

Background: CblC processes cobalamins entering a cell to a common intermediate.
Results: Pathogenic mutations at Arg-161 weaken glutathione binding to CblC and stabilize cob(II)alamin.
Conclusion: The R161Q/G mutations impair the dealkylation but not the decyanation activity of CblC.
Significance: Increased redox cycling by the CblC mutants explains the observed cellular oxidative stress associated with this disorder.
Keywords: Adenosylcobalamin (AdoCbl), Chaperone, Enzyme Kinetics, Oxidation-Reduction (Redox), Trafficking, Cofactor
Abstract
Human CblC catalyzes the elimination of the upper axial ligand in cobalamin or B12 derivatives entering the cell from circulation. This processing step is critical for assimilation of dietary cobalamin into the active cofactor forms that support the B12-dependent enzymes, methionine synthase and methylmalonyl-CoA mutase. Using a modified nitroreductase scaffold tailored to bind cobalamin and glutathione, CblC exhibits versatility in the mechanism by which it removes cyano versus alkyl ligands in cobalamin. In this study, we have characterized the effects of two pathogenic missense mutations at the same residue, R161G and R161Q, which are associated with early and late onset of the CblC disorder, respectively. We find that the R161Q and R161G CblC mutants display lower protein stability and decreased dealkylation but not decyanation activity, suggesting that cyanocobalamin might be therapeutically useful for patients carrying mutations at Arg-161. The mutant proteins also exhibit impaired glutathione binding. In the presence of physiologically relevant glutathione concentrations, stabilization of the cob(II)alamin derivative is observed, which occurs at the expense of increased oxidation of glutathione. Futile redox cycling, which is suppressed in wild-type human CblC, explains the reported increase in oxidative stress levels associated with the CblC disorder.
Introduction
Mammals utilize B12 or cobalamin derivatives as cofactors for two enzymes, methionine synthase and methylmalonyl-CoA mutase (1). Methionine synthase is essential for freeing the circulating form of the vitamin, 5-methyltetrahydrofolate, to the reduced folate pool, which in turn is needed to support various synthetic reactions necessary for cellular proliferation and homeostasis. Methylmalonyl-CoA mutase is a clearing house for methylmalonyl-CoA generated during breakdown of cholesterol, branched-chain amino acids, and odd-chain fatty acids, and it converts it to succinyl-CoA, which supports energy metabolism (2). Because mammals lack the ability to synthesize cobalamins de novo, they have evolved strategies to assimilate dietary B12 into the two biologically active cofactor forms, methylcobalamin (MeCbl)2 and 5′-deoxyadenosylcobalamin (AdoCbl) (3–5). Insights into the complex pathway for transport and intracellular processing of B12 have emerged from clinical genetics studies on patients exhibiting derangements in cobalamin metabolism and biochemical studies on the affected proteins (3–6). Cobalamin disorders are classified into nine distinct complementation groups: cblA–G, cblJ, and mut (5). In addition, an X-linked cobalamin disorder (cblX) has been reported in which mutations localize to a transcriptional coregulator of the CblC gene (7).
Mutations in CblC (also referred to as MMACHC for methylmalonic aciduria type C and homocystinuria) represent the most common inborn error of cobalamin metabolism. Cobalamin disorders (OMIM 277400) are rare and inherited in an autosomal recessive manner (6). Heterogeneity within cblC patients has been observed, and the most severe presentation of the multisystemic disease is associated with its early onset with affected individuals displaying neurologic, hematologic, and ophthalmologic abnormalities in the first years of life (8–10). The late onset form of the cblC disorder has been reported in a smaller number of cases and is associated with a better response to treatment (10). So far, >75 mutations have been identified in the gene encoding CblC, and >700 affected alleles have been sequenced (8, 11, 12).
Human CblC exhibits relaxed substrate specificity and binds a variety of B12 derivatives (13). The structure of human CblC (14) reveals a commodious binding site for cobalamin where a variety of upper or β-axial ligands can be accommodated including cyano- (15), aquo-, methyl-, and 5′-deoxyadenosine (13, 16) in addition to a variety of unnatural cobalamins (16, 17). The substrate ambiguity of CblC is consistent with its varied processing functions following receipt of the B12 cargo from the lysosomal transporters, LMBD1 and ABCD4 (18, 19). Functionally, CblC catalyzes the removal of the upper or β-axial ligand to generate a common intermediate that can be partitioned into the synthesis of the active cofactors, MeCbl and AdoCbl. When CblC binds CNCbl, electron transfer from reduced flavin, which is free or bound to an oxidoreductase (14, 15, 20), or from GSH (21) facilitates cleavage of the cobalt-carbon bond and elimination of the cyanide ion. In contrast, when CblC binds alkylcobalamins, it catalyzes the nucleophilic displacement of the alkyl group by a tightly bound GSH (16). The products of the decyanation and dealkylation reactions are cob(II)alamin and cob(I)alamin, respectively. Both are subsequently oxidized under aerobic conditions, to aquocobalamin (OH2Cbl) (22). When CblC binds glutathionylcobalamin (GSCbl), it catalyzes the elimination of the glutathionyl group by GSH, forming GSSG (23).
The three-dimensional structure of CblC in the apo-, MeCbl- and AdoCbl-bound forms reveals the repurposing of a scaffold from the nitroreductase superfamily for housing the decyanation and dealkylation reactions (14, 24). Arginine residues frame the cavity around the upper axial face of the corrin ring, and Arg-161 and Arg-230 have been proposed to be important for GSH binding (Fig. 1, A and B) (24). Arg-161 is a highly conserved residue and does not make direct contact with the B12 cofactor in any of the available CblC structures. On the other hand, Arg-161 is involved in electrostatic interactions with citrate, a putative proxy for GSH that was co-crystallized in the CblC structure with AdoCbl bound (24). Mutations at Arg-161 are among the most common of the missense mutations leading to the cblC disorder, and depending on the nature of the substitution, these mutations are associated with either early (R161G) or late (R161Q) onset of the disease (20). The R161Q mutant has been characterized minimally, reported to decrease the Tm of CblC by 2 °C (25) and the MeCbl dealkylation activity ∼10-fold (24).
FIGURE 1.
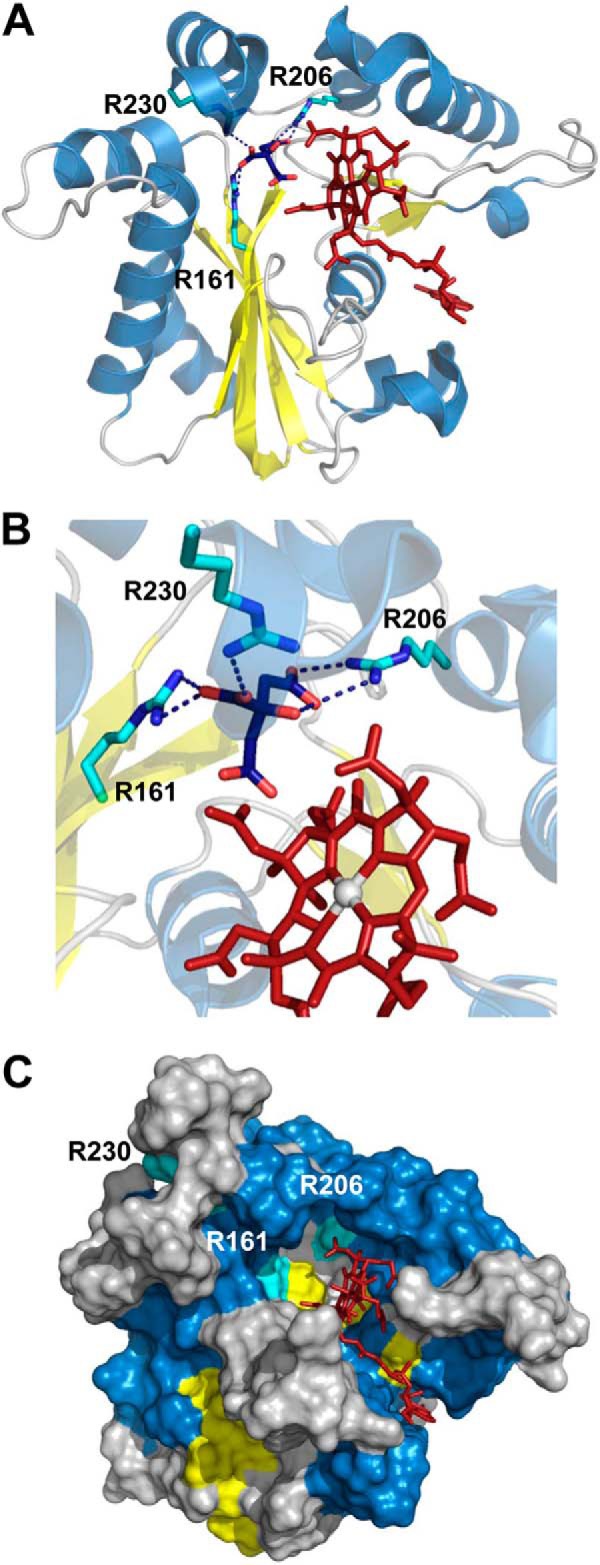
Structure of human CblC with AdoCbl bound (Protein Data Bank (PDB) 3SOM) showing the locations of Arg-161, Arg-206 and Arg-230 predicted to be involved in GSH-binding. A, cartoon representation of CblC structure (PDB: 3SOM) with helixes, β-sheets, and loops colored in blue, yellow, and gray, respectively. AdoCbl is shown in stick representation in red, and Arg-161, Arg-206, and Arg-230 are shown in cyan. Citrate bound to the active site is represented in blue. B, close-up of the hydrogen-bonding interactions between Arg-161, Arg-206, and Arg-230 and citrate bound at the active site. C, surface representation of the CblC structure indicating accessibility of the upper and lower faces of the corrin ring in the absence of bound GSH.
In this study, we describe the detailed characterization of a pair of disease-associated mutations at Arg-161. Our study demonstrates weakened GSH binding and unexpectedly, stabilization of cob(II)alamin under aerobic conditions, which is accompanied by enhanced GSSG production. Characterization of the Arg-161 mutants reveals a latent mechanism for protecting against reactive oxygen species (ROS) production by wild-type human CblC by stabilizing OH2Cbl. Corruption of this protective strategy by the Arg-161 mutations promotes futile redox cycling at physiologically relevant concentrations of GSH, between oxidized and reduced cobalamins, and leads to enhanced generation of ROS, a hallmark of the cblC disease (26).
EXPERIMENTAL PROCEDURES
Materials
AdoCbl, CNCbl, OH2Cbl, MeCbl, GSH, and NADPH were purchased from Sigma-Aldrich. Isopropyl β-d-thiogalactopyranoside, dl-DTT, and Tris (2-carboxyethyl) phosphine hydrochloride were from Gold Biotechnology. All other chemicals and reagents were purchased from Fisher Scientific unless otherwise stated.
Generation of Arg-161 Mutants of Human CblC
Construction of the mutants was performed using the QuikChange site-directed mutagenesis kit according to the manufacturer's instructions (Agilent Technologies Inc.). The pet28b vector encoding the human CblC spanning residues 1–244 was used as a template with the following sense primers for generating R161G and R161Q, respectively: 5′-GGCTGGTTTGCCATCGGAGGGGTAGTGCGCTG-3′ and 5′-GGCTGGTTTGCCATCCAAGGGTAGTGCTGCTG-3′. The mutagenic codons specifying glycine and glutamine are underlined. The presence of the mutations was confirmed by nucleotide sequencing at the University of Michigan DNA Sequencing Core.
Expression and Purification of Wild-type and R161Q/G CblC
Recombinant wild-type and mutant CblC were expressed in Escherichia coli BL21(DE3) as described previously with minor modifications (14). Briefly, cells harvested from 6 liters of Luria Bertani medium were suspended in 150 ml of 50 mm Tris, pH 8.0, 300 mm NaCl, 20 mm imidazole containing 0.2 mg ml−1 lysozyme and one Complete EDTA-free protease inhibitor tablet. Cell lysates were cleared of debris by centrifugation (38,000 × g for 60 min), and CblC was isolated by affinity chromatography using a Ni-nitrilotriacetic acid-agarose column (Qiagen). The protein was further purified by size-exclusion chromatography using a HiLoad 16/60 Superdex 200 prep grade column (GE Healthcare) pre-equilibrated with 0.1 m HEPES, pH 7.4, 150 mm KCl, 10% glycerol (Buffer A). Fractions containing highly pure wild-type and mutant CblC were concentrated using Centricon filters (YM-10), flash-frozen in liquid nitrogen, and stored at −80 °C.
Thermal Denaturation Assays
The stability of R161G and R161Q relative to wild-type CblC was assessed in thermal denaturation experiments as described (14). Briefly, 0.5 mg ml−1 wild-type or R161Q/G CblC in Buffer A was incubated in a cuvette, and the temperature was gradually increased from 25 to 75 °C. The change in optical density at 600 nm was recorded in a spectrophotometer equipped with a temperature-controlled water bath (Fisher Scientific). The Tm was determined using Equation 1.
Dealkylation and Decyanation Activities of Wild-type and R161Q/G CblC
Dealkylation assays were performed in Buffer A at 25 °C and monitored by UV-visible spectroscopy. Reactions were initiated by the addition of GSH (1 μm–10 mm) to the aerobic assay mixtures (200 μl final volume) containing 30 μm CblC (wild type or R161Q/G) and 25 μm MeCbl, respectively. Kinetic parameters for the GSH-dependent dealkylation (kobs, Kact) were determined from the dependence of the reaction rate on GSH concentration at 355 nm (corresponding to OH2Cbl formation). Decyanation of CNCbl was monitored at 530 and 475 nm as described previously (15). Briefly, an anaerobic mixture of methionine synthase reductase (4 μm) was reduced with NADPH (200 μm), and the absorbance baseline was adjusted after 5 min of incubation. The decyanation reaction was initiated by the addition of a mixture containing 50 μm CblC and 20 μm CNCbl in Buffer A (200 μl final volume). Spectra were recorded at 25 °C under anaerobic conditions on a Cary100 spectrophotometer equipped with a temperature-controlled water bath.
EPR Spectroscopy
Wild-type and mutant CblC samples were prepared aerobically for EPR spectroscopy. Samples containing 100 μm wild-type or R161Q/G CblC, 75 μm MeCbl, and 10 mm GSH in Buffer A were incubated at 25 °C for 10 min. Reaction mixtures were then transferred to EPR tubes and frozen in liquid nitrogen. A base-off cob(II)alamin reference sample was prepared by photolysis of 100 μm AdoCbl in 1% H3PO4 as described previously (27). EPR spectra were recorded on a Bruker EMX spectrometer (Bruker Corp.) equipped with an Oxford ITC4 temperature controller, a Bruker gaussmeter, and a Hewlett-Packard model 5340 automatic frequency counter. The parameters used for spectral acquisition were: temperature, 100 K; microwave power, 20 milliwatt; microwave frequency, 9.38 GHz; receiver gain, 1 × 105; modulation amplitude, 10.0 G; modulation frequency, 100 kHz.
GSH-dependent Reduction of CblC-bound OH2Cbl
OH2Cbl (20 μm) was incubated with wild-type or R161Q/G CblC (50 μm) in Buffer A, and unbound OH2Cbl was removed using a Microcon YM-10 centrifugal filtration unit. Then, 10 mm GSH was added to initiate the reduction reaction. The rate of OH2Cbl reduction was determined by the increase in absorbance at 475 nm due to cob(II)alamin formation at 25 °C.
HPLC Analysis of GSH and GSSG
The concentrations of GSH and GSSG in assay mixtures were determined at the start and end of the OH2Cbl reduction reactions using an HPLC method as described (21, 28). Briefly, OH2Cbl (20 μm) bound to wild-type, R161Q, and R161G CblC proteins (50 μm) was treated with 10 mm GSH, and the reaction was allowed to proceed at 25 °C for 1 h. Proteins were precipitated using 1:1 (v/v) 0.21 m metaphosphoric acid solution containing 5.3 mm EDTA and 150 mm NaCl, and thiols in the supernatant were alkylated with monoiodoacetic acid (37 mm final concentration). Then, the pH of the samples was adjusted to ∼8 by the addition of 3 μl of saturated K2CO3, and the samples were incubated for 1 h at room temperature in the dark. The samples were further derivatized by the addition of an equal volume of 2,4-dinitrofluorobenzene (1.5% v/v in ethanol) to the reaction mixture, and the reaction was allowed to proceed for 4 h at room temperature in the dark. The derivatized GSH and GSSG samples were separated by HPLC on a μBondapak NH2 column (Waters, 300 × 3.9 mm, 10 μm) using an 18–60% (v/v) gradient of Solvent B with a flow rate of 1 ml min−1. Solvent A contained 80% (v/v) methanol, and Solvent B contained 1.25 m ammonium acetate, 25% acetic acid, and 50% methanol. Under these conditions, the retention times for GSH and GSSG were 17.4 and 22.5 min, respectively.
Oxygen Consumption Analysis
The rate of oxygen consumption accompanying GSH-dependent decyanation of CNCbl or reduction of OH2Cbl by CblC was determined using a polarographic assay. The reaction mixture containing wild-type or mutant CblC (50 μm) and either CNCbl or OH2Cbl (20 μm) in Buffer A was incubated with 10 mm GSH in a Gilson type chamber (1.5 ml total volume) equipped with a Clark oxygen electrode and a magnetic stirrer. Oxygen consumption was monitored at room temperature on a Kipp & Zonen BD single channel chart recorder in the absence or presence of GSH. Oxygen consumption was expressed as μmol of O2 min−1 mg−1 protein at 22 °C.
Isothermal Titration Calorimetry (ITC)
Calorimetric studies were performed using a VP-ITC microcalorimeter (MicroCal Inc., Northampton, MA). Binding of GSH (0.5–10 mm) to wild-type or mutant CblC (20–200 μm) was monitored at 25 °C in Buffer A by titrating aliquots from a 300-μl syringe loaded with the ligand into a 1.43-ml sample cell containing the protein. Samples were prepared by filtration through a 0.2-μm filter and then degassed under vacuum at 4 °C using a ThermoVac sample degasser. GSH (10-μl injections of 5 μm GSH for wild type and 50 μm GSH for the Arg-161 mutants) was added to 20–35 μm CblC. The data were analyzed using a single-site binding model using the MicroCal ORIGIN program. Titrations were performed in triplicate.
RESULTS
Role of Arginines in CblC
Arginine residues lining the active site of CblC have been suggested to be involved in GSH binding (24). To test this hypothesis, we used phenylglyoxal, an arginine-reactive reagent (29). Incubation of CblC with 4 mm phenylglyoxal at 37 °C resulted in ∼80% inhibition of the GSH-dependent MeCbl dealkylation in 30 min (Fig. 2A). Higher phenylglyoxal concentrations induced protein precipitation.
FIGURE 2.
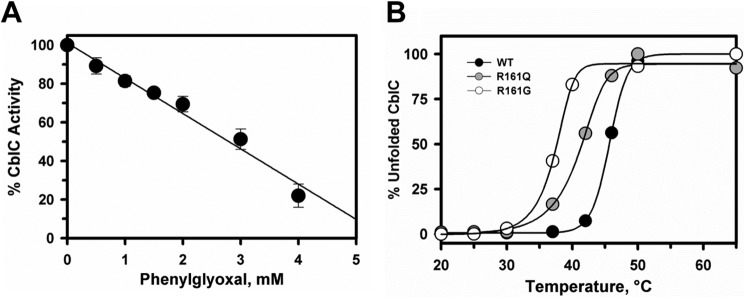
Effect of arginines on the activity and stability of CblC. A, phenylglyoxal inhibits dealkylation of MeCbl by CblC. CblC (30 μm) and MeCbl (20 μm) in Buffer A were incubated with 10 mm GSH and increasing concentrations of phenylglyoxal (0–4 mm) for 30 min, and its effect on the demethylation activity was determined as described under “Experimental Procedures.” Plots were normalized to denote maximum activity in the absence of phenylglyoxal. The data represent the mean ± S.D. of three independent experiments. B, the effect of R161Q/G substitutions on the stability of CblC as monitored at 600 nm. Plots for the temperature-dependent unfolding of R161Q (gray circles) and R161G (open circles) mutants are compared with wild-type (black circles) CblC. The plots were normalized to the starting absorbance at 600 nm representing folded protein. The Tm values were determined as described under “Experimental Procedures.”
Relative Stabilities of R161Q and R161G CblC
As the nature of the substitution at Arg-161 dictates early (R161G) versus late (R161Q) onset of the cblC disease, we determined the impact of these mutations on protein stability (Fig. 2B). Comparison of the thermal denaturation profiles demonstrated that the R161G mutant is considerably less stable (Tm = 38.1 ± 0.7 °C) than wild-type CblC (Tm = 46.6 ± 1.5 °C), whereas R161Q showed intermediate stability (Tm = 43.4 ± 0.2 °C).
Effect of the R161G/Q Mutants on the Dealkylation Activity of CblC
The alkyltransferase activities of R161Q and R161G CblC were tested in the presence of MeCbl and 1 mm GSH (Fig. 3, A–C). Both mutants exhibited diminished activity: ∼6-fold (R161Q) and 10-fold (R161G) lower than wild-type CblC (Fig. 3D). The dependence of the kobs of the demethylase activity on GSH concentration was determined under aerobic conditions yielding a Kact for GSH = 0.025 ± 0.004 mm (wild-type CblC), 1.03 ± 0.09 mm (R161Q CblC), and 0.62 ± 0.07 mm (R161G CblC), respectively (Fig. 3, E and F). Thus, the Arg-161 mutants exhibit a 25–40-fold reduced affinity for GSH as compared with wild-type CblC.
FIGURE 3.

Dealkylation of MeCbl by wild-type and R161Q/G CblC. A–C, time-dependent changes in the UV-visible absorption spectra of MeCbl (25 μm) bound to wild-type (A), R161Q (B), or R161G (C) CblC (30 μm) in Buffer A following the addition of 1 mm GSH under aerobic conditions, at 25 °C. The initial CblC-bound MeCbl spectrum has an absorption maxima at 460 nm (blue trace), whereas the product spectra recorded at 30 min shows features characteristic for OH2Cbl at 353 and 525 nm (red traces). The insets in in A–C show the concentration of the MeCbl consumed and OH2Cbl formed during the reactions. D, comparison of the MeCbl dealkylation rates for wild-type (black circles), R161Q (gray circles), and R161G CblC (open circles). E and F, dependence of the initial rate of dealkylation of MeCbl bound to wild-type (E) or R161Q/G (F) CblC on GSH concentration. Aerobic reaction mixtures containing 30 μm CblC, 25 μm MeCbl, and 0–5 mm GSH in Buffer A were monitored at 25 °C at 353 nm. The data are the mean ± S.D. of at least three independent experiments.
Stabilization of Cob(II)alamin by the Arg-161 CblC Mutants
Because GSH concentrations can vary from 1 to 10 mm depending on the cell type (30, 31), the reduced affinity of the CblC mutants for GSH could be masked. Hence, we decided to examine the dealkylation reaction at higher GSH concentrations. Surprisingly, in the presence of 5–10 mm GSH, cob(II)alamin rather than OH2Cbl was observed as evidenced by the increase in absorption at 473 nm (Fig. 4A). Stabilization of the cob(II)alamin species by CblC is unexpected because wild-type CblC stabilizes cob(III)alamin with a water ligand, i.e. OH2Cbl (16), even in the presence of 10 mm GSH (Fig. 4B). Under anaerobic conditions, the cob(I)alamin product of the dealkylation reaction was observed with the R161Q/G mutants consistent with nucleophilic displacement of the methyl group of MeCbl by the thiolate of GSH (data not shown) as reported previously for wild-type CblC (16). Further evidence for stabilization of the paramagnetic cob(II)alamin species by the mutants was obtained by EPR spectroscopy (Fig. 4C). The product of R161G/Q-catalyzed dealkylation of MeCbl under aerobic conditions was clearly base-off cob(II)alamin. Base-off refers to the conformation of cobalamin in which the endogenous dimethylbenzimidazole base, which is a component of the corrin ring structure, is not coordinated to the cobalt. The eight-line hyperfine splitting and the 140 Gauss spacing between the singlets in the high-field region of the EPR spectra are also seen in the spectrum of authentic base-off cobalamin (Fig. 4C, lower spectrum). The eight-line hyperfine splitting arises from coupling of the unpaired electron with the cobalt nucleus (I = 7/2).
FIGURE 4.

Stabilization of cob(II)alamin by R161Q/G CblC at high GSH concentrations. UV-visible spectra were recorded to monitor dealkylation of MeCbl. A and B, the reaction mixtures contained R161Q CblC (A) or wild-type CblC (30 μm) and MeCbl (25 μm) (B) in aerobic Buffer A at 25 °C. The initial spectra are shown in blue, and spectra were recorded every 3 min following the addition of 10 mm GSH (only a few spectra are shown for clarity). The spectral changes were consistent with formation of cob(II)alamin with an absorption maximum at 473 nm with R161Q CblC (red trace, A) and OH2Cbl with absorption maxima at 353 and 525 nm with wild-type CblC (red trace, B). C, EPR spectra of authentic cob(II)alamin (lower) generated as described under “Experimental Procedures” compared with cob(II)alamin formed during dealkylation of MeCbl catalyzed by R161Q (upper) and R161G (middle) CblC. The samples were prepared as described under “Experimental Procedures.”
Effect of the R161G/Q Mutants on the Decyanation Activity of CblC
Decyanation of CNCbl by R161Q and R161G CblC was monitored anaerobically in the presence of NADPH/methionine synthase reductase (Fig. 5). In contrast to the 6–10-fold effects on dealkylation activity and as reported previously for the R161Q mutant (20), a negligible effect of the mutations was observed on the decyanation activity. The kobs values for R161Q and R161G were 0.11 ± 0.01 and 0.12 ± 0.01 min−1, respectively, which is comparable with the value for wild-type CblC (0.13 ± 0.01 min−1).
FIGURE 5.
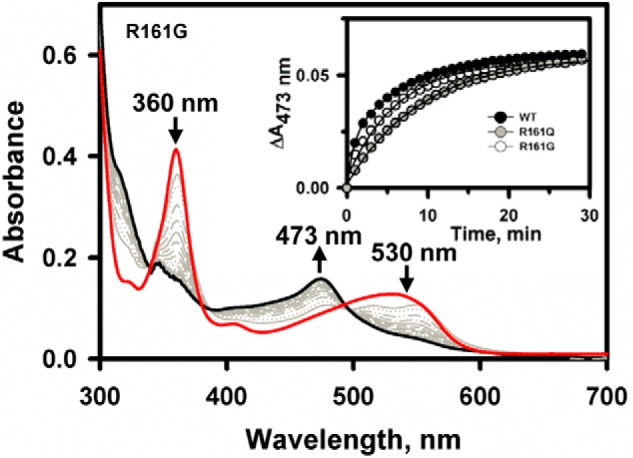
Decyanation kinetics of wild-type and mutant CblCs. Shown are changes in the UV-visible absorption spectrum of CNCbl (20 μm) bound to R161G CblC (50 μm) in Buffer A at 25 °C following addition to a mixture of methionine synthase reductase/NADPH (4 μm/200 μm) under anaerobic conditions. The initial spectrum is in red, selected traces over the reaction course are in gray, and the final spectrum at 60 min is in black. The inset shows the reaction kinetics monitored at 473 nm for wild-type (black circles), R161Q (gray circles), and R161G (open circles) CblC.
Reduction of CblC-bound OH2Cbl by GSH
The kinetics of GSH-dependent reduction of OH2Cbl bound to wild-type CblC versus R161Q/G CblC were compared. In the presence of 10 mm GSH, cob(II)alamin formation was observed under both aerobic (Fig. 6A) and anaerobic (Fig. 6B) conditions with the R161Q/G mutants. In contrast, the addition of 10 mm GSH to wild-type CblC with bound OH2Cbl induced spectral changes consistent with base-off OH2Cbl, but detectable cob(II)alamin was only observed under anaerobic conditions (not shown). The kinetics of cob(II)alamin appearance were monitored at 475 nm and yielded kobs values of 0.0046 ± 0.0005 and 0.027 ± 0.003 min−1 for the R161Q and R161G mutants, respectively (Fig. 6A, inset). Under anaerobic conditions, the kobs for cob(II)alamin formation by wild-type, R161Q, and R161G CblC were 0.015 ± 0.002, 0.017 ± 0.002, and 0.018 ± 0.003 min−1, respectively (Fig. 6B and inset). GSSG generation was enhanced in the presence of R161Q (0.9 ± 0.1 mm in 60 min) and R161G (1.9 ± 0.2 mm) CblC as compared with wild-type (0.47 ± 0.04 mm) CblC (Fig. 6C). The kinetics of oxygen consumption paralleled the difference in GSSG accumulation by the wild type versus the Arg-161 mutants (Fig. 6D).
FIGURE 6.

GSH-dependent reduction of OH2Cbl by wild-type and R161Q/G CblC. A and B, changes in the UV-visible absorption spectra of OH2Cbl (20 μm) bound to R161G CblC (50 μm) in Buffer A were monitored at 25 °C under aerobic (A) and anaerobic conditions (B). Reactions were initiated by the addition of 10 mm GSH. The initial spectra are in red, selected traces over the reaction course time are in gray, and the final spectra at 60 min are in black. Following the addition of GSH to the aerobic sample (A), the absorption maximum shifts from 525 to 516 nm (red dashed trace) before cob(II)alamin formation (473 nm maximum) is observed. The insets in A and B show the reaction kinetics monitored at 475 nm for wild-type (black circles), R161Q (gray circles), and R161G (open circles) CblC. The slight decrease in absorbance at 475 nm over time observed with the more active R161G CblC suggests slow oxidation of cob(II)alamin to OH2Cbl. C, HPLC analysis of GSSG formed after 60 min in the assay mixtures during the reaction shown in A. The data represent the mean ± S.D. of three independent experiments. D, oxygen consumption kinetics associated with GSH-dependent reduction of OH2Cbl in the presence of wild-type (black circles), R161Q (gray circles), and R161G (open circles) CblC.
DISCUSSION
The repurposing of the nitroreductase scaffold during evolution gave rise to CblC, an enzyme that exhibits substrate ambiguity and impressive catalytic versatility. The structures of two recently described cobalamin-dependent dehalogenases reveal that within the nitroreductase superfamily, they most closely resemble CblC (32, 33), suggesting that the ancestry of CblC, described thus far only in higher organisms, is related to these bacterial dehalogenases. The broad range of catalytic functions exhibited by CblC includes decyanation, dealkylation, and OH2Cbl reduction (15, 16, 21). The structure of human CblC shows a cavernous active site (14, 24), which can accommodate cobalamins with a variety of upper axial ligands (13). Cobalamin is bound in a base-off conformation, and the lower or α-face of the corrin ring is vacant and solvent-accessible in the structure of human CblC (14, 24). Unlike the Caenorhabditis elegans CblC, cob(I)alamin and cob(II)alamin bound to wild-type human CblC are subject to facile oxidation forming OH2Cbl (Fig. 7). The structure of human CblC suggests that access to oxidants could occur from the upper or lower axial face of cobalamin (Fig. 1C).
FIGURE 7.
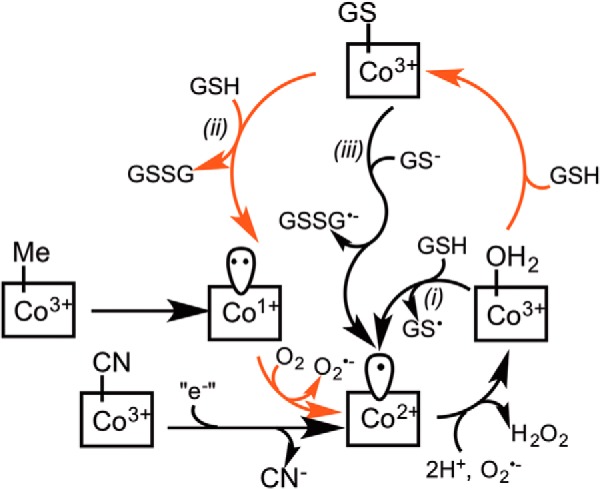
Alternative mechanisms for redox cycling and cob(II)alamin stabilization by the R161Q/G mutants. Demethylation of MeCbl by CblC leads to cob(I)alamin formation, which is labile and is readily oxidized to form cob(II)alamin, and subsequently, to OH2Cbl, which is stabilized by wild-type CblC. Enhanced formation of GSSG accompanying cob(II)alamin stabilization by the R161 mutants could occur by one of three routes denoted i–iii. In path i, OH2Cbl is reduced by GSH to cob(II)alamin. In paths ii and iii, OH2Cbl is initially converted to GSCbl. The latter is either displaced by a second mole of GSH (path ii) or reduced, leading to cob(II)alamin (path iii). The GS• radical formed in path i is rapidly quenched by GSH, giving GSSG•−, which is also formed in path iii, and the GSSG•− radical reacts rapidly with O2 to form O2⨪, further enhancing ROS production. The reactions that are predicted to be accelerated in the Arg-161 mutants are depicted by red arrows.
The ability of CblC to displace the glutathionyl moiety of GSCbl with GSH (21, 23) indicates that the active site can accommodate two equivalents of GSH. Although a structure of CblC with GSH has been elusive, the binding site for one of the GSH moieties is suggested by the active site location of citrate, derived from the crystallization solution (24). Multiple electrostatic interactions pin the citrate in the arginine-rich pocket on the upper face of the corrin ring, and these interactions include Arg-161 (Fig. 1B). An arginine residue is also involved in GSH binding in some glutathione S-transferase superfamily members (34), and their mutation affects catalytic activity (35–37).
Modulation of CblC activity by Arg-161 is of interest because it is a common locus for missense mutations in patients with the cblC class of cobalamin defects (9). Arg-161 is highly conserved, and disease severity and age of onset are correlated with whether it is mutated to glutamine or glycine. Furthermore, cblC patients exhibit high levels of oxidative damage biomarkers, and human cblC fibroblasts have elevated levels of ROS (26, 38). Furthermore, an imbalance in GSH metabolism with a significant decrease in GSH and an increase in GSSG concentration has been reported in CblC-deficient individuals (40). Although limited qualitative analyses of R161Q CblC have revealed impaired GSH-dependent MeCbl dealkylation activity (24), they have not provided insights into elevated ROS formation nor the phenotypic differences associated with a glycine versus glutamine substitution at this location.
In this study, we report that mutations at Arg-161 are destabilizing and reduce the Tm of CblC by ∼3 °C (R161Q) and 8.5 °C (R161G), respectively, as compared with the wild-type protein (Fig. 2B). With a Tm of 38.1 ± 0.7 °C, the R161G mutant would be very susceptible to aggregation at the normal body temperature of 37 °C, which is highly likely to be a contributing factor to early disease onset. Mutations at Arg-161 also decrease the affinity of CblC for GSH by 25- (R161G) and 40-fold (R161Q), respectively, consistent with a role for this residue in substrate binding, as postulated (24). The kobs of the MeCbl dealkylation reaction is diminished 6–10-fold by the R161Q and R161G mutations (Fig. 3). Interestingly, and in contrast to the dealkylation activity of CblC, the Arg-161 mutants do not significantly affect the decyanation activity in the presence of NADPH and the flavoprotein, methionine synthase reductase (Fig. 5). These results suggest that unlike a number of cblC patients who do not respond well to CNCbl (or vitamin B12) therapy (41, 42), patients carrying the R161Q/G mutants might. Given their lower Tm values, however, the efficacy of CNCbl therapy would be predicated by the steady-state levels of the R161Q/G mutants.
Unexpectedly, the R161Q and R161G mutations stabilize cob(II)alamin in the presence of high but not low GSH and are thereby distinct from wild-type CblC, which stabilizes OH2Cbl (Figs. 3 and 4) and binds it with higher affinity. Depending on the cell type, both 1 mm GSH and 10 mm GSH concentrations are physiologically relevant. The preferential stabilization by the Arg-161 mutants of cob(II)alamin with a concomitant increase in GSSG production offers insights into ROS production, which is relevant to disease (38, 39).
In the dealkylation and decyanation reactions catalyzed by wild-type human CblC, the initially formed cob(I)alamin and cob(II)alamin products are oxidized to OH2Cbl (Fig. 7). Although the electron acceptors during each of these oxidation reactions are not known, it is presumably O2 during oxidation of cob(I)alamin and the superoxide anion (O2⨪) during oxidation of cob(II)alamin based on thermodynamic considerations. Thus, the redox potentials for the O2/O2⨪ and O2⨪/H2O2 couples at pH 7.0 are −330 mV and +890 mV, respectively (43). The redox potentials for the base-off cob(II)alamin/cob(I)alamin and base-off OH2Cbl/cob(II)alamin couples are −500 mV and +510 mV, respectively (44). In solution, cob(II)alamin is an efficient O2⨪ scavenger, reacting at rates approaching that of superoxide dismutase (45). The accessibility of the upper axial face of the corrin ring to oxidants is not known because a structure of CblC with GSH is not available. The lower axial corrin face appears to be solvent-exposed (Fig. 1C), allowing access to oxidants. In principle, the Arg-161 mutants could stabilize cob(II)alamin by limiting access of oxidant to the cobalt or by increasing futile redox cycling. The observed formation of GSSG under aerobic conditions where cob(II)alamin is apparently stabilized is indicative of cryptic cycling of cobalt between the 2+ and 3+ oxidation states (Fig. 7).
Stabilization of cob(II)alamin and oxidation of GSH to GSSG has been observed previously with the C. elegans CblC (21) and occurs via a futile redox cycle (Fig. 7), which is suppressed in human CblC. OH2Cbl-dependent oxidation of thiols to their disulfides in solution has been reported previously (46–48). In principle, the redox cycle could proceed via one of three pathways. In the first, OH2Cbl is reduced by GSH to form cob(II)alamin, and the resulting thiyl radical is rapidly quenched in reactions with a second mole of GSH and O2 to give GSSG and O2⨪. The redox potential for the GS•/GSH couple is +920 mV at pH 7.4 (49) and considerably more positive than for the base-off OH2Cbl/cob(II)alamin couple (+510 mV). Kinetic coupling of the unfavorable GSH oxidation reaction to formation of GSSG•− from GS• and GSH followed by quenching of the radical anion with oxygen could conceivably provide the driving force for this reaction.
In the second pathway, OH2Cbl is converted to GSCbl, but only at high concentrations of GSH, and the resulting GSCbl is cleaved by GSH in a nucleophilic displacement reaction to yield GSSG. The product of this reaction is the highly reactive cob(I)alamin species, which reacts rapidly with O2 to form cob(II)alamin and O2⨪. It is possible that mutation of the Arg-161 residue results in poorer stabilization of O2⨪, promoting its loss from the active site and thus contributing to the stabilization of cob(II)alamin. Stabilization of O2⨪ produced in situ via autoxidation has been observed in the heme pocket in the β-chain of hemoglobin (50). Path ii invokes binding of a second equivalent of GSH to CblC (Fig. 7). We know that the first equivalent of GSH binds tightly to human CblC (KD = 30 ± 1 μm (16)) and is positioned to promote nucleophilic chemistry and to suppress sulfur coordination to the cobalt because conversion of wild-type CblC-bound OH2Cbl to GSCbl is not observed (21, 22). Stabilization of cob(II)alamin at high GSH concentrations suggests unmasking of a second lower affinity GSH binding site by mutations at the Arg-161 position. Accommodation of two GSH equivalents in the active site is consistent with the conversion of GSCbl to cob(I)alamin in the presence of GSH by bovine and C. elegans CblC (21, 23). In the third pathway, electron transfer from the second equivalent of GSH yields cob(II)alamin and GSSG•−. This third mechanism is supported by solution studies demonstrating reduction of thiolate-liganded cob(III)alamin to cob(II)alamin by a peripherally bound thiol, presumed to be present at the lower axial corrin face (39).
CONCLUSION
We demonstrate that at physiologically relevant GSH concentrations, cob(II)alamin is the predominant species bound to CblC in patients harboring the R161G and R161Q mutations. Both mutations decrease the Tm of CblC, and the differences in the magnitude of destabilization likely contribute to early versus late onset of the disease. Furthermore, we show that the mutations impair the dealkylation but not the decyanation activity of CblC, suggesting that in patients with mutations at Arg-161, CNCbl might be therapeutically useful. Finally, based on precedence for deglutathionylation of GSCbl by GSH, we favor path ii as an explanation for how the Arg-161 mutants stabilize cob(II)alamin. We predict that loss of the positively charged Arg-161 residue destabilizes O2⨪ binding in the corrin pocket and thus stabilizes cob(II)alamin against further oxidation. On the other hand, mutation of Arg-161 to the smaller glutamine and glycine residues promotes binding of a second equivalent of GSH, which forms a transient GSCbl species by displacing the water ligand in OH2Cbl. Because GSCbl is not observed even at high concentrations of GSH, it suggests that its formation is slow, whereas its conversion to cob(II)alamin (via oxidation of the initially formed cob(I)alamin) is rapid. By promoting futile cycling, the Arg-161 mutants lead to enhanced O2⨪ and GSSG formation, contributing to oxidative stress observed in fibroblasts (26) and increased GSSG levels seen in lymphocytes from cblC patients (40).
This work was supported by National Institutes of Health Grant DK45776 (to R. B.).
- MeCbl
- methylcobalamin
- AdoCbl
- 5′-deoxy-adenosylcobalamin
- CNCbl
- cyanocobalamin
- GSCbl
- glutathionyl-cobalamin
- OH2Cbl
- aquocobalamin
- ROS
- reactive oxygen species.
REFERENCES
- 1. Banerjee R., Ragsdale S. W. (2003) The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Annu. Rev. Biochem. 72, 209–247 [DOI] [PubMed] [Google Scholar]
- 2. Banerjee R. (2001) Radical peregrinations catalyzed by coenzyme B12-dependent enzymes. Biochemistry 40, 6191–6198 [DOI] [PubMed] [Google Scholar]
- 3. Banerjee R., Gherasim C., Padovani D. (2009) The tinker, tailor, soldier in intracellular B12 trafficking. Curr. Opin. Chem. Biol. 13, 484–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gherasim C., Lofgren M., Banerjee R. (2013) Navigating the B12 road: assimilation, delivery and disorders of cobalamin. J. Biol. Chem. 288, 13186–13193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Watkins D., Rosenblatt D. S. (2012) Update and new concepts in vitamin responsive disorders of folate transport and metabolism. J. Inherit. Metab. Dis. 35, 665–670 [DOI] [PubMed] [Google Scholar]
- 6. Shevell M. I., Rosenblatt D. S. (1992) The neurology of cobalamin. Can. J. Neurol. Sci. 19, 472–486 [PubMed] [Google Scholar]
- 7. Yu H. C., Sloan J. L., Scharer G., Brebner A., Quintana A. M., Achilly N. P., Manoli I., Coughlin C. R., 2nd, Geiger E. A., Schneck U., Watkins D., Suormala T., Van Hove J. L., Fowler B., Baumgartner M. R., Rosenblatt D. S., Venditti C. P., Shaikh T. H. (2013) An X-linked cobalamin disorder caused by mutations in transcriptional coregulator HCFC1. Am. J. Hum. Genet. 93, 506–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lerner-Ellis J. P., Tirone J. C., Pawelek P. D., Doré C., Atkinson J. L., Watkins D., Morel C. F., Fujiwara T. M., Moras E., Hosack A. R., Dunbar G. V., Antonicka H., Forgetta V., Dobson C. M., Leclerc D., Gravel R. A., Shoubridge E. A., Coulton J. W., Lepage P., Rommens J. M., Morgan K., Rosenblatt D. S. (2006) Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type. Nat. Genet. 38, 93–100 [DOI] [PubMed] [Google Scholar]
- 9. Lerner-Ellis J. P., Anastasio N., Liu J., Coelho D., Suormala T., Stucki M., Loewy A. D., Gurd S., Grundberg E., Morel C. F., Watkins D., Baumgartner M. R., Pastinen T., Rosenblatt D. S., Fowler B. (2009) Spectrum of mutations in MMACHC, allelic expression, and evidence for genotype-phenotype correlations. Hum. Mutat. 30, 1072–1081 [DOI] [PubMed] [Google Scholar]
- 10. Rosenblatt D. S., Aspler A. L., Shevell M. I., Pletcher B. A., Fenton W. A., Seashore M. R. (1997) Clinical heterogeneity and prognosis in combined methylmalonic aciduria and homocystinuria (cblC). J. Inherit. Metab. Dis. 20, 528–538 [DOI] [PubMed] [Google Scholar]
- 11. Liu M. Y., Yang Y. L., Chang Y. C., Chiang S. H., Lin S. P., Han L. S., Qi Y., Hsiao K. J., Liu T. T. (2010) Mutation spectrum of MMACHC in Chinese patients with combined methylmalonic aciduria and homocystinuria. J. Hum. Genet. 55, 621–626 [DOI] [PubMed] [Google Scholar]
- 12. Gündüz M., Ekici F., Özaydın E., Ceylaner S., Perez B. (2014) Reversible pulmonary arterial hypertension in cobalamin-dependent cobalamin C disease due to a novel mutation in the MMACHC gene. Eur. J. Pediatr. 173, 1707–1710 [DOI] [PubMed] [Google Scholar]
- 13. Hannibal L., Kim J., Brasch N. E., Wang S., Rosenblatt D. S., Banerjee R., Jacobsen D. W. (2009) Processing of alkylcobalamins in mammalian cells: a role for the MMACHC (cblC) gene product. Mol. Genet. Metab. 97, 260–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koutmos M., Gherasim C., Smith J. L., Banerjee R. (2011) Structural basis of multifunctionality in a vitamin B12-processing enzyme. J. Biol. Chem. 286, 29780–29787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim J., Gherasim C., Banerjee R. (2008) Decyanation of vitamin B12 by a trafficking chaperone. Proc. Natl. Acad. Sci. U.S.A. 105, 14551–14554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim J., Hannibal L., Gherasim C., Jacobsen D. W., Banerjee R. (2009) A human vitamin B12 trafficking protein uses glutathione transferase activity for processing alkylcobalamins. J. Biol. Chem. 284, 33418–33424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ruetz M., Gherasim C., Gruber K., Fedosov S., Banerjee R., Kräutler B. (2013) Access to organometallic arylcobaltcorrins through radical synthesis: 4-ethylphenylcobalamin, a potential “antivitamin B12”. Angew. Chem. Int. Ed. Engl. 52, 2606–2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rutsch F., Gailus S., Miousse I. R., Suormala T., Sagné C., Toliat M. R., Nürnberg G., Wittkampf T., Buers I., Sharifi A., Stucki M., Becker C., Baumgartner M., Robenek H., Marquardt T., Höhne W., Gasnier B., Rosenblatt D. S., Fowler B., Nürnberg P. (2009) Identification of a putative lysosomal cobalamin exporter altered in the cblF defect of vitamin B12 metabolism. Nat. Genet. 41, 234–239 [DOI] [PubMed] [Google Scholar]
- 19. Coelho D., Kim J. C., Miousse I. R., Fung S., du Moulin M., Buers I., Suormala T., Burda P., Frapolli M., Stucki M., Nürnberg P., Thiele H., Robenek H., Höhne W., Longo N., Pasquali M., Mengel E., Watkins D., Shoubridge E. A., Majewski J., Rosenblatt D. S., Fowler B., Rutsch F., Baumgartner M. R. (2012) Mutations in ABCD4 cause a new inborn error of vitamin B12 metabolism. Nat. Genet. 44, 1152–1155 [DOI] [PubMed] [Google Scholar]
- 20. Froese D. S., Zhang J., Healy S., Gravel R. A. (2009) Mechanism of vitamin B12-responsiveness in cblC methylmalonic aciduria with homocystinuria. Mol. Genet. Metab. 98, 338–343 [DOI] [PubMed] [Google Scholar]
- 21. Li Z., Gherasim C., Lesniak N. A., Banerjee R. (2014) Glutathione-dependent one-electron transfer reactions catalyzed by a B12 trafficking protein. J. Biol. Chem. 289, 16487–16497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jeong J., Ha T. S., Kim J. (2011) Protection of aquo/hydroxocobalamin from reduced glutathione by a B12 trafficking chaperone. BMB Rep. 44, 170–175 [DOI] [PubMed] [Google Scholar]
- 23. Jeong J., Park J., Park J., Kim J. (2014) Processing of glutathionylcobalamin by a bovine B12 trafficking chaperone bCblC involved in intracellular B12 metabolism. Biochem. Biophys. Res. Commun. 443, 173–178 [DOI] [PubMed] [Google Scholar]
- 24. Froese D. S., Krojer T., Wu X., Shrestha R., Kiyani W., von Delft F., Gravel R. A., Oppermann U., Yue W. W. (2012) Structure of MMACHC reveals an arginine-rich pocket and a domain-swapped dimer for its B12 processing function. Biochemistry 51, 5083–5090 [DOI] [PubMed] [Google Scholar]
- 25. Froese D. S., Healy S., McDonald M., Kochan G., Oppermann U., Niesen F. H., Gravel R. A. (2010) Thermolability of mutant MMACHC protein in the vitamin B12-responsive cblC disorder. Mol. Genet. Metab. 100, 29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Richard E., Jorge-Finnigan A., Garcia-Villoria J., Merinero B., Desviat L. R., Gort L., Briones P., Leal F., Pérez-Cerdá C., Ribes A., Ugarte M., Pérez B., and MMACHC Working Group (2009) Genetic and cellular studies of oxidative stress in methylmalonic aciduria (MMA) cobalamin deficiency type C (cblC) with homocystinuria (MMACHC). Hum. Mutat. 30, 1558–1566 [DOI] [PubMed] [Google Scholar]
- 27. Yamanishi M., Labunska T., Banerjee R. (2005) Mirror “base-off” conformation of coenzyme B12 in human adenosyltransferase and its downstream target, methylmalonyl-CoA mutase. J. Am. Chem. Soc. 127, 526–527 [DOI] [PubMed] [Google Scholar]
- 28. Mosharov E., Cranford M. R., Banerjee R. (2000) The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry 39, 13005–13011 [DOI] [PubMed] [Google Scholar]
- 29. Takahashi K. (1968) The reaction of phenylglyoxal with arginine residues in proteins. J. Biol. Chem. 243, 6171–6179 [PubMed] [Google Scholar]
- 30. Meister A. (1995) Glutathione metabolism. Methods Enzymol. 251, 3–7 [DOI] [PubMed] [Google Scholar]
- 31. Vitvitsky V., Dayal S., Stabler S., Zhou Y., Wang H., Lentz S. R., Banerjee R. (2004) Perturbations in homocysteine-linked redox homeostasis in a murine model for hyperhomocysteinemia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287, R39–R46 [DOI] [PubMed] [Google Scholar]
- 32. Payne K. A., Quezada C. P., Fisher K., Dunstan M. S., Collins F. A., Sjuts H., Levy C., Hay S., Rigby S. E., Leys D. (2015) Reductive dehalogenase structure suggests a mechanism for B12-dependent dehalogenation. Nature 517, 513–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bommer M., Kunze C., Fesseler J., Schubert T., Diekert G., Dobbek H. (2014) Structural basis for organohalide respiration. Science 346, 455–458 [DOI] [PubMed] [Google Scholar]
- 34. Armstrong R. N. (1997) Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem. Res. Toxicol. 10, 2–18 [DOI] [PubMed] [Google Scholar]
- 35. Wang R. W., Newton D. J., Johnson A. R., Pickett C. B., Lu A. Y. (1993) Site-directed mutagenesis of glutathione S-transferase YaYa: mapping the glutathione-binding site. J. Biol. Chem. 268, 23981–23985 [PubMed] [Google Scholar]
- 36. Pal A., Gu Y., Herzog C., Srivastava S. K., Zimniak P., Ji X., Singh S. V. (2001) Role of arginine 216 in catalytic activity of murine Alpha class glutathione transferases mGSTAl-1 and mGSTA2–2 toward carcinogenic diol epoxides of polycyclic aromatic hydrocarbons. Carcinogenesis 22, 1301–1305 [DOI] [PubMed] [Google Scholar]
- 37. Stenberg G., Board P. G., Carlberg I., Mannervik B. (1991) Effects of directed mutagenesis on conserved arginine residues in a human Class Alpha glutathione transferase. Biochem. J. 274, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mc Guire P. J., Parikh A., Diaz G. A. (2009) Profiling of oxidative stress in patients with inborn errors of metabolism. Mol. Genet. Metab. 98, 173–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ramasamy S., Kundu T. K., Antholine W., Manoharan P. T., Rifkind J. M. (2012) Internal spin trapping of thiyl radical during the complexation and reduction of cobalamin with glutathione and dithiothrietol. J. Porphyr. Phthalocyanines 16, 25–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pastore A., Martinelli D., Piemonte F., Tozzi G., Boenzi S., Di Giovamberardino G., Petrillo S., Bertini E., Dionisi-Vici C. (2014) Glutathione metabolism in cobalamin deficiency type C (cblC). J. Inherit. Metab. Dis. 37, 125–129 [DOI] [PubMed] [Google Scholar]
- 41. Bartholomew D. W., Batshaw M. L., Allen R. H., Roe C. R., Rosenblatt D., Valle D. L., Francomano C. A. (1988) Therapeutic approaches to cobalamin-C methylmalonic acidemia and homocystinuria. J. Pediatr. 112, 32–39 [DOI] [PubMed] [Google Scholar]
- 42. Andersson H. C., Shapira E. (1998) Biochemical and clinical response to hydroxocobalamin versus cyanocobalamin treatment in patients with methylmalonic acidemia and homocystinuria (cblC). J. Pediatr. 132, 121–124 [DOI] [PubMed] [Google Scholar]
- 43. Wood P. M. (1988) The potential diagram for oxygen at pH 7. Biochem. J. 253, 287–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lexa D., Saveant J.-M. (1983) The electrochemistry of vitamin B12. Acc. Chem. Res. 16, 235–243 [Google Scholar]
- 45. Suarez-Moreira E., Yun J., Birch C. S., Williams J. H., McCaddon A., Brasch N. E. (2009) Vitamin B12 and redox homeostasis: cob(II)alamin reacts with superoxide at rates approaching superoxide dismutase (SOD). J. Am. Chem. Soc. 131, 15078–15079 [DOI] [PubMed] [Google Scholar]
- 46. Peel J. L. (1962) Vitamin B12 derivatives and the CO2-pyruvate exchange reaction: a reappraisal. J. Biol. Chem. 237, PC263–PC265 [PubMed] [Google Scholar]
- 47. Aronovitch J., Grossowicz N. (1962) Cobalamin catalyzed oxidation of sulfhydryl groups. Biochem. Biophys. Res. Commun. 8, 416–420 [DOI] [PubMed] [Google Scholar]
- 48. Schrauzer G. N., Sibert J. W. (1969) Electron transfer reactions catalyzed by vitamin B12 and related compounds: the reduction of dyes and of riboflavin by thiols. Arch. Biochem. Biophys. 130, 257–266 [DOI] [PubMed] [Google Scholar]
- 49. Madej E., Wardman P. (2007) The oxidizing power of the glutathione thiyl radical as measured by its electrode potential at physiological pH. Arch. Biochem. Biophys. 462, 94–102 [DOI] [PubMed] [Google Scholar]
- 50. Balagopalakrishna C., Abugo O. O., Horsky J., Manoharan P. T., Nagababu E., Rifkind J. M. (1998) Superoxide produced in the heme pocket of the β-chain of hemoglobin reacts with the β-93 cysteine to produce a thiyl radical. Biochemistry 37, 13194–13202 [DOI] [PubMed] [Google Scholar]