

Background: Methionine sulfoxide post-translational modifications have an important new signaling role in cells.
Results: Methionine sulfoxide reductase MsrA of the pathogenic actinomycete Corynebacterium diphtheriae (Cd-MsrA) uses a unique intramolecular redox relay mechanism coupled to mycothiol.
Conclusion: For methionine sulfoxide control, Cd-MsrA is flexible in receiving electrons from both the thioredoxin and the mycothiol pathways.
Significance: C. diphtheriae MsrA is a redox regulator for methionine sulfoxide signaling.
Keywords: Disulfide, Enzyme Mechanism, Kinetics, Oxidation-Reduction (Redox), Redox Regulation, Thiol, X-ray Crystallography, Redox, Thiol-Disulfide Exchange
Abstract
Methionine sulfoxide reductases are conserved enzymes that reduce oxidized methionines in proteins and play a pivotal role in cellular redox signaling. We have unraveled the redox relay mechanisms of methionine sulfoxide reductase A of the pathogen Corynebacterium diphtheriae (Cd-MsrA) and shown that this enzyme is coupled to two independent redox relay pathways. Steady-state kinetics combined with mass spectrometry of Cd-MsrA mutants give a view of the essential cysteine residues for catalysis. Cd-MsrA combines a nucleophilic cysteine sulfenylation reaction with an intramolecular disulfide bond cascade linked to the thioredoxin pathway. Within this cascade, the oxidative equivalents are transferred to the surface of the protein while releasing the reduced substrate. Alternatively, MsrA catalyzes methionine sulfoxide reduction linked to the mycothiol/mycoredoxin-1 pathway. After the nucleophilic cysteine sulfenylation reaction, MsrA forms a mixed disulfide with mycothiol, which is transferred via a thiol disulfide relay mechanism to a second cysteine for reduction by mycoredoxin-1. With x-ray crystallography, we visualize two essential intermediates of the thioredoxin relay mechanism and a cacodylate molecule mimicking the substrate interactions in the active site. The interplay of both redox pathways in redox signaling regulation forms the basis for further research into the oxidative stress response of this pathogen.
Introduction
The pathogenic actinomycete Corynebacterium diphtheriae causes diphtheria, a toxin-mediated disease, by outsmarting the host immune system (1). One way to fight off pathogenic bacteria is by producing reactive oxygen species. Proteins sensitive to reactive oxygen species are particularly those with sulfurous amino acids like cysteine (2) and methionine (3). Upon methionine oxidation, these proteins may get damaged or show an altered local conformation and subsequently act as regulatory switches that specifically initiate signaling pathways within various biological processes (3).
The reaction of oxygen species with methionine leads to the formation of methionine sulfoxide (Met-SO),4 which comes in two stereospecific types, Met-S-SO and Met-R-SO. Methionine sulfoxides are not only the result of a nonspecific reaction but are also formed after a catalyzed reaction by oxygen-consuming NADPH-dependent enzymes or by the newly identified oxidase activity of MsrA (4–6), a methionine sulfoxide reductase with stereoselective reductase (7) and oxidase activity (4) toward the S isomer. The R isomer is specifically reduced by another methionine sulfoxide reductase, MsrB, whose active site is the mirror of that of MsrA (8) and for which no oxidase activity has been described. The oxidase and reductase activity of MsrA makes post-translational methionine sulfoxide regulation appealing because it may have an important signaling role, akin to protein phosphorylation. As a consequence, reversible methionine oxidation is now being acknowledged as a powerful mode for triggering protein activity (9). Recent examples are the transcription factor HypT (10) and the kinase CaMKII (11), which underscore the relevance and the importance of methionine oxidation and the action of methionine sulfoxide reductases in the regulation of protein activity.
We focus here on MsrA from the pathogenic actinomycete C. diphtheriae because we want to understand the mechanism that this MsrA is using to reduce Met-S-SO and determine to which redox pathway it is coupled. In general, MsrA enzymes from different species employ a similar catalytic mechanism (12). A nucleophilic Cys attacks the sulfur of a methionine sulfoxide, resulting in the formation of a sulfenic acid (-SOH) and the concomitant release of Met. The regeneration of the reduction activity via the reduction of the sulfenic acid on MsrA can occur through an intra- or intermolecular thiol disulfide exchange mechanism, linked to the thioredoxin (Trx) (13) or the glutathione (GSH)/glutaredoxin electron transfer pathways (14), driven by the reducing power of NADPH. Actinomycetes, however, do not have the GSH/glutaredoxin system but rather the homologous mycothiol (MSH)/mycoredoxin-1 (Mrx1) system (15). Recently, we found MsrA to be S-mycothiolated in Corynebacterium glutamicum under hypochloric stress (16). The question remains whether and how this reduction system can act as part of the catalytic mechanism of MsrA in C. diphtheriae.
With steady-state kinetics, mass spectrometry, and x-ray crystallography, we present the essential catalytic cysteines of Cd-MsrA for the reduction of l-Met-SO, investigate the use of the Trx/Trx reductase (TrxR) pathway as a reducing system, and show how it uses a previously undescribed MSH relay mechanism to couple to the MSH/Mrx1/mycothione reductase (Mtr) reduction pathway.
EXPERIMENTAL PROCEDURES
Cloning of Cd-msrA and Site-directed Mutagenesis
The Cd-msrA gene was amplified from the total DNA of C. diphtheriae (strain NCTC13129) by PCR, using forward (5′-TTACATATGGGATGGTTATTTGGCGCACC-3′) and reverse (5′-TTAGGATCCCTACGCCTCTGGGATTCCGC-3′) primers. The amplified PCR product was inserted into pET-28a(+), resulting in the Cd-msrA plasmid.
Site-directed mutagenesis was performed on the Cd-msrA plasmid using the QuikChangeTM site-directed mutagenesis protocol (Stratagene). Forward primers 5′-TATATAGGTATCGGCAGTTACTGGGGAGCAGAA-3′, 5′-ACCTACCGTGAAACCAGCACCGGACGCACCAAC-3′, 5′-AACCCACTAGGATACAGCCCGCACCACTCCACC-3′, and 5′-TCCACCGGAGTGGCCAGCGGAATCCCAGAGGCG-3′, and reverse primers 5′-TTCTGCTCCCCAGTAACTGCCGATACCTATATA-3′, 5′-GTTGGTGCGTCCGGTGCTGGTTTCACGGTAGGT-3′, 5′-GGTGGAGTGGTGCGGGCTGTATCCTAGTGGGTT-3′, and 5′-CGCCTCTGGGATTCCGCTGGCCACTCCGGTGGA-3′ were used to generate the Cd-msrA mutants C52S, C87S, C206S, and C215S, respectively. The C87S/C206S double mutant was generated by performing site-directed mutagenesis, using the primers to insert the C206S mutation and the C87S plasmid as a template.
Purification of Mycothiol
MSH was purified from Mycobacterium smegmatis mc2155, based on the protocol described previously (17).
Expression and Purification of the Cd-MsrA Constructs
A single colony of Rosetta (DE3) (Cd-msrA) was grown overnight in lysogeny broth medium, supplemented with 25 μg/ml kanamycin. Subsequently, 1 liter of Terrific broth cultures were inoculated with a 1:100 dilution of this preculture and grown at 37 °C until an A600 nm of 0.7 was reached. After induction with 0.5 mm isopropyl β-d-1-thiogalactopyranoside, the cells were grown for 3 h at 30 °C. The cells were harvested by centrifugation (15 min at 5,000 rpm, at 4 °C; Beckman JLA8.1000 rotor), and the pellet was resuspended in 50 mm Tris, pH 8.0, 500 mm NaCl, 1 mm tris(2-carboxyethyl) phosphine, 50 μg/ml deoxyribonluclease I (Sigma-Aldrich), 20 mm MgCl2, 0.1 mg/ml 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride, and 1 μg/ml leupeptin. The cells were lysed using French press disruption (Constant Systems) at 20,000 p.s.i. and centrifuged at 18,000 rpm for 40 min to remove cell debris. The cell lysate was loaded onto an Ni2+-Sepharose column, equilibrated in 50 mm Tris, pH 8.0, 500 mm NaCl, and 1 mm tris(2-carboxyethyl) phosphine. MsrA was eluted using a linear gradient to 0.5 m imidazole in the same buffer. Depending on the level of purity, the MsrA constructs were either dialyzed to 25 mm Tris, pH 8.0, 1 mm tris(2-carboxyethyl) phosphine, and 1 mm EDTA or further purified on a Superdex75 16/60 preparation grade size exclusion chromatography column (GE Healthcare) equilibrated in the same buffer.
Reversed Phase Chromatography Analysis of l-Met Formation
A reaction mixture containing 25 μm Cd-MsrA (WT or the mutants C52S, C87S, C206S, or C215S), 1 mm l-Met-SO, and 10 mm DTT was incubated for 10 min at 25 °C. The reaction was stopped by adding 1% TFA, and the sample was diluted 5 times in 0.1% TFA, 15% acetonitrile. 100 μl of the diluted sample was injected on an ACE 5 C18 AR column (Achrom), equilibrated in 0.1% TFA, 15% acetonitrile, and eluted isocratically at 0.5 ml/min. Met formation was followed at 215 nm.
Kinetics of the l-Met-S-SO Reduction by Cd-MsrA Coupled to the Trx/TrxR Pathway
The coupled enzyme assay for the Trx/TrxR pathway described by Van Laer et al. (15) was adapted to include Cd-MsrA. Briefly, before use, Cd-MsrA was incubated with 10 mm DTT for 30 min at room temperature. DTT was removed by size exclusion chromatography on a Superdex75 HR 10/30 column, equilibrated in PBS.
A reaction mixture of 500 μm NADPH, 6 μm C. glutamicum TrxR, 3 μm C. glutamicum Trx, and 300 nm Cd-MsrA was incubated for 10 min at 37 °C in a PBS buffer solution. Following the incubation, l-Met-SO (Sigma) was added to start the reaction, and the decrease in NADPH absorbance at 340 nm was monitored using a SpectraMax 340PC spectrophotometer (Molecular Devices). At the first instance, using a concentration of 1 mm l-Met-SO, progress curves were recorded using varying concentrations of Cd-MsrA (100, 200, 300, and 400 nm) or increasing the Trx/TrxR concentrations (3/6 and 5/10 μm) to ensure that the rate-limiting step within the coupled assay is the reduction of l-Met-SO by Cd-MsrA. Subsequently, the progress curves were recorded using varying substrate concentrations (0–500 μm). The initial velocity (vi) for each substrate concentration was measured, and the vi/E0 versus [l-Met-S-SO] values were plotted and fitted with the Michaelis-Menten equation to obtain the kinetic parameters Km, kcat, and Vmax. Because the substrate, l-Met-SO, is a 50/50% mixture of the S and R epimeric forms, the substrate concentrations were halved to plot the [l-Met-S-SO] in the Michaelis-Menten curve. Three independent replicates of vi were measured for each substrate concentration. Trx and TrxR were cloned and purified as described (17).
Coupled Assay with the MSH/Mrx1/Mtr Pathway
The coupled enzyme assay the MSH/Mrx1/Mtr pathway described by Van Laer et al. (15) was adapted to include Cd-MsrA, as described above. A reaction mixture of 500 μm NADPH, 5 μm C. glutamicum Mtr, 2 μm C. glutamicum Mrx1, and 350 μm MSH was incubated at 37 °C, and the absorbance of NADPH at 340 nm was monitored until a stable baseline was reached. To the reaction mixture, 15 μm Cd-MsrA was added, and again the mixture was incubated until a stable baseline was obtained before adding 500 μm l-Met-SO. The initial velocities of the reaction were determined by monitoring the NADPH consumption. The concentrations of C. glutamicum Mrx1, MSH, and Mtr were determined to be non-rate-limiting, as described (15, 17). Briefly, progress curves were recorded using a concentration of 1 mm l-Met-SO and varying concentrations of Cd-MsrA (7.5, 15, and 30 μm) or varying concentrations of C. glutamicum Mrx1 (1, 2, and 4 μm). Subsequently, progress curves were recorded using varying substrate concentrations (0–650 μm). The vi for each substrate concentration was measured, and the vi/E0 versus [l-Met-S-SO] values were plotted and fitted with the Michaelis-Menten equation to obtain the kinetic parameters Km, kcat, and Vmax. C. glutamicum Mrx1 and Mtr were cloned and purified as described (17).
Stopped Flow Analysis of Free Thiol Content
Free thiol content was monitored by following the DTNB consumption. As described above, Cd-MsrA (WT or its Cys mutants) was incubated with 10 mm DTT for 30 min at room temperature. DTT was removed by size exclusion chromatography on a Superdex75 HR 10/30 column, equilibrated in 100 mm sodium phosphate buffer, pH 7.4, 0.1 mm DTPA. Reduced Cd-MsrA (WT or its Cys mutants, 2.5 μm), was mixed with DTNB (300 μm) using a stopped-flow apparatus (1.1-ms mixing time) coupled with an absorbance and fluorescence detector (Applied Photophysics, SV20). The reaction was followed at λ = 412 nm, where 2-nitro-5-thiobenzoate, formed upon DTNB reduction, absorbs maximally. An absorption coefficient of 14,150 m−1 cm−1 (18) was used to quantify the 2-nitro-5-thiobenzoate formed. Experiments were performed in the size exclusion chromatography buffer at 25 °C. In order to determine the free thiol content upon l-Met-SO reduction, Cd-MsrA (WT or its Cys mutants) was incubated with l-Met-SO (0.5 mm) for 10 min, before mixing with DTNB in the stopped flow apparatus.
Mass Spectrometric Analysis of Cd-MsrA Mycothiolation
A reaction mixture of 600 μm MSH and 50 μm Cd-MsrA (wild type (WT) or C52S mutant) was incubated in the presence or absence of 2 mm l-Met-SO or 0.5 mm H2O2 (only for WT) for 10 min at room temperature, in the absence of any reducing agent. The samples were then alkylated with 5 mm N-ethylmaleimide and incubated for 10 min in the dark. S-Mycothiolation of the Cd-MsrA (WT or C52S mutant) Cys residues was assessed by mass spectrometry. Briefly, the proteins were desalted and concentrated on a C-18 spin column (Pierce) prior to an overnight proteolytic digestion with trypsin or chymotrypsin in 50 mm NH4HCO3 at 30 °C. The reaction was stopped by adding 0.1% TFA.
The peptides were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) in an ion trap mass spectrometer (LTQ XL, Thermo Scientific) as described (19). The mass spectrometer was operated in the data-dependent mode and switched automatically between MS, Zoom Scan for charge state determination, and MS/MS at a collision energy of 35% for sequence information. Multistage activation was enabled to promote richer fragmentation of daughter ions resulting from neutral loss of inositol from S-mycothiolated peptides. This process was repeated for the five most abundant ions, and dynamic exclusion was turned on to allow analysis of co-eluting peptides.
For peptide identification, peak lists were generated using the application spectrum selector in the Proteome Discoverer version 1.4 package. The resulting peak lists were searched using Sequest against a target-decoy C. diphtheriae protein database downloaded from Uniprot and comprising 2,267 forward entries. The following parameters were used. Trypsin or chymotrypsin was selected with specific cleavage only at one end of the peptide sequence; the number of internal cleavage sites was set to 1; the mass tolerance for precursor and fragment ions was 1.1 and 1.0 Da, respectively; and the considered dynamic modifications were +16.0 Da for oxidized methionine, +125.0 Da for N-ethylmaleimide, and +484.0 Da for MSH addition to cysteine. Peptide matches were filtered using the q value, and the posterior error probability was calculated by the percolator algorithm, ensuring an estimated false positive rate below 5% for the S-mycothiolated peptide. MS/MS fragmentation was validated manually.
MsrA Crystallization, X-ray Data Collection, and Structure Solution
Purified MsrA was concentrated to 50 mg/ml using 10-kDa cut-off Vivaspin concentrators and crystallized using hanging-drop vapor diffusion at 20 °C, in 0.1 m sodium cacodylate, pH 7.0, 0.35 m ammonium sulfate, and 20% PEG 8000. Drops were composed of 1 μl of Cd-MsrA and 1 μl of precipitant solution.
For x-ray data collection, the Cd-MsrA crystals were cryoprotected with 20% ethylene glycol. X-ray data were collected at 100 K at the Proxima 1 beamline of the Soleil synchrotron facility. X-ray data were processed using XDS (20). The structure of MsrA was solved by molecular replacement using Phaser (21) from the Phenix suite (22), and the E. coli MsrA as a search model (Protein Data Bank code 1FF3, 49% sequence identity). The initial MsrA model was built using AutoBuild (23) from the Phenix suite, and the structure was further completed manually using Coot (24) and refined using Phenix.refine from the Phenix suite. Data collection statistics and refinement parameters are summarized in Table 1.
TABLE 1.
Free thiol content of Cd-MsrA upon l-Met-SO reduction, as determined by reaction with DTNB
Stopped flow was used to determine the thiol content of WT, C206S, C215S, and C87S/C206S mutants in the presence and absence of l-Met-SO.
| Without l-Met-SO |
With l-Met-SO |
|||
|---|---|---|---|---|
| Expected value | Obtained value | Expected value | Obtained value | |
| WT | 4 | 3.5 | 1 | 0.6 |
| C206S | 3 | 2.8 | 1 | 1.2 |
| C215S | 3 | 2.8 | 1 | 1.1 |
| C87S/C206S | 2 | 1.7 | 0 | 0.3 |
RESULTS
Only Cys52 Is Essential for Reduction of l-Met-SO by Cd-MsrA
After purifying recombinant Cd-MsrA, we first verified whether it reduces l-Met-SO, by monitoring the conversion of l-Met-SO to l-Met on reversed phase chromatography (Fig. 1). We first injected l-Met alone and observed that l-Met had an elution peak at 7.3 min. This elution peak was not observed when l-Met-SO was injected. When we injected a preincubated mixture of Cd-MsrA, DTT, and l-Met-SO, we observed a peak at exactly the same retention time as for the l-Met. We used DTT to recycle Cd-MsrA, so detectable amounts of l-Met would be formed. To rule out the possible contribution of DTT toward l-Met-SO reduction, a preincubated mixture of DTT and l-Met-SO was injected, which does not show l-Met formation. These results indicate that the purified recombinant Cd-MsrA is an active methionine sulfoxide reductase.
FIGURE 1.
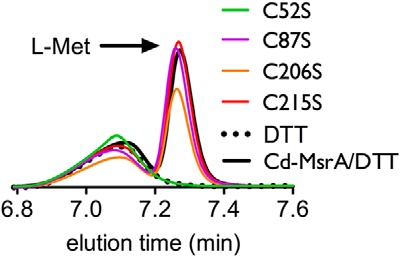
Cys52 is essential for the reduction of l-Met-SO to l-Met. The reversed phase chromatography HPLC chromatograms following the conversion of l-Met-SO to l-Met (peak at retention time 7.3 min) by Cd-MsrA WT and the four Cys to Ser mutants are shown.
In order to reveal which Cys residues are important for the reduction of l-Met-SO, we constructed four Cys to Ser Cd-MsrA mutants (C52S, C87S, C206S, and C215S) by site-directed mutagenesis. These were expressed in Rosetta Escherichia coli cells and purified to homogeneity. We followed the l-Met formation after incubation of l-Met-SO with these Cd-MsrA mutants using reversed phase chromatography. C52S showed no l-Met formation (Fig. 1). On the other hand, l-Met formation was observed for C87S, C206S, and C215S (Fig. 1). This experiment clearly shows that Cys52 is essential for the reduction of l-Met-SO and can be identified as the nucleophilic Cys.
Cys52, Cys206, and Cys215 Are Essential for the Recycling of Cd-MsrA through the Trx/TrxR Pathway
Next, we wanted to check whether Cd-MsrA uses the Trx/TrxR pathway as a reducing system, as shown for other MsrAs (12). Therefore, we coupled the reduction of l-Met-SO by Cd-MsrA to the Trx/TrxR pathway and followed the NADPH consumption at 340 nm (Fig. 2). It turned out that Cd-MsrA indeed uses the Trx/TrxR pathway as a reducing system. The conditions used to acquire the progress curves were rate-limiting for the reduction of l-Met-SO by Cd-MsrA, as assessed by the doubling of the initial velocities when doubling the Cd-MsrA concentration (Fig. 2A). Similarly, the concentrations of Trx and TrxR were shown not to be rate-limiting, as assessed by unchanged initial velocities upon changes in Trx and TrxR concentrations (Fig. 2B). The kinetic parameters for the catalyzed reduction of l-Met-S-SO by Cd-MsrA were obtained by fitting the vi/E0 versus substrate concentration with the Michaelis-Menten equation, which resulted in a Km of 85.2 ± 13.8 μm, a kcat of 0.1 ± 0.007 s−1, and a catalytic efficiency (kcat/Km) of 1.2 × 103 m−1 s−1 (Fig. 2C).
FIGURE 2.

Cys52, Cys206, and Cys215 are essential for the recycling of Cd-MsrA through the Trx/TrxR pathway. A, in the presence of l-Met-SO and the Trx/TrxR pathway, consumption of NADPH at varying concentrations of Cd-MsrA is shown. Cd-MsrA concentrations of 100, 200, 300, and 400 nm result in initial velocities of 4.2, 7.7, 9.9, and 15 milliabsorbance units/min, respectively. B, increasing the Trx/TrxR concentrations from 3/6 μm to 5/10 μm had no effect on initial velocities. C, Cd-MsrA follows Michaelis-Menten kinetics when coupled to the Trx/TrxR pathway. The plot of vi/E0 versus substrate concentration is shown. The data are presented as a mean ± S.D. (error bars) of at least two independent experiments. D, progress curves obtained using the WT and four Cys to Ser mutants of Cd-MsrA coupled to the Trx/TrxR pathway are shown.
To determine the essential Cys residues for coupling to the Trx/TrxR pathway, we coupled l-Met-SO reduction by Cd-MsrA to the Trx/TrxR pathway and monitored the NADPH consumption at 340 nm in function of time for the Cd-MsrA mutants C52S, C87S, C206S, and C215S. NADPH consumption was absent for the C52S, C206S, and C215S mutants because progress curves were similar to the one obtained in the absence of enzyme (Fig. 2D). On the other hand, the C87S mutant showed a clear decrease in the absorbance of NADPH, similar to the progress curve for WT Cd-MsrA. We concluded that aside from Cys52, Cys206 and Cys215 are also essential for the recycling of Cd-MsrA coupled to the Trx/TrxR pathway.
Based on these results and in analogy with the reduction system for other MsrAs (12, 25), we hypothesized that Cys206 and Cys215 are the recycling Cys residues, performing a nucleophilic attack on the sulfenic acid formed on Cys52 upon l-Met-SO reduction (Fig. 3, step 1), forming either a Cys52–Cys206 or Cys52–Cys215 intramolecular disulfide (Fig. 3, step 2). This disulfide is then attacked by the second resolving cysteine, which forms the intramolecular Cys206-Cys215 disulfide (Fig. 3, step 3). The latter will be exposed to the Trx/TrxR pathway to recycle Cd-MsrA (Fig. 3, step 4). Neither the Cys52–Cys206 nor the Cys52–Cys215 disulfide bond is reduced by Trx, as indicated by the lack of NADPH consumption in the coupled assays (Fig. 2D).
FIGURE 3.

Proposed disulfide relay mechanism of Cd-MsrA coupled to the Trx/TrxR pathway. The reaction starts with a nucleophilic attack of the Cd-MsrA Cys52 on the sulfoxide of l-Met-SO, which results in the formation of a sulfenic acid (-SOH) on Cys52, while reduced l-Met is released (step 1). Next, either Cys206 or Cys215 attacks the sulfur of the -SOH on Cys52, causing the release of a water molecule and the formation of a Cys52–Cys206 or Cys52–Cys215 disulfide (step 2). The second resolving Cys (either Cys206 or Cys215) then attacks the sulfur of the first resolving Cys and forms a Cys206–Cys215 disulfide (step 3), which is exposed to thioredoxin for regeneration (step 4).
In order to determine which recycling Cys performs the nucleophilic attack on Cys52, we determined the number of free thiols in Cd-MsrA, WT or the C206S, C215S, and C87S/C206S mutants, upon reduction of l-Met-SO, in the absence of any reducing agent (Table 1). In the WT, three thiols are lost upon reduction of l-Met-SO, consistent with the formation of the Cys206–Cys215 disulfide bond and a sulfenic acid on Cys52 upon reduction of a second molecule of l-Met-SO. This leaves only Cys87 in the free thiol state. The fact that only one free thiol (Cys87) is found in both the C206S and C215S mutants upon l-Met-SO reduction indicates that both Cys206 and Cys215 can perform the nucleophilic attack on Cys52. This is confirmed by the result for the C87S/C206S double mutant.
The X-ray Structure Visualizes Reduced Cd-MsrA and the First Step of the Thiol Disulfide Relay Mechanism
To obtain insights into the molecular details of this proposed recycling mechanism of Cd-MsrA, we decided to crystallize Cd-MsrA using the hanging drop vapor diffusion method. At 20 °C and at a concentration of 50 mg/ml, Cd-MsrA forms orthorhombic crystals (C2221, a = 86.40, b = 140.17, c = 140.55, α = β = γ = 90°), containing three monomers per asymmetric unit (AU). All x-ray data collection statistics and structure refinement parameters are summarized in Table 2. Overall, Cd-MsrA shows an α/β roll conformation, as seen for other MsrAs (26–31). Cd-MsrA shows 32–49% sequence identity to other MsrAs for which the structure has been solved. When comparing the root mean square deviations with the E. coli (Protein Data Bank entry 1FF3, 49% sequence identity) and bovine (Protein Data Bank entry 1FVG, 46% sequence identity) MsrAs, we obtained values of 1.82 Å for 197 Cαs, and 1.13 Å for 189 Cαs, respectively.
TABLE 2.
Data collection and refinement statistics
| Parameter | Value |
|---|---|
| Data set | Cd-MsrA |
| Processing | |
| Space group | C2221 |
| Cell parameters (Å) (α = β = γ = 90°) | |
| a | 86.40 |
| b | 140.17 |
| c | 140.55 |
| Resolution (Å)a | 50-1.89 (2.01-1.89) |
| Total reflections | 598,855 (90,743) |
| Unique reflections | 67,677 (90,743) |
| Completeness (%) | 99.7 (98.1) |
| Multiplicity | 8.85 (8.50) |
| CC1/2 (%) | 99.7 (77.4) |
| Rmeas (%) | 15.4 (89.7) |
| 〈I/σ(I)〉 | 11.79 (2.27) |
| Mosaicity (degrees) | 0.077 |
| Refinement | |
| Resolution range (Å) | 41.30-1.89 |
| Percentage observed (%) | 99.7 |
| Rcryst (%)b | 15.4 |
| Rfree (%)c | 19.0 |
| Root mean square deviation | |
| Bonds (Å) | 0.010 |
| Angles (degrees) | 1.229 |
| Ramachandran plot | |
| Most favored (%) | 96.2 |
| Additionally allowed (%) | 3.7 |
| Disallowed (%) | 0.2 |
| Protein Data Bank code | 4D7L |
a Data in parentheses are for the highest resolution shell.
b Rcryst = Σ‖Fo| − |Fc‖/Σ|Fo|, where Fo and Fc are observed and calculated structure factor amplitude, respectively.
c Rfree is the same as Rcryst, but using a random subset of 5% of the data excluded from the refinement.
In the active site of all three chains of the AU, the nucleophilic Cys52 and the resolving Cys206 are partially in a disulfide bond and partially in the reduced form (Fig. 4, A and B). After refinement, the reduced form of Cys52 shows an occupancy ranging from 53 to 60% over the three copies in the AU. The occupancy of the oxidized form ranges from 40 to 47% over the three chains in the AU. The Cys52–Cys206 disulfide is one of the intramolecular disulfides within the proposed thiol relay mechanism coupled to Trx.
FIGURE 4.
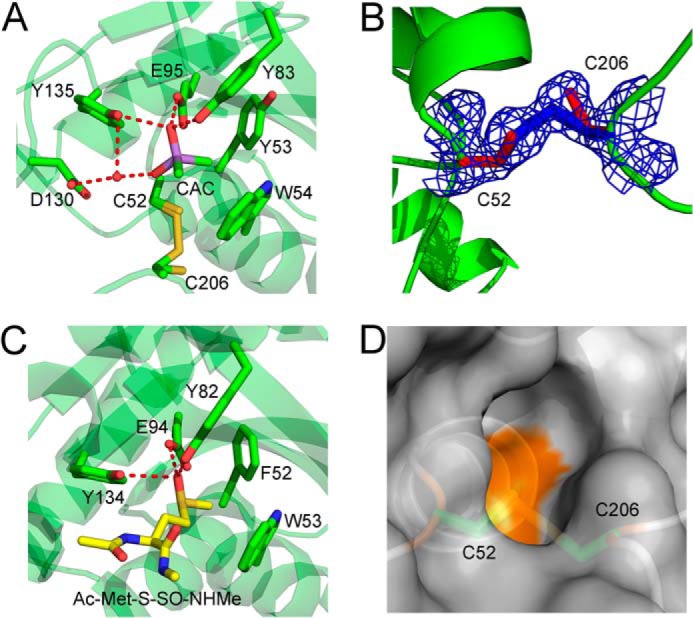
The Cd-MsrA active site shows the first disulfide bond of the relay mechanism and a cacodylate molecule mimicking the substrate. A, the Cd-MsrA active site shows that Cys52 and Cys206 can both be in the reduced form, as in a disulfide bond state. Furthermore, a cacodylate molecule, shown in a purple stick representation, forms hydrogen bonds with Tyr83, Glu95, and Tyr135 and interacts with Asp130 via a water molecule. The two methyl groups of the cacodylate molecule are oriented toward the hydrophobic pocket composed of Tyr53 and Trp54, on the other side of the active site. The Cys52, Tyr53, Trp54, Tyr83, Glu95, Asp130, Tyr135, and Cys206 residues are shown in stick representation. B, the electron density around Cys52 and Cys206 is shown at 1.2σ contour level. C, the N. meningitidis MsrA (Protein Data Bank code 3BQF) active site shows stabilization interactions between the substrate, Ac-Met-S-SO-NHMe (shown in a yellow stick representation), and the conserved hydrogen bond donors Tyr82, Glu94, and Tyr134. These residues, as well as Phe52 and Trp53, are shown in a green stick representation. D, the disulfide bond between Cys52 and Cys206 is not surface-exposed. Cd-MsrA is shown in a gray surface representation, whereas Cys52 and Cys206 are shown in a green stick representation. The figures were generated using MacPyMol (Schroedinger, LLC).
In addition to the intramolecular Cys52–Cys206 disulfide bond, a disulfide bond between Cys87 of one monomer and Cys215 of a symmetry-related molecule is formed (not shown). This intermolecular disulfide does not seem to have any mechanistic relevance because the C87S mutant still showed consumption of NADPH when coupled to the Trx/TrxR pathway (Fig. 2D). This intermolecular disulfide has also been observed in the crystal structure of E. coli MsrA (28).
A Cacodylate Molecule in the Active Site Mimics the l-Met-SO-bound State
The active site of Cd-MsrA is conserved, showing a hydrophobic pocket composed of Tyr53 and Trp54 and a number of hydrogen bond-donating residues (Tyr83, Glu94, Asp130, and Tyr135) (Fig. 4A). The indole ring of Trp54 is stabilized through a hydrogen bond between the NH group of the indole and the Nδ of His194 and by a stacking interaction with the phenyl ring of Tyr197. Tyr53 is stabilized through a hydrogen bond between the oxygen atom of Tyr53 and the Nδ of His194. His194 belongs to the second conserved region of MsrAs, which contains amino acids involved in stabilizing the active site residues (12).
It is important to note that the active site reveals a bound molecule of cacodylate, which has two methyl groups oriented toward the hydrophobic pocket (Fig. 4A). Hydrogen bonds are also formed between the cacodylate oxygen and the conserved hydrogen bond donors in the active site; the oxygen atom of cacodylate is stabilized via hydrogen bond interactions with the ϵ-oxygen atom of Glu95 and the hydroxyl group of both Tyr83 and Tyr135. Asp130 interacts via a water molecule with the second oxygen atom of cacodylate. With these interactions, the cacodylate mimics the l-Met-SO substrate; the oxygen atom of the cacodylate is located at the position of the sulfoxide oxygen of the Met-SO, and the two methyl groups of cacodylate are directed toward the hydrophobic pocket, as is the ϵ-methyl of Met-SO, as can be seen in the structure of Neisseria meningitidis MsrA bound to Ac-Met-S-SO-NHMe (Fig. 4C) (27).
In the Presence of l-Met-SO, Cd-MsrA Is in Vitro Mycothiolated on All Three Catalytic Cys Residues through a Unique MSH Relay Mechanism
We have previously shown in vivo that in the non-pathogenic actinomycete C. glutamicum, the Cys86 of C. glutamicum MsrA (the equivalent of Cys87 in Cd-MsrA) was S-mycothiolated under oxidative stress (16). S-mycothiolation is a protection mechanism of vulnerable cysteines against overoxidation. Aside from the use of MSH as a protection system, Cd-MsrA might also use the MSH/Mrx1/Mtr pathway for catalytic recycling (32). We decided to test this hypothesis by incubating WT Cd-MsrA with the substrate l-Met-SO in the presence of MSH for 10 min at room temperature, in the absence of any reducing agent, and to analyze the sample with mass spectrometry. To our surprise, we not only found the nucleophilic Cys52 to be S-mycothiolated (Table 3), similar to the S-glutathionylation of the nucleophilic cysteine of poplar MsrA2 (14), but also the two resolving cysteines Cys206 and Cys215 (Table 3 and Fig. 5). This either could be the result of an independent mycothiolation of all three Cys residues, upon sulfenylation, or could suggest a relay mechanism involving the transfer of MSH from Cys52 to the resolving cysteines Cys206 and Cys215. If this MSH transfer hypothesis is true, then removing the nucleophilic cysteine should block the MSH transfer. Therefore, we incubated the mutant C52S with l-Met-SO in the presence of MSH under the same experimental conditions and analyzed the sample with mass spectrometry, demonstrating that the two resolving cysteines Cys206 and Cys215 indeed did not show S-mycothiolation (Table 3).
TABLE 3.
In the presence of l-Met-SO, Cd-MsrA is mycothiolated on all three catalytic Cys residues
Mass spectrometry analysis shows S-mycothiolation on WT Cd-MsrA Cys52, Cys206, and Cys215, only in the presence of l-Met-SO. Mutation of Cys52 to Ser shows absence of S-mycothiolation on all Cys residues, and the addition of H2O2 shows mycothiolation on Cys87 as well as the other Cys residues. Cysteine-containing peptides were detected by mass spectrometry either as reduced (N-ethylmaleimide) or S-mycothiolated with their corresponding peptide spectral matches, which reflect their relative abundance. Subsequently, peptide spectral match values were expressed as percentages. ND, not detected; —, not determined; NEM, N-ethylmaleimide; MSH, S-mycothiolated.
| Residue-3′ | WT-3′ control | WT + l-Met-SO | WT + H2O2 | C52S-3′ control | C52S + l-Met-SO |
|---|---|---|---|---|---|
| Cys52 | NEM (100%) | NEM (44%) | NEM (37%) | — | — |
| MSH (56%) | MSH (63%) | ||||
| Cys87a | ND | ND | MSH (100%) | NEM (100%) | NEM (100%) |
| Cys206 | NEM (100%) | NEM (44%) | NEM (88%) | NEM (100%) | NEM (100%) |
| MSH (56%) | MSH (12%) | ||||
| Cys215 | NEM (100%) | NEM (37%) | NEM (75%) | NEM (100%) | NEM (100%) |
| MSH (63%) | MSH (25%) |
a The Cys87 peptide could only be detected after chymotrypsin digestion in the WT+ H2O2 and C52S samples.
FIGURE 5.

In the presence of l-Met-SO, Cd-MsrA is mycothiolated on all three catalytic Cys residues. Identification of S-mycothiolation on Cys206 is shown. The multistage activation LC-MSn spectrum shows MS2 data obtained from a +2 parent ion with m/z 1314.8 combined with MS3 data obtained from a daughter ion of m/z 1224.8 resulting from a neutral loss of inositol (180 Da). The y- and b-series of ions allow exact localization of the mixed disulfide between mycothiol and the Cys residue. &, daughter ions obtained after the neutral loss.
In contrast to the in vivo result upon hydrochloric acid stress of C. glutamicum, Cys87 was not found to be mycothiolated in the presence of l-Met-SO (Table 3). This indicates that mycothiolation of this Cys is not part of the catalytic process but instead occurs upon oxidation by a strong oxidant. In order to confirm this, we performed the mass spectrometric analysis of mycothiolation in the presence of H2O2 instead of l-Met-SO. Indeed, in this case Cys87 was found to be mycothiolated (Table 3).
Transfer of Mycothiol to Cys206 Is Essential for Cd-MsrA to Couple to the MSH/Mrx1/Mtr Reduction Pathway
In order to determine which mycothiolated Cys is substrate for Mrx1, we monitored the NADPH consumption for the catalyzed l-Met-SO reduction coupled to the MSH/Mrx1/Mtr pathway (Fig. 6). Progress curves of the WT and the four Cys mutants were recorded under rate-limiting conditions for substrate reduction, as assessed by the doubling of the initial velocities when doubling the Cd-MsrA concentration in the reaction mixture (Fig. 6A). Similarly, the concentration of Mrx-1 was shown not to be rate-limiting, as assessed by unchanged initial velocities upon changes in Mrx-1 concentration (Fig. 6B). Whereas the C87S mutant showed a similar progress curve as the WT, no NADPH consumption was observed for the C52S mutant (Fig. 6C), clearly highlighting the importance of sulfenylated Cys52 in the first step of the MSH redox relay mechanism. Although Cys215 was shown to be mycothiolated by mass spectrometry, the C206S mutant did not show any NADPH consumption. This shows that mycothiolated Cys215, Cys52, or potentially even mycothiolated Cys87 is not reduced by Mrx1. Thus, transfer of MSH to Cys206 is required for coupling to the MSH/Mrx1/Mtr pathway. The C215S mutant showed a steeper progress curve compared with the WT (Fig. 6C), confirming that the formation of S-mycothiolated Cys215 is not required for coupling to the MSH/Mrx1/Mtr pathway and indicating competition between the formation of Cys206-MSH and the Cys206-Cys215 disulfide bond in the WT, which would make the WT less efficient in coupling to the MSH/Mrx1/Mtr pathway.
FIGURE 6.

Cys206 is essential for Cd-MsrA to couple to the MSH/Mrx1/Mtr reduction pathway. A, in the presence of l-Met-SO and the MSH/Mrx1/Mtr pathway, Cd-MsrA shows consumption of NADPH. Doubling of the Cd-MsrA concentration resulted in doubling of the initial velocities. Cd-MsrA concentrations of 7.5, 15, and 30 μm resulted in initial velocities of 4.9, 10, and 22 milliabsorbance units/min. B, doubling of the Mrx1 concentration had no effect on the initial velocity. C, MSH/Mrx1/Mtr-coupled assay progress curves obtained using the WT and four Cys mutants of Cd-MsrA are shown. D, steady-state kinetics for the WT and C215S Cd-MsrA mutant. The plot of vi/E0 versus substrate concentration is shown. The data are presented as a mean ± S.D. (error bars) of three independent experiments.
To verify this, we determined the steady-state kinetics for l-Met-SO reduction by both the WT and C215S mutant enzymes coupled to the MSH/Mrx1/Mtr pathway (Fig. 6D). From the Michaelis-Menten plots, we determined the kinetic parameters for WT (Km = 17.8 ± 2.2 μm and kcat = 0.002 ± 0.00005 s−1, resulting in a catalytic efficiency kcat/Km = 99.8 m−1 s−1) and the C215S mutant (Km = 34.6 ± 4.5 μm and kcat = 0.004 ± 0.0002 s−1, resulting in a catalytic efficiency kcat/Km = 115.6 m−1 s−1). These results indeed confirm that, in the mutant, the mycothiolation on Cys206 is not disturbed through disulfide bond formation with Cys215.
Based on these results, we propose an alternative recycling mechanism for Cd-MsrA (Fig. 7). The reaction starts in the same way as described for the Trx/TrxR recycling pathway (Fig. 3); Cys52 gets sulfenylated, whereas the l-Met product is released (Fig. 7, step 1). In the next step, the first step of the MSH relay mechanism, MSH attacks the sulfenic acid on Cys52, and a Cys52-MSH mixed disulfide is formed (Fig. 7, step 2). In the second step, either Cys206 or Cys215 can attack this mixed disulfide, making Cys52 ready to start another cycle (Fig. 7, step 3 or 4). MSH bound to Cys215 needs to be transferred to Cys206 (Fig. 7, step 5) because only mycothiolated Cys206 can be reduced by the MSH/Mrx1/Mtr pathway (Fig. 7, step 6).
FIGURE 7.

Proposed mycothiol relay mechanism of Cd-MsrA using the MSH/Mrx1/Mtr pathway. The reaction starts with a nucleophilic attack of Cys52 on the sulfoxide of l-Met-SO, which results in the formation of a sulfenic acid (-SOH) on the Cys52, while reduced l-Met is released (step 1). MSH then attacks the sulfenic acid and forms a mixed disulfide with the sulfur of Cys52 (step 2). Then an MSH relay mechanism starts with a nucleophilic attack of either Cys206 or Cys215 on the mixed disulfide, forming a new mixed disulfide between the sulfur of Cys206 or Cys215 and MSH (steps 3 and 4). MSH needs to be transferred from Cys215 to Cys206 (step 5) to become a substrate for the MSH/Mrx1/Mtr pathway (step 6).
DISCUSSION
Understanding the details of how C. diphtheriae MsrA (Cd-MsrA) rescues possible damaged proteins after an oxidative stress attack of the host innate immune system or how it is involved in methionine sulfoxide signaling is especially interesting because this might lead to new strategies to weaken the defense system of the pathogenic C. diphtheriae. Both MsrA and MsrB have been shown to play a role in the oxidative stress response and pathogenicity of microorganisms. For example, a decrease in infectivity of Streptococcus pneumoniae has been observed after the deletion of the msrAB1 gene (33). Similarly, a study on the pathogenic actinomycete Mycobacterium tuberculosis, which can survive major reactive oxygen species and reactive nitrogen species attacks during the host immune response within macrophages, has shown that MsrA and MsrB single and double deletion strains were sensitive for oxidative stress (34). Here, we used x-ray crystallography, kinetics, and mass spectrometry to enlarge our knowledge of the redox pathways and the cysteine residues of Cd-MsrA involved in the reduction of methionine sulfoxide.
MsrAs stereospecifically reduce both free and protein Met-SO to Met, leaving the MsrA in its oxidized form. To regain their reductase activity, MsrAs use reducing pathways (35), such as the Trx/TrxR pathway, which is used by most organisms to maintain a reducing environment within cells. Highlighting the importance of the Trx reducing pathway, Trx deletion mutants of Met auxotrophs of E. coli (36) and yeast (37) are incapable of using Met-SO as a Met source. In this study, we have shown that Cd-MsrA is also coupled to the Trx/TrxR pathway, by monitoring NADPH consumption in a coupled enzyme assay. To determine the essential Cys residues involved in catalyzing l-Met-SO reduction, we constructed four Cys to Ser mutants of Cd-MsrA. Whereas the C87S mutant showed the same activity as the WT enzyme, the C52S, C206S, and C215S mutants showed complete absence of NADPH consumption linked to the Trx/TrxR pathway (Fig. 2D). On the other hand, looking at the l-Met-SO reduction (Fig. 1), only the C52S mutant was unable to reduce l-Met-SO, suggesting that Cys52 is the nucleophilic cysteine and that Cys206 and Cys215 are essential to couple the MsrA to the Trx/TrxR pathway. Through similarity with E. coli MsrA (35) and Bos taurus MsrA (29), we propose an intramolecular disulfide cascade mechanism, in which Cys52 is the nucleophilic Cys necessary for the reduction of l-Met-SO to l-Met, and Cys206 and Cys215 are suggested to be the resolving Cys residues (Fig. 3). Thiol content determinations using DTNB indicate that both Cys206 and Cys215 can perform a nucleophilic attack on Cys52 (Table 1). The fourth cysteine, Cys87, is not required for l-Met-SO reduction or for coupling to the Trx/TrxR system. To compare with other MsrAs that use three Cys residues in their catalytic mechanism, we determined the catalytic efficiency for the reduction of l-Met-SO by Cd-MsrA. With a value of 1.2 × 103 m−1 s−1, the catalytic efficiency is in the same range as the kcat/Km values reported for E. coli (2 × 103 m−1 s−1) (35) and Populus trichocarpa (1.2 × 103 m−1 s−1) (31) MsrAs.
To obtain further insights into the molecular details of this proposed recycling mechanism of Cd-MsrA, we determined its crystal structure. The overall structure shows an α/β roll conformation, as is the case for other MsrAs (26–31). The crystal structure revealed a snapshot of two intermediates of the thiol disulfide relay mechanism used to couple to the Trx/TrxR system, whereas a cacodylate molecule in the active site mimics the substrate-bound state. Cys52 and Cys206 are found both in the reduced and disulfide-bonded forms in the crystal structure. After refinement, the occupancy of the reduced form of Cys52 ranges from 53 to 60% over the three chains in the AU, whereas the occupancy of the oxidized form ranges from 40 to 47% (Fig. 4, A and B). X-ray and NMR studies on E. coli and N. meningitidis MsrA showed that the formation of the first disulfide bond in the thiol disulfide relay mechanism requires large conformational flexibilities (27, 28, 30). Obtaining a crystal structure of fully reduced and/or oxidized Cd-MsrA would be required to assess whether this conformational change is also required for Cd-MsrA.
In E. coli MsrA, the conformational changes upon oxidation were necessary to expose the first disulfide bond (Cys51–Cys198), making it accessible for Trx (28). However, Trx reduces this disulfide bond with a lower catalytic efficiency than the second disulfide (Cys198–Cys206) of the catalytic mechanism (13).
In the case of Cd-MsrA, Trx is not able to reduce the Cys52–Cys206 disulfide bond (Fig. 2D). Inspection of the crystal structure indeed shows that the Cys52–Cys206 disulfide bond is not surface-exposed (Fig. 4D). Structural details of the Cys206–Cys215 disulfide form of Cd-MsrA, which is reduced by Trx, remain to be determined. This disulfide form of the enzyme has not been shown for any other MsrAs either. Although the formation of this disulfide bond is fast (13), the NMR structure of E. coli MsrA indicates a higher flexibility of the C terminus of the enzyme and shows that Cys198 and Cys206 are 24 Å apart (28), thus indicating that an additional conformational change would be required to form this Cys198–Cys206 disulfide. In the crystal structure of Cd-MsrA, despite the fact that the C terminus is stabilized through crystal contacts, the distance between Cys206 and Cys215 sulfurs is 19 Å, confirming the need for a conformational change.
The active site of Cd-MsrA shows the typical MsrA signature, with on one side a hydrophobic pocket (composed of Tyr53 and Trp54) and on the other side a number of hydrogen bond-donating residues (Tyr83, Glu94, Asp130, and Tyr135) (Fig. 4A). Cd-MsrA is unique in having a Tyr at position 53, whereas the other MsrAs have a Phe at this position (Fig. 8). As in all other MsrA crystal structures available, except for the reduced form of N. meningitidis MsrA, the Cd-MsrA active site is occupied by a ligand (26, 27, 29–31). In Cd-MsrA, a cacodylate molecule mimics the l-Met-SO substrate with its two methyl groups oriented toward the hydrophobic pocket and hydrogen-bonding interactions with the conserved hydrogen bond donors in the active site (Fig. 4A). These stabilization interactions were also observed in the x-ray structure of N. meningitidis MsrA bound to Ac-Met-S-SO-NHMe (27) (Fig. 4C).
FIGURE 8.

Multiple sequence alignment of MsrA from different organisms. Shown are C. glutamicum, C. diphtheriae, M. tuberculosis, M. smegmatis, and E. coli MsrA (accession numbers WP_011015498, WP_021336192, KEP38594, WP_011729814, and ADT77859, respectively). MsrAs were aligned using ClustalW2 and presented using Jalview. Intensity of the blue color gradient is based on 50% identity. The Cys residues are numbered based on the Cd-MsrA sequence numbering, along with conserved residues present in the hydrophobic and hydrophilic pocket of the MsrA active site. The red color indicates non-conserved Cys residues.
We have previously shown in vivo that in the non-pathogenic actinomycete C. glutamicum, the Cys86 of MsrA (the equivalent of Cys87 in Cd-MsrA) was S-mycothiolated under oxidative stress (16). MSH is a low molecular weight thiol analog of GSH found in actinomycetes (38–40). Based on this observation, we investigated the role of S-mycothiolation as a possible catalytic mechanism of Cd-MsrA in vitro. We checked whether, aside from protecting vulnerable cysteines, as was shown for C. glutamicum thiol peroxidase (16), MSH might play a role as a catalytic electron transfer low molecular weight thiol during the reduction of l-Met-SO by Cd-MsrA. In the presence of the substrate l-Met-SO, we observed not only the nucleophilic Cys52 of the WT Cd-MsrA to be S-mycothiolated (Table 3), as was shown for S-glutathionylation of poplar MsrA2 (14), but also Cys206 and Cys215 (Fig. 5). The S-mycothiolation of Cys52, Cys206, and Cys215 indicates either that these three Cys residues are directly S-mycothiolated through three independent sulfenylation actions or, alternatively, that there might be a possible relay mechanism in which MSH is transferred from Cys52 to Cys206 and Cys215. Mass spectrometry analysis for the C52S mutant confirmed that Cys52 is the nucleophilic cysteine that initiates the mycothiol transfer reaction. An MSH/Mrx1/Mtr-coupled enzyme assay with the Cys to Ser mutants of Cd-MsrA showed that only Cys52 and Cys206 are essential for catalyzing l-Met-SO reduction (Fig. 6C). Although Cys215 was also shown to be S-mycothiolated in our mass spectrometry results, it cannot be reduced by Mrx1, as shown by the coupled assay for the C206S mutant. Furthermore, its presence seemed to reduce the efficiency of the reaction (Fig. 6C). This might be explained by competition between S-mycothiolation of Cys206 and Cys206–Cys215 disulfide bond formation in the WT Cd-MsrA, so a fraction of Cd-MsrA would not be reduced through the MSH pathway. Steady-state kinetics for the l-Met-SO reduction by the C215S mutant, coupled to the MSH/Mrx1/Mtr pathway, indeed shows that the catalytic efficiency is higher than that of the WT (kcat/Km = 115.6 and 99.8 m−1 s−1, respectively) (Fig. 6D). In this respect, the M. tuberculosis and M. smegmatis MsrAs only contain two cysteines and, at the position of the Cd-MsrA Cys215, a serine (Fig. 8), suggesting that if these MsrAs also use the proposed MSH relay mechanism, they could do so more efficiently.
It is important to note that in Cd-MsrA, the S-mycothiolated nucleophilic cysteine (Cys52) cannot be reduced by Mrx1. Transfer of MSH to Cys206 is required so that S-mycothiolated Cd-MsrA becomes a substrate of Mrx1. This mechanism is different from the GSH pathway described for poplar MsrA2, in which, following l-Met-SO reduction and formation of a sulfenic acid on the nucleophilic Cys, only the nucleophilic Cys gets S-glutathionylated, and this S-thiolated nucleophilic cysteine is directly reduced by glutaredoxin (14).
In contrast to the in vivo result upon hydrochloric acid stress of C. glutamicum, Cys87 was not found to be mycothiolated in the presence of l-Met-SO (Table 3). This indicates that mycothiolation of this Cys is not part of the catalytic process but occurs upon oxidation by a strong oxidant. Mass spectrometry indeed confirms the mycothiolation of Cys87 in the presence of the strong oxidant H2O2 instead of the substrate l-Met-SO (Table 3).
In conclusion, we have shown that Cd-MsrA is capable of using the Trx/TrxR pathway as a reducing system following l-Met-SO reduction. We have identified the Cys residues that are catalytically essential for the l-Met-SO reduction and coupling to the Trx/TrxR pathway. Most importantly, we have shown that Cd-MsrA uses a unique MSH relay mechanism to couple to the MSH/Mrx1/Mtr reduction pathway, as an alternative pathway. Because the kcat/Km values determined demonstrate that the Trx-based system is 12-fold more efficient than the MSH-based system (∼1,200 versus 100 m−1 s−1), the biological relevance of the competition between the pathways will require further experiments with, for example, knock-out strains. Our findings form the basis for further investigation into the interplay of these two systems and their link to oxidative stress response and pathogenicity.
Acknowledgments
We thank the beamline scientists of the Proxima 1 beamline at the Soleil synchrotron facility for technical support.
Addendum
While this manuscript was under review, Si et al. (41) also showed that the C. glutamicum MsrA can use both the Trx/TrxR and MSH/Mrx1 pathways as reducing system.
This work was supported by Agentschap voor Innovatie door Wetenschap en Technologie (IWT), Vlaams Instituut voor Biotechnologie (VIB), the HOA project of the onderzoeksraad of the Vrije Universiteit Brussel (VUB), an Omics@VIB Marie Curie COFUND fellowship, a VUB bridging grant, VUB Strategic Research Program Grant SRP34, and Hercules Foundation Equipment Grants HERC16 and UABR/09/005.
- Met-SO
- methionine sulfoxide
- MsrA and -B
- methionine sulfoxide reductase A and B, respectively
- Cd-MsrA
- C. diphtheriae MsrA
- Trx
- thioredoxin
- MSH
- mycothiol
- Mrx1
- mycoredoxin-1
- TrxR
- thioredoxin reductase
- Mtr
- mycothione reductase
- AU
- asymmetric unit
- DTNB
- 5,5′-dithiobis(nitrobenzoic acid)
- TFA
- trifluoroacetic acid.
REFERENCES
- 1. Bell C. E., Eisenberg D. (1996) Crystal structure of diphtheria toxin bound to nicotinamide adenine dinucleotide. Biochemistry 35, 1137–1149 [DOI] [PubMed] [Google Scholar]
- 2. Roos G., Messens J. (2011) Protein sulfenic acid formation: from cellular damage to redox regulation. Free Radic. Biol. Med. 51, 314–326 [DOI] [PubMed] [Google Scholar]
- 3. Drazic A., Winter J. (2014) The physiological role of reversible methionine oxidation. Biochim. Biophys. Acta 1844, 1367–1382 [DOI] [PubMed] [Google Scholar]
- 4. Lim J. C., You Z., Kim G., Levine R. L. (2011) Methionine sulfoxide reductase A is a stereospecific methionine oxidase. Proc. Natl. Acad. Sci. U.S.A. 108, 10472–10477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lim J. C., Kim G., Levine R. L. (2013) Stereospecific oxidation of calmodulin by methionine sulfoxide reductase A. Free Radic. Biol. Med. 61, 257–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kriznik A., Boschi-Muller S., Branlant G. (2014) Kinetic evidence that methionine sulfoxide reductase A can reveal its oxidase activity in the presence of thioredoxin. Arch. Biochem. Biophys. 548, 54–59 [DOI] [PubMed] [Google Scholar]
- 7. Sharov V. S., Ferrington D. A., Squier T. C., Schöneich C. (1999) Diastereoselective reduction of protein-bound methionine sulfoxide by methionine sulfoxide reductase. FEBS Lett. 455, 247–250 [DOI] [PubMed] [Google Scholar]
- 8. Grimaud R., Ezraty B., Mitchell J. K., Lafitte D., Briand C., Derrick P. J., Barras F. (2001) Repair of oxidized proteins: identification of a new methionine sulfoxide reductase. J. Biol. Chem. 276, 48915–48920 [DOI] [PubMed] [Google Scholar]
- 9. Lee B. C., Gladyshev V. N. (2011) The biological significance of methionine sulfoxide stereochemistry. Free Radic. Biol. Med. 50, 221–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Drazic A., Miura H., Peschek J., Le Y., Bach N. C., Kriehuber T., Winter J. (2013) Methionine oxidation activates a transcription factor in response to oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 110, 9493–9498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Erickson J. R., Joiner M.-L., Guan X., Kutschke W., Yang J., Oddis C. V., Bartlett R. K., Lowe J. S., O'Donnell S. E., Aykin-Burns N., Zimmerman M. C., Zimmerman K., Ham A.-J., Weiss R. M., Spitz D. R., Shea M. A., Colbran R. J., Mohler P. J., Anderson M. E. (2008) A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133, 462–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boschi-Muller S., Gand A., Branlant G. (2008) The methionine sulfoxide reductases: catalysis and substrate specificities. Arch. Biochem. Biophys. 474, 266–273 [DOI] [PubMed] [Google Scholar]
- 13. Boschi-Muller S., Olry A., Antoine M., Branlant G. (2005) The enzymology and biochemistry of methionine sulfoxide reductases. Biochim. Biophys. Acta 1703, 231–238 [DOI] [PubMed] [Google Scholar]
- 14. Couturier J., Vignols F., Jacquot J.-P., Rouhier N. (2012) Glutathione- and glutaredoxin-dependent reduction of methionine sulfoxide reductase A. FEBS Lett. 586, 3894–3899 [DOI] [PubMed] [Google Scholar]
- 15. Van Laer K., Buts L., Foloppe N., Vertommen D., Van Belle K., Wahni K., Roos G., Nilsson L., Mateos L. M., Rawat M., van Nuland N. A. J., Messens J. (2012) Mycoredoxin-1 is one of the missing links in the oxidative stress defence mechanism of Mycobacteria. Mol. Microbiol. 86, 787–804 [DOI] [PubMed] [Google Scholar]
- 16. Chi B. K., Busche T., Van Laer K., Bäsell K., Becher D., Clermont L., Seibold G. M., Persicke M., Kalinowski J., Messens J., Antelmann H. (2014) Protein S-mycothiolation functions as redox-switch and thiol protection mechanism in Corynebacterium glutamicum under hypochlorite stress. Antioxid. Redox Signal. 20, 589–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ordóñez E., Van Belle K., Roos G., De Galan S., Letek M., Gil J. A., Wyns L., Mateos L. M., Messens J. (2009) Arsenate reductase, mycothiol, and mycoredoxin concert thiol/disulfide exchange. J. Biol. Chem. 284, 15107–15116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Riener C. K., Kada G., Gruber H. J. (2002) Quick measurement of protein sulfhydryls with Ellman's reagent and with 4,4′-dithiodipyridine. Anal. Bioanal. Chem. 373, 266–276 [DOI] [PubMed] [Google Scholar]
- 19. Pyr Dit Ruys S., Wang X., Smith E. M., Herinckx G., Hussain N., Rider M. H., Vertommen D., Proud C. G. (2012) Identification of autophosphorylation sites in eukaryotic elongation factor-2 kinase. Biochem. J. 442, 681–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallog. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Zwart P. H., Hung L.-W., Read R. J., Adams P. D. (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 25. Boschi-Muller S., Branlant G. (2014) Methionine sulfoxide reductase: chemistry, substrate binding, recycling process and oxidase activity. Bioorg. Chem. 57, 222–230 [DOI] [PubMed] [Google Scholar]
- 26. Taylor A. B., Benglis D. M., Jr., Dhandayuthapani S., Hart P. J. (2003) Structure of Mycobacterium tuberculosis methionine sulfoxide reductase A in complex with protein-bound methionine. J. Bacteriol. 185, 4119–4126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ranaivoson F. M., Antoine M., Kauffmann B., Boschi-Muller S., Aubry A., Branlant G., Favier F. (2008) A structural analysis of the catalytic mechanism of methionine sulfoxide reductase A from Neisseria meningitidis. J. Mol. Biol. 377, 268–280 [DOI] [PubMed] [Google Scholar]
- 28. Coudevylle N., Antoine M., Bouguet-Bonnet S., Mutzenhardt P., Boschi-Muller S., Branlant G., Cung M.-T. (2007) Solution structure and backbone dynamics of the reduced form and an oxidized form of E. coli methionine sulfoxide reductase A (MsrA): structural insight of the MsrA catalytic cycle. J. Mol. Biol. 366, 193–206 [DOI] [PubMed] [Google Scholar]
- 29. Lowther W. T., Brot N., Weissbach H., Matthews B. W. (2000) Structure and mechanism of peptide methionine sulfoxide reductase, an “anti-oxidation” enzyme. Biochemistry 39, 13307–13312 [DOI] [PubMed] [Google Scholar]
- 30. Tête-Favier F., Cobessi D., Boschi-Muller S., Azza S., Branlant G., Aubry A. (2000) Crystal structure of the Escherichia coli peptide methionine sulphoxide reductase at 1.9 Å resolution. Structure 8, 1167–1178 [DOI] [PubMed] [Google Scholar]
- 31. Rouhier N., Kauffmann B., Tete-Favier F., Palladino P., Gans P., Branlant G., Jacquot J.-P., Boschi-Muller S. (2007) Functional and structural aspects of poplar cytosolic and plastidial type a methionine sulfoxide reductases. J. Biol. Chem. 282, 3367–3378 [DOI] [PubMed] [Google Scholar]
- 32. Hugo M., Van Laer K., Reyes A. M., Vertommen D., Messens J., Radi R., Trujillo M. (2014) Mycothiol/mycoredoxin 1-dependent reduction of the peroxiredoxin AhpE from Mycobacterium tuberculosis. J. Biol. Chem. 289, 5228–5239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wizemann T. M., Moskovitz J., Pearce B. J., Cundell D., Arvidson C. G., So M., Weissbach H., Brot N., Masure H. R. (1996) Peptide methionine sulfoxide reductase contributes to the maintenance of adhesins in three major pathogens. Proc. Natl. Acad. Sci. U.S.A. 93, 7985–7990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee W. L., Gold B., Darby C., Brot N., Jiang X., de Carvalho L. P. S., Wellner D., St John G., Jacobs W. R., Jr., Nathan C. (2009) Mycobacterium tuberculosis expresses methionine sulphoxide reductases A and B that protect from killing by nitrite and hypochlorite. Mol. Microbiol. 71, 583–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Boschi-Muller S., Azza S., Sanglier-Cianferani S., Talfournier F., Van Dorsselear A., Branlant G. (2000) A sulfenic acid enzyme intermediate is involved in the catalytic mechanism of peptide methionine sulfoxide reductase from Escherichia coli. J. Biol. Chem. 275, 35908–35913 [DOI] [PubMed] [Google Scholar]
- 36. Jacob C., Kriznik A., Boschi-Muller S., Branlant G. (2011) Thioredoxin 2 from Escherichia coli is not involved in vivo in the recycling process of methionine sulfoxide reductase activities. FEBS Lett. 585, 1905–1909 [DOI] [PubMed] [Google Scholar]
- 37. Mouaheb N., Thomas D., Verdoucq L., Monfort P., Meyer Y. (1998) In vivo functional discrimination between plant thioredoxins by heterologous expression in the yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 95, 3312–3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Masip L., Veeravalli K., Georgiou G. (2006) The many faces of glutathione in bacteria. Antioxid. Redox Signal. 8, 753–762 [DOI] [PubMed] [Google Scholar]
- 39. Newton G. L., Arnold K., Price M. S., Sherrill C., Delcardayre S. B., Aharonowitz Y., Cohen G., Davies J., Fahey R. C., Davis C. (1996) Distribution of thiols in microorganisms: mycothiol is a major thiol in most actinomycetes. J. Bacteriol. 178, 1990–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Van Laer K., Hamilton C. J., Messens J. (2013) Low-molecular-weight thiols in thiol-disulfide exchange. Antioxid. Redox Signal. 18, 1642–1653 [DOI] [PubMed] [Google Scholar]
- 41. Si M., Zhang L., Chaudry M.T., Dinge W., Xu Y., Chen C., Akbar A., Shen X., Liu S.-J. (2015). Corynebacterium glutamicum methionine sulfoxide reductase A uses both mycoredoxin and thioredoxin for regeneration and oxidative stress resistance. Appl. Environment. Microbiol. 10.1128/AEM.04221-14 [DOI] [PMC free article] [PubMed] [Google Scholar]