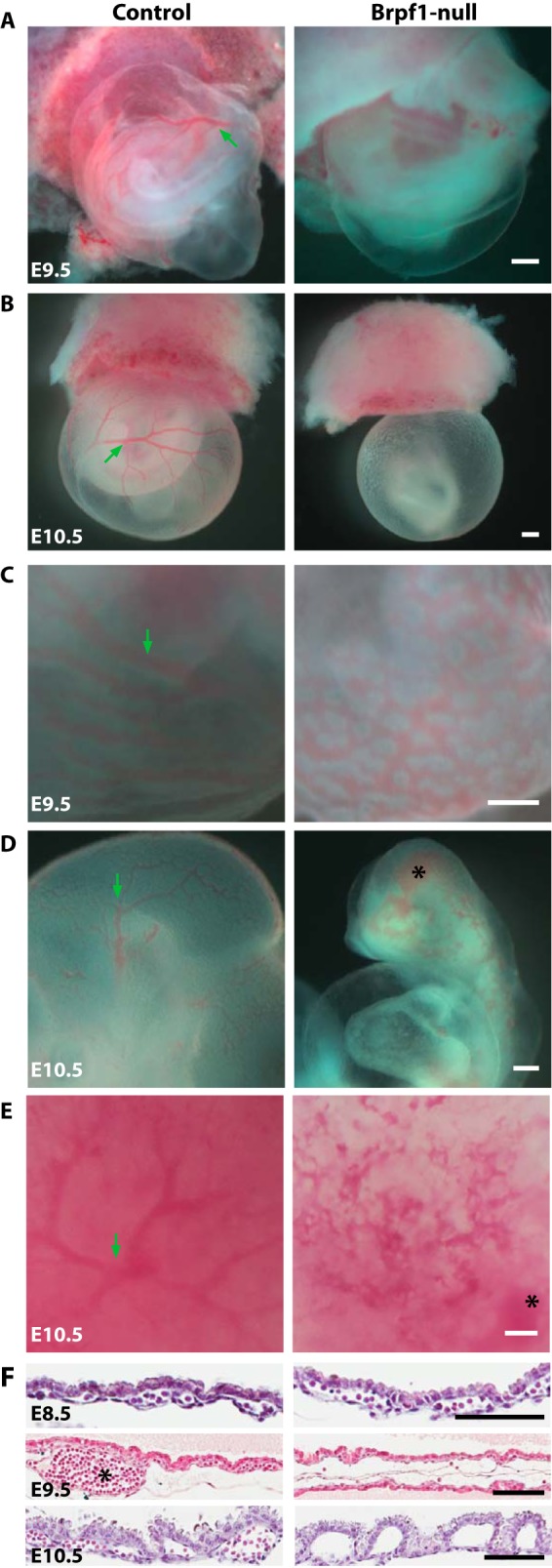
Aberrant blood vessels in Brpf1-deficient conceptuses. A and B, gross morphology of control and knockout conceptuses at E9.5 and E10.5. Note that the mutant yolk sacs were pale, without any visible vasculature. At E8.75, mutant conceptuses appeared normal (data not shown). C, in a less severe mutant yolk sac, a nonhierarchial vasculature was observed at E9.5, compared with the large vessels (indicated by a green arrow) and small branches of the control yolk sac. D and E, arrested vasculogenesis also occurred in the embryo proper (D) and chorionic plate (E) at E10.5. Note the hemorrhage in the mutant cephalic region and chorionic plate, marked with black asterisks in D and E, respectively. F, H&E staining of control and mutant yolk sac sections at E8.5 (top), E9.5 (middle), and E10.5 (bottom). At E8.5, nucleated red blood cells were found in both control and mutant yolk sacs. At E9.5, in those severely affected mutant yolk sacs, only a few erythrocytes could be found in serial sections, whereas in the control yolk sacs, large vessels (marked with a asterisk) were frequently seen. At E10.5, no erythrocytes existed in the mutant yolk sacs. Scale bars, 400 μm (A–E) and 100 μm (F).
To further characterize the vascular defects, we performed whole-mount immunohistochemical analysis with an antibody against CD31 (also known as platelet endothelial cell adhesion molecule 1 (PECAM1)), a well known and widely used marker for vasculature (50). As shown in Fig. 2, A and B, CD31+ vasculature was well formed in the control but not mutant embryo at E9.5. Similar to what observed in the embryo proper, the vascular network was well organized in the control but not mutant yolk sac or chorionic plate (Fig. 2, C and D). Benzidine staining was used to detect hemoglobin in erythrocytes and visualize blood vessels. As shown in Fig. 2E, vasculogenesis was arrested at the primitive plexus stage in the mutant yolk sac as opposed to the hierarchical vascular feature observed in the control yolk sac. In addition, staining was weaker in the mutant vasculature, suggesting lower expression of hemoglobin and/or fewer erythrocytes. Together, these results support that Brpf1 is required for vasculogenesis in the yolk sac and embryo proper.
FIGURE 2.
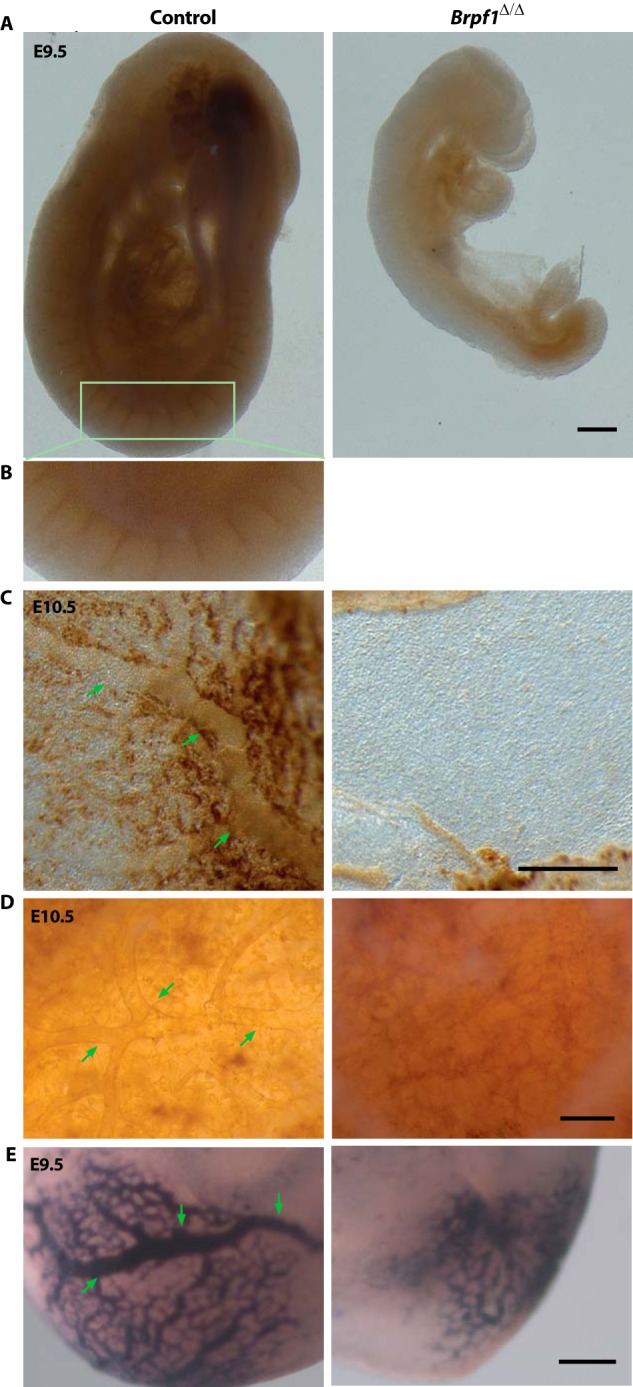
Analysis of Brpf1-null conceptuses by immunohistochemistry and benzidine staining. A, vascular networks in control and Brpf1Δ/Δ embryos at E9.5 were visualized by whole-mount anti-CD31 immunohistochemistry. The vascular network was hierarchically organized in the control but not mutant embryos. B, enlargement of the region boxed in A). C and D, similarly at E10.5, branching vessels were detected in the control but not mutant yolk sac (C) and chorionic plate (D). As in A, vasculature networks were visualized by whole-mount anti-CD31 immunostaining. E, benzidine staining of erythrocytes in the E9.5 control and mutant yolk sacs. Scale bars, 200 μm.
Brpf1 Regulates Vasculature Formation in the Placenta
In placental mammals, the placenta is an extraembryonic tissue essential for gas and nutrient exchange between the embryonic and maternal circulation systems (51–53). In mice, placental development begins after fusion of the chorionic plate with allantois around E8.5. Buds of allantoic cells including blood vessel precursors invade the chorionic plate to initiate branching morphogenesis. With expansion of the trophoblast compartment, the labyrinthine space is established for gas and nutrient exchange between the embryonic and maternal circulation systems. The mutant placenta was smaller at E10.5. H&E staining of sagittal sections at E9.5 and E10.5 revealed that there was no branching of fetal blood vessels in the mutant labyrinthine layer, in contrast to the extensive intermingling of maternal and embryonic blood spaces in the control placenta (Fig. 3, A and B). The mutant labyrinth appeared devoid of fetal blood vessels, and the labyrinthine layer was much thinner. Maternal erythrocytes were still present in the blood sinus (Fig. 3C, right, black arrows). Closer examination of the mutant placenta revealed formation of initial buds (Fig. 3C, right, green arrows), but further invasion into the labyrinth failed to occur. By contrast, no such defects were found in the mutant placenta lacking Moz (data not shown), an acetyltransferase known to interact with Brpf1 in vitro (14, 16), suggesting that Brpf1 may not act through Moz during labyrinth formation.
FIGURE 3.
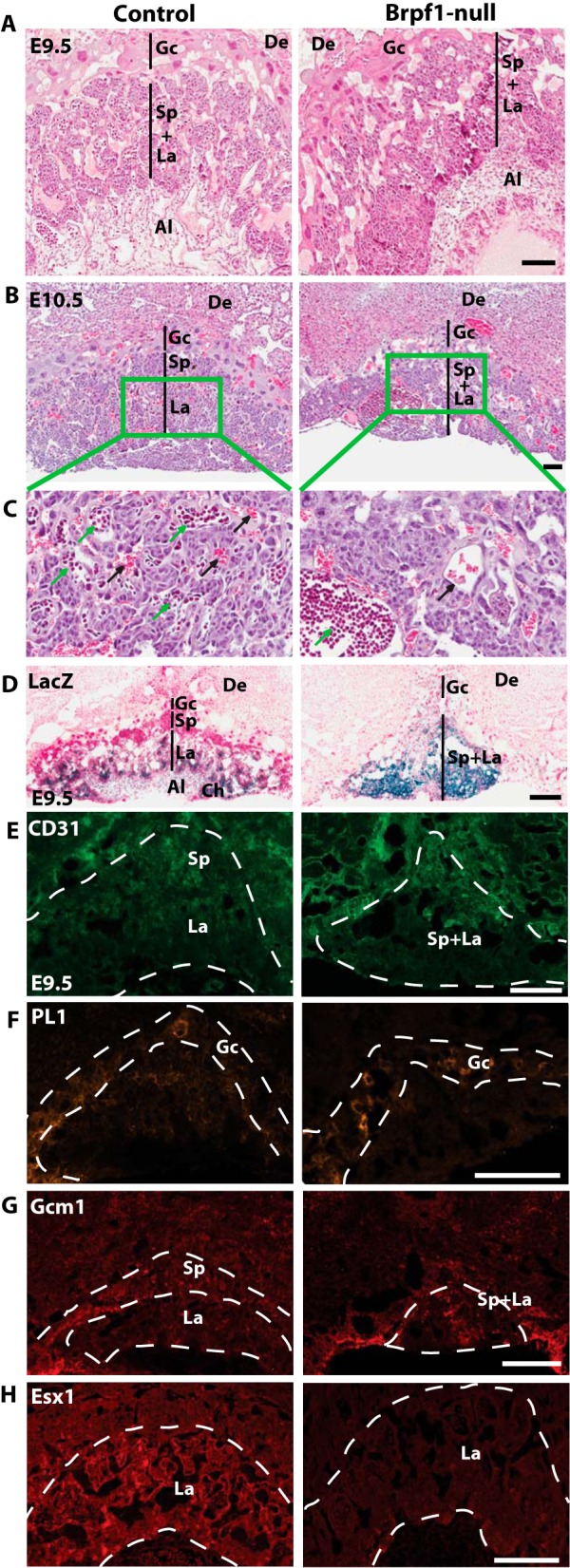
Vascular defects in Brpf1-null placenta. A and B, H&E-stained sagittal sections of control and mutant placentae at E9.5 and E10.5. At E9.5 (A), different layers were not so clearly separated in the mutant placenta as those in the control. At E10.5 (B), the mutant placenta collapsed, containing sparse giant cells and a thin layer of spongiotrophoblast cells. C, magnified views of the areas boxed in B. In the control placenta (left), fetal blood vessels (green arrows) and maternal blood sinuses (black arrows) intermingled with each other, whereas in the mutant placenta (right), no blood vessels branched out even though large buds of the fetal blood formed near the chorionic plate (green arrow) and the maternal blood sinuses looked normal (black arrow). D, β-galactosidase staining of E9.5 placental sections. In the Brpf1l/+ placenta, β-galactosidase activity (blue) was high in the labyrinth and chorionic plate but moderate in the allantois. In the Brpf1Δ/Δ placenta, the labyrinth and spongiotrophoblast layers shrank and were disorganized. E, CD31 immunostaining of E9.5 placental sections. The mutant labyrinth was structurally abnormal and lacked fetal blood vessels. F, PL1 immunostaining of E9.5 placental sections. The giant trophoblast layer (PL1+) appeared roughly normal in both control and mutant placenta. G and H, immunostaining of E9.5 placental sections for the labyrinth markers, Gcm1, and Esx1. In the mutant labyrinth, Gcm1 expression was abnormally condensed (G, right), and Esx1 expression was almost abolished (H, right). Extraembryonic structures were identified according to a published atlas (51, 89). Al, allantois; Ch, chorionic plate; De, decidua; Gc, giant trophoblast cells; La, labyrinth; Sp, spongiotrophoblast layer. Scale bars, 100 μm (A–D) and 200 μm (E–H).
Periodic acid Schiff staining is often used to detect glycogen-rich trophoblasts (52, 53). Such staining of wild-type and mutant placental sections also confirmed the defects in the labyrinth (data not shown). Consistent with the defective labyrinthine structure in the homozygous mutant (Fig. 3A), Brpf1 was highly expressed in the labyrinth of the placenta from the heterozygous conceptuses (Fig. 3D, left). The β-galactosidase-positive trophoblasts were well distributed within the labyrinth. By contrast, in the placenta from homozygous mutant conceptuses, β-galactosidase-positive trophoblasts shrank severely in the labyrinth (Fig. 3D, right). To gain mechanistic insights into the observed placental defects, we performed immunofluorescence microscopy. Consistent with the vascular defects (Figs. 1 and 2), staining with the anti-CD31 antibody revealed defective vasculature within the mutant labyrinth (Fig. 3E). Placental lactogen 1 (PL1) marks giant trophoblasts (53). As shown in Fig. 3F, the giant trophoblast layer (PL1+) remained roughly normal in the mutant placenta, indicating that the placental defect is specific to the labyrinth. The transcription factors Gcm1 and Esx1 are important for placental development (54, 55). We thus analyzed their expression and distribution. As reported (54), Gcm1 expression was enriched in the spongiotrophoblast layer of the control placenta (Fig. 3G, left). This distribution pattern was altered in the mutant placenta (Fig. 3G, right). Esx1 was relatively uniform in the control placenta, but its level deceased dramatically in the mutant placenta (Fig. 3H). Therefore, Brpf1 inactivation disrupts labyrinthine organization in the placenta and deregulates expression of Gcm1 and Esx1.
Brpf1 Loss Generates Abnormal Embryos with Severe Neural Tube Defects
We next examined abnormalities in the mutant embryos. From gross appearance, the mutant embryos began to show subtle growth retardation at the head fold as early as E8.75 (Fig. 4, A and B). At E9.5, the defect in the cephalic region was dramatic, and the neural tube failed to close, as opposed to that of the control embryos (Fig. 4, C and D). The neurodevelopmental defect was the most obvious phenotype, including forebrain hypoplasia and an open neural tube in the cephalic region but not in the trunk region (Fig. 4, C and D). At E10.5, the mutant embryos exhibited a significantly reduced size and were characterized by balloon-like pericardial sacs and severe hemorrhage (Fig. 4, E and F). To further characterize the neural tube defects, we carried out histological analysis of control and mutant embryos. H&E staining indicated that despite some slight differences, the neural tube was relatively normal at E8.75 (Fig. 4H). However, at E9.5, the cranial neural folds did not converge at the dorsal midline, indicating an open neural tube (Fig. 4I). At E10.5, the neural tube was still open, and the brain was largely missing (Fig. 4J).
FIGURE 4.

Neural tube defects in Brpf1-null embryos. A and B, mutant embryos had underdeveloped head folds (HF) but were otherwise relatively normal at E8.75. C and D, at E9.5, the mutant embryos began to show obvious abnormalities. One feature with high penetrance was the open neural tube, especially in the cephalic region. E and F, in addition to the neural tube defect, the mutant embryo at E10.5 showed other phenotypes, including pericardial effusion and hemorrhage. No viable mutant embryos could be recovered at E11.5 (G). H–J, H&E staining of control and Brpf1 mutant embryos from E8.75 to E10.5. At E8.75 (H), the neural fold in the cephalic region was still open in both control and mutant frontal sections. At E9.5 (I), the open neural tube defect became obvious as seen in transverse sections. Although they were closed in the control, the forebrain and hindbrain of the mutant embryo were still open. At E10.5 (J), no compartmentation of the forebrain, midbrain, and hindbrain was observed in the mutant embryo (bottom). The mutant brain only had some residual tissues. Embryonic structures were identified according to a published atlas (51, 89). Fb, forebrain; Mb, midbrain; Hb, hindbrain; HF, head fold; NE, neuroepithelium. Scale bars, 200 μm.
Neural tube defects could be due to several reasons, including birth and migration of neurons. To examine this issue, we first detected neuronal progenitors with an antibody specific to Sox2 (56). As shown in Fig. 5A, there were two major differences between control and mutant neural tubes: 1) the population of Sox2+ stem cells or progenitors was obviously smaller due to the reduced size of the neural tube, and 2) the relative distribution appeared to be uneven in the mutant neural tube as compared with the uniform distribution of wild-type Sox2+ cells. These differences suggest defective generation and/or migration of these cells. To gain further insights we analyzed embryo sections with an antibody against Tuj1, the neuron-specific class III β-tubulin that marks post-mitotic neurons (57). This analysis revealed a striking defect in neuron number and migration pattern. As shown in Fig. 5, B and E, Tuj1+ neurons were concentrated at the outer rim of the wild-type neural tube after migrating from the inner surface of the neural tube where they were born. In the mutant sections there were fewer Tuj1+ neurons, and some of them migrated abnormally, indicating that birth and migration of neurons are compromised in the mutant embryos.
FIGURE 5.

Defective cell proliferation and migration in the mutant neural tube. A, E9.5 embryo sections were immunostained to detect Sox2+ neural precursors. Different from the wild type, the mutant neural tube exhibited uneven distribution of Sox2+ cells. The green arrow marks the loss of Sox2 expression in the ventral part of the neural tube. B and E, anti-Tuj1 immunostaining was performed on transverse sections of control and mutant embryos at E9.5. Boxed regions in B and D are shown in magnified views in C and E, respectively. In the mutant neural tube there were fewer Tuj1+ post mitotic neurons in the cephalic (B and C) and trunk (D and E) regions. In addition, these neurons failed to arrive in the outer epithelium of the neural tube. F, the control and mutant embryos at E9.5 were immunostained to detect Ser-10 phosphorylation of histone H3 (H3S10P). Compared with the control, the mutant embryo contained fewer H3S10P+ nuclei. G, higher magnification images of the hindbrain regions boxed in F. 1B, first branchial arch; DA, dorsal aorta; Fb, forebrain; H, heart; Hb, hindbrain; Mb, midbrain; NC, neural crest; NT, neural tube in the trunk; T, tail; YS, yolk sac. Scale bars, 200 μm in A and F; 100 μm for B and D.
To delineate the underlying cellular mechanisms, we analyzed cell proliferation by immunofluorescence microscopic analysis with an antibody recognizing histone H3 phosphorylated at Ser-10, a well known marker for mitotic cells (58). As shown in Fig. 5, F and G, in the mutant embryo there were fewer cells positive for the mark than those in the control embryo, indicating that Brpf1 inactivation affects cell proliferation. We also assessed whether Brpf1 inactivation induces apoptosis. For this, we performed immunofluorescence microscopic analysis of embryo sections with an antibody specific to the activated form of caspase 3. As shown in Fig. 6, A–C, no evident apoptosis was observed in the mutant embryo at E8.5 or E9.5, but marked apoptosis was present at E10.5. As morphologic defects were obvious at E9.5 (Fig. 4), we concluded that apoptosis is not the reason for the neural tube defect observed at E9.5. In addition, the control and mutant embryos showed no evident difference in the number of cells positive for phosphorylation of H2AX at Ser-139 (or the γH2AX form) (Fig. 6D), a well -known histone mark for DNA damage and genomic instability (59). These results indicate that neural tube defects caused by Brpf1 deficiency may be due to compromised cell proliferation rather than apoptosis or DNA damage.
FIGURE 6.

Fluorescence microscopy of control and mutant embryos. A–C, analysis of apoptosis in control and mutant embryos by immunostaining with an antibody against cleaved caspase 3. In the E8.5 frontal sections (A) and the E9.5 sagittal sections (B), apoptosis was almost undetectable in control and Brpf1 null embryos. However, in E10.5 transverse sections (C), evident apoptosis (marked with green arrows) was detected in the forebrain, hindbrain, and tail region of the mutant embryo. D, immunostaining of E9.5 embryonic sections to detect Ser-139 phosphorylation of histone H2AX (γH2AX). γH2AX foci are indicated with green arrows. E, immunostaining to detect Hbo1 expression in E9.5 embryonic sections. 1B, first branchial arch; Fb, forebrain; H, heart; Hb, hindbrain; HF, head fold; L, limb bud; Mb, midbrain; NE, neuroepithelium; NT, neural tube in the trunk region; OV, optic vesicle; S, somite; T, tail; Y, yolk sac. Scale bars, 200 μm in A–C and 100 μm for D.
Brpf1 Controls Primitive Hematopoiesis and Embryonic Fibroblast Proliferation
H&E staining of control and mutant yolk sac sections revealed striking defects in erythrocytes at E9.5 and E10.5 (Fig. 1F). To determine whether hematopoietic progenitors within the mutant yolk sac vasculature are still functional, we performed colony formation assays in vitro using methylcellulose-based media. As shown in Fig. 7A, mutant hematopoietic progenitors formed fewer colonies than the control. The mutant colonies were also much smaller (data not shown), suggesting defective cell growth and/or proliferation. Thus, Brpf1 loss causes defective hematopoiesis.
FIGURE 7.
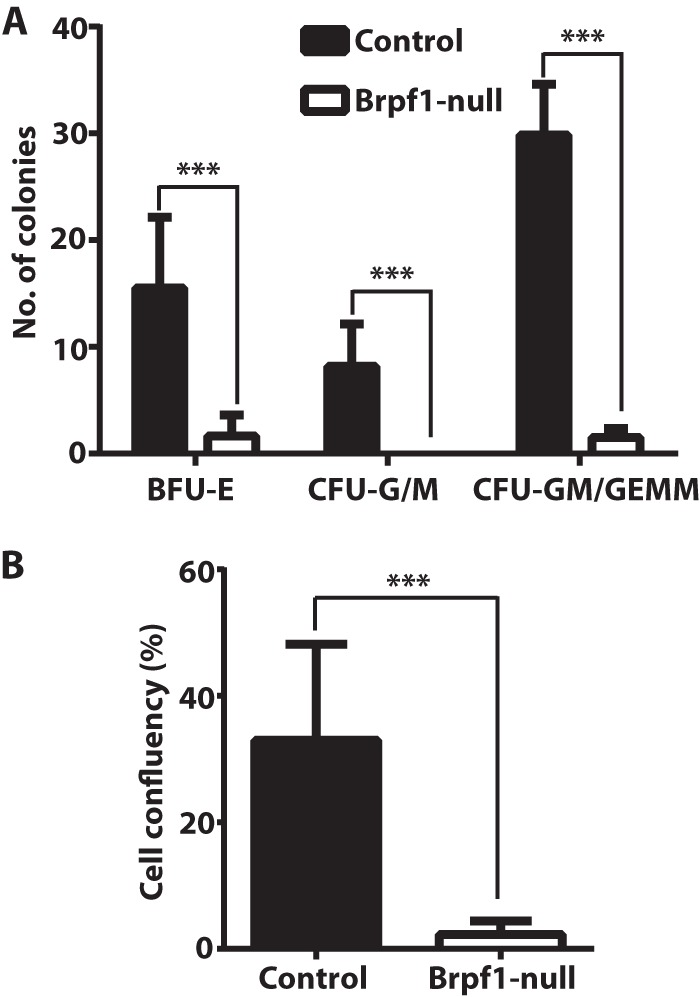
Importance of Brpf1 for proliferation of hematopoietic progenitors and embryonic fibroblasts. A, colony formation assay performed on four pairs of control and mutant yolk sacs at E9.5. Burst forming unit-erythroid (BFU-E), colony forming unit-granulocyte/monocyte (CFU-G/M), and granulocyte, monocyte/granulocyte, erythrocyte, monocyte, and megakaryocyte (CFU-GM/GEMM) were enumerated on day 8 after culturing in Methocult M3434 media. B, MEFs were isolated from control and mutant embryos at E9.5 and cultured for 1 week to monitor confluency by IncuCyte. The values on day 7 were calculated and are shown here (n = 9 for control and n = 4 for mutant). ***, p < 0.001.
Like hematopoietic progenitors, fibroblasts isolated from mutant embryos at E9.5 did not proliferate in vitro (Fig. 7B). To substantiate this, we sought to circumvent the failure to derive MEFs from the mutant embryos. We thus prepared MEFs from Brpf1f/f;ER-Cre embryos for inducible inactivation of the Brpf1 gene in vitro by treatment with 4-hydroxytamoxifen. Importantly, the induced inactivation inhibited growth (Fig. 8A) and cell cycle progression (Fig. 8, B and C) of embryonic fibroblasts. To examine the underlying molecular mechanisms, we performed immunoblotting with various antibodies. As shown in Fig. 8D, induced inactivation of the Brpf1 gene also decreased acetylation of histone H3 at Lys-9 and Lys-14. Because loss of Hbo1, but not Moz, yields a similar decrease in histone H3 acetylation (17, 44), we assessed Hbo1 expression. According to fluorescence signal intensity, the Hbo1 protein level decreased in the mutant embryos (Fig. 6E). However, different from this, induced inactivation of the Brpf1 gene in vitro did not alter the levels of different Hbo1 isoforms in MEFs (Fig. 8D). Brpf1 stimulates acetyltransferase activity of Hbo1 (10), so it remains possible that its enzymatic activity is lower in mutant MEFs.
FIGURE 8.

Induced Brpf1 inactivation causes defective cell growth, cell cycle control and histone H3 acetylation. Brpf1f/f;ER-Cre MEFs derived from E15.5 embryos were treated with vehicle and 4-hydroxytamoxifen to prepare Brpf1f/f and Brpf1−/− MEFs, respectively. A, MEF growth was monitored by IncuCyte for 7 days. The difference between Brpf1f/f and Brpf1−/− MEFs became significant at day 4. B and C, Brpf1f/f and Brpf1−/− MEFs were stained with propidium iodide (PI) and analyzed by flow cytometry. Representative graphs were shown (B), and the ratio of different cell cycle phases was quantified (C). Experiments in A–C were repeated with three batches of MEFs isolated from three independent litters. *, p < 0.05; **, p < 0.01. D, immunoblotting analysis of Brpf1f/f and Brpf1−/− MEF extracts with antibodies specific to histone H3, its acetylated forms (H3K14ac, H3K9ac, and H3ac), and Hbo1 as indicated. Histone H4 acetylation at Lys-16 and the γH2AX level remained unaltered in mutant MEF extracts (data not shown).
Brpf1 Regulates Gene Expression
The acetyltransferases Moz, Morf, and Hbo1 interact efficiently with Brpf1 in cell-based assays (10, 14, 16). Brpf1 is paralogous to Brpf2 and Brpf3 (14, 16). We thus analyzed whether Brpf1 affects transcript levels of these five proteins. As shown in Fig. 9, A and B, the levels were not affected in mutant embryos or MEFs. Because cell growth and proliferation were impaired in Brpf1-deficient mutant embryos and MEFs (Figs. 7 and 8), we measured levels of different cell cycle inhibitors. The transcript levels of p16 and p27 were affected in mutant MEFs (Fig. 9B). We also performed microarray-based gene expression analysis of control and mutant embryos at E8.75, when morphological defects were still subtle (Fig. 4A). Among the genes selected for validation by RT-qPCR (Table 1), we confirmed that Rpl10l (ribosomal protein L10-like) mRNA decreased, whereas Scp3l (synaptonemal complex protein 3-like; or Gm773, for predicted gene 773) mRNA increased in Brpf1-null embryos (Fig. 9A). A smaller increase of Scp3l mRNA was also detected in mutant MEFs (Fig. 9B). Thus, microarray analysis and RT-qPCR validation indicate that Brpf1 loss perturbs expression of genes such as Rpl10l, Scp3l, p16, and p27.
FIGURE 9.
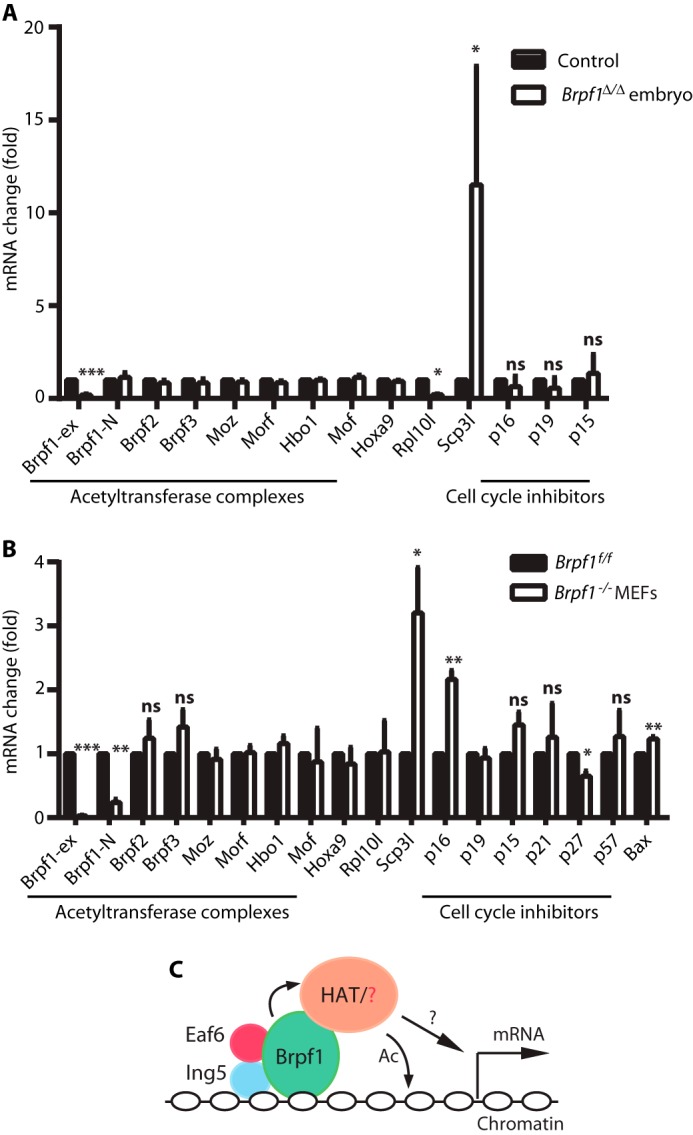
Brpf1 regulates gene expression. A, RT-qPCR analysis in control and Brpf1-deficient embryos at E8.75. Three pairs of embryos were used. B, RT-qPCR analysis in Brpf1f/f and Brpf1−/− MEFs. The experiment was performed in triplicate with three batches of MEFs isolated from three independent litters as in Fig. 8, A–C. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not statistically significant. C, a simplified model illustrates how the Brpf1-Ing5-Eaf6 trimeric complex may recruit histone acetyltransferases (HAT) such as Moz/Morf and Hbo1 to regulate gene expression. In addition to multiple histone binding domains that Brpf1 possesses, Ing5 has its own PHD finger to recognize methylated histone H3. Brpf1 may also act through an unknown partner(s) as indicated by the red question mark. The black question mark denotes that the recruitment may lead to either transcriptional activation or repression. Ac, acetylation.
DISCUSSION
Brpf1 Regulates Mouse Embryogenesis
To extend previous molecular studies (10, 14, 16) and determine the biological functions of Brpf1, we have recently analyzed its spatiotemporal expression during mouse development (38). This analysis revealed that Brpf1 is dynamically expressed in different extra-embryonic and embryonic tissues (38). Consistent with this, inactivation of the gene led to embryonic lethality around E9.5 (38). Here, we have systematically analyzed the mutant animals and identified severe vascular defects in the placenta, yolk sac, and embryo proper (Figs. 1–3). Neural tube closure (Figs. 4–6) and hematopoietic progenitors (Fig. 7A) were also compromised. Related to the underlying cellular mechanisms, Brpf1 inactivation deregulates different cell programs, e.g. inhibiting cell proliferation (Figs. 5, F and G, 7, and 8), hindering neuron migration (Fig. 5, B and E) and reducing histone H3 acetylation (Fig. 8D). In addition, erythrocytes and their progenitors were affected (Figs. 1F and 7A). This may be an important factor contributing to defective vasculature formation (Fig. 1) as primitive erythroid cells are critical for vascular remodeling (for review, see Ref. 60).
As for the underlying molecular mechanisms for the observed defects, the microarray-based gene expression analysis revealed that Brpf1 inactivation reduced the Rpl10l mRNA level (Fig. 9A and supplemental Table S1). This was specific to mutant embryos at E8.75 as no such reduction was observed in cultured MEFs isolated from E15.5 embryos (Fig. 9B). Related to this, specific inactivation of the Brpf1 gene in the forebrain does not affect the Rpl10l mRNA level (39). The reduced transcription of Rpl10l in mutant embryos at E8.75 is consistent with the role of Brpf1 in promoting histone acetylation and stimulating transcriptional coactivation in vitro (16). Rpl10l is highly homologous to Rpl10 (61), so we can speculate about Rpl10l functions from various studies of Rpl10, known to be important for nuclear export and allosteric movement of the 60 S ribosomal subunit (62, 63). Human RPL10 mutations have been detected in leukemia (64, 65) and implicated in abnormal brain development leading to autism (66), intellectual disability (67, 68), and microcephaly (69). Moreover, faulty translation resulting from loss of other ribosomal proteins constitutes an important pathogenic mechanism (69, 70) and causes diverse developmental defects (71–73).
In addition, dramatically elevated transcription of Scp3l was also observed (Fig. 9A and supplemental Table S1). This elevation was also detected in mutant MEFs (Fig. 9B). With a COR (meiotic chromosome core) domain sharing 49% sequence identity to that of Scp3 (74), Scp3l has been referred to as Xlr6 (X-linked lymphocyte-regulated protein 6) (75) and Slx2 (Scp3-like X-linked 2) (76, 77). Scp3 is a meiosis-specific protein critical for meiotic chromosome segregation and embryo survival (74). Some evidence suggests that Scp3l has a similar function in meiotic chromosome segregation (75, 77). In addition, Scp3 is part of the Xlr family whose members also regulate the immune system (78). It is presently unclear whether Scp3l is involved in immune regulation. In light of its potential importance in chromosome segregation, up-regulation of Scp3l mRNA may be detrimental to Brpf1-deficient embryos and MEFs.
At the sequence level, BRPF proteins are conserved from Caenorhabditis elegans to humans. There are no orthologs in lower metazoans, yeasts, or plants. Although the biological function of Drosophila Brpf remains elusive, C. elegans Lin-49 regulates neuron asymmetry, hindgut development, and fecundity (79, 80). Deletion of zebrafish Brpf1 alters pharyngeal segmental identity (81), and inactivation of medaka Brpf1 affects craniofacial skeletons (82), so Brpf1 regulates skeletal development in fish. Based on these genetic studies, it will be interesting to investigate whether mammalian Brpf1 also plays a role in skeletogenesis. However, these genetic studies would not predict our findings about the important roles of mouse Brpf1 in the placenta, vasculature, and neural tube. Thus, the results described herein are unexpected and provide novel insights into biological functions of mammalian BRPF1.
In mammals, BRPF1 is paralogous to BRPF2 and BRPF3 (14, 16). Loss of mouse Brpf2 leads to embryonic lethality at E15.5, with abnormal eye development and faulty erythropoiesis (17), indicating that Brpf2 regulates mouse embryogenesis at a later stage than Brpf1. No genetic studies have been reported for mouse Brpf3, but the severe embryonic lethality of Brpf1 or Brpf2 inactivation supports that both have unique functions during mouse embryogenesis. It will be important to investigate whether Brpf3 also plays a role in mouse embryos.
Cooperation of Brpf1 with Moz, Morf, and Hbo1 during Embryo Development
Molecular studies have established that human BRPF1 interacts with MOZ, MORF, and HBO1 to govern their acetyltransferase activities and substrate specificity (10, 14, 16). Decreased Rpl10l expression is consistent with the transcriptional activation ability observed for Moz, Morf, and Brpf1 in vitro (Fig. 9C) (16, 83). Related to the elevated level of Scp3l mRNA in Brpf1−/− embryos and MEFs, we have recently reported that forebrain-specific loss of Brpf1 causes transcriptional up-regulation of Hox genes and various other transcription factors (39). Interestingly, Drosophila Hbo1 is required for repression of Hox genes (84), suggesting interaction of Brpf1 with Hbo1 in silencing gene expression under certain developmental contexts (Fig. 9C).
To gain insights into how Moz, Morf, and Hbo1 may mediate the effects of Brpf1 on mouse embryo development, we need to compare results from related genetic studies. Three mutant strains have been engineered for mouse Moz. In one of them the first coding exon was replaced with a neo cassette, and no protein was expressed (41). Homozygous mutant embryos survive until E14.5. In another, the neo coding sequence is fused to that for the N-terminal two-thirds of Moz (42). Although no Moz-neo fusion protein is detectable, lethality occurs at birth (42). A third strain contains a point mutation to abrogate acetyltransferase activity (85). This mutation causes shortened lifespan; 40% of the mutant mice die at 6 months of age, with lower body weight, smaller thymus and spleen, and defective hematopoiesis (85). In addition to Moz, Morf was investigated in a gene-trap strain possessing ∼10% residual mRNA. The mutant mice die at weaning and display dwarfism, craniofacial abnormalities, and cerebral defects (43). Thus, the phenotypes of the Moz and Morf mutant mice do not predict the severe phenotypes observed in Brpf1 knockouts.
There are several possible explanations. Moz and Morf are paralogous (83) and may have overlapping functions in vivo, so their double knockouts should lead to more severe phenotypes than single knockouts. Moreover, Morf-deficient mice still carry residual transcripts (43), and the total knock-out may yield more dramatic phenotypes. As Brpf1 interacts with both Moz and Morf, as suggested by cell-based studies (10, 14, 16), its loss may affect the function of both to yield the severe phenotypes observed. Alternatively, there are other binding partners that may contribute to these defects.
One such candidate is Hbo1, whose loss leads to embryonic lethality at E10.5, with developmental delay starting at ∼E8.5 (44). This lethality window is similar to that of Brpf1−/− embryos (38). In addition, there are other similarities such as neural tube and vascular defects between Brpf1 and Hbo1 mutants (Figs. 1 and 4) (44). Moreover, histone H3K14 acetylation is dramatically decreased in both Brpf1 and Hbo1 mutants (Fig. 8D) (44). Along with the molecular interaction between Brpf1 and Hbo1 (10), it is tempting to speculate that Brpf1 targets Hbo1 during mouse embryogenesis (Fig. 9C). However, Brpf1 and Hbo1 mutants also display clear distinctions in placental and embryonic defects, indicating that Hbo1 may not be the sole target in vivo. In support of this, Hbo1 expression is ubiquitous (Fig. 6E) (44), whereas spatiotemporal expression of Brpf1 is specific to different tissues and dynamic at different developmental stages (38). Furthermore, Hbo1 is dispensable for MEF growth and proliferation (44), but Brpf1 mutant MEFs were difficult to derive at E9.5 (Fig. 7B), and induced Brpf1 inactivation in vitro led to proliferative defects (Fig. 8). Of relevance, the human BRPF1 gene is mutated in pediatric cancers (36) and adult medulloblastoma (37); no such mutations have been identified in the HBO1 gene.
From the above discussions, it is clear that genetic studies of mouse Moz, Morf, and Hbo1 (41–44) indicate that loss of none of them alone faithfully phenocopies the Brpf1 gene inactivation. One scenario is that all three contribute to Brpf1 function during embryo development (Fig. 9C). Alternatively, Brpf1 may also act independently of them (Fig. 9C). Related to this, three other acetyltransferases, Gcn5, p300, and CBP, are worthy of consideration because their loss also causes neural tube defects (86, 87). It should be noted, however, that neural tube defects described for Gcn5−/−, p300−/−, or Cbp−/− embryos (86, 87) are different from those described here (Fig. 4). Interestingly, as described here for Brpf1 (Figs. 1, 7, and 8), p300 loss causes severe defects in vasculature and cell proliferation (87). Thus, the relationship with different acetyltransferases in vivo may not be as simple as demonstrated in vitro (10, 14, 16). Further studies are needed to investigate how Brpf1 interacts with various histone acetyltransferases during embryo development (Fig. 9C).
In conclusion, we have analyzed mouse embryos lacking Brpf1 and identified its crucial role in vasculature formation and neural tube development. The results also demonstrate that Brpf1 loss deregulates cellular and gene expression programs. Together with recent reports on its function in forebrain development (39, 40), these new results reiterate the importance of mouse Brpf1 in different developmental processes. Of relevance, Brpf2 (or Brd1, for bromodomain protein 1) regulates embryogenesis around E13.5 and early thymocyte development (17, 88). Therefore, despite their high sequence similarity (14, 16), Brpf1 and Brpf2 are not redundant in vivo, and both have important roles during mouse development.
Supplementary Material
Acknowledgment
We thank Maxime Bouchard for comments on the manuscript.
The work was supported by operating grants from Canadian Institutes of Health Research (CIHR) and Natural Sciences and Engineering Research Council of Canada (NSERC) (to X.-J. Y.).

This article contains supplemental Table 1.
Original microarray data and normalized datasets were deposited in the GEO database under the accession number GSE63908.
- PHD
- plant homeodomain-linked
- MOZ
- monocytic leukemia zinc finger
- MORF
- MOZ-related factor
- BRPF1
- bromodomain- and PHD finger-containing protein 1
- MEF
- mouse embryonic fibroblast
- qPCR
- quantitative PCR
- PL1
- placental lactogen 1
- Gcm1
- glial cell missing 1
- Esx1
- extraembryonic-spermatogenesis-homebox protein 1.
REFERENCES
- 1. Strahl B. D., Allis C. D. (2000) The language of covalent histone modifications. Nature 403, 41–45 [DOI] [PubMed] [Google Scholar]
- 2. Berger S. L. (2007) The complex language of chromatin regulation during transcription. Nature 447, 407–412 [DOI] [PubMed] [Google Scholar]
- 3. Bonasio R., Tu S., Reinberg D. (2010) Molecular signals of epigenetic states. Science 330, 612–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Suganuma T., Workman J. L. (2011) Signals and combinatorial functions of histone modifications. Annu. Rev. Biochem. 80, 473–499 [DOI] [PubMed] [Google Scholar]
- 5. Taverna S. D., Li H., Ruthenburg A. J., Allis C. D., Patel D. J. (2007) How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 14, 1025–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Latham J. A., Dent S. Y. (2007) Cross-regulation of histone modifications. Nat. Struct. Mol. Biol. 14, 1017–1024 [DOI] [PubMed] [Google Scholar]
- 7. Musselman C. A., Lalonde M. E., Côté J., Kutateladze T. G. (2012) Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 19, 1218–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang X. J., Ullah M. (2007) MOZ and MORF, two large MYSTic HATs in normal and cancer stem cells. Oncogene 26, 5408–5419 [DOI] [PubMed] [Google Scholar]
- 9. Qin S., Jin L., Zhang J., Liu L., Ji P., Wu M., Wu J., Shi Y. (2011) Recognition of unmodified histone H3 by the first PHD finger of bromodomain-PHD finger protein 2 provides insights into the regulation of histone acetyltransferases monocytic leukemic zinc-finger protein (MOZ) and MOZ-related factor (MORF). J. Biol. Chem. 286, 36944–36955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lalonde M. E., Avvakumov N., Glass K. C., Joncas F. H., Saksouk N., Holliday M., Paquet E., Yan K., Tong Q., Klein B. J., Tan S., Yang X. J., Kutateladze T. G., Côté J. (2013) Exchange of associated factors directs a switch in HBO1 acetyltransferase histone tail specificity. Genes Dev. 27, 2009–2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lubula M. Y., Eckenroth B. E., Carlson S., Poplawski A., Chruszcz M., Glass K. C. (2014) Structural insights into recognition of acetylated histone ligands by the BRPF1 bromodomain. FEBS Lett. 588, 3844–3854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vezzoli A., Bonadies N., Allen M. D., Freund S. M., Santiveri C. M., Kvinlaug B. T., Huntly B. J., Göttgens B., Bycroft M. (2010) Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1. Nat. Struct. Mol. Biol. 17, 617–619 [DOI] [PubMed] [Google Scholar]
- 13. Wu H., Zeng H., Lam R., Tempel W., Amaya M. F., Xu C., Dombrovski L., Qiu W., Wang Y., Min J. (2011) Structural and histone binding ability characterizations of human PWWP domains. PLoS ONE 6, e18919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Doyon Y., Cayrou C., Ullah M., Landry A. J., Côté V., Selleck W., Lane W. S., Tan S., Yang X. J., Côté J. (2006) ING tumor suppressors are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol. Cell 21, 51–64 [DOI] [PubMed] [Google Scholar]
- 15. Perry J. (2006) The Epc-N domain: a predicted protein-protein interaction domain found in select chromatin associated proteins. BMC Genomics 7, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ullah M., Pelletier N., Xiao L., Zhao S. P., Wang K., Degerny C., Tahmasebi S., Cayrou C., Doyon Y., Goh S. L., Champagne N., Côté J., Yang X. J. (2008) Molecular architecture of quartet MOZ/MORF histone acetyltransferase complexes. Mol. Cell Biol. 28, 6828–6843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mishima Y., Miyagi S., Saraya A., Negishi M., Endoh M., Endo T. A., Toyoda T., Shinga J., Katsumoto T., Chiba T., Yamaguchi N., Kitabayashi I., Koseki H., Iwama A. (2011) The Hbo1-Brd1/Brpf2 complex is responsible for global acetylation of H3K14 and required for fetal liver erythropoiesis. Blood 118, 2443–2453 [DOI] [PubMed] [Google Scholar]
- 18. Lalonde M. E., Cheng X., Côté J. (2014) Histone target selection within chromatin: an exemplary case of teamwork. Genes Dev. 28, 1029–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Borrow J., Stanton V. P., Jr., Andresen J. M., Becher R., Behm F. G., Chaganti R. S., Civin C. I., Disteche C., Dubé I., Frischauf A. M., Horsman D., Mitelman F., Volinia S., Watmore A. E., Housman D. E. (1996) The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nat. Genet. 14, 33–41 [DOI] [PubMed] [Google Scholar]
- 20. Chinen Y., Taki T., Tsutsumi Y., Kobayashi S., Matsumoto Y., Sakamoto N., Kuroda J., Horiike S., Nishida K., Ohno H., Uike N., Taniwaki M. (2014) The leucine twenty homeobox (LEUTX) gene, which lacks a histone acetyltransferase domain, is fused to KAT6A in therapy-related acute myeloid leukemia with t(8;19)(p11;q13). Genes Chromosomes Cancer 53, 299–308 [DOI] [PubMed] [Google Scholar]
- 21. Dulak A. M., Stojanov P., Peng S., Lawrence M. S., Fox C., Stewart C., Bandla S., Imamura Y., Schumacher S. E., Shefler E., McKenna A., Carter S. L., Cibulskis K., Sivachenko A., Saksena G., Voet D., Ramos A. H., Auclair D., Thompson K., Sougnez C., Onofrio R. C., Guiducci C., Beroukhim R., Zhou Z., Lin L., Lin J., Reddy R., Chang A., Landrenau R., Pennathur A., Ogino S., Luketich J. D., Golub T. R., Gabriel S. B., Lander E. S., Beer D. G., Godfrey T. E., Getz G., Bass A. J. (2013) Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat. Genet. 45, 478–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moore S. D., Herrick S. R., Ince T. A., Kleinman M. S., Dal Cin P., Morton C. C., Quade B. J. (2004) Uterine leiomyomata with t(10;17) disrupt the histone acetyltransferase MORF. Cancer Res. 64, 5570–5577 [DOI] [PubMed] [Google Scholar]
- 23. Panagopoulos I., Gorunova L., Bjerkehagen B., Heim S. (2015) Novel KAT6B-KANSL1 fusion gene identified by RNA sequencing in retroperitoneal leiomyoma with t(10;17)(q22;q21). PLoS ONE 10, e0117010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lynch H., Wen H., Kim Y. C., Snyder C., Kinarsky Y., Chen P. X., Xiao F., Goldgar D., Cowan K. H., Wang S. M. (2013) Can Unknown predisposition in familial breast cancer be family-specific? Breast. J. 19, 520–528 [DOI] [PubMed] [Google Scholar]
- 25. Grasso C. S., Wu Y. M., Robinson D. R., Cao X., Dhanasekaran S. M., Khan A. P., Quist M. J., Jing X., Lonigro R. J., Brenner J. C., Asangani I. A., Ateeq B., Chun S. Y., Siddiqui J., Sam L., Anstett M., Mehra R., Prensner J. R., Palanisamy N., Ryslik G. A., Vandin F., Raphael B. J., Kunju L. P., Rhodes D. R., Pienta K. J., Chinnaiyan A. M., Tomlins S. A. (2012) The mutational landscape of lethal castration-resistant prostate cancer. Nature 487, 239–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zack T. I., Schumacher S. E., Carter S. L., Cherniack A. D., Saksena G., Tabak B., Lawrence M. S., Zhang C. Z., Wala J., Mermel C. H., Sougnez C., Gabriel S. B., Hernandez B., Shen H., Laird P. W., Getz G., Meyerson M., Beroukhim R. (2013) Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 45, 1134–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sheikh B. N., Lee S. C., El-Saafin F., Vanyai H. K., Hu Y., Pang S. H., Grabow S., Strasser A., Nutt S. L., Alexander W. S., Smyth G. K., Voss A. K., Thomas T. (2015) MOZ regulates B cell progenitors and, consequently, Moz haploinsufficiency dramatically retards MYC-induced lymphoma development. Blood 125, 1910–1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kraft M., Cirstea I. C., Voss A. K., Thomas T., Goehring I., Sheikh B. N., Gordon L., Scott H., Smyth G. K., Ahmadian M. R., Trautmann U., Zenker M., Tartaglia M., Ekici A., Reis A., Dörr H. G., Rauch A., Thiel C. T. (2011) Disruption of the histone acetyltransferase MYST4 leads to a Noonan syndrome-like phenotype and hyperactivated MAPK signaling in humans and mice. J. Clin. Invest. 121, 3479–3491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Clayton-Smith J., O'Sullivan J., Daly S., Bhaskar S., Day R., Anderson B., Voss A. K., Thomas T., Biesecker L. G., Smith P., Fryer A., Chandler K. E., Kerr B., Tassabehji M., Lynch S. A., Krajewska-Walasek M., McKee S., Smith J., Sweeney E., Mansour S., Mohammed S., Donnai D., Black G. (2011) Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the say-barber-biesecker variant of Ohdo syndrome. Am. J. Hum. Genet. 89, 675–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Campeau P. M., Kim J. C., Lu J. T., Schwartzentruber J. A., Abdul-Rahman O. A., Schlaubitz S., Murdock D. M., Jiang M. M., Lammer E. J., Enns G. M., Rhead W. J., Rowland J., Robertson S. P., Cormier-Daire V., Bainbridge M. N., Yang X. J., Gingras M. C., Gibbs R. A., Rosenblatt D. S., Majewski J., Lee B. H. (2012) Mutations in KAT6B, encoding a histone acetyltransferase, cause genitopatellar syndrome. Am. J. Hum. Genet. 90, 282–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Simpson M. A., Deshpande C., Dafou D., Vissers L. E., Woollard W. J., Holder S. E., Gillessen-Kaesbach G., Derks R., White S. M., Cohen-Snuijf R., Kant S. G., Hoefsloot L. H., Reardon W., Brunner H. G., Bongers E. M., Trembath R. C. (2012) De novo mutations of the gene encoding the histone acetyltransferase KAT6B cause genitopatellar syndrome. Am. J. Hum. Genet. 90, 290–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Szakszon K., Salpietro C., Kakar N., Knegt A. C., Olah E., Dallapiccola B., Borck G. (2013) De novo mutations of the gene encoding the histone acetyltransferase KAT6B in two patients with Say-Barber/Biesecker/Young-Simpson syndrome. Am. J. Med. Genet. A 161A, 884–888 [DOI] [PubMed] [Google Scholar]
- 33. Yu H. C., Geiger E. A., Medne L., Zackai E. H., Shaikh T. H. (2014) An individual with blepharophimosis-ptosis-epicanthus inversus syndrome (BPES) and additional features expands the phenotype associated with mutations in KAT6B. Am. J. Med. Genet. A 164A, 950–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Arboleda V. A., Lee H., Dorrani N., Zadeh N., Willis M., Macmurdo C. F., Manning M. A., Kwan A., Hudgins L., Barthelemy F., Miceli M. C., Quintero-Rivera F., Kantarci S., Strom S. P., Deignan J. L., UCLA Clinical Genomics Center, Grody W. W., Vilain E., Nelson S. F. (2015) De novo nonsense mutations in KAT6A, a lysine acetyl-transferase gene, cause a syndrome including microcephaly and global developmental delay. Am. J. Hum. Genet. 96, 498–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tham E., Lindstrand A., Santani A., Malmgren H., Nesbitt A., Dubbs H. A., Zackai E. H., Parker M. J., Millan F., Rosenbaum K., Wilson G. N., Nordgren A. (2015) Dominant mutations in KAT6A cause intellectual disability with recognizable syndromic features. Am. J. Hum. Genet. 96, 507–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huether R., Dong L., Chen X., Wu G., Parker M., Wei L., Ma J., Edmonson M. N., Hedlund E. K., Rusch M. C., Shurtleff S. A., Mulder H. L., Boggs K., Vadordaria B., Cheng J., Yergeau D., Song G., Becksfort J., Lemmon G., Weber C., Cai Z., Dang J., Walsh M., Gedman A. L., Faber Z., Easton J., Gruber T., Kriwacki R. W., Partridge J. F., Ding L., Wilson R. K., Mardis E. R., Mullighan C. G., Gilbertson R. J., Baker S. J., Zambetti G., Ellison D. W., Zhang J., Downing J. R. (2014) The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat. Commun. 5, 3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kool M., Jones D. T., Jäger N., Northcott P. A., Pugh T. J., Hovestadt V., Piro R. M., Esparza L. A., Markant S. L., Remke M., Milde T., Bourdeaut F., Ryzhova M., Sturm D., Pfaff E., Stark S., Hutter S., Seker-Cin H., Johann P., Bender S., Schmidt C., Rausch T., Shih D., Reimand J., Sieber L., Wittmann A., Linke L., Witt H., Weber U. D., Zapatka M., König R., Beroukhim R., Bergthold G., van Sluis P., Volckmann R., Koster J., Versteeg R., Schmidt S., Wolf S., Lawerenz C., Bartholomae C. C., von Kalle C., Unterberg A., Herold-Mende C., Hofer S., Kulozik A. E., von Deimling A., Scheurlen W., Felsberg J., Reifenberger G., Hasselblatt M. (2014) Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 25, 393–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. You L., Chen L., Penney J., Miao D., Yang X. J. (2014) Expression atlas of the epigenetic regulator Brpf1 and its requirement for survival of mouse embryos. Epigenetics 9, 860–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. You L., Zou J., Zhao H., Bertos N. R., Park M., Wang E., Yang X. J. (2015) Deficiency of the chromatin regulator Brpf1 causes abnormal brain development. J. Biol. Chem. 290, 7114–7129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. You L., Yan K., Zhou J., Zhao H., Bertos N. R., Park M., Wang E., Yang X. J. (2015) The lysine acetyltransferase activator Brpf1 governs dentate gyrus development through neural stem cells and progenitors. PLoS Genet. 11, e1005034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Katsumoto T., Aikawa Y., Iwama A., Ueda S., Ichikawa H., Ochiya T., Kitabayashi I. (2006) MOZ is essential for maintenance of hematopoietic stem cells. Genes Dev. 20, 1321–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Thomas T., Corcoran L. M., Gugasyan R., Dixon M. P., Brodnicki T., Nutt S. L., Metcalf D., Voss A. K. (2006) Monocytic leukemia zinc finger protein is essential for the development of long-term reconstituting hematopoietic stem cells. Genes Dev. 20, 1175–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thomas T., Voss A. K., Chowdhury K., Gruss P. (2000) Querkopf, a MYST family histone acetyltransferase, is required for normal cerebral cortex development. Development 127, 2537–2548 [DOI] [PubMed] [Google Scholar]
- 44. Kueh A. J., Dixon M. P., Voss A. K., Thomas T. (2011) HBO1 is required for H3K14 acetylation and normal transcriptional activity during embryonic development. Mol. Cell Biol. 31, 845–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Orkin S. H., Harosi F. I., Leder P. (1975) Differentiation in erythroleukemic cells and their somatic hybrids. Proc. Natl. Acad. Sci. U.S.A. 72, 98–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Canault M., Certel K., Schatzberg D., Wagner D. D., Hynes R. O. (2010) The lack of ADAM17 activity during embryonic development causes hemorrhage and impairs vessel formation. PLoS ONE 5, e13433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim G. W., Li L., Gorbani M., You L., Yang X. J. (2013) Mice lacking α-tubulin acetyltransferase 1 are viable but display α-tubulin acetylation deficiency and dentate gyrus distortion. J. Biol. Chem. 288, 20334–20350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vindelov L. L. (1977) Flow microfluorometric analysis of nuclear DNA in cells from solid tumors and cell suspensions. A new method for rapid isolation and straining of nuclei. Virchows Arch. B Cell Pathol. 24, 227–242 [PubMed] [Google Scholar]
- 49. Walkinshaw D. R., Weist R., Kim G. W., You L., Xiao L., Nie J., Li C. S., Zhao S., Xu M., Yang X. J. (2013) The tumor suppressor kinase LKB1 activates the downstream kinases SIK2 and SIK3 to stimulate nuclear export of class IIa histone deacetylases. J. Biol. Chem. 288, 9345–9362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Horak E. R., Leek R., Klenk N., LeJeune S., Smith K., Stuart N., Greenall M., Stepniewska K., Harris A. L. (1992) Angiogenesis, assessed by platelet/endothelial cell adhesion molecule antibodies, as indicator of node metastases and survival in breast cancer. Lancet 340, 1120–1124 [DOI] [PubMed] [Google Scholar]
- 51. Kaufman M. H., Bard J. B. L. (1999) The Anatomical Basis of Mouse Development, Academic Press, San Diego. CA [Google Scholar]
- 52. Rossant J., Cross J. C. (2001) Placental development: lessons from mouse mutants. Nat. Rev. Genet. 2, 538–548 [DOI] [PubMed] [Google Scholar]
- 53. Simmons D. G., Cross J. C. (2005) Determinants of trophoblast lineage and cell subtype specification in the mouse placenta. Dev. Biol. 284, 12–24 [DOI] [PubMed] [Google Scholar]
- 54. Anson-Cartwright L., Dawson K., Holmyard D., Fisher S. J., Lazzarini R. A., Cross J. C. (2000) The glial cells missing-1 protein is essential for branching morphogenesis in the chorioallantoic placenta. Nat. Genet. 25, 311–314 [DOI] [PubMed] [Google Scholar]
- 55. Li Y., Behringer R. R. (1998) Esx1 is an X-chromosome-imprinted regulator of placental development and fetal growth. Nat. Genet. 20, 309–311 [DOI] [PubMed] [Google Scholar]
- 56. Graham V., Khudyakov J., Ellis P., Pevny L. (2003) SOX2 functions to maintain neural progenitor identity. Neuron 39, 749–765 [DOI] [PubMed] [Google Scholar]
- 57. Memberg S. P., Hall A. K. (1995) Dividing neuron precursors express neuron-specific tubulin. J. Neurobiol. 27, 26–43 [DOI] [PubMed] [Google Scholar]
- 58. Hans F., Dimitrov S. (2001) Histone H3 phosphorylation and cell division. Oncogene 20, 3021–3027 [DOI] [PubMed] [Google Scholar]
- 59. Paull T. T., Rogakou E. P., Yamazaki V., Kirchgessner C. U., Gellert M., Bonner W. M. (2000) A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 10, 886–895 [DOI] [PubMed] [Google Scholar]
- 60. Baron M. H., Vacaru A., Nieves J. (2013) Erythroid development in the mammalian embryo. Blood Cells Mol. Dis. 51, 213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Uechi T., Maeda N., Tanaka T., Kenmochi N. (2002) Functional second genes generated by retrotransposition of the X-linked ribosomal protein genes. Nucleic Acids Res. 30, 5369–5375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sengupta J., Bussiere C., Pallesen J., West M., Johnson A. W., Frank J. (2010) Characterization of the nuclear export adaptor protein Nmd3 in association with the 60 S ribosomal subunit. J. Cell Biol. 189, 1079–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sulima S. O., Gülay S. P., Anjos M., Patchett S., Meskauskas A., Johnson A. W., Dinman J. D. (2014) Eukaryotic rpL10 drives ribosomal rotation. Nucleic Acids Res. 42, 2049–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. De Keersmaecker K., Atak Z. K., Li N., Vicente C., Patchett S., Girardi T., Gianfelici V., Geerdens E., Clappier E., Porcu M., Lahortiga I., Lucà R., Yan J., Hulselmans G., Vranckx H., Vandepoel R., Sweron B., Jacobs K., Mentens N., Wlodarska I., Cauwelier B., Cloos J., Soulier J., Uyttebroeck A., Bagni C., Hassan B. A., Vandenberghe P., Johnson A. W., Aerts S., Cools J. (2013) Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat. Genet. 45, 186–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sulima S. O., Patchett S., Advani V. M., De Keersmaecker K., Johnson A. W., Dinman J. D. (2014) Bypass of the pre-60 S ribosomal quality control as a pathway to oncogenesis. Proc. Natl. Acad. Sci. U.S.A. 111, 5640–5645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chiocchetti A., Pakalapati G., Duketis E., Wiemann S., Poustka A., Poustka F., Klauck S. M. (2011) Mutation and expression analyses of the ribosomal protein gene RPL10 in an extended German sample of patients with autism spectrum disorder. Am. J. Med. Genet. A 155A, 1472–1475 [DOI] [PubMed] [Google Scholar]
- 67. Vandewalle J., Van Esch H., Govaerts K., Verbeeck J., Zweier C., Madrigal I., Mila M., Pijkels E., Fernandez I., Kohlhase J., Spaich C., Rauch A., Fryns J. P., Marynen P., Froyen G. (2009) Dosage-dependent severity of the phenotype in patients with mental retardation due to a recurrent copy-number gain at Xq28 mediated by an unusual recombination. Am. J. Hum. Genet. 85, 809–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Piton A., Redin C., Mandel J. L. (2013) XLID-causing mutations and associated genes challenged in light of data from large-scale human exome sequencing. Am. J. Hum. Genet. 93, 368–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Brooks S. S., Wall A. L., Golzio C., Reid D. W., Kondyles A., Willer J. R., Botti C., Nicchitta C. V., Katsanis N., Davis E. E. (2014) A novel ribosomopathy caused by dysfunction of RPL10 disrupts neurodevelopment and causes X-linked microcephaly in humans. Genetics 198, 723–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Narla A., Ebert B. L. (2011) Translational medicine: ribosomopathies. Blood 118, 4300–4301 [DOI] [PubMed] [Google Scholar]
- 71. Kondrashov N., Pusic A., Stumpf C. R., Shimizu K., Hsieh A. C., Xue S., Ishijima J., Shiroishi T., Barna M. (2011) Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell 145, 383–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhang Y., Duc A. C., Rao S., Sun X. L., Bilbee A. N., Rhodes M., Li Q., Kappes D. J., Rhodes J., Wiest D. L. (2013) Control of hematopoietic stem cell emergence by antagonistic functions of ribosomal protein paralogs. Dev. Cell 24, 411–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Signer R. A., Magee J. A., Salic A., Morrison S. J. (2014) Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509, 49–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yuan L., Liu J. G., Hoja M. R., Wilbertz J., Nordqvist K., Höög C. (2002) Female germ cell aneuploidy and embryo death in mice lacking the meiosis-specific protein SCP3. Science 296, 1115–1118 [DOI] [PubMed] [Google Scholar]
- 75. Tsutsumi M., Kogo H., Kowa-Sugiyama H., Inagaki H., Ohye T., Kurahashi H. (2011) Characterization of a novel mouse gene encoding an SYCP3-like protein that relocalizes from the XY body to the nucleolus during prophase of male meiosis I. Biol. Reprod. 85, 165–171 [DOI] [PubMed] [Google Scholar]
- 76. Shi Y. Q., Zhuang X. J., Xu B., Hua J., Liao S. Y., Shi Q., Cooke H. J., Han C. (2013) SYCP3-like X-linked 2 is expressed in meiotic germ cells and interacts with synaptonemal complex central element protein 2 and histone acetyltransferase TIP60. Gene 527, 352–359 [DOI] [PubMed] [Google Scholar]
- 77. Zhuang X. J., Shi Y. Q., Xu B., Chen L., Tang W. H., Huang J., Lian Y., Liu P., Qiao J. (2014) SLX2 interacting with BLOS2 is differentially expressed during mouse oocyte meiotic maturation. Cell Cycle 13, 2231–2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Siegel J. N., Turner C. A., Klinman D. M., Wilkinson M., Steinberg A. D., MacLeod C. L., Paul W. E., Davis M. M., Cohen D. I. (1987) Sequence analysis and expression of an X-linked, lymphocyte-regulated gene family (XLR). J. Exp. Med. 166, 1702–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chamberlin H. M., Thomas J. H. (2000) The bromodomain protein LIN-49 and trithorax-related protein LIN-59 affect development and gene expression in Caenorhabditis elegans. Development 127, 713–723 [DOI] [PubMed] [Google Scholar]
- 80. Chang S., Johnston R. J., Jr., Hobert O. (2003) A transcriptional regulatory cascade that controls left/right asymmetry in chemosensory neurons of C. elegans. Genes Dev. 17, 2123–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Laue K., Daujat S., Crump J. G., Plaster N., Roehl H. H., Tübingen 2000 Screen Consortium, Kimmel C. B., Schneider R., Hammerschmidt M. (2008) The multidomain protein Brpf1 binds histones and is required for Hox gene expression and segmental identity. Development 135, 1935–1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hibiya K., Katsumoto T., Kondo T., Kitabayashi I., Kudo A. (2009) Brpf1, a subunit of the MOZ histone acetyl transferase complex, maintains expression of anterior and posterior Hox genes for proper patterning of craniofacial and caudal skeletons. Dev. Biol. 329, 176–190 [DOI] [PubMed] [Google Scholar]
- 83. Champagne N., Bertos N. R., Pelletier N., Wang A. H., Vezmar M., Yang Y., Heng H. H., Yang X. J. (1999) Identification of a human histone acetyltransferase related to monocytic leukemia zinc finger protein. J. Biol. Chem. 274, 28528–28536 [DOI] [PubMed] [Google Scholar]
- 84. Grienenberger A., Miotto B., Sagnier T., Cavalli G., Schramke V., Geli V., Mariol M. C., Berenger H., Graba Y., Pradel J. (2002) The MYST domain acetyltransferase Chameau functions in epigenetic mechanisms of transcriptional repression. Curr. Biol. 12, 762–766 [DOI] [PubMed] [Google Scholar]
- 85. Perez-Campo F. M., Borrow J., Kouskoff V., Lacaud G. (2009) The histone acetyl transferase activity of monocytic leukemia zinc finger is critical for the proliferation of hematopoietic precursors. Blood 113, 4866–4874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bu P., Evrard Y. A., Lozano G., Dent S. Y. (2007) Loss of Gcn5 acetyltransferase activity leads to neural tube closure defects and exencephaly in mouse embryos. Mol. Cell. Biol. 27, 3405–3416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yao T. P., Oh S. P., Fuchs M., Zhou N. D., Ch'ng L. E., Newsome D., Bronson R. T., Li E., Livingston D. M., Eckner R. (1998) Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 93, 361–372 [DOI] [PubMed] [Google Scholar]
- 88. Mishima Y., Wang C., Miyagi S., Saraya A., Hosokawa H., Mochizuki-Kashio M., Nakajima-Takagi Y., Koide S., Negishi M., Sashida G., Naito T., Ishikura T., Onodera A., Nakayama T., Tenen D. G., Yamaguchi N., Koseki H., Taniuchi I., Iwama A. (2014) Histone acetylation mediated by Brd1 is crucial for Cd8 gene activation during early thymocyte development. Nat. Commun. 5, 5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kaufmann M. H. (1994) The Atlas of Mouse Development, Revised 1994 Ed., Elsevier, Boston [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.