

Background: Photolyases use light energy to repair UV-damaged DNA.
Results: The crystal structure of a “class III CPD” photolyase reveals a new binding pocket for an “5,10-methenyltetrahydrofolate” antenna chromophore.
Conclusion: Related plant cryptochromes might use the same antenna chromophore binding pocket.
Significance: The photolyase crystal structure allows a better understanding of the evolutionary transition of photolyases to plant cryptochromes.
Keywords: Cryptochrome, Crystal Structure, DNA Repair, Flavin Adenine Dinucleotide (FAD), Molecular Evolution, Mutagenesis in Vitro, Photobiology, Photoreceptor, Site-directed Mutagenesis, Photoreduction
Abstract
Photolyases are proteins with an FAD chromophore that repair UV-induced pyrimidine dimers on the DNA in a light-dependent manner. The cyclobutane pyrimidine dimer class III photolyases are structurally unknown but closely related to plant cryptochromes, which serve as blue-light photoreceptors. Here we present the crystal structure of a class III photolyase termed photolyase-related protein A (PhrA) of Agrobacterium tumefaciens at 1.67-Å resolution. PhrA contains 5,10-methenyltetrahydrofolate (MTHF) as an antenna chromophore with a unique binding site and mode. Two Trp residues play pivotal roles for stabilizing MTHF by a double π-stacking sandwich. Plant cryptochrome I forms a pocket at the same site that could accommodate MTHF or a similar molecule. The PhrA structure and mutant studies showed that electrons flow during FAD photoreduction proceeds via two Trp triads. The structural studies on PhrA give a clearer picture on the evolutionary transition from photolyase to photoreceptor.
Introduction
Photolyases are light-dependent enzymes that repair UV-induced DNA lesions, which are either cyclobutane pyrimidine dimers (CPD)5 or (6-4)-photoproducts (1–3). The related plant cryptochromes regulate growth, development, and circadian rhythms in a blue light-dependent manner. Recent phylogenetic studies divided the cryptochrome/photolyase family (CPF) into seven major classes, three classes of CPD photolyases termed CPD class I to III, CRY-DASH proteins, (6-4)-photolyases together with animal cryptochromes, plant cryptochromes, and FeS-BCPs (4) that contain bacterial (6-4)-photolyases and photoreceptors with an iron-sulfur cluster (Fig. 1). Cry-DASH proteins have been denominated according to their proposed photoreceptor function, but it has later been found that they repair CPD lesions on single-stranded DNA (5).
FIGURE 1.

Phylogenetic tree with selected members of the CPF. Seven phylogenetic classes are indicated on the right and distinguished by different colors. CryP (written in black) appears separate and cannot be assigned to one of the classes. Abbreviations: Arath, A. thaliana; Agrtu, A. tumefaciens; Azoca, Azorhizobium caulinodans ORS 571; Braja, Bradyrhizobium japonicum USDA 110; Caucr, Caulobacter crescentus; Drome, D. melanogaster; Escco, E. coli; Homsa, Homo sapiens; Phatr, Phaeodactylum tricornutum; Oceal, Oceanocaulix alexandrii; Orysa, Oryza sativa; Rhosp, Rhodobacter sphaeroides 2.4.1; Rhopa, Rhodopseudomonas palustris; Sphsp, Sphingomonas sp. SKA58; Synel, Synechococcus elongatus PCC 6301, S6803: Synechocystis sp. PCC 6803; Theth, Thermus thermophilus HB8, Vibch, Vibrio cholerae; Xenla, Xenopus laevis; Cry, cryptochrome; DASH, DASH cryptochrome; PL64, (6-4)photolyase; Phr, photolyase; Plr, photolyase-related protein; CPD, cyclobutane pyrimidine dimer; FeS-BCP, iron-sulfur cluster containing bacterial cryptochromes and photolyases.
All CPF members incorporate flavin adenine dinucleotide (FAD) as spectrally and redox-active chromophore. Photolyases employ fully reduced flavin adenine dinucleotide (FAD), FADH−, for UV-damaged DNA repair in vivo and in vitro. The repair is initialized by direct light absorption of FADH− or by energy transfer from the excited second chromophore, the antenna chromophore. An electron is subsequently transmitted from FADH− to the DNA lesion, leading to an unstable CPD or (6–4)-photoproduct radical, which finally splits into individual pyrimidines (1, 6). An electron is transferred back to the FADH semiquinone, which becomes fully reduced again. Cryptochromes have typically lost the DNA binding and repair activity (3). In vitro, the FAD of photolyases and cryptochromes typically converts to the oxidized form (7). Irradiation in the presence of reducing agents converts oxidized FAD of photolyases and typical cryptochromes into the reduced or semiquinone form, respectively. A series of three tryptophans (Trp-306/359/382 in Escherichia coli photolyase) is typically involved in this electron transfer. This classical Trp triad (1) is conserved in all CPFs except CPD class II photolyases (8, 9) and FeS-BCP (10, 11). However, site-directed mutagenesis experiments showed that these residues are not necessary for in vivo function of E. coli photolyase (12) or plant Arabidopsis thaliana cryptochrome 2 (Arath-Cry2) (13). Recent studies revealed alternative electron pathways (7, 14, 15); in E. coli photolyase, electrons can either flow via Trp-384 or via Trp-316 and Trp-382 to FAD. Alternative pathways could explain the functionality of CPF proteins in which the classical photoreduction pathway is blocked.
Photolyases also contain a second chromophore with a proposed function as antenna in the N-terminal α/β domain that transfers the energy of absorbed light to FAD by Förster resonance energy transfer. In this way, light capture is increased severalfold over that of FADH− alone, which has a low extinction coefficient in the visible range. The nature of the antenna chromophore and its position within the protein structure varies between photolyases: flavin mononucleotide (FMN) (17), 8-hydroxy-5-deazaflavin (18), FAD (19), and 6,7-dimethyl-8-ribityllumazine (10, 11) bind to a homologous position inside the protein at the C-terminal edge of a β-sheet, whereas MTHF binds to a groove at the outside of the protein (20, 21). Whether cryptochromes carry an antenna chromophore is still unclear: an action spectrum for degradation of recombinant Arath-Cry2 in living cells has a peak at 380 nm, providing strong evidence for a pterin antenna chromophore in Cry2 (22). Spectra of purified recombinant Arabidopsis and mustard cryptochromes provided evidence for an MTHF chromophore (23). However, in other preparations of plant and animal cryptochromes no antenna chromophores were found (24–26) and in the crystal structure of plant cryptochromes the typical MTHF binding pocket is blocked by amino acid side chains of the protein (25).
With the exception of the class III photolyases, crystal structures of all CPF classes have been determined (11, 18–21, 25, 27, 28), in some cases also in complex with DNA (8, 29, 30). These studies have greatly advanced the understanding of the catalytic mechanism of photolyases and the action of cryptochromes. Although all members show high conservation in their overall structure, there are significant variations in structural details such as electron pathways, protein surface charges, modes of interaction with DNA, chromophore binding, length and conformation of the C-terminal domain, and inter-domain loops or key amino acids. Here we present the crystal structure of the CPD class III photolyase PhrA (4) from Agrobacterium tumefaciens at 1.67-Å resolution. The structure reveals a unique 5,10-methenyltetrahydrofolate (MTHF) binding site in which the chromophore is stabilized by double π-stacking interaction with the protein. Besides the classical Trp triad, an alternative Trp triad was found. Both topics were further investigated by site-directed mutagenesis analyses. CPD class III photolyases are the phylogenetic sister group of plant cryptochromes (4, 31). The structural studies on PhrA are thus also relevant for the evolutionary transition from photolyase to photoreceptor.
MATERIALS AND METHODS
Protein Purification and Crystallization
The PhrA protein was expressed in E. coli and purified by affinity chromatography as described (4). Before crystallization, additional size exclusion chromatography was performed with a 200-ml Superdex 200 HR 10/30 column (GE Healthcare) using 12.5 mm Tris, 1.25 mm EDTA, 2.5% (w/v) glycerol, 75 mm sodium chloride, pH 7.8, as chromatography buffer. For crystallization, 4 μl of PhrA protein (11 mg/ml in the same buffer) was mixed with an equal volume of reservoir solution (3 m ammonium sulfate and 10% (w/v) glycerol) and equilibrated against 1 ml of reservoir solution by the sitting drop vapor diffusion method at 277 K in darkness. Yellow tabular crystals appeared within 1 week and grew to a size of about (0.08 × 0.1 × 0.2) mm3 after 3 weeks.
Data Collection and Structure Analysis
Diffraction data collection was performed at 100 K using synchrotron x-ray sources at BESSY II, Berlin, Germany, and ESRF, Grenoble, France (32). Best diffraction data were collected at beamline BL 14.1 (33) at BESSY II, at a wavelength of λ = 0.91842 Å. All images were indexed, integrated, and scaled using the XDS program package (34) and the CCP4 program SCALA (35, 36). The PhrA crystals belonged to the trigonal space group P3221 (a = b = 81.87 Å, c = 195.95 Å, α = β = 90°, γ = 120°). Table 1 summarizes the statistics for crystallographic data collection and structural refinement. Initial phases for PhrA were obtained by the conventional molecular replacement protocol (rotation, translation, and rigid-body fitting) using the A. thaliana cryptochrome 1 (Arath-Cry1) crystal structure (PDB entry 1U3C) (25) without ligands as the initial search model. Molecular replacement was achieved using the CCP4 program PHASER (37) by placing the Arath-Cry1 monomer (rotation function (RFZ): Z = 8.9; translation function (TFZ): Z = 17.9 for PhrA; RFZ and TFZ as defined by PHASER). An AutoBuild/RESOLVE (with the correct PhrA sequence) and a simulated annealing procedure with the resulting model were performed using a slow-cooling protocol and a maximum likelihood target function, energy minimization, and B-factor refinement by the program PHENIX (38), which was carried out in the resolution range of 34.30 - 1.67 Å for the PhrA crystal structure. After the first rounds of refinement, the FAD and MTHF in the ligand binding pockets were clearly visible in the electron density of both σA-weighted 2Fo − Fc maps, as well as in the σA-weighted simulated annealing omitted density maps. PhrA was modeled with TLS refinement (39) using anisotropic temperature factors for all atoms. Restrained, individual B-factors were refined, and the crystal structure was finalized by the CCP4 program REFMAC5 (40) and other programs of the CCP4 suite (41, 42). The final model has agreement factors Rfree and Rwork of 18.0 and 15.6%, respectively. Manual rebuilding of the PhrA model and electron density interpretation were performed after each refinement cycle using the program COOT (41). The side chain atoms of Lys-42, -183, -193, -430, -440, -441, -444, -452, and -477 (atoms: CG, CD, CE, NZ) and Asp-441 (atoms: CG, OD1, and OD2) on the protein surface were removed from the PhrA structure due to not well defined electron density. Structure validation was performed with programs PHENIX (43), SFCHECK (44), PROCHECK (45), WHAT_CHECK/WHAT IF (46, 47), and RAMPAGE (48). Potential hydrogen bonds and van der Waals contacts were analyzed using programs HBPLUS (49) and LigPlot+ (50). All crystal structure superpositions of backbone α-carbon traces were performed using the CCP4 program LSQKAB (35). All molecular graphics representations in this work were created using PyMol (51).
TABLE 1.
Data collection and refinement statistics
| PhrA – class III photolyase (PDB entry 4U63) | |
|---|---|
| Data collectiona (wavelength) | BESSY II, BL14.1 |
| λ = 0.91842 Å | |
| Space group | P3221 |
| Cell dimensions | |
| a, b, c (Å) | 81.87 81.87 195.95 |
| α, β, γ (°) | 90.0, 90.0, 120.0 |
| Resolution (Å) | 34.40-1.67 (1.76-1.67)b |
| Rmerge | 0.085 (0.932) |
| Rpim | 0.030 (0.327) |
| 〈I/σ(I)〉 | 13.7 (2.3) |
| Completeness (%) | 100.0 (100.0) |
| Redundancy | 9.2 (9.0) |
| Wilson B factor (Å2) | 21.5 |
| Refinement | |
| Resolution (Å) | 34.40-1.67 |
| No. Reflections | 84,703 |
| Rwork/Rfree (%) | 15.6/18.0 |
| Overall B factor (Å2) | 30.1 |
| No. atoms/residues (1 monomer per asymmetric unit) | 4650/482 |
| Protein (PhrA) | 3913/478 |
| Expression tag | 25/4 |
| Others | |
| Water | 548/547 |
| Flavin-adenine dinucleotide (FAD) | 53/1 |
| 5,10-Methenyl-6,7,8-trihydrofolic acid (MTHF) | 33/1 |
| Sulfate ion (SO4) | 70/14 |
| 2-Amino-2-hydroxymethyl-propane-1,3-diol (Tris-buffer) | 8/1 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 1.23 |
| Ramachandran plotc | |
| % Favored | 97.5 |
| Allowed | 2.5 |
| Outlier | 0.0 |
a One crystal was used.
b Highest resolution shell is shown in parentheses.
c Ramachandran plot created by RAMPAGE (48) using the data from Richardson and co-workers.
Sequence Analyses
For conservation studies, representative sequences for CPD class III photolyases, plant cryptochromes, Cry-DASH proteins, and CPD class I photolyases were selected according to the phylogenetic study in which the CPD class III was described (31). Multiple sequence alignments were performed with ClustalX 2.0.12 (52).
For phylogenetic studies, a selection of CPF protein sequences according to Ref. 4 and the CryP sequence were combined and aligned with MUSCLE (53). Tree construction was performed with the maximum likelihood function of MEGA 6 (54) and 100 bootstrap iterations.
Site-directed Mutagenesis
Site-directed mutagenesis was carried out according to the QuikChange protocol (Stratagene, La Jolla, CA) as described by the supplier. The correctness of mutant sequences was confirmed by DNA sequencing. The expression and purification protocols were the same as for the wild-type PhrA except that size exclusion chromatography was omitted.
UV-Visible Absorption Spectroscopy and Protein Photoreduction
UV-visible absorption spectra were measured with a Jasco V550 photometer. For photoreduction measurements, each sample was incubated in ground buffer (50 mm Tris/Cl, 5 mm EDTA, 10% glycerol (w/v), 300 mm NaCl, pH 7.8) for 24 h in darkness at 277 K to oxidize FAD. Thereafter, DTT was added to a final concentration of 10 mm. The light source for photoreduction consisted of blue light-emitting diodes HLMP-HB57-LMC (Avago Technologies, Böblingen, Germany) with a maximum emission wavelength of 470 nm and a light intensity of 55 μmol m−2 s−1. Spectra were recorded 1, 2, 3, 5, 7, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 70, 80, 90, 100, 110, and 120 min after onset of the light. UV-visible absorption spectra of the crystals were measured offline before and after x-ray irradiation at 100 K using a HR2000 (OceanOptics) on a microspectrophotometer setup at beamline ID29S-Cryobench (ESRF, Grenoble, France) (55).
RESULTS AND DISCUSSION
Crystallization and Overall Structure of PhrA
Recombinant expression, purification, crystallization screening, and optimization yielded PhrA crystals with a size of (0.08 × 0.1 × 0.2) mm3. UV-visible spectra of crystals measured with a microspectrophotometer show that FAD is initially in the oxidized form, whereas it becomes reduced by the x-ray beam during data collection (Fig. 2), an effect that has been described for Anacystis nidulans photolyase (56). The best crystals diffracted to a resolution of 1.67 Å and the structure of PhrA was solved by molecular replacement using Arath-Cry1 (PDB entry 1U3C) as the initial search model. The crystal structure contains 482 residues (residues 2–478 of PhrA plus 5 residues of the N-terminal tag) and reveals an overall-fold that is typical for CPF proteins (Fig. 3). The N-terminal α/β domain (residues 2–130) and a C-terminal helical domain (residues 208–478) are connected by a loop of 77 residues (131–207), and the U-shaped catalytic chromophore FAD is deeply buried in the center of the helical domain (Fig. 3, inset a). The crystal structure of the closest relative Arath-Cry1 (PDB entry 1U3C) matches with an r.m.s. deviations of 1.8 Å. The second and third best matches were found for Anani-PL (PDB entry 1TEZ, r.m.s. deviations = 1.9 Å) and E. coli photolyase (Escco-PL, PDB entry 1DNP, r.m.s. deviations = 2.2 Å), structures of other CPF members (PDB entries 1OWL, 1NP7, 4I6G, 2VTB, 4JZY, 4DJA, and 3ZXS) yielded r.m.s. deviations values between 2.2 and 3.8 Å (all r.m.s. deviations calculated by DALI (57)).
FIGURE 2.

UV-visible absorption spectra measured in the PhrA crystal before (black straight line) and after (gray dotted line) x-ray exposure. Oxidized FAD has an absorption maximum at 450 nm, this absorbance is lost upon reduction.
FIGURE 3.

Overall structure and cofactor arrangement of PhrA. The ribbon representation shows the α/β-domain (N-terminal domain; green) and the helical domain (catalytic domain; blue) connected by a long inter-domain linker (orange). The cofactors MTHF (colored in pink) and FAD (colored in yellow) are illustrated as sticks model. FAD/MTHF binding sites: inset a, FAD chromophore and inset b, MTHF ligand are shown as stick models with σA-weighted 2Fo − Fc electron density maps contoured at 1.2 σ (blue mesh).
DNA Binding
The DNA binding groove and the FAD access cavity of photolyases have an extended positive charged surface area for interaction with the negatively charged DNA (2). The same feature was found for the PhrA structure (Fig. 4). The more negatively charged surface of plant cryptochromes is probably one cause for their missing DNA binding capacity (2). Six residues are conserved in the cavities of PhrA, Escco-PL, and Anani-PL but are different in Arath-Cry1; only one amino acid (Lys-276) is identical in PhrA and Arath-Cry1 (Table 2). Thus, despite the close relationship between PhrA and Arath-Cry1, the FAD access cavity and the surface charge of the DNA binding region of PhrA are more similar to other photolyases. The transition of photolyase to cryptochrome is characterized by a rapid evolution in this region.
FIGURE 4.
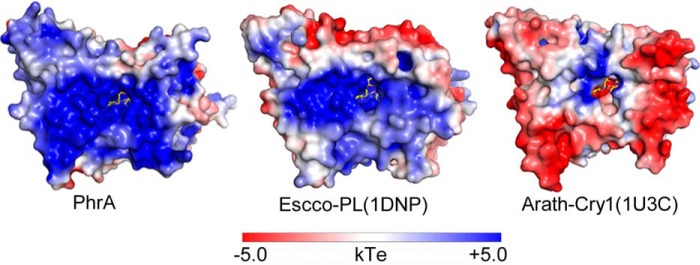
Electrostatic surface potentials of PhrA and comparison with E. coli photolyase and A. thaliana cryptochrome 1. Electrostatic surface potentials were calculated using the programs PyMol, APBS (67), and PDB code 2PQR with the non-linear Poisson-Boltzmann equation contoured at ± 5 kT/e. Negatively and positively charged surface areas are colored in red and blue, respectively.
TABLE 2.
Residues forming the FAD access cavity
Residues that are conserved among CPD photolyases are underlined, whereas the Lys residues identical in PhrA and ArathCry1 are printed in italics.
| PDB numbers | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Arath-Cry1, 1U3C | Ser-236 | Arg-240 | Lys-292 | Leu-296 | Ser-300 | Asp-359 | Val-363 | Leu-398 | Tyr-402 | Arg-411 | Asp-417 | Glu-422 |
| Escco-PL, 1DNP | Glu-223 | Asp-227 | Asn-273 | Trp-277 | Tyr-281 | Asn-341 | Met-345 | Gly-380 | Trp-384 | Ala-393 | Phe-399 | Gln-404 |
| PhrA, 4U63 | Glu-227 | Phe-232 | Lys-276 | Trp-280 | Cys-284 | Asn-343 | Met-347 | Ala-382 | Trp-386 | Ser-395 | Phe-401 | Gln-406 |
| Anani-PL, 1TEZ | Asp-229 | Asn-233 | Glu-282 | Trp-286 | Tyr-290 | Asn-349 | Met-353 | Gly-388 | Trp-392 | Lys-K401 | he-406 | Gln-411 |
| Arath-Cry3, 2VTB | Lys-273 | Asn-277 | Phe-324 | Trp-328 | Phe-332 | Asn-391 | Gln-395 | Gly-430 | Tyr-434 | Arg-446 | Phe-448 | Gln-453 |
In the CPD class I photolyase Anani-PL, six conserved residues interact directly with the CPD lesion (Fig. 5) (29). These amino acids are identical in Anani-PL and PhrA (Arg-230, Glu-277, Trp-280, Asn-343, Met-347, and Trp-386) and all except Trp-386 have the same spatial orientation. The aromatic side chain of Trp-386 is rotated away from the putative CPD lesion (Fig. 5B); its NE1 atom interacts with Arg-147 and the highly conserved Asp-393 via a water molecule, whereas Trp-392 of Anani-PL forms a van der Waals contact with the methyl group of one of the lesion thymines. Importantly, when the homologous Trp in Escco-PL was mutated, the catalytic activity decreased 100-fold (12). The Cry-DASH protein Arath-Cry3 has a Tyr (Tyr-434) at the homologous position. The side chain of this Tyr is also moved away from the lesion site and interacts with Asp-441 (21, 58), the homolog of Asp-393 in PhrA. In the presence of damaged DNA, this side chain rotates toward the lesion and forms a hydrogen bonding contact (59). The side chain of Trp-386 in PhrA is located at the same position as that of Tyr-434 in Arath-Cry3 although both aromatic planes are almost perpendicular to one another. Nevertheless, Trp-386 of PhrA could rotate in different rotameric states to a functional lesion binding position upon DNA interaction (Fig. 5B).
FIGURE 5.
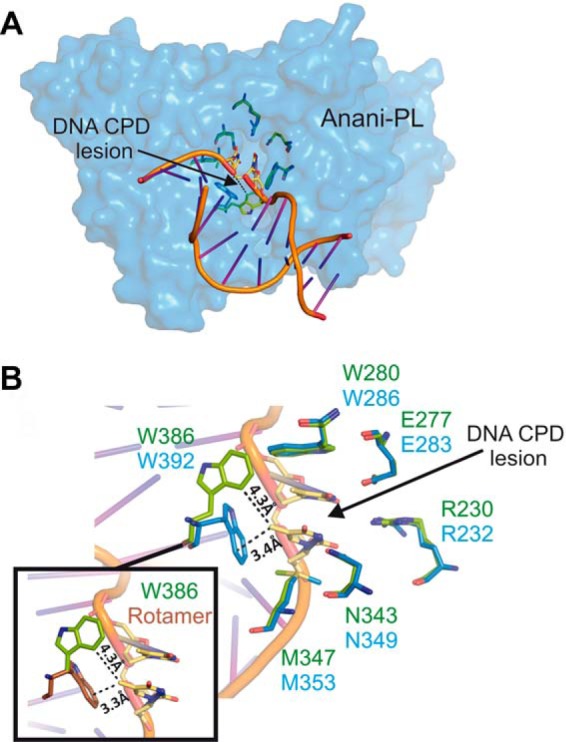
Putative CPD lesion binding of PhrA. A, surface presentation of the Anani-PL crystal structure (PDB entry 1TEZ) and bound DNA with CPD; the six lesion-interacting amino acids are shown in blue, homologous amino acids of PhrA in green. B, enlarged view of the lesion contact, color code as in A; note the different orientation of Trp-386/Trp-392 in PhrA/Anani-PL, respectively. Inset, Trp-386 of PhrA and a potential rotamer of Trp-386 (brown) that could interact with the CPD lesion in the same manner as Trp-392 of Anani-PL.
FAD Binding and Stabilization
In plant cryptochromes, FAD is either in the oxidized or semi-reduced state in vivo, whereas photolyases in the active state have a fully reduced FADH− chromophore. Among the 25 amino acids that interact with FAD in PhrA, 17 (68%) are strictly conserved with Arath-Cry1. This contrasts with the 38% identity of both full-length sequences. Four replacements are conservative and three of the other replacements are not specific, as the corresponding amino acids are not conserved in class III photolyases or plant cryptochromes. However, one important specific difference between the FAD binding pockets in both classes of proteins is the substitution of Asn-380 in PhrA, which forms a hydrogen bond with the N5 atom of FAD, by Asp-396 in Cry1. The possible effects of these two amino acid residues on the redox properties of FAD have been explained based on the fact that only Asp can act as an acid or base as required for FAD protonation/deprotonation and only Asp carries a negative charge that destabilizes FADH− (26, 60). It has indeed been shown that Asp-396 of Arath-Cry1 stabilizes FAD in the semiquinone state (61). One of the late steps in the transition of photolyases to plant cryptochromes regarding the FAD binding site is thus the conversion of this Asn to Asp, linked to the stabilization of semireduced FAD in the cryptochromes. A diatom cryptochrome CryP (62) that is phylogenetically related to CPD class III photolyases and plant cryptochromes (Fig. 1) has an Asn homologous to Asn-380 of PhrA, indicating that it might also function as photolyase.
MTHF Binding in PhrA
By UV-visible spectroscopy it has been found that PhrA incorporates MTHF as antenna chromophore (4), which is present in many other photolyases and the Cry-DASH protein Arath-Cry3. The identity of MTHF in PhrA was confirmed by the crystal structure: the electron density of the second organic cofactor next to FAD matches exactly with MTHF (Fig. 3, inset b). However, the PhrA crystal structure reveals a unique MTHF binding site different from other CPF structures: in Escco-PL and Arath-Cry3, MTHF is coordinated by amino acids from helices α2, α5, and α14 (20, 21, 58), and the distance between MTHF-N10 and FAD-N5 is 15–17 Å. In PhrA, MTHF is located on the opposite side relative to the isoalloxazine ring of FAD (Fig. 6A) and the distance between MTHF-N10 and FAD-N5 is 18.9 Å. The angle that is formed between the conjugated ring systems of MTHF and the isoalloxazine ring of FAD is similar to that found in Escco-PL. Thus, MTHF of PhrA is well suited for Förster energy transfer to FAD(H). The binding pocket in PhrA is formed by nine amino acids within helices α2, α7, α15, and α17 and one loop that connects helix α6 with α7 (Fig. 6B and Table 3). Leu-335, Trp-336, Asp-370, and Thr-371 shape a perfect pocket for the MTHF pterin ring system, which is stabilized by a hydrogen bonding network with backbone carbonyl groups of these residues. Trp-196 and Trp-336 form a double π-stacking sandwich with MTHF: their ring systems are located at 3.5-Å distances on each side of the four ring system of MTHF, with the conjugated systems of the three molecules were aligned in a parallel manner. The Trp/MTHF interactions could be the cause for a slightly red-shifted absorption maximum of MTHF in wild-type PhrA, which is at 390 nm (Fig. 7A) (4) versus that of Escco-PL, which is at 384 nm (63). Ala-50, Phe-200, and Gly-339 form additional hydrophobic interactions with the ring system of MTHF. The benzoyl-glutamic acid end of MTHF reaches out of the pocket, as in other photolyases, and is stabilized from one side by a hydrophobic interaction with Pro-194.
FIGURE 6.

MTHF binding sites of PhrA and other MTHF binding CPFs. A, MTHF chromophores of PhrA, E. coli CPD photolyase (Escco-PL, PDB entry 1DNP), and A. thaliana Cry3 (Arath-Cry3, PDB entry 2VTB). Distances between Asn-10 of MTHF and N5 of FAD are indicated. B, MTHF binding in PhrA. Potential hydrogen bonds or van der Waals contacts are shown as dashed lines. C, UV-visible spectra of wild type PhrA and the mutants W196A and W336A. D, putative MTHF cavity of plant cryptochrome, superposition of PhrA and A. thaliana Cry1 (Arath-Cry1, PDB entry 1U3C) with protein backbones drawn in green/blue/orange and gray, respectively. Relevant amino acids of Cry1 and the potential MTHF interacting amino acids of Arath-Cry1 are drawn as sticks; dashed lines indicated the same types of contacts as in C.
TABLE 3.
MTHF interacting amino acids of PhrA in CPD III, plant Cry, CryDASH, and CPD I subfamilies and diatom CryP
35 CPD III members, 22 plant Cry members, 85 CPD I and 15 CryDASH members were used for conservation analysis. For CryP (accession code EEC49202) (62), no other group members are known. A dash indicates that there is no counterpart residue at the certain position. Amino acids are shown by the three letter abbreviations, the numbers in the first line give the position within the PhrA sequence. Percentages show the fraction of identical amino acids. Amino acids found in other sequences are given in parentheses.
| Amino acids | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| PhrA | Ala-50 | Pro-194 | Trp-196 | Phe-200 | Leu-335 | Trp-336 | Gly-339 | Asn-370 | Thr-371 |
| CPDIII | Ala 97% (Val) | Pro 71% (Gly, Asn, Ile) | Trp 94% (Arg) | Leu 51% | Leu 94% (Met) | Trp 91% (Asn, Phe, Tyr) | Gly 100% | Asn 85% (Glu, Asn, Lys) | Thr 80% (Cys, Ala, His) |
| PlantCry | Val 72% | Lys 77% | Ser 100% | Leu 100% | Leu 90% (Ile, Ala) | Trp 95% (Leu) | Gly 100% | Asp 100% | Thr 80% (Ala, Val) |
| CryP | Ala | Thr | Trp | Ile | Leu | Tyr | Gly | Tyr | Thr |
| CPDI | – | – | Phe 29% | – | Leu 81% | Asn 47% | – | Gln 22% | His 24% |
| CryDASH | – | – | Val 20% | – | Leu 100% | Ala 26% | – | Thr 46% | Leu 20% |
FIGURE 7.
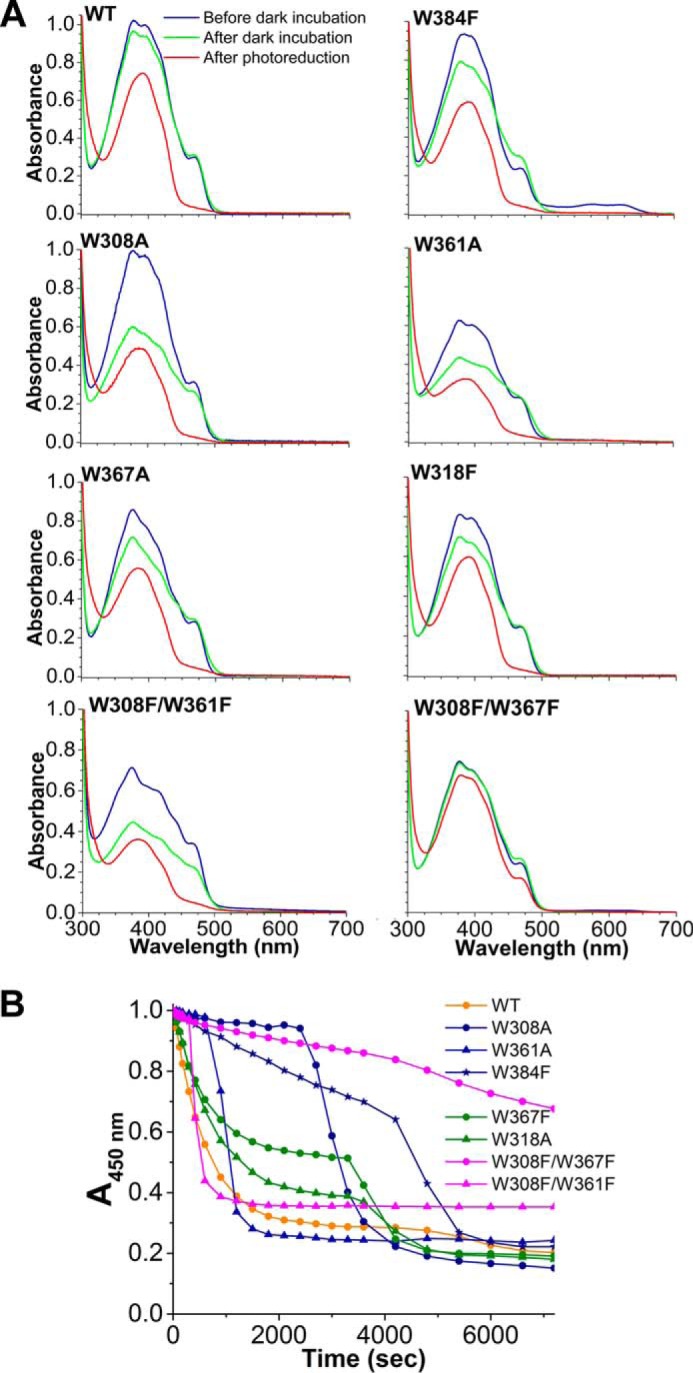
Photoreduction of PhrA. UV-visible spectral properties (A) and photoreduction of PhrA wild-type (WT) and mutated proteins (B). Absorption values at 450 nm were taken from UV-visible spectra measured at indicated time points upon onset of blue-light irradiation. For each protein, these values were normalized against the value measured at t = 0 min.
The strong double π-stacking interaction is exceptional for chromophores in the CPF. We therefore tested the role of either Trp by site-directed mutagenesis. The spectra of purified W196A and W336A mutants are devoid of the strong 390-nm absorption characteristic for MTHF and resemble those of free oxidized FAD (Fig. 6C). Thus, both Trp residues are required for efficient MTHF binding.
MTHF Binding in Other CPD Class III Photolyases
To investigate the conservation of MTHF binding amino acids in CPD class III photolyases, we checked sequences of those 35 members that led to the definition of this class (31). All MTHF coordinating residues except Phe-200 and Pro-194 are conserved in 80–97% of the proteins (Table 3). The two π-stacking residues, Trp-196 and Trp-336, are conserved in 33 and 32 of the 35 sequences, respectively. The results obtained with W196A or W336A mutants of PhrA suggests that CPD class III photolyases with one replacement of a Trp bind MTHF with a lower affinity or not at all. Taken together, we propose that the other 31 CPD class III photolyases incorporate MTHF within the same interaction region and similar interacting residues as PhrA.
Potential MTHF Binding Site in Plant and Diatom Cryptochrome
Plant cryptochromes probably bind MTHF or a similar molecule as antenna chromophore, but this chromophore is not present in the Arath-Cry1 crystal structure (25). The classical MTHF binding site that is found in e.g. E. coli photolyase, is completely blocked by internal amino acid side chains in the Cry1 structure (25). Because of their close relationship, plant cryptochromes might use the same MTHF binding site as PhrA. Indeed, a groove of proper size is found in Arath-Cry1 at the position of the PhrA-MTHF binding pocket. All four hydrogen bond forming residues including Trp-336 are highly conserved in plant cryptochromes and the hydrogen bond forming atoms are at the same positions in Arath-Cry1. The ring system of the Trp-336 homolog of Arath-Cry1 (Trp-352) is rotated out of the prospective binding pocket but one of the allowed side chain rotamers would be in the same position as in PhrA (Fig. 6D). The other π-stacking Trp of PhrA is replaced by a highly conserved Ser in plant cryptochromes (Ser-205 of Arath-Cry1) (Table 3). Again, we conclude that either MTHF may bind to the homologous binding pocket in plant cryptochromes but with weaker interaction than in PhrA, or that this cofactor does not bind at all to these proteins. Alternatively, the transition from the hydrophobic Trp in PhrA to a polar Ser in Cry1 (Ser-205 in Arath-Cry1, Fig. 6D) could be linked to a transition of the ligand from unpolar MTHF to another but similar ligand with a polar counterpart.
The diatom cryptochrome CryP carries an MTHF antenna chromophore (62). According to a homology model, the classical MTHF binding pocket of CryP is also blocked in CryP, whereas a cavity is found at the position of the PhrA MTHF binding pocket that is lined by several residues that are homologous to PhrA (Fig. 8). Of the two tryptophan residues that form the double π-stacking sandwich with MTHF in PhrA, Trp-196, is conserved in CryP, whereas Trp-336 is replaced by a tyrosine (Tyr-373 in CryP) that could serve a similar function in stabilizing the antenna chromophore in CryP. Thus, CryP could have the same MTHF binding site as PhrA.
FIGURE 8.
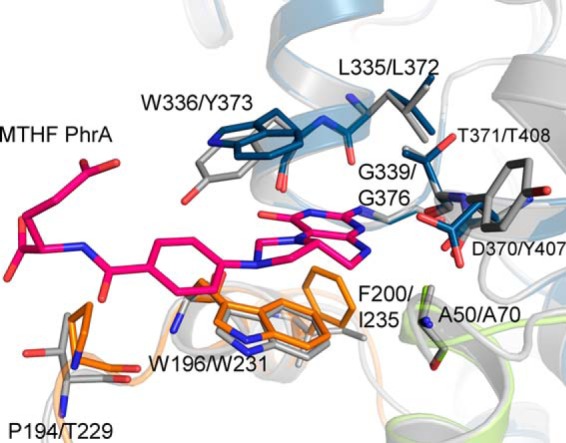
Proposed MTHF pocket of Phaeodactylum tricornutum CryP. The homology model, constructed with the Swissmodel server (16) using PhrA as template, is drawn in gray, the PhrA structure is drawn in green/blue/orange. Relevant amino acids are drawn as sticks and labeled; the first label refers to PhrA, the second to CryP.
Two Photoreduction Trp Triads in PhrA
In PhrA, the transition of FAD from the oxidized to the reduced form has been confirmed as a blue light-dependent process in vitro (4). PhrA has all three Trp residues of the classical pathway at positions 308, 361, and 384, which are located at edge-to-edge distances of 4.4, 4.8, and 4.0 Å to each other and to the isoalloxazine ring of FAD, respectively. The PhrA crystal structure revealed also a second putative electron pathway with Trp residues at positions 367, 318, and 384 (Fig. 9A) and edge-to-edge distances of 3.8, 3.8, and 4.0 Å, respectively. Remarkably, Trp-384 is shared by both triads and is at the same time the nearest one to FAD. To verify whether both Trp triads function as electron pathways for FAD photoreduction, we mutated each of the involved Trp to Ala or Phe and analyzed photoreduction through UV-visible spectroscopy.
FIGURE 9.
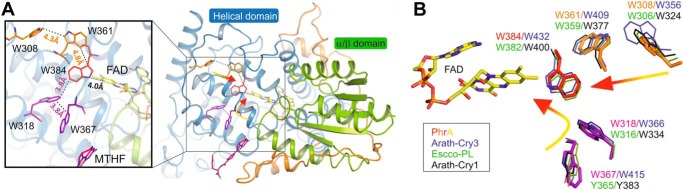
Trp triads of PhrA and comparison with other CPF structures. A, two Trp triads in PhrA. The classical Trp triad is presented in red and orange, Trp of the alternative pathway are drawn in magenta. B, superposition of the residues of PhrA Trp triads (color code as in A) with Escco-PL (green lines), Arath-Cry1 (black lines), and Arath-Cry3 (blue lines).
Whereas the wild-type and almost all mutant proteins had a bright yellow color after expression and purification under aerobic conditions, the color of the W384F mutant was greenish. This results from a strong absorption in the 510–560 nm range, which is indicative of semi-reduced FAD. Thus, the oxidation of FADH− is incomplete in W384F at this stage of purification. All other proteins showed insignificant absorption in this spectral range and have spectral characteristics of fully oxidized FAD (Fig. 7A). Electron flow during FAD oxidation is less well characterized than during photoreduction. The mutant spectra imply that Trp-384 plays a central role in the oxidation of FAD, possibly a structural role, but the reason for the slower oxidation of the mutant is unknown. Before the photoreduction measurements, all proteins were incubated overnight at 277 K. Thereafter, FAD of W384F was also in the oxidized state (Fig. 7A). In most mutants, this incubation also resulted in a partial decrease of the MTHF absorbance, which we regard as MTHF loss.
Photoreduction of the wild-type as characterized by the absorbance decrease at 450 nm was almost complete after 50 min irradiation. All proteins with a single mutation also showed such a decrease in absorbance (Fig. 7B), but the reduction was slower than in the wild-type and the reduction patterns had two kinetic components. The mutants of the classical pathway, W308A and W361A, have an initial delay phase of 45 and 15 min, respectively. The initial absorbance decrease of the mutants of the alternative pathway, W318F and W367A, was slightly slower than in the wild-type, whereas the initial decrease was very slow in W384F, a mutant in which according to our hypothesis both pathways are affected. The double mutant W308F/W361F (both amino acids within the classical pathway) is also characterized by an initial delay, which lasts, however, only a few minutes. Thereafter, a very rapid reduction was observed. The double mutant W308F/W367F in which the surface Trp residues of both triads were exchanged showed the slowest photoreduction. However, the rate was still not zero and the curve was again complex. In PhrA mutants, the second component could result from a light-induced protein conformational change that facilitates electron flow. Although these complex photoreduction curves are not clearly understood, our results show that both triads are required for normal photoreduction. The results also suggest that possibly a third pathway exists, because the W384F mutant and the W308F/W367F double mutant still undergo photoreduction. It has been reported that electrons can flow between the isoalloxazine and the adenine moiety of FAD of E. coli photolyase (15). Our structure shows that Trp-280, which is conserved among many CPD class I and class III photolyases as well as in FeS-bacterial cryptochromes and photolyase proteins, has a 5.4-Å edge to edge distance to the adenine ring system. Trp-280 forms a network with four other Trp residues (153, 256, 299, and 386) with edge to edge distances between 3.4 and 7.8 Å. All five Trp residues are located close to the surface. To what extent the residues of this network are possibly involved in light-activated electron transfer from a soluble reductant via the adenine moiety to electronically excited FADH− remains to be answered by further photoreduction studies on PhrA in combination with site-directed mutagenesis.
The Alternative Trp Triad in Other Classes of CPF
The classical photoreduction pathway is conserved in all class I photolyases, Cry-DASH proteins, cryptochromes, eukaryotic (6-4)-photolyases, and class III photolyases (64). We found Trp-367 and Trp-318 of the alternative electron pathway of PhrA conserved in 34 of 35 investigated class III photolyases. In the remaining sequence, the surface located Trp-367 is substituted by a Tyr. The alternative Trp triad is also conserved in 9 of 15 analyzed Cry-DASH members, including Arath-Cry3 (Fig. 9B) (PDB entry 2VTB, Trp-415-Trp-366-Trp-432). In Escco-PL and Arath-Cry1, Trp-318 of PhrA is conserved (Trp-316 and Trp-366, respectively), whereas Trp-367 of PhrA is replaced by a Tyr (Tyr-365 and Tyr-383, respectively). Of the 85 and 22 investigated CPD class I and plant Cry members, 57 and 95% have a Tyr counterpart, respectively. Given that Trp as well as Tyr can serve as electron transmitters in photolyases (65, 66), a large number of CPF members could have a second functional electron pathway. In Escco-PL, Trp-316 has indeed been shown to transmit electrons via the central Trp-382 to FAD (15), although the role of Tyr-365 has not been investigated so far.
Photolyase mutants in which Trp residues of the classical pathway are replaced are often characterized by a reduced or completely blocked in vitro photoreduction (12). These results speak for one pathway only. However, alternative pathways could be active under differing pH/redox conditions.
Conclusions
Although crystal structures of many photolyases and cryptochromes are available, structural information of CPD class III photolyases has been lacking. The PhrA structure closes this gap. Our data show that CPD class III photolyases harbor a unique antenna chromophore binding site and a Y-shaped Trp pathway for photoreduction. The antenna chromophore is sandwiched between two Trp residues with which it interacts by π-stacking. The close relationship with plant cryptochromes and structural coincidence suggests that these cryptochromes use the same binding site for their antenna chromophores. Thus, the PhrA structure provides a clue to the yet open question of the identity and binding site of plant cryptochrome antenna chromophores. The alternative photoreduction pathway of PhrA has homologs in many other photolyases and cryptochromes, either with Tyr or Trp residues at the surface.
Acknowledgments
We gratefully acknowledge Sybille Wörner, Jennifer Welschhof, and Ernst Heene for technical assistance. We thank Isabel Molina and Benjamin Zienicke for fruitful discussions. We are grateful to the scientific staff of the European Synchrotron Radiation Facility (ESRF, Grenoble) at beamlines ID14-1, ID23-1, ID23-2, ID29, and ID 14-4, and Uwe Müller, Manfred Weiss, and the scientific staff of the BESSY-MX/Helmholtz Zentrum Berlin für Materialien und Energie at beamlines BL14.1, BL14.2, and BL14.3 operated by the Joint Berlin MX-Laboratory at the BESSY II electron storage ring (Berlin-Adlershof, Germany), where the data were collected, for continuous support. We thank Antoine Royant for extensive support with the usage of the microspectrophotometer on the crystals at the ID29S-Cryobench (ESRF, Grenoble).
This work was supported by Deutsche Forschungsgemeinschaft (DFG) Grant 799/8-1 (to T. L.), and SFB 740-B6 and SFB 1078-B6 (to P. S.), and grants from the China Scholarship Council (to F. Z.), Karlsruhe House of Young Scientists (to F. Z.), and DFG Cluster of Excellence “Unifying Concepts in Catalysis” Research Field D3/E3-1 (to P. S.).
The atomic coordinates and structure factors (code 4U63) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CPD
- cyclobutane pyrimidine dimer
- MTHF
- 5,10-methenyltetrahydrofolate
- CPF
- cryptochrome/photolyase family
- r.m.s.
- root mean square
- Arath-Cry1
- A. thaliana cryptochrome 1
- Arath-Cry2
- A. thaliana cryptochrome 2
- Anani-PL
- A. nidulans photolyase
- Escco-PL
- E. coli photolyase
- FeS-BCP
- FeS bacterial cryptochrome and photolyases.
REFERENCES
- 1. Sancar A. (2003) Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem. Rev. 103, 2203–2237 [DOI] [PubMed] [Google Scholar]
- 2. Müller M., Carell T. (2009) Structural biology of DNA photolyases and cryptochromes. Curr. Opin. Struct. Biol. 19, 277–285 [DOI] [PubMed] [Google Scholar]
- 3. Chaves I., Pokorny R., Byrdin M., Hoang N., Ritz T., Brettel K., Essen L. O., van der Horst G. T., Batschauer A., Ahmad M. (2011) The cryptochromes: blue light photoreceptors in plants and animals. Annu. Rev. Plant Biol. 62, 335–364 [DOI] [PubMed] [Google Scholar]
- 4. Oberpichler I., Pierik A. J., Wesslowski J., Pokorny R., Rosen R., Vugman M., Zhang F., Neubauer O., Ron E. Z., Batschauer A., Lamparter T. (2011) A photolyase-like protein from Agrobacterium tumefaciens with an iron-sulfur cluster. Plos One 6, e26775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Selby C. P., Sancar A. (2006) A cryptochrome/photolyase class of enzymes with single-stranded DNA-specific photolyase activity. Proc. Natl. Acad. Sci. U.S.A. 103, 17696–17700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu Z., Tan C., Guo X., Kao Y. T., Li J., Wang L., Sancar A., Zhong D. (2011) Dynamics and mechanism of cyclobutane pyrimidine dimer repair by DNA photolyase. Proc. Natl. Acad. Sci. U.S.A. 108, 14831–14836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hoang N., Schleicher E., Kacprzak S., Bouly J. P., Picot M., Wu W., Berndt A., Wolf E., Bittl R., Ahmad M. (2008) Human and Drosophila cryptochromes are light activated by flavin photoreduction in living cells. PLoS Biol. 6, e160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kiontke S., Geisselbrecht Y., Pokorny R., Carell T., Batschauer A., Essen L. O. (2011) Crystal structures of an archaeal class II DNA photolyase and its complex with UV-damaged duplex DNA. EMBO J. 30, 4437–4449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hitomi K., Arvai A. S., Yamamoto J., Hitomi C., Teranishi M., Hirouchi T., Yamamoto K., Iwai S., Tainer J. A., Hidema J., Getzoff E. D. (2012) Eukaryotic class II cyclobutane pyrimidine dimer photolyase structure reveals basis for improved ultraviolet tolerance in plants. J. Biol. Chem. 287, 12060–12069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Geisselbrecht Y., Frühwirth S., Schroeder C., Pierik A. J., Klug G., Essen L. O. (2012) CryB from Rhodobacter sphaeroides: a unique class of cryptochromes with new cofactors. EMBO Rep. 13, 223–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang F., Scheerer P., Oberpichler I., Lamparter T., Krauß N. (2013) Crystal structure of a prokaryotic (6–4) photolyase with an Fe-S cluster and a 6,7-dimethyl-8-ribityllumazine antenna chromophore. Proc. Natl. Acad. Sci. U.S.A. 110, 7217–7222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li Y. F., Heelis P. F., Sancar A. (1991) Active site of DNA photolyase: tryptophan-306 is the intrinsic hydrogen atom donor essential for flavin radical photoreduction and DNA repair in vitro. Biochemistry 30, 6322–6329 [DOI] [PubMed] [Google Scholar]
- 13. Li X., Wang Q., Yu X., Liu H., Yang H., Zhao C., Liu X., Tan C., Klejnot J., Zhong D., Lin C. (2011) Arabidopsis cryptochrome 2 (CRY2) functions by the photoactivation mechanism distinct from the tryptophan (trp) triad-dependent photoreduction. Proc. Natl. Acad. Sci. U.S.A. 108, 20844–20849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Biskup T., Paulus B., Okafuji A., Hitomi K., Getzoff E. D., Weber S., Schleicher E. (2013) Variable electron transfer pathways in an amphibian cryptochrome: tryptophan versus tyrosine-based radical pairs. J. Biol. Chem. 288, 9249–9260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu Z., Tan C., Guo X., Li J., Wang L., Sancar A., Zhong D. (2013) Determining complete electron flow in the cofactor photoreduction of oxidized photolyase. Proc. Natl. Acad. Sci. U.S.A. 110, 12966–12971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Cassarino T. G., Bertoni M., Bordoli L., Schwede T. (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Klar T., Kaiser G., Hennecke U., Carell T., Batschauer A., Essen L. O. (2006) Natural and non-natural antenna chromophores in the DNA photolyase from Thermus thermophilus. Chembiochem 7, 1798–1806 [DOI] [PubMed] [Google Scholar]
- 18. Tamada T., Kitadokoro K., Higuchi Y., Inaka K., Yasui A., de Ruiter P. E., Eker A. P., Miki K. (1997) Crystal structure of DNA photolyase from Anacystis nidulans. Nat. Struct. Biol. 4, 887–891 [DOI] [PubMed] [Google Scholar]
- 19. Fujihashi M., Numoto N., Kobayashi Y., Mizushima A., Tsujimura M., Nakamura A., Kawarabayasi Y., Miki K. (2007) Crystal structure of archaeal photolyase from Sulfolobus tokodaii with two FAD molecules: implication of a novel light-harvesting cofactor. J. Mol. Biol. 365, 903–910 [DOI] [PubMed] [Google Scholar]
- 20. Park H. W., Kim S. T., Sancar A., Deisenhofer J. (1995) Crystal structure of DNA photolyase from Escherichia coli. Science 268, 1866–1872 [DOI] [PubMed] [Google Scholar]
- 21. Huang Y., Baxter R., Smith B. S., Partch C. L., Colbert C. L., Deisenhofer J. (2006) Crystal structure of cryptochrome 3 from Arabidopsis thaliana and its implications for photolyase activity. Proc. Natl. Acad. Sci. U.S.A. 103, 17701–17706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hoang N., Bouly J. P., Ahmad M. (2008) Evidence of a light-sensing role for folate in Arabidopsis cryptochrome blue-light receptors. Mol. Plant 1, 68–74 [DOI] [PubMed] [Google Scholar]
- 23. Malhotra K., Kim S. T., Batschauer A., Dawut L., Sancar A. (1995) Putative blue-light photoreceptors from Arabidopsis thaliana and Sinapis alba with a high degree of sequence homology to DNA photolyase contain the two photolyase cofactors but lack DNA repair activity. Biochemistry 34, 6892–6899 [DOI] [PubMed] [Google Scholar]
- 24. Selby C. P., Sancar A. (2012) The second chromophore in Drosophila photolyase/cryptochrome family photoreceptors. Biochemistry 51, 167–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brautigam C. A., Smith B. S., Ma Z., Palnitkar M., Tomchick D. R., Machius M., Deisenhofer J. (2004) Structure of the photolyase-like domain of cryptochrome 1 from Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 101, 12142–12147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kottke T., Batschauer A., Ahmad M., Heberle J. (2006) Blue-light-induced changes in Arabidopsis cryptochrome 1 probed by FTIR difference spectroscopy. Biochemistry 45, 2472–2479 [DOI] [PubMed] [Google Scholar]
- 27. Komori H., Masui R., Kuramitsu S., Yokoyama S., Shibata T., Inoue Y., Miki K. (2001) Crystal structure of thermostable DNA photolyase: pyrimidine-dimer recognition mechanism. Proc. Natl. Acad. Sci. U.S.A. 98, 13560–13565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maul M. J., Barends T. R., Glas A. F., Cryle M. J., Domratcheva T., Schneider S., Schlichting I., Carell T. (2008) Crystal structure and mechanism of a DNA (6–4) photolyase. Angew. Chem. Int. Ed. Engl. 47, 10076–10080 [DOI] [PubMed] [Google Scholar]
- 29. Mees A., Klar T., Gnau P., Hennecke U., Eker A. P., Carell T., Essen L. O. (2004) Crystal structure of a photolyase bound to a CPD-like DNA lesion after in situ repair. Science 306, 1789–1793 [DOI] [PubMed] [Google Scholar]
- 30. Glas A. F., Schneider S., Maul M. J., Hennecke U., Carell T. (2009) Crystal structure of the T(6–4)C lesion in complex with a (6–4) DNA photolyase and repair of UV-induced (6–4) and Dewar photolesions. Chemistry 15, 10387–10396 [DOI] [PubMed] [Google Scholar]
- 31. Oztürk N., Kao Y. T., Selby C. P., Kavakli I. H., Partch C. L., Zhong D., Sancar A. (2008) Purification and characterization of a type III photolyase from Caulobacter crescentus. Biochemistry 47, 10255–10261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. de Sanctis D., Beteva A., Caserotto H., Dobias F., Gabadinho J., Giraud T., Gobbo A., Guijarro M., Lentini M., Lavault B., Mairs T., McSweeney S., Petitdemange S., Rey-Bakaikoa V., Surr J., Theveneau P., Leonard G. A., Mueller-Dieckmann C. (2012) ID29: a high-intensity highly automated ESRF beamline for macromolecular crystallography experiments exploiting anomalous scattering. J. Synchrotron Radiat. 19, 455–461 [DOI] [PubMed] [Google Scholar]
- 33. Mueller U., Darowski N., Fuchs M. R., Förster R., Hellmig M., Paithankar K. S., Pühringer S., Steffien M., Zocher G., Weiss M. S. (2012) Facilities for macromolecular crystallography at the Helmholtz-Zentrum Berlin. J. Synchrotron Radiat. 19, 442–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kabsch W. (2010) Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Collaborative Computational Project, Number 4. (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 36. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 37. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Winn M. D., Isupov M. N., Murshudov G. N. (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57, 122–133 [DOI] [PubMed] [Google Scholar]
- 40. Vagin A. A., Steiner R. A., Lebedev A. A., Potterton L., McNicholas S., Long F., Murshudov G. N. (2004) REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr. 60, 2184–2195 [DOI] [PubMed] [Google Scholar]
- 41. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cowtan K., Emsley P., Wilson K. S. (2011) From crystal to structure with CCP4 introduction. Acta Crystallogr. D Biol. Crystallogr. 67, 233–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 44. Vaguine A. A., Richelle J., Wodak S. J. (1999) SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr. D Biol. Crystallogr. 55, 191–205 [DOI] [PubMed] [Google Scholar]
- 45. Laskowski R. A., Moss D. S., Thornton J. M. (1993) Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol. 231, 1049–1067 [DOI] [PubMed] [Google Scholar]
- 46. Hooft R. W., Vriend G., Sander C., Abola E. E. (1996) Errors in protein structures. Nature 381, 272. [DOI] [PubMed] [Google Scholar]
- 47. Vriend G. (1990) WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 8, 52–6, 29 [DOI] [PubMed] [Google Scholar]
- 48. Lovell S. C., Davis I. W., Arendall W. B., 3rd, de Bakker P. I., Word J. M., Prisant M. G., Richardson J. S., Richardson D. C. (2003) Structure validation by Cα geometry: φ,ψ and Cβ deviation. Proteins 50, 437–450 [DOI] [PubMed] [Google Scholar]
- 49. McDonald I. K., Thornton J. M. (1994) Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238, 777–793 [DOI] [PubMed] [Google Scholar]
- 50. Laskowski R. A., Swindells M. B. (2011) LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model 51, 2778–2786 [DOI] [PubMed] [Google Scholar]
- 51. DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific, San Carlos, CA [Google Scholar]
- 52. Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F., Higgins D. G. (1997) The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Royant A., Carpentier P., Ohana J., McGeehan J., Paetzold B., Noirclerc-Savoye M., Vernede X., Adam V., Bourgeois D. (2007) Advances in spectroscopic methods for biological crystals: 1. fluorescence lifetime measurements. J. Appl. Crystallogr. 40, 1105–1112 [Google Scholar]
- 56. Kort R., Komori H., Adachi S., Miki K., Eker A. (2004) DNA apophotolyase from Anacystis nidulans: 1.8-Å structure, 8-HDF reconstitution and x-ray-induced FAD reduction. Acta Crystallogr. D Biol. Crystallogr. 60, 1205–1213 [DOI] [PubMed] [Google Scholar]
- 57. Holm L., Sander C. (1993) Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233, 123–138 [DOI] [PubMed] [Google Scholar]
- 58. Klar T., Pokorny R., Moldt J., Batschauer A., Essen L. O. (2007) Cryptochrome 3 from Arabidopsis thaliana: structural and functional analysis of its complex with a folate light antenna. J. Mol. Biol. 366, 954–964 [DOI] [PubMed] [Google Scholar]
- 59. Pokorny R., Klar T., Hennecke U., Carell T., Batschauer A., Essen L. O. (2008) Recognition and repair of UV lesions in loop structures of duplex DNA by DASH-type cryptochrome. Proc. Natl. Acad. Sci. U.S.A. 105, 21023–21027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Balland V., Byrdin M., Eker A. P., Ahmad M., Brettel K. (2009) What makes the difference between a cryptochrome and DNA photolyase? a spectroelectrochemical comparison of the flavin redox transitions. J. Am. Chem. Soc. 131, 426–427 [DOI] [PubMed] [Google Scholar]
- 61. Burney S., Wenzel R., Kottke T., Roussel T., Hoang N., Bouly J. P., Bittl R., Heberle J., Ahmad M. (2012) Single amino acid substitution reveals latent photolyase activity in Arabidopsis cry1. Angew. Chem. Int. Ed. Engl. 51, 9356–9360 [DOI] [PubMed] [Google Scholar]
- 62. Juhas M., von Zadow A., Spexard M., Schmidt M., Kottke T., Büchel C. (2014) A novel cryptochrome in the diatom Phaeodactylum tricornutum influences the regulation of light-harvesting protein levels. FEBS J. 281, 2299–2311 [DOI] [PubMed] [Google Scholar]
- 63. Payne G., Sancar A. (1990) Absolute action spectrum of E-FADH2 and E-FADH2-MTHF forms of Escherichia coli DNA photolyase. Biochemistry 29, 7715–7727 [DOI] [PubMed] [Google Scholar]
- 64. Liu B., Liu H., Zhong D., Lin C. (2010) Searching for a photocycle of the cryptochrome photoreceptors. Curr. Opin. Plant Biol. 13, 578–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Aubert C., Mathis P., Eker A. P., Brettel K. (1999) Intraprotein electron transfer between tyrosine and tryptophan in DNA photolyase from Anacystis nidulans. Proc. Natl. Acad. Sci. U.S.A. 96, 5423–5427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Aubert C., Vos M. H., Mathis P., Eker A. P., Brettel K. (2000) Intraprotein radical transfer during photoactivation of DNA photolyase. Nature 405, 586–590 [DOI] [PubMed] [Google Scholar]
- 67. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]