

Background: Viral suppressors of RHIM-dependent activation of pro-necrotic RIP3 kinase are crucial for successful infection in mice.
Results: Human CMV blocks TNF-induced and murine CMV-induced necroptosis after RIP3 activation.
Conclusion: Necrotic membrane leakage is blocked in infected cells despite the activation of MLKL.
Significance: Viral inhibition of necroptosis will facilitate understanding of the final steps in this pathway.
Keywords: Caspase, Herpesvirus, Innate Immunity, Necrosis (Necrotic Death), Tumor Necrosis Factor (TNF), Human CMV, MCMV, Programmed Necrosis, Regulated Cell Death
Abstract
Necroptosis is an alternate programmed cell death pathway that is unleashed by caspase-8 compromise and mediated by receptor-interacting protein kinase 3 (RIP3). Murine cytomegalovirus (CMV) and herpes simplex virus (HSV) encode caspase-8 inhibitors that prevent apoptosis together with competitors of RIP homotypic interaction motif (RHIM)-dependent signal transduction to interrupt the necroptosis. Here, we show that pro-necrotic murine CMV M45 mutant virus drives virus-induced necroptosis during nonproductive infection of RIP3-expressing human fibroblasts, whereas WT virus does not. Thus, M45-encoded RHIM competitor, viral inhibitor of RIP activation, sustains viability of human cells like it is known to function in infected mouse cells. Importantly, human CMV is shown to block necroptosis induced by either TNF or M45 mutant murine CMV in RIP3-expressing human cells. Human CMV blocks TNF-induced necroptosis after RIP3 activation and phosphorylation of the mixed lineage kinase domain-like (MLKL) pseudokinase. An early, IE1-regulated viral gene product acts on a necroptosis step that follows MLKL phosphorylation prior to membrane leakage. This suppression strategy is distinct from RHIM signaling competition by murine CMV or HSV and interrupts an execution process that has not yet been fully elaborated.
Introduction
Regulated cell death is important for eliminating cells during development and to guard the host from infection with microbial or viral pathogens (1–3). The mitochondrial pathway of apoptosis, mediated by the Bcl2 family proteins, contributes to both development and host defense. In humans, Casp84 and Casp10 initiate apoptosis triggered through TNF family death receptors, although this pathway may switch to an alternate receptor-interacting protein kinase 3 (RIP3)-dependent programmed necrosis outcome, called necroptosis, in cells with adequate levels of this pro-necrotic kinase (2, 4–7). In mice, Casp8-dependent apoptosis and RIP3-dependent necroptosis contribute directly to host defense (6) and are together dispensable for development (8). Programmed necrosis was initially demonstrated in cultured cells subjected to Casp8 compromise after death receptor activation (9, 10). The RIP3-mediated pathway was implicated in the innate immune response to vaccinia infection (11) and was shown to be physiologically relevant against the natural mouse pathogen murine cytomegalovirus (CMV). This betaherpesvirus specifically blocks necroptosis to sustain infection (12, 13). In addition to death receptors and virus infection, Toll-like receptor (TLR) activation and interferon-dependent innate immune signaling unleash necroptosis (14–16). Although there are strong parallels in all of these settings (17), most of the mechanistic understanding of this pathway derives from studies on TNF receptor 1 (TNFR1) signaling (8, 11, 18–23).
Although the host defense contribution of necroptosis first emerged from studies of vaccinia (11, 24) as well as reovirus (25), it was the elaboration of murine CMV M45-encoded viral inhibitor of RIP activation (vIRA) that provided unambiguous evidence showing the key role of necroptosis in mammalian host defense (12, 13). vIRA blocks RIP homotypic interaction motif (RHIM)-dependent interactions (26, 27) between RIP3 and other RHIM-containing adapters, RIP1 (11, 22, 23), DNA inducer of interferon (DAI) (12, 13), and TIR domain-containing adapter-inducing interferon β (TRIF) (17, 28). Casp8 is the well established mediator of apoptosis downstream of death receptors, such as TNFR1, and TLRs, such as TLR3 (11, 22, 23, 29). Necroptosis is mediated by a RHIM-dependent RIP1-RIP3 interaction activated downstream of TNFR1 (11, 22, 23), DAI-RIP3 interaction during murine CMV infection (12, 13), and TRIF-RIP3 interaction following TLR3 engagement (17, 28). A Casp8-containing ripoptosome forms from preexisting cytosolic components (14) to regulate extrinsic apoptosis and RIP3 kinase-dependent phosphorylation events necessary for the execution of necroptosis (17). This signaling complex has recently been implicated in a novel form of RIP3 RHIM-dependent apoptosis (30). In necroptosis, RIP3 kinase activity drives phosphorylation of and interaction with mixed lineage kinase domain-like (MLKL) pseudokinase (31, 32) to form a necrosome (33). These events commit cells to membrane leakage and death, although the mechanistic details of execution remain open (34–36). Casp8 also contributes to Casp1 activation and proinflammatory IL-1β production (37–39). Altogether, three important points have emerged from recent studies on necroptosis (2, 4–7). (i) Large DNA viruses sensitize cells to necroptosis by encoding inhibitors of Casp8 (8, 11, 24, 40). (ii) Murine CMV encodes vIRA, a natural RHIM signaling competitor to prevent necroptosis by blocking DAI-RIP3 complex formation independent of RIP1 in mice (12, 13). (iii) The human pathogens herpes simplex virus (HSV1) and HSV2 encode inhibitors of Casp8 activation that also suppress RHIM signaling in human cells (41). All known RIP3 kinase-dependent necrotic death pathways require RHIM-mediated signal transduction to converge on common phosphorylation of the pseudokinase MLKL (17, 34, 42), and all are blocked by vIRA (6).
Murine CMV M45-encoded vIRA also suppresses RHIM-dependent apoptosis (27, 43, 44) and regulates NF-κB activation (26) independent of virus infection; however, the physiological importance of vIRA during infection is directed at necroptosis (12, 13), which would otherwise eliminate infected host cells and halt infection (45, 46). Thus, M45mutRHIM virus infection fails to infect either immunocompetent or immunodeficient mice (12, 47) but proceeds in Rip3−/− or Dai−/− (also called Zbp1−/−) mice where other immune control mechanisms remain operational. M45 carries out activities that are independent of RHIM function, increasing RIP1-dependent NF-κB activation immediately following infection and preventing NEMO-mediated activation of NF-κB later in infection (43, 48, 49). Recently, the RHIM-targeted suppressor strategy has been extended to HSV, where the large subunit of ribonucleotide reductase (R1) suppresses necroptosis in human cells (41) in addition to its role in suppressing Casp8-dependent apoptosis (50). Surprisingly, HSV triggers necroptosis in mouse cells, and this vulnerability is influenced by R1 RHIM signaling (51, 52). In contrast, murine CMV vIRA suppresses cell death in both mouse and human cells (41). Expression of the HSV R1 during infection blocks Casp8 (50) and exposes cells to necroptosis (41), a dramatic interplay between one viral gene product and cognate-regulated cell death pathways (6, 47). Human CMV UL36 (53) and murine CMV M36 (54, 55) encode the viral inhibitor of Casp8 activation (vICA) (56, 57). Casp8 suppression prevents apoptosis and is necessary for successful infection of monocyte-derived cell lineages (58) that control viral dissemination (59). In addition, both murine and human CMV encode evolutionarily conserved suppressors that prevent activation of pro-apoptotic Bcl2 family members Bax and Bak (60–66). These viral countermeasures reinforce the central role of cell death pathways in host defense against infection (6, 58, 67, 68). In addition, the human CMV major immediate early 1 (IE1) gene product has been associated with cell death suppression (69) and plays a crucial role regulating levels of viral and host cell gene expression (70). An evolutionary dialogue seems to have played out over hundreds of millions of years between successful herpesviruses and their hosts such that perturbation of Casp8 and RIP3 may contribute to inflammatory disease driven by infectious and genetic insults (71, 72).
Here, we demonstrate that human CMV blocks necroptosis induced by either TNFR1 activation or murine CMV infection in permissive human fibroblasts transduced with human RIP3 (hRIP3). RIP3 transduction confers sensitivity to necroptosis that is lost during cell propagation in culture, a condition that is reminiscent of cultured mouse fibroblasts (11, 12). Human CMV confers IE1-dependent suppression of necroptosis at a step that follows RIP3 phosphorylation and activation of MLKL. This death is independent of the viral UL138 modulation of TNF signaling (73, 74). Thus, human CMV prevents necroptosis through a mechanism distinct from either murine CMV or HSV. We establish the core parameters through which this alternate form of extrinsic cell death contributes to host defense in humans and add to the list of evolutionarily ancient, common cell signaling pathways against which human CMV deploys countermeasures.
EXPERIMENTAL PROCEDURES
Reagents
Dimethyl sulfoxide (DMSO), cycloheximide, and phosphonoformate were purchased from Sigma. Necrostatin-1 and MG132 were purchased from Calbiochem. Recombinant human TNF and caspase inhibitor Z-VAD-fmk were purchased from R&D Systems and Enzo Life Sciences, respectively. λ-Protein phosphatase was purchased from New England Biolabs. IAP antagonist/Smac mimetic BV6 was a gift from D. Vucic (Genentech, Inc.). RIP3 kinase inhibitor GSK′840 was a kind gift from P. Gough (GlaxoSmithKline). The following antibodies were used in cell death assays, immunoblot (IB), and immunofluorescence analysis (IFA): anti-Fas antibody (clone 7C11, Beckman Coulter), rabbit anti-RIP3 (ab72106, Abcam), rabbit anti-MLKL (clone 58-70, Sigma), rabbit anti-MLKL(phospho-Ser-358) (ab187091, Abcam), mouse anti-RIP1 (clone 38, BD Biosciences), mouse anti-FLAG (clone M2, Sigma), anti-Myc (clone 9E10, Santa Cruz Biotechnology), mouse anti-human CMV IE1/IE2 (clone 8B1.2, Millipore), mouse anti-pp65 (clone CH12, Virusys), mouse anti-murine CMV IE1 (Croma 101, gift from S. Jonjic, University of Rijeka, Croatia), rabbit anti-M45 (gift from David Lembo, University of Turin, Italy), mouse anti-β-actin (clone AC-74, Sigma-Aldrich), peroxidase-labeled horse anti-mouse or anti-rabbit IgG (Vector Laboratories), Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen), and Alexa Fluor 568-conjugated goat anti-mouse IgG (Invitrogen). Anti-FLAG M2 affinity gel (Sigma) was used for IP experiments.
Plasmids, Transfection, and Transduction
To create hRIP3-expressing lentiviral vector, hRIP3 open reading frame (ORF) was inserted into pLV-EF1α-MCS-IRES-Puro lentiviral vector (Biosettia). Three-tandem FLAG epitope-tagged hRIP3 expression plasmid was constructed by inserting hRIP3 ORF into p3xFLAG-CMV10 vector (Sigma). Overlap extension PCR was employed to generate expression constructs of hRIP3 mutants, tetra-Ala RHIM domain (amino acids 459–462) substitution (mutRHIM), K50A, S227A, and K50A plus tetra-Ala RHIM domain substitution (K50mutRHIM). For hMLKL knockdown experiments, puromycin cassettes in pLKO.1-hMLKL (32) or the pLKO.1 control scramble shRNA expressing lentiviral vector (75) were swapped with the existing hygromycin cassette. All plasmids were verified by DNA sequencing. The M45-Myc and mRIP3 expression constructs in pTag5A (Stratagene) and p3xFLAG-CMV10 (Sigma), respectively, were reported previously (27). Transient transfections were performed with Lipofectamine LTX with Plus reagent (Invitrogen). Lentivirus stock was prepared from 293T cells that were transfected with pLV-hRIP3 or pLKO.1 constructs along with psPAX2 (76) and VSV-G-expressing plasmids. Low passage newborn foreskin fibroblasts (HFs) were transduced with lentiviral vector and selected with 0.5 μg/ml puromycin and/or 25 μg/ml hygromycin (Invitrogen).
Cells and Viruses
HFs and ihf-ie1.3, 293T, and 3T3-SA cells were cultured as described (12, 70, 77). HT-29, IMR-90, and human microvascular endothelial (HMEC-1) cells were maintained in DMEM containing 4.5 g/ml glucose, 10% fetal bovine serum (Atlanta Biologicals), 2 mm l-glutamine, 100 units/ml penicillin, and 100 units/ml streptomycin (Invitrogen). THP-1 cells from A. Kowalczyk (Emory University) were cultured and induced differentiation to macrophages as described previously (57). Human CMV strains were chosen to represent known genetic variants that arise during propagation in cell culture. Laboratory strain Towne-BAC, TowneΔUL36, IE1-null virus (CR208), and its repaired virus (CRQ208) were described previously, as was the parental virus TownevarRIT3 used in their preparation (57, 70, 78). Laboratory strain AD169varATCC and low passage strain Merlin strain were obtained from American Type Culture Collection, variant AD169-BAC was from J. Munger (Rochester University, New York, USA), variant AD169varDE was from M. Mach (Institute for Virologie, Erlangen, Germany), low passage strain Toledo was from S. Plotkin (Philadelphia, Pennsylvania, USA), and low passage strain endotheliotropic TB40E-BAC4 was from C. Sinzger (Institute for Virology, Tubingen, Germany). In experiments utilizing UV-irradiated virus, the viral suspension was exposed to 254-nm light at 360 mJ/cm2 with a model XL-1500 Spectrolinker UV cross-linker (Spectronics Corp.) prior to infection. Virus titers were determined by a plaque assay on HFs or ihf-ie1.3 cells. BAC-derived parental murine CMV K181-BAC and M45mutRHIM viruses were described previously (12). All human or murine CMV infections were performed at an MOI of 3 or 10, and cells were incubated for 1 h with virus before changing the medium.
Cell Viability Assay
Cells (5,000 cells/well) were seeded into Corning 96-well tissue culture plates. 16–24 h postseeding, medium was replaced with 50 μl of viral inoculum. Alternatively, cells were treated with the indicated reagents, and solvent, DMSO, was kept constant for all experiments. Unless otherwise indicated, 30 ng/ml TNF, 5 μm BV6, and/or 25 μm Z-VAD-fmk were used. The concentration of each reagent was optimized to exhibit specific efficacy without detectable cytotoxicity. Cell viability was assessed by measuring the intracellular levels of ATP using the Cell Titer-Glo luminescent cell viability assay kit (Promega) according to the manufacturer's instructions. Luminescence was measured on a Synergy HT multidetection microplate reader (BioTek) (12). For real-time imaging of cell permeability with IncuCyte Zoom (Essen Bioscience), cells (15,000 cells/well) were seeded into Corning 48-well tissue culture plates and cultured in medium containing 50 nm SYTOX Green (Molecular Probes) or 5 μm propidium iodide (PI) (Sigma) (15).
Immunoblot and Immunoprecipitation
IB and IP were performed as described previously (77, 79). Clarified cell lysates were incubated overnight with anti-FLAG M2 affinity gel and washed four times prior to analysis. λ-Phosphatase treatment was done according to the manufacturer's instructions. Cell lysates and IP samples were separated on an SDS-polyacrylamide gel, followed by transfer to a polyvinylidene difluoride membrane (Immobilon, Millipore), probed with primary antibodies, incubated with the HRP-conjugated secondary antibody, and detected with ECL Western blotting detection reagent (GE Healthcare).
Microscopy
IFA and transmission electron microscopy (TEM) were performed as described previously (77, 79). For IFA, cells were fixed in 3.7% formaldehyde and incubated in blocking buffer containing 0.5% bovine serum albumin and 5% goat serum. After incubation with rabbit anti-RIP3 or mouse anti-murine CMV IE1, goat anti-rabbit IgG conjugated to Alexa Fluor 488 or goat anti-mouse IgG conjugated to Alexa Fluor 568 was added, respectively. The cells were incubated with DAPI (Roche Applied Science) to stain nuclei. Images were acquired on an LSM 510 Meta confocal fluorescence microscope (Carl Zeiss). For TEM, cells were fixed in 2.5% glutaraldehyde in 0.1 m cacodylate buffer (pH 7.2). The cells were washed in 0.1 m cacodylate buffer, postfixed with the same buffer with 1% osmium tetroxide, washed, and dehydrated through a graded series of ethanol to 100% and embedded in epoxy resin. The sections were counterstained with uranyl acetate and lead citrate. Images were acquired on a JEOL JEM-1210 transmission electron microscope operated at 75 kV.
Statistical Analyses
Statistical comparisons employed parametric evaluation using Student's t test (GraphPad Prism software). All experiments were repeated at least three times with similar results, and data are represented as the mean ± S.D.
RESULTS
Necroptosis Sensitivity of Human CMV-susceptible Cells
We initially sought to identify human CMV-susceptible cells capable of supporting RIP3-dependent death. Human colon carcinoma HT-29 cells showed the expected pattern of apoptosis when treated with TNF (T) together with BV6 (S), an IAP antagonist (also called a SMAC mimetic) that reduces polyubiquitination of RIP1 and increases sensitivity to cell death (80), and the death pattern switched to necroptosis, as expected, when the broad caspase inhibitor Z-VAD-fmk was employed (V) (22, 41). When evaluated under similar conditions, CMV-susceptible HFs, HMEC-1 cells, and THP-1-derived macrophage-like cells were insensitive to T alone, T + S, or T + S + V (Fig. 1A). Like HFs, CMV-susceptible, commercially available IMR-90 fetal lung fibroblasts were also insensitive to treatment with T, T + V, T + S, or the combination of all three (data not shown). Undifferentiated THP-1 cells, which are not susceptible to CMV, were sensitive to necroptosis (Fig. 1A), although these cells resisted S alone, S + V, or T + S across a broad range of IAP antagonist concentrations from 0.01 to 3 μm (data not shown). THP-1-derived macrophages were insensitive to death elicited by any treatment combination (Fig. 1A). The pattern of cell death in undifferentiated and differentiated THP-1 cells was similar to the behavior of primary macrophages (81). HMEC-1 cells were sensitive to T + S apoptosis, and death was blocked by caspase inhibition. IB analyses were used to assess levels of hRIP1, hRIP3, and hMLKL in the different cell types (Fig. 1B). HFs or IMR-90 fibroblasts had readily detectable hRIP1 but low levels of hRIP3 and hMLKL compared with HT-29 cells, suggesting that resistance may have been dictated by inadequate levels of key mediators (11, 12). hRIP3 and hMLKL levels were low in HMEC-1 cells, consistent with the inability of these cells to support necroptosis. Undifferentiated, necroptosis-sensitive THP-1 cells had readily detectable hRIP3 but low levels of hRIP1 and hMLKL. The observation that THP-1 monocytes support necroptosis even though levels of hRIP1 and MLKL are low suggested that hRIP3 may be limiting in HFs. MLKL levels increased as THP-1 monocytes were differentiated into macrophages, where a slower migrating hRIP3 species was observed but was not investigated further. Although THP-1-derived macrophages support human CMV replication (57), these cells resist necroptosis. Although both HT-29 and undifferentiated THP-1 cells are sensitive to necroptosis, neither supports human CMV replication. These data show that available necrosis-sensitive cells are unsuitable for human CMV studies, leading us to modify CMV-permissive HFs using a strategy applied in previous investigations of the necroptosis pathway in mouse cells (12, 13).
FIGURE 1.

Susceptibility of human CMV-susceptible cell types to TNFR1-dependent necroptosis. A, relative viability of the indicated cells treated for 18 h with TNF (T; 30 ng/ml), IAP antagonist/SMAC mimetic, BV6 (S; 5 μm), and/or caspase inhibitor, Z-VAD-fmk (V; 25 μm), alone or in the combinations shown, except that BV6 was used at 1 μm for THP-1 cells and THP-1-derived macrophages. The viability of four replicate HT-29, HF, and HMEC-1 cultures (n = 4) and three replicate THP-1 cultures (n = 3) was determined by measuring intracellular ATP levels as a percentage of vehicle (0.1% DMSO)-treated controls (untreated cells). B, IB analysis of the indicated cell types to detect hRIP3, hRIP1, and hMLKL, with β-actin as a loading control. Error bars, S.D.
Transduction of HFs with hRIP3 Confers Sensitivity to TNFR1-dependent Necroptosis
HFs were transduced to express either WT, ATP binding site K50A mutant, or MLKL-interaction site S227A of hRIP3 (Fig. 2A). All resulting cells remained healthy despite increased levels of transduced protein (Fig. 2B). Empty vector (EV)-transduced cells showed similar low levels of endogenous hRIP3 as nontransduced cells (compare with Fig. 1B). As expected from prior studies with mouse fibroblasts (12), HFs transduced with a tetra-Ala RHIM substitution mutant (mutRHIM) exhibited 10–20-fold higher levels of mutant protein in a heterogeneous pattern distinct from WT hRIP3. Cells transduced with kinase-inactive K50A mutant or a dual K50A/mutRHIM mutant hRIP3 showed reduced levels of hRIP1 (Fig. 2C), indicating that RIP1 levels were influenced by kinase-inactive RIP3 independent of RHIM interactions. Furthermore, levels of hRIP1 did not increase markedly in the presence of the proteasome inhibitor MG132, although the RIP3 kinase-inactive mutant itself became modified as a result of this treatment (Fig. 2D). From this evaluation, proteasome degradation does not appear to contribute to the reduced RIP1 levels in cells carrying kinase-inactive RIP3.
FIGURE 2.

Transduction of HFs with hRIP3 confers sensitivity to TNFR1-dependent necroptosis. A, schematic representation of the hRIP3 protein, depicting wild type (WT), tetra-Ala RHIM domain (amino acids 459–462) substitution (mutRHIM) (85), ATP binding site mutant (K50A) known to be kinase inactive (22, 85), and hMLKL interaction site (31) mutant (S227A), constructed in lentiviral vectors. B, IB of hRIP3 and hRIP1 in control (Empty) and recombinant hRIP3-expressing lentivirus-transduced HFs, with β-actin loading control. C, IB of HFs transduced with empty vector, K50A, or combined K50A and tetra-Ala RHIM domain substitution (K50mutRHIM) mutant hRIP3 lentiviruses. D, IB of HFs transduced with K50A mutant hRIP3 treated with proteasome inhibitor MG132 (20 μm) for 6 h. E, relative viability of hRIP3-expressing HF cell cultures (n = 3) treated with increasing concentrations of S alone or together with T and/or V. F, relative viability of indicated lentivirus-transduced HFs (n = 4) under assay conditions described in Fig. 1A. G, relative viability of hRIP3-WT-expressing HFs exposed to vehicle (0.1% DMSO), RIP3 kinase inhibitor, GSK′840 (3 μm), or RIP1 kinase inhibitor, necrostatin-1 (Nec-1) (30 μm), respectively (n = 3). H, relative viability of hRIP3-WT-expressing HFs expressing control (scramble) or hMLKL-specific shRNA (n = 3). I, IB depicts the level of hRIP3 and hMLKL in transduced cells prior to treatment, with β-actin loading control. Error bars, S.D.; *, p < 0.05; **, p < 0.001.
RIP3 levels are known to be limiting in cultured cells (11, 12, 22, 23), although cells in mouse tissues are susceptible to necroptosis (12, 13). Once transduced with WT hRIP3, HFs became sensitive to death induced by treatment with T alone, T + V, T + S, or T + S + V (Fig. 2, E and F). The sensitivity of cells to TNFR1-dependent death aligns with the host defense contribution of signal transduction through this death receptor. Cells resisted S alone or S + V across a broad range of IAP antagonist concentrations from 0.01 to 3 μm (Fig. 2E). Treatment with either T + S or T + S + V resulted in the greatest levels of death, indicating that sensitization to T was markedly enhanced by inhibition of polyubiquitination even in the absence of caspase inhibition. Parenthetically, although RIP1 and RIP3 cleavage may be the mechanism through which Casp8 short circuits necroptotic machinery (82–84), cleavage products for either RIP1 or RIP3 were not detected during treatment. Consistent with the expected signaling requirements, HFs transduced with RIP3 RHIM (85), kinase-inactive (K50A) (22, 85), or MLKL binding site (S227A) (31) mutants did not support necroptosis (Fig. 2F). Furthermore, treatment with RIP3 inhibitor GSK′840 (3 μm) (30) or RIP1 kinase inhibitor necrostatin-1 (30 μm) (21) reversed the pattern of death (Fig. 2G). In addition to the requirement for pro-necrotic kinases (17, 22), knockdown of hMLKL prevented death (Fig. 2, H and I), demonstrating the key contribution (31, 32) of this recognized RIP3 kinase target to execution (31, 32). These data indicate that HFs support necroptosis as long as RIP3 levels are increased above a critical threshold level. hRIP3-transduced HFs, like control cells transduced with EV, exhibited modest sensitivity to apoptosis induced by cycloheximide (C) alone or T + C (60). Under these conditions, the addition of caspase inhibitor potentiated necroptosis only in hRIP3-transduced HFs and not in cells expressing K50A mutant (data not shown). In a pattern that was distinct from TNFR1 activation, anti-Fas antibody (F) activation of death receptor CD95/Fas alone did not sensitize to death. F + C induced apoptosis in EV- or K50A mutant-transduced cells and this death was reversed by the addition of caspase inhibitor; however, in hRIP3-transduced HFs, F + C + V induced the expected necroptosis pattern of death (data not shown). In all, HFs became sensitive to death receptor-initiated hRIP3-dependent necroptosis once engineered to express sufficient hRIP3, as observed previously with other human as well as mouse cells (11, 12, 22, 23).
Cell Morphology, hRIP3 Localization, and hRIP3 Phosphorylation during TNFR1-dependent Necroptosis in HFs
Because necroptosis proceeds via cell swelling and plasma membrane permeabilization (2, 4), we employed time lapse analysis with cell-impermeable dye to interrogate plasma membrane integrity in hRIP3-expressing HFs (Fig. 3, A and B). Cells treated with T + S + V developed increased SYTOX Green fluorescence by 2 h post-treatment (hpt) and affected nearly 80% of cells by 24 hpt. Although fewer than 20% of EV-transduced HFs stained positive with SYTOX Green over a similar time course, this level of death was eliminated in K50A mutant-expressing HFs, most likely due to the reduction in RIP1 as well as dominant-negative inhibition of endogenous hRIP3. When death of WT hRIP3-expressing cells was evaluated by TEM, membrane disruption and other features characteristic of necroptosis were observed (Fig. 3C). By IFA, hRIP3 showed a diffuse pattern in the absence of treatment that developed into discrete foci within 4 h of treatment (Fig. 3D). The proportion of the cells with a disrupted membrane and discrete foci of hRIP3 was similar to the number of SYTOX Green-positive cells.
FIGURE 3.

Characterization of hRIP3 modification during TNFR1-dependent necroptosis in HFs. A, bright field micrographs of SYTOX Green-positive HFs with endogenous hRIP3 (Empty) compared with HFs expressing recombinant hRIP3-WT and hRIP3-K50A, evaluated at the indicated time points of T + S + V treatment as described in the legend to Fig. 1A. Original magnification was ×200. B, time course evaluation of death of HFs in A over 24 h in the presence of T + S + V by SYTOX Green uptake on an IncuCyte instrument (n = 3). C, TEM photomicrographs of HFs transduced with EV, hRIP3-WT, or hRIP3-K50A at 4 or 24 hpt with vehicle (DMSO) or T + S + V. White or black arrowheads point to cell swelling or plasma membrane leakage, respectively. Scale bars, 10 μm. D, IFA analysis of hRIP3 in HFs treated with T + S + V for 4 h. White arrows, discrete punctate foci formed during necroptosis. Scale bars, 20 μm. E, IB of hRIP3 and hRIP1 in lentivirus-transduced HFs before and at the indicated time points after T + S + V treatment, with β-actin loading control. F, IB of hRIP3 and hRIP1 in hRIP3-WT-expressing HFs at 4 hpt with the indicated agents, with β-actin loading control. G, IB of hRIP3 in WT-expressing HFs at 4 hpt with T + S + V. Cell lysate was incubated with or without λ-phosphatase (λPPase) prior to IB, with β-actin loading control. *, modified hRIP3 in E–G. Error bars, S.D.
Next, we employed IB analysis to interrogate the impact of treatment on hRIP3. hRIP3-expressing HFs undergoing necroptosis showed the expected slower migrating pro-necrotic hRIP3 forms within 2 hpt (Fig. 3E). These species increased by 4 hpt as RIP1 levels declined, changes that did not occur in hRIP3-K50A-transduced HFs, where low levels of RIP1 remained constant. Consistent with the behavior of necroptosis-sensitive HT-29 cells (22, 41), treatment with T + S was sufficient to induce modification of hRIP3 that was modestly enhanced in the presence of caspase inhibitor (Fig. 3F), and this slow migrating hRIP3 band was eliminated by λ-phosphatase treatment (Fig. 3G), affirming a modification of hRIP3 in HFs as observed previously in HT-29 cells (22, 41). Taken together, these data demonstrate that hRIP3-expressing HFs are highly sensitive to TNFR1-dependent necroptosis as long as IAPs and/or Casp8 are compromised.
Murine CMV M45mutRHIM-induced Necroptosis in HFs
Murine CMV M45mutRIM virus induces rapid DAI-RIP3-dependent necroptosis in mouse cells independent of death receptor signaling and RIP1 (12, 13). Although the virus-induced pathway is distinct from TNFR1-dependent death (6, 7), M45-encoded vIRA blocks RHIM signaling in either pathway (12, 13). Like all CMVs, murine CMV replication is species-restricted but is able to efficiently enter HFs and express early genes without progressing into viral DNA replication (86–88). hRIP3-transduced HFs became sensitive to M45mutRHIM-induced death, assessed by either ATP levels (Fig. 4A) or permeability to SYTOX Green (Fig. 4, B and C), following a course that developed more slowly than in permissive mouse cells (12, 13). The elaboration of murine CMV vIRA by parental K181 blocked necroptosis at times when M45mutRHIM virus induced death. Similar to mouse cells, the addition of IAP antagonist (Fig. 4D) or caspase inhibitor (Fig. 4E) failed to increase the sensitivity of infected cells to death. Murine CMV encodes the Casp8 inhibitor, vICA, an immediate early product expressed in HFs that may sensitize cells to necroptosis (8). hRIP3 RHIM, kinase-inactive and hMLKL interaction site mutants all failed to support virus-induced necroptosis (Fig. 4F), consistent with the expected role of hRIP3 in phosphorylation and activation of hMLKL (17, 31, 32). When WT and mutant M45 protein were subjected to IB, levels were found to be comparable in virus-infected HFs (Fig. 4G). Unexpectedly, protein levels in infected human cells were similar to mouse fibroblasts despite the fact that HFs do not support murine CMV replication. Importantly, M45-encoded vIRA interacted with hRIP3 in a pattern that was similar to mouse cells (Fig. 4H) shown in previous studies (12, 13). The interaction depended on the RHIM and kinase activity of RIP3. Thus, M45mutRHIM-infected HFs are sensitive to necroptosis, and WT murine CMV prevents this pathway through the elaboration of vIRA although HFs are nonpermissive for this mouse virus.
FIGURE 4.

Murine CMV M45 RHIM-dependent suppression of virus-induced necrosis in HFs. A, relative viability of hRIP3-WT-expressing HFs following infection with parental WT (K181-BAC) or M45mutRHIM virus (MOI of 10) with ATP levels assessed as described in the legend to Fig. 1A (n = 6). B, time course (IncuCyte) analysis of hRIP3-WT-expressing HFs following infection with K181-BAC or M45mutRHIM virus showing cell viability assessed by the uptake of SYTOX Green (50 nm) in the cultures (n = 3). C, bright field photomicrographs of SYTOX Green-positive cells in hRIP3-WT-expressing cells at 24 and 48 hpi with K181-BAC or M45mutRHIM viruses (original magnification, ×200). D, relative viability of hRIP3-WT-expressing HFs at 24 and 48 hpi with parental WT or M45mutRHIM viruses (MOI of 10) in the absence (DMSO) or presence of BV6 (5 μm) (n = 3). E, relative viability of hRIP3-WT-expressing HFs at 24 and 48 hpi with parental WT or M45mutRHIM viruses (MOI of 10) in the absence (DMSO) or the presence of Z-VAD-fmk (25 μm) (n = 3). F, relative viability control HFs (Empty) or cells transduced with the indicated WT or mutant recombinant hRIP3 at 48 hpi with parental WT or M45mutRHIM virus (n = 4). Error bars, S.D.; **, p < 0.001. G, IB analysis for detection of M45 at the indicated times in hRIP3-WT-expressing HFs following parental WT or M45mutRHIM virus infection (MOI of 10), with permissive mouse 3T3-SA cells as a positive control and β-actin loading control. H, IB of Myc-tagged M45 protein following IP with anti-FLAG-conjugated agarose from 293T cell extracts 24 h post-transfection with hRIP3-WT, hRIP3-K50A, hRIP3mutRHIM, or mRIP3-WT 3FLAG-tagged expression constructs, with empty control and β-actin loading control. *, modified hRIP3. A nonspecific (NS) band is shown to the right of the lanes. WCL, whole cell lysate.
Human CMV Inhibits TNFR1-dependent Necroptosis
Next, we investigated the impact of human CMV on necroptosis induced by treatment with T + S + V. Following infection for 24 h with Towne-BAC, TownevarRIT3, TB40E-BAC4, Toledo, Merlin, AD169-BAC, AD169varDE, or AD169varATCC (MOI of 3), hRIP3-expressing HFs were treated for an additional 24 h to trigger TNFR1-dependent necroptosis. All tested human CMV strains inhibited cell death (Fig. 5A). Towne-BAC, TownevarRIT3, TB40E-BAC4, Toledo, and Merlin strains exhibited the greatest resistance, and AD169-BAC, AD169varDE, and AD169varATCC showed intermediate levels of resistance (p < 0.05 compared with mock). TownevarRIT3 and AD169varATCC protected to a different extent, although neither encodes a functional vICA (53), so we employed Towne-BAC-derived TowneΔUL36 virus to assess the contribution of Casp8 inhibition to death patterns. RIP3-expressing HFs infected with TowneΔUL36 virus (57) were more sensitive than Towne-BAC to T alone or T + S. The addition of caspase inhibitor blocked death (Fig. 5B). Thus, elaboration of vICA did not impact necroptosis but revealed its expected contribution to suppression of apoptosis (53). In order to exclude any impact of CMV on generation of ATP by modifying mitochondrial respiration (89), we assessed uptake of PI as direct measures of membrane permeability. RIP3-expressing HFs infected with Towne-BAC and treated with T + S + V resisted PI uptake (Fig. 5, C and D) compared with mock-infected cells. Surprisingly, a slower migrating, modified form of RIP3 previously associated with necroptosis (22) remained detectable, although cells remained viable and expressed IE1-p72 at levels similar to untreated cells (Fig. 5E). Interestingly, phosphorylated MLKL levels were readily detected, suggesting that the virus-imposed block occurs downstream of this executioner. Treatment with λ-phosphatase affirmed the phosphorylation of hRIP3 (see Fig. 3G), consistent with the characterization in HT-29 cells (22). Thus, inhibition of necroptosis by human CMV influences steps that follow RHIM signal transduction as well as RIP3 autophosphorylation and phosphorylation-dependent activation of MLKL, a pattern that is distinct from either murine CMV (12, 13) or HSV (41).
FIGURE 5.

Human CMV inhibits TNFR1-dependent necroptosis. A, relative viability of hRIP3-WT-expressing HFs infected for 24 h with indicated human CMV strains (MOI of 3) and treated for an additional 24 h with T + S + V before being assessed as described in the legend to Fig. 1A (n = 3–6). B, relative viability of hRIP3-WT-expressing HFs infected for 24 h with Towne-BAC or TowneΔUL36 virus and treated for an additional 24 h with the indicated combination of T, S, and/or V (n = 3). C, death of Towne-BAC virus-infected hRIP3-WT-expressing HFs, compared with uninfected (Mock), after either vehicle (DMSO) or T + S + V treatment for the indicated times (starting at 24 hpi) with cell death assessed in real time by uptake of PI using an IncuCyte instrument (n = 3). D, photomicrographs of mock and Towne-BAC virus-infected hRIP3-WT-expressing HFs at 24 hpi plus 4 hpt with vehicle (DMSO) or T + S + V, showing levels of PI+ fluorescence without and with phase contrast overlay. Original magnification was ×200. E, IB of hRIP1, hRIP3, and phospho-hMLKL (pMLKL) in Towne-BAC virus-infected hRIP3-WT-expressing HFs treated for 4 h with T + S + V at 24 hpi, with IE1 infection and β-actin loading controls. *, modified forms of hRIP1 and hRIP3. F, single-step growth curves of Towne-BAC, AD169-BAC, or TB40E-BAC4 viruses on HFs with endogenous hRIP3 (Empty) or expressing recombinant hRIP3-WT or hRIP3-K50A. Viral titers were determined on culture supernatants by plaque assay at the indicated time points (n = 3). The 0 dpi time point indicates the input virus inoculum, and the detection limit of the plaque assay is indicated by a dashed line. G, virus titers in HFs with endogenous hRIP3 (Empty) or expressing recombinant hRIP3-WT or hRIP3-K50A infected with Towne-BAC virus followed at 24 hpi by treatment with vehicle (DMSO), T, T + S, or T + S + V, with virus titers determined on culture supernatants by plaque assay at 4 dpi (n = 3). All human CMV infections were performed at an MOI of 3. Error bars, S.D. #, not significant (p > 0.05).
We evaluated the impact of RIP3 expression on viral replication levels and found that both hRIP3-WT and hRIP3-K50A-expressing HFs supported Towne-BAC, AD169-BAC, or TB40E-BAC4 (MOI of 3) replication to levels observed in control HFs (Fig. 5F). In addition, treatment with T, T + S, or T + S + V failed to compromise viral replication when assessed at 4 dpi, although overall titers were modestly reduced in cells with combined treatment (Fig. 5G). These data indicate that viral suppression of TNFR1-mediated, hRIP3-dependent necroptosis sustains cell viability to support production of viral progeny.
Human CMV Infection Inhibits M45mutRHIM Virus-induced Programmed Necrosis
We employed superinfection experiments with murine CMV in order to determine whether human CMV blocks virus-induced necroptosis. hRIP3-expressing HFs were infected with Towne, AD169, or Toledo strains (MOI of 3) followed 24 h later by murine CMV K181-BAC or M45mutRHIM virus (MOI of 10). After an additional 48 h, cell viability was assessed. Whereas only 50% of HFs exposed to M45mutRHIM virus survived these conditions, 80–90% of M45mutRHIM-infected cells survived when HFs were infected with human CMV independent of strain (Fig. 6A). The proportion of murine CMV-derived IE1-expressing cells was similar in Towne-BAC virus and mock-infected hRIP3-WT-expressing HFs, indicating that murine CMV was not compromised by prior human CMV infection (Fig. 6B). Furthermore, murine 3T3-SA cells exposed to Towne or AD169 infection resisted M45mutRHIM-induced necroptosis to equivalent levels (Fig. 6C). Although human CMV is species-restricted, both of these strains efficiently enter and express early gene products in mouse fibroblasts (90). The ability of human CMV to block virus-induced necroptosis as well as TNFR1-dependent necroptosis was most consistent with the function of a virion component or early gene product acting on necroptosis.
FIGURE 6.
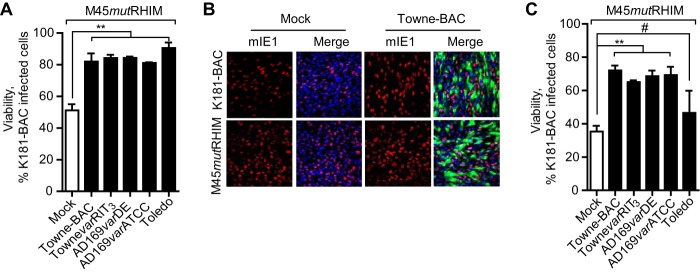
Human CMV inhibits murine CMV M45mutRHIM-induced necroptosis. A, relative viability of human CMV-infected hRIP3-WT-expressing HFs (MOI of 3) exposed to M45mutRHIM virus (MOI of 10) at 24 hpi assessed at 72 hpi with human CMV, expressed as a percentage relative to a parallel set of cultures exposed to parental WT (K181-BAC) murine CMV (n = 3). B, human CMV-infected hRIP3-WT-expressing HFs exposed to K181-BAC or M45mutRHIM virus as described in A and assessed by IFA with murine CMV IE1 (mIE1) stained (red) at 48 hpi with human CMV. The merged images include Towne-BAC-encoded GFP fluorescence (green) and DAPI staining to identify nuclei (blue). Original magnification was ×200. C, relative viability of the indicated human CMV-infected 3T3-SA cells (MOI of 3) exposed to M45mutRHIM virus (MOI of 10) at 24 hpi assessed at 48 hpi with human CMV and expressed as a percentage relative to a parallel set of cultures exposed to K181-BAC murine CMV (n = 3). Error bars, S.D.; *, p < 0.05; **, p < 0.001; #, not significant (p > 0.05).
Human CMV IE1 Function Is Required for Suppression of Necroptosis
In order to further evaluate the contribution of human CMV functions to suppression of necroptosis, we first conducted a time-of-addition experiment to determine when during infection HFs became resistant to necroptosis. The viability of HFs transduced with WT, K50A, and EV was determined by treating cells for 24 h with T + S + V starting at 6, 24, or 48 hpi with Towne-BAC virus (MOI of 3) (Fig. 7A). Cells remained sensitive to necrosis at 6 hpi but resisted death initiated later, and, as expected, neither EV control nor K50A mutant showed sensitivity. Inhibition of viral DNA synthesis with the polymerase inhibitor phosphonoformate (300 μg/ml) did not sensitize to necroptosis (Fig. 7B), indicating that suppression develops independent of viral DNA synthesis. To determine the impact of input virion or newly synthesized proteins, we tested UV-inactivated virus. Virus particles failed to confer resistance to cells at a time when viable virus suppressed necroptosis (Fig. 7C). As expected, IB analysis revealed that UV-irradiated virus delivered virion tegument protein (pp65) but blocked expression of IE1 (Fig. 7D). Together with the time course (Fig. 7A), these data implicate a newly synthesized viral or cellular gene product in viral suppression of TNFR1-dependent necroptosis.
FIGURE 7.

Human CMV IE1 function contributes to suppression of necrosis. A, relative viability of Towne-BAC virus-infected HFs (MOI of 3) with endogenous hRIP3 (Empty) or expressing recombinant hRIP3-WT or hRIP3-K50A and subjected to T + S + V treatment beginning at 6, 24, or 48 hpi with assessment 24 h after the start of treatment as described in the legend to Fig. 5A (n = 3). B, relative viability of Towne-BAC virus-infected hRIP3-WT-expressing HFs (MOI of 3) cultured for 24 h treated with T + S + V in the presence or absence of phosphonoformate (PFA) (300 μg/ml) (n = 3). C, relative viability of HFs with endogenous hRIP3 (Empty) compared with hRIP3-WT- or hRIP3-K50A-expressing HFs exposed to Towne-BAC virus (UV (−)) (MOI of 3) or an equivalent dose of UV-irradiated Towne-BAC stock (UV (+)) and treated with T + S + V for 24 h (n = 4). D, IB of pp65 and IE1 in hRIP3-WT-expressing HFs exposed to medium (Mock), Towne-BAC virus (MOI of 3), or UV-irradiated Towne-BAC at 6 and 24 hpi, with β-actin loading control. E, relative viability of HFs with endogenous hRIP3 (Empty) compared with hRIP3-WT- or hRIP3-K50A-expressing HFs exposed to IE1-null virus (CR208) or rescue IE1-expressing virus (CRQ208) (MOI of 3) treated with T + S + V for 24 h (n = 4). F, IB of pp65 and IE1 in Mock-, CR208- and CRQ208-infected hRIP3-WT-expressing HFs (MOI of 3) at 24 hpi, with β-actin loading control. G, single-step growth curves of CR208 or CRQ208 viruses on HFs with endogenous hRIP3 (Empty) or expressing recombinant hRIP3-WT or hRIP3-K50A (MOI of 3). Viral titers were determined on culture supernatants by plaque assay at the indicated time points (n = 3). The 0 dpi time point indicates the input virus inoculum, and the detection limit of the plaque assay is indicated by a dashed line. H, virus titers in HFs with endogenous hRIP3 (Empty) or expressing recombinant hRIP3-WT or hRIP3-K50A infected with CR208 or CRQ208 viruses (MOI of 3) followed at 24 hpi by treatment with T + S + V, with virus titers determined on culture supernatants by plaque assay at 4 dpi (n = 3). Error bars, S.D.; *, p < 0.05; **, p < 0.001; #, not significant (p > 0.05).
We next evaluated the contribution of the viral regulatory protein IE1 (91), which dictates gene expression levels and modulates innate immune signaling early after infection (92). IE1-null virus (CR208) and repaired virus (CRQ208) (70) were employed to test whether IE1 influenced suppression. CR208-infected cells were insensitive to T + S + V, whereas IE1-expressing CRQ208-infected cells resisted necroptosis (Fig. 7, E and F) as efficiently as parental TownevarRIT3 (see Fig. 5A). As expected, EV- and hRIP3-K50A-transduced HFs resisted necroptosis. The levels of replication, assessed in supernatants of CRQ208- or CR208-infected (MOI of 3) cells, were not influenced by the higher levels of hRIP3-WT or K50A mutant expression (Fig. 7G), although induction of necroptosis at 1 dpi significantly reduced CR208 virus yields in the supernatant at 4 dpi (Fig. 7H). Thus, human CMV-encoded IE1 protein is necessary for the virus to block necroptosis.
DISCUSSION
Programmed cell death is an ancient means of eliminating infected cells for purposes of host defense (1–3, 6). Large DNA viruses encode inhibitors of apoptosis that sensitize cells to necroptosis (2, 7, 41). The rodent herpesvirus murine CMV (12, 13) and the primate alphaherpesviruses HSV1 and HSV2 UL39 (41) encode R1 homologs that inhibit necroptosis by competing with RHIM signaling partners of the pro-necrotic kinase, RIP3. In addition, HSV1 ICP6 and HSV2 ICP10 directly bind the death effector domain of Casp8 (50), thereby unleashing necroptosis during infection (41). The betaherpesviruses murine CMV and human CMV encode a conserved inhibitor of Casp8 activation, vICA (54, 56, 57), with a capacity to sensitize to necroptosis independent of virus infection (8). This alternate cell death pathway is completely suppressed by M45-dependent disruption of RHIM signaling (12, 13). It has remained a mystery whether suppression of necroptosis plays an important role in human CMV pathogenesis as it does for the biologically similar rodent virus. Neither human CMV UL45 nor any other viral protein has a RHIM (93), and UL45 mutant virus fails to exhibit any phenotypic similarity to M45 mutants (94, 95). We have shown here that human CMV blocks necroptosis but employs a different strategy than murine CMV.
By making use of primary human fibroblasts transduced with RIP3 to increase levels of this key pro-necrotic kinase, we generated human CMV permissive primary cells that support necroptosis. Like transduced NIH3T3 cells (11, 12), the resultant hRIP3-expressing HFs became susceptible to TNFR1-dependent necroptosis. Sensitivity to either TNFR1- of M45mutRHIM-induced necroptosis was eliminated during infection with human CMV, providing direct evidence of virus-specific inhibition of this alternate death pathway. The mechanism of inhibition was distinct from MCMV and appeared to target a step downstream of RHIM signaling, after RIP3 kinase-dependent phosphorylation of MLKL (29, 72). This execution phase of necroptosis, which drives the disruption of the plasma membrane, remains to be completely dissected. The mechanism of human CMV suppression is likely to provide additional insights into the precise steps driving execution of human cells. Therefore, this work, together with our previous identification of M45 and UL39 as RHIM-dependent inhibitors of necroptosis (12, 13, 27, 41), establishes the core parameters through which cell death pathways function in pathogenesis as well as the diverse ways herpesviruses infecting rodents and humans successfully counter these host defense pathways.
Human CMV IE1 expression is necessary for the suppression of TNFR1-dependent necroptosis. The IE1 gene product, IE1-p72 is primarily involved in regulating viral transcription during productive replication (70) as an accessory to IE2-p86 (91). IE1-p72 also impacts the host cell response to infection and modulates the activation of interferon (92). IE1-p72 is a suppressor of TNFR1-dependent apoptosis in HeLa cells (69), although phenotypic evaluation of IE1 mutant viruses has not revealed any impact on cell survival before our current study. In addition to the work shown, IE1-p72-expressing ihf-ie1.3 cells were tested and failed to resist necroptosis, suggesting that the requirement here during infection is for optimal expression of an early gene (70). Using permissive cells engineered to carry adequate RIP3, IE1 mutant virus fails to protect from TNFR1-dependent signal transduction, whereas parental virus effectively protects cells from insult.
Death receptor signaling contributes to CMV biology, during both productive infection and reactivation from latency. Human CMV strains down-modulate expression of TNFR1 starting between 18 and 24 hpi (96–98). Up to that time, the UL138-encoded viral promoter of TNFR1 signaling sustains the stability of TNFR1 and confers increased signal transduction that may also contribute to reactivation from latency (73, 74). In our hands, viral strains that lack UL138 (Towne and AD169 variants) exhibit varying sensitivity to TNFR1-dependent signaling, indicating that the viral promoter of TNFR1 signaling does not impact necroptosis in the assays we employed here. Furthermore, strains that encode UL138 (TB40E-BAC4, Toledo, and Merlin) all showed resistance to necroptosis similar to UL138-deficient Towne. These results demonstrate a crucial contribution of IE1-p72 in establishing a cellular environment that prevents necroptosis during viral infection, acting either directly (92) or indirectly (91). Such countermeasures certainly may be important during productive infection as well as during latency.
Observations that murine CMV M45 protects and M45mutRHIM fails to protect human fibroblasts extend findings made in necroptosis-sensitive HT-29 cells (41) and are consistent with other work showing vIRA suppression of RHIM signal transduction in both mouse and human cells (12, 17, 26, 27). It is important to note that the cellular processes leading to M45mutRHIM activation of necroptosis in human cells has not been the focus of this study, leaving the investigation of similarities between human and mouse cells (13) for future investigation. In mouse cells infected with M45mutRHIM, the cytosolic dsDNA sensor, DAI, recruits RIP3 to trigger RHIM-dependent necroptosis. Human CMV infection is already known to be sensed by DAI, and this contributes to the stimulation of an innate immune response (99). The activation of interferon by DAI depends on RHIM signal transduction (100). The apparent mechanism of IE1-dependent suppression of necroptosis downstream of RHIM signal transduction as well as downstream of RIP3 kinase-dependent activation of MLKL aligns with the ability of infected cells to sustain DAI-dependent interferon-like responses (99). Further characterization of the triggers and outcomes affecting necroptosis will require the use of transduced cells similar to those described here to unveil collaborating viral and host factors. Future studies will seek to identify the precise viral or host cell inhibitors as well as to characterize the behavior of potential sensitizers, such as the Casp8 inhibitor, vICA, as well as the mitochondrial cell death suppressors. It is important to recognize that the detection of virus-induced necroptosis emerged from the finding that murine CMV M45 is a powerful suppressor of RHIM signal transduction (12, 17, 26, 27). Whereas mouse studies have supported the broad importance of necrotic death as an alternate pathway of host defense, our findings here bring to light the central role that RIP3 plays in human cells and reveals the existence of machinery that may contribute to viral pathogenesis as well as inflammation and associated tissue damage.
Acknowledgments
We thank Domagoj Vucic (Genentech/Roche, Inc.) for providing BV6, Peter Gough (GlaxoSmithKline) for RIP1 and RIP3 kinase inhibitors, and Zheng-Gang Liu (National Institutes of Health) for providing hMLKL shRNA constructs. Microscopy was performed at the Robert P. Apkarian Integrated Electron Microscopy Core and the Cell Imaging and Microscopy Core of the Winship Cancer Institute at Emory University.
Footnotes
- Casp
- caspase
- CMV
- cytomegalovirus
- HSV
- herpes simplex virus
- RIP
- receptor-interacting protein kinase
- hRIP
- human RIP
- TLR
- Toll-like receptor
- TNFR1
- TNF receptor 1
- vIRA
- viral inhibitor of RIP activation
- RHIM
- RIP homotypic interaction motif
- DAI
- DNA inducer of interferon
- TRIF
- TIR domain-containing adapter-inducing interferon β
- MLKL
- mixed lineage kinase domain-like
- hMLKL
- human MLKL
- vICA
- viral inhibitor of Casp8 activation
- Z
- benzyloxycarbonyl
- fmk
- fluoromethyl ketone
- IAP
- inhibitor of apoptosis protein
- IB
- immunoblot
- IP
- immunoprecipitation
- HF
- newborn foreskin fibroblast
- MOI
- multiplicity of infection
- PI
- propidium iodide
- IFA
- immunofluorescence analysis
- TEM
- transmission electron microscopy
- T
- TNF
- S
- BV6 (SMAC mimetic)
- V
- Z-VAD-fmk
- C
- cycloheximide
- HMEC-1 cell
- human microvascular endothelial cell
- EV
- empty vector
- hpt
- h post-treatment
- hpi
- hours post infection
- dpi
- days postinfection
- ORF
- open reading frame.
REFERENCES
- 1. Lamkanfi M., Dixit V. M. (2010) Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 8, 44–54 [DOI] [PubMed] [Google Scholar]
- 2. Mocarski E. S., Upton J. W., Kaiser W. J. (2012) Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat. Rev. Immunol. 12, 79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sridharan H., Upton J. W. (2014) Programmed necrosis in microbial pathogenesis. Trends Microbiol. 22, 199–207 [DOI] [PubMed] [Google Scholar]
- 4. Moquin D., Chan F. K. (2010) The molecular regulation of programmed necrotic cell injury. Trends Biochem. Sci. 35, 434–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hedrick S. M., Ch'en I. L., Alves B. N. (2010) Intertwined pathways of programmed cell death in immunity. Immunol. Rev. 236, 41–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kaiser W. J., Upton J. W., Mocarski E. S. (2013) Viral modulation of programmed necrosis. Curr. Opin. Virol. 3, 296–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Upton J. W., Chan F. K. (2014) Staying alive: cell death in antiviral immunity. Mol. Cell 54, 273–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kaiser W. J., Upton J. W., Long A. B., Livingston-Rosanoff D., Daley-Bauer L. P., Hakem R., Caspary T., Mocarski E. S. (2011) RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471, 368–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vercammen D., Beyaert R., Denecker G., Goossens V., Van Loo G., Declercq W., Grooten J., Fiers W., Vandenabeele P. (1998) Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 187, 1477–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kawahara A., Ohsawa Y., Matsumura H., Uchiyama Y., Nagata S. (1998) Caspase-independent cell killing by Fas-associated protein with death domain. J. Cell Biol. 143, 1353–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cho Y. S., Challa S., Moquin D., Genga R., Ray T. D., Guildford M., Chan F. K. (2009) Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Upton J. W., Kaiser W. J., Mocarski E. S. (2010) Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 7, 302–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Upton J. W., Kaiser W. J., Mocarski E. S. (2012) DAI (ZBP1/DLM-1) complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Feoktistova M., Geserick P., Panayotova-Dimitrova D., Leverkus M. (2012) Pick your poison: The ripoptosome, a cell death platform regulating apoptosis and necroptosis. Cell Cycle 11, 460–467 [DOI] [PubMed] [Google Scholar]
- 15. Kaiser W. J., Daley-Bauer L. P., Thapa R. J., Mandal P., Berger S. B., Huang C., Sundararajan A., Guo H., Roback L., Speck S. H., Bertin J., Gough P. J., Balachandran S., Mocarski E. S. (2014) RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc. Natl. Acad. Sci. U.S.A. 111, 7753–7758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thapa R. J., Nogusa S., Chen P., Maki J. L., Lerro A., Andrake M., Rall G. F., Degterev A., Balachandran S. (2013) Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc. Natl. Acad. Sci. U.S.A. 110, 3109–3118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kaiser W. J., Sridharan H., Huang C., Mandal P., Upton J. W., Gough P. J., Sehon C. A., Marquis R. W., Bertin J., Mocarski E. S. (2013) Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 288, 31268–31279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Holler N., Zaru R., Micheau O., Thome M., Attinger A., Valitutti S., Bodmer J. L., Schneider P., Seed B., Tschopp J. (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489–495 [DOI] [PubMed] [Google Scholar]
- 19. Micheau O., Thome M., Schneider P., Holler N., Tschopp J., Nicholson D. W., Briand C., Grütter M. G. (2002) The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J. Biol. Chem. 277, 45162–45171 [DOI] [PubMed] [Google Scholar]
- 20. Micheau O., Tschopp J. (2003) Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190 [DOI] [PubMed] [Google Scholar]
- 21. Degterev A., Huang Z., Boyce M., Li Y., Jagtap P., Mizushima N., Cuny G. D., Mitchison T. J., Moskowitz M. A., Yuan J. (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119 [DOI] [PubMed] [Google Scholar]
- 22. He S., Wang L., Miao L., Wang T., Du F., Zhao L., Wang X. (2009) Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. Cell 137, 1100–1111 [DOI] [PubMed] [Google Scholar]
- 23. Zhang D. W., Shao J., Lin J., Zhang N., Lu B. J., Lin S. C., Dong M. Q., Han J. (2009) RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336 [DOI] [PubMed] [Google Scholar]
- 24. Chan F. K., Shisler J., Bixby J. G., Felices M., Zheng L., Appel M., Orenstein J., Moss B., Lenardo M. J. (2003) A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J. Biol. Chem. 278, 51613–51621 [DOI] [PubMed] [Google Scholar]
- 25. Berger A. K., Danthi P. (2013) Reovirus activates a caspase-independent cell death pathway. mBio 4, e00178–00113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rebsamen M., Heinz L. X., Meylan E., Michallet M. C., Schroder K., Hofmann K., Vazquez J., Benedict C. A., Tschopp J. (2009) DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-κB. EMBO Rep. 10, 916–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Upton J. W., Kaiser W. J., Mocarski E. S. (2008) Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J. Biol. Chem. 283, 16966–16970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. He S., Liang Y., Shao F., Wang X. (2011) Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. U.S.A. 108, 20054–20059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li J., McQuade T., Siemer A. B., Napetschnig J., Moriwaki K., Hsiao Y. S., Damko E., Moquin D., Walz T., McDermott A., Chan F. K., Wu H. (2012) The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150, 339–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mandal P., Berger S. B., Pillay S., Moriwaki K., Huang C., Guo H., Lich J. D., Finger J., Kasparcova V., Votta B., Ouellette M., King B. W., Wisnoski D., Lakdawala A. S., DeMartino M. P., Casillas L. N., Haile P. A., Sehon C. A., Marquis R. W., Upton J., Daley-Bauer L. P., Roback L., Ramia N., Dovey C. M., Carette J. E., Chan F. K., Bertin J., Gough P. J., Mocarski E. S., Kaiser W. J. (2014) RIP3 induces apoptosis independent of pro-necrotic kinase activity. Mol. Cell 56, 481–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sun L., Wang H., Wang Z., He S., Chen S., Liao D., Wang L., Yan J., Liu W., Lei X., Wang X. (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 [DOI] [PubMed] [Google Scholar]
- 32. Zhao J., Jitkaew S., Cai Z., Choksi S., Li Q., Luo J., Liu Z. G. (2012) Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. U.S.A. 109, 5322–5327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vandenabeele P., Declercq W., Van Herreweghe F., Vanden Berghe T. (2010) The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci. Signal. 3, re4. [DOI] [PubMed] [Google Scholar]
- 34. Murphy J. M., Czabotar P. E., Hildebrand J. M., Lucet I. S., Zhang J. G., Alvarez-Diaz S., Lewis R., Lalaoui N., Metcalf D., Webb A. I., Young S. N., Varghese L. N., Tannahill G. M., Hatchell E. C., Majewski I. J., Okamoto T., Dobson R. C., Hilton D. J., Babon J. J., Nicola N. A., Strasser A., Silke J., Alexander W. S. (2013) The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453 [DOI] [PubMed] [Google Scholar]
- 35. Cai Z., Jitkaew S., Zhao J., Chiang H. C., Choksi S., Liu J., Ward Y., Wu L. G., Liu Z. G. (2014) Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 16, 55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen X., Li W., Ren J., Huang D., He W. T., Song Y., Yang C., Li W., Zheng X., Chen P., Han J. (2014) Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 24, 105–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Antonopoulos C., El Sanadi C., Kaiser W. J., Mocarski E. S., Dubyak G. R. (2013) Pro-apoptotic chemotherapeutic drugs induce non-canonical processing and release of IL-1β via caspase-8 in dendritic cells. J. Immunol. 191, 4789–4803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weng D., Marty-Roix R., Ganesan S., Proulx M. K., Vladimer G. I., Kaiser W. J., Mocarski E. S., Pouliot K., Chan F. K., Kelliher M. A., Harris P. A., Bertin J., Gough P. J., Shayakhmetov D. M., Goguen J. D., Fitzgerald K. A., Silverman N., Lien E. (2014) Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc. Natl. Acad. Sci. U.S.A. 111, 7391–7396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Philip N. H., Dillon C. P., Snyder A. G., Fitzgerald P., Wynosky-Dolfi M. A., Zwack E. E., Hu B., Fitzgerald L., Mauldin E. A., Copenhaver A. M., Shin S., Wei L., Parker M., Zhang J., Oberst A., Green D. R., Brodsky I. E. (2014) Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-κB and MAPK signaling. Proc. Natl. Acad. Sci. U.S.A. 111, 7385–7390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li M., Beg A. A. (2000) Induction of necrotic-like cell death by tumor necrosis factor alpha and caspase inhibitors: novel mechanism for killing virus-infected cells. J. Virol. 74, 7470–7477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guo H., Omoto S., Harris P. A., Finger J. N., Bertin J., Gough P. J., Kaiser W. J., Mocarski E. S. (2015) Herpes simplex virus suppression of necroptosis in human cells. Cell Host Microbe 17, 243–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xie T., Peng W., Yan C., Wu J., Gong X., Shi Y. (2013) Structural insights into RIP3-mediated necroptotic signaling. Cell Rep. 5, 70–78 [DOI] [PubMed] [Google Scholar]
- 43. Mack C., Sickmann A., Lembo D., Brune W. (2008) Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1-interacting protein. Proc. Natl. Acad. Sci. U.S.A. 105, 3094–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brune W., Ménard C., Heesemann J., Koszinowski U. H. (2001) A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science 291, 303–305 [DOI] [PubMed] [Google Scholar]
- 45. Brune W. (2011) Inhibition of programmed cell death by cytomegaloviruses. Virus Res. 157, 144–150 [DOI] [PubMed] [Google Scholar]
- 46. Lembo D., Donalisio M., Hofer A., Cornaglia M., Brune W., Koszinowski U., Thelander L., Landolfo S. (2004) The ribonucleotide reductase R1 homolog of murine cytomegalovirus is not a functional enzyme subunit but is required for pathogenesis. J. Virol. 78, 4278–4288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mocarski E. S., Kaiser W. J., Livingston-Rosanoff D., Upton J. W., Daley-Bauer L. P. (2014) True grit: programmed necrosis in antiviral host defense, inflammation, and immunogenicity. J. Immunol. 192, 2019–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fliss P. M., Jowers T. P., Brinkmann M. M., Holstermann B., Mack C., Dickinson P., Hohenberg H., Ghazal P., Brune W. (2012) Viral mediated redirection of NEMO/IKKγ to autophagosomes curtails the inflammatory cascade. PLoS Pathog. 8, e1002517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Krause E., de Graaf M., Fliss P. M., Dölken L., Brune W. (2014) Murine cytomegalovirus virion-associated protein M45 mediates rapid NF-κB activation after infection. J. Virol. 88, 9963–9975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dufour F., Sasseville A. M., Chabaud S., Massie B., Siegel R. M., Langelier Y. (2011) The ribonucleotide reductase R1 subunits of herpes simplex virus types 1 and 2 protect cells against TNFalpha- and FasL-induced apoptosis by interacting with caspase-8. Apoptosis 16, 256–271 [DOI] [PubMed] [Google Scholar]
- 51. Wang X., Li Y., Liu S., Yu X., Li L., Shi C., He W., Li J., Xu L., Hu Z., Yu L., Yang Z., Chen Q., Ge L., Zhang Z., Zhou B., Jiang X., Chen S., He S. (2014) Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc. Natl. Acad. Sci. U.S.A. 111, 15438–15443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huang Z., Wu S.-Q., Liang Y., Zhou X., Chen W., Li L., Wu J., Zhuang Q., Chen C., Li J., Zhong C., Xia W., Zhou R., Zheng C., Han J. (2015) Targeting HSV-1 protein ICP6 by RIP1 and RIP3 initiates necroptosis to restrict HSV-1 propagation in mice. Cell Host Microbe 17, 229–242 [DOI] [PubMed] [Google Scholar]
- 53. Skaletskaya A., Bartle L. M., Chittenden T., McCormick A. L., Mocarski E. S., Goldmacher V. S. (2001) A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc. Natl. Acad. Sci. U.S.A. 98, 7829–7834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McCormick A. L., Skaletskaya A., Barry P. A., Mocarski E. S., Goldmacher V. S. (2003) Differential function and expression of the viral inhibitor of caspase 8-induced apoptosis (vICA) and the viral mitochondria-localized inhibitor of apoptosis (vMIA) cell death suppressors conserved in primate and rodent cytomegaloviruses. Virology 316, 221–233 [DOI] [PubMed] [Google Scholar]
- 55. Ménard C., Wagner M., Ruzsics Z., Holak K., Brune W., Campbell A. E., Koszinowski U. H. (2003) Role of murine cytomegalovirus US22 gene family members in replication in macrophages. J. Virol. 77, 5557–5570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cicin-Sain L., Ruzsics Z., Podlech J., Bubic I., Menard C., Jonjić S., Reddehase M. J., Koszinowski U. H. (2008) Dominant-negative FADD rescues the in vivo fitness of a cytomegalovirus lacking an antiapoptotic viral gene. J. Virol. 82, 2056–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McCormick A. L., Roback L., Livingston-Rosanoff D., St Clair C. (2010) The human cytomegalovirus UL36 gene controls caspase-dependent and -independent cell death programs activated by infection of monocytes differentiating to macrophages. J. Virol. 84, 5108–5123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McCormick A. L., Mocarski E. S. (2013) Cell death pathways controlled by cytomegaloviruses. in Cytomegaloviruses: From Molecular Pathogenesis to Intervention (Reddehase M. J., ed) pp. 263–276, Caister Scientific Press, Norfolk, UK [Google Scholar]
- 59. Daley-Bauer L. P., Mocarski E. S. (2013) Myeloid cell recruitment and function in cytomegalovirus immunity and pathogenesis. in Cytomegaloviruses: From Molecular Pathogenesis to Intervention (Reddehase M. J., ed) pp. 363–373, Caister Scientific Press, Norfolk, UK [Google Scholar]
- 60. Goldmacher V. S., Bartle L. M., Skaletskaya A., Dionne C. A., Kedersha N. L., Vater C. A., Han J. W., Lutz R. J., Watanabe S., Cahir McFarland E. D., Kieff E. D., Mocarski E. S., Chittenden T. (1999) A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc. Natl. Acad. Sci. U.S.A. 96, 12536–12541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Karbowski M., Norris K. L., Cleland M. M., Jeong S. Y., Youle R. J. (2006) Role of Bax and Bak in mitochondrial morphogenesis. Nature 443, 658–662 [DOI] [PubMed] [Google Scholar]
- 62. Cam M., Handke W., Picard-Maureau M., Brune W. (2010) Cytomegaloviruses inhibit Bak- and Bax-mediated apoptosis with two separate viral proteins. Cell Death Differ. 17, 655–665 [DOI] [PubMed] [Google Scholar]
- 63. Manzur M., Fleming P., Huang D. C., Degli-Esposti M. A., Andoniou C. E. (2009) Virally mediated inhibition of Bax in leukocytes promotes dissemination of murine cytomegalovirus. Cell Death Differ. 16, 312–320 [DOI] [PubMed] [Google Scholar]
- 64. Crosby L. N., McCormick A. L., Mocarski E. S. (2013) Gene products of the embedded m41/m41.1 locus of murine cytomegalovirus differentially influence replication and pathogenesis. Virology 436, 274–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Handke W., Luig C., Popovic B., Krmpotic A., Jonjic S., Brune W. (2013) Viral inhibition of BAK promotes murine cytomegalovirus dissemination to salivary glands. J. Virol. 87, 3592–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fleming P., Kvansakul M., Voigt V., Kile B. T., Kluck R. M., Huang D. C., Degli-Esposti M. A., Andoniou C. E. (2013) MCMV-mediated inhibition of the pro-apoptotic Bak protein is required for optimal in vivo replication. PLoS Pathog. 9, e1003192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. McCormick A. L. (2008) Control of apoptosis by human cytomegalovirus. Curr. Top. Microbiol. Immunol. 325, 281–295 [DOI] [PubMed] [Google Scholar]
- 68. McCormick A. L., Roback L., Wynn G., Mocarski E. S. (2013) Multiplicity-dependent activation of a serine protease-dependent cytomegalovirus-associated programmed cell death pathway. Virology 435, 250–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhu H., Shen Y., Shenk T. (1995) Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J. Virol. 69, 7960–7970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Greaves R. F., Mocarski E. S. (1998) Defective growth correlates with reduced accumulation of a viral DNA replication protein after low multiplicity infection by a human cytomegalovirus ie1 mutant. J. Virol. 72, 366–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Green D. R., Victor B. (2012) The pantheon of the fallen: why are there so many forms of cell death? Trends Cell Biol. 22, 555–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Moriwaki K., Chan F. K. (2013) RIP3: a molecular switch for necrosis and inflammation. Genes Dev. 27, 1640–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Le V. T., Trilling M., Hengel H. (2011) The cytomegaloviral protein pUL138 acts as potentiator of tumor necrosis factor (TNF) receptor 1 surface density to enhance ULb′-encoded modulation of TNF-α signaling. J. Virol. 85, 13260–13270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Montag C., Wagner J. A., Gruska I., Vetter B., Wiebusch L., Hagemeier C. (2011) The latency-associated UL138 gene product of human cytomegalovirus sensitizes cells to tumor necrosis factor α (TNF-α) signaling by upregulating TNF-α receptor 1 cell surface expression. J. Virol. 85, 11409–11421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 76. Naldini L., Blömer U., Gage F. H., Trono D., Verma I. M. (1996) Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. U.S.A. 93, 11382–11388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Omoto S., Mocarski E. S. (2013) Cytomegalovirus UL91 is essential for transcription of viral true late (γ2) genes. J. Virol. 87, 8651–8664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Marchini A., Liu H., Zhu H. (2001) Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J. Virol. 75, 1870–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kaiser W. J., Offermann M. K. (2005) Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J. Immunol. 174, 4942–4952 [DOI] [PubMed] [Google Scholar]
- 80. Varfolomeev E., Blankenship J. W., Wayson S. M., Fedorova A. V., Kayagaki N., Garg P., Zobel K., Dynek J. N., Elliott L. O., Wallweber H. J., Flygare J. A., Fairbrother W. J., Deshayes K., Dixit V. M., Vucic D. (2007) IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 131, 669–681 [DOI] [PubMed] [Google Scholar]
- 81. Müller-Sienerth N., Dietz L., Holtz P., Kapp M., Grigoleit G. U., Schmuck C., Wajant H., Siegmund D. (2011) SMAC mimetic BV6 induces cell death in monocytes and maturation of monocyte-derived dendritic cells. PLoS One 6, e21556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lin Y., Devin A., Rodriguez Y., Liu Z. G. (1999) Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 13, 2514–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Feng S., Yang Y., Mei Y., Ma L., Zhu D. E., Hoti N., Castanares M., Wu M. (2007) Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell. Signal. 19, 2056–2067 [DOI] [PubMed] [Google Scholar]
- 84. Oberst A., Dillon C. P., Weinlich R., McCormick L. L., Fitzgerald P., Pop C., Hakem R., Salvesen G. S., Green D. R. (2011) Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sun X., Yin J., Starovasnik M. A., Fairbrother W. J., Dixit V. M. (2002) Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J. Biol. Chem. 277, 9505–9511 [DOI] [PubMed] [Google Scholar]
- 86. Manning W. C., Murphy J. E., Jolly D. J., Mento S. J., Ralston R. O. (1998) Use of a recombinant murine cytomegalovirus expressing vesicular stomatitis virus G protein to pseudotype retroviral vectors. J. Virol. Methods 73, 31–39 [DOI] [PubMed] [Google Scholar]
- 87. Walker D., Hudson J. (1987) Analysis of immediate-early and early proteins of murine cytomegalovirus in permissive and nonpermissive cells. Arch. Virol. 92, 103–119 [DOI] [PubMed] [Google Scholar]
- 88. Jurak I., Brune W. (2006) Induction of apoptosis limits cytomegalovirus cross-species infection. EMBO J. 25, 2634–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kaarbø M., Ager-Wick E., Osenbroch P. Ø., Kilander A., Skinnes R., Müller F., Eide L. (2011) Human cytomegalovirus infection increases mitochondrial biogenesis. Mitochondrion 11, 935–945 [DOI] [PubMed] [Google Scholar]
- 90. Lafemina R. L., Hayward G. S. (1988) Differences in cell-type-specific blocks to immediate early gene expression and DNA replication of human, simian and murine cytomegalovirus. J. Gen. Virol. 69, 355–374 [DOI] [PubMed] [Google Scholar]
- 91. Stinski M. F., Meier J. L. (2007) Immediate-early CMV gene regulation and function. in Human Herpesviruses: Biology, Therapy and Immunoprophylaxis, 2011/02/25 Ed (Arvin A. M., Campadelli-Fiume G., Mocarski E. S., Moore P. S., Roizman B., Whitley R., Yamanishi K., eds) pp. 241–263, Cambridge Press, Cambridge, UK: [PubMed] [Google Scholar]
- 92. Paulus C., Nevels M. (2009) The human cytomegalovirus major immediate-early proteins as antagonists of intrinsic and innate antiviral host responses. Viruses 1, 760–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lembo D., Brune W. (2009) Tinkering with a viral ribonucleotide reductase. Trends Biochem. Sci. 34, 25–32 [DOI] [PubMed] [Google Scholar]
- 94. Hahn G., Khan H., Baldanti F., Koszinowski U. H., Revello M. G., Gerna G. (2002) The human cytomegalovirus ribonucleotide reductase homolog UL45 is dispensable for growth in endothelial cells, as determined by a BAC-cloned clinical isolate of human cytomegalovirus with preserved wild-type characteristics. J. Virol. 76, 9551–9555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Patrone M., Percivalle E., Secchi M., Fiorina L., Pedrali-Noy G., Zoppé M., Baldanti F., Hahn G., Koszinowski U. H., Milanesi G., Gallina A. (2003) The human cytomegalovirus UL45 gene product is a late, virion-associated protein and influences virus growth at low multiplicities of infection. J. Gen. Virol. 84, 3359–3370 [DOI] [PubMed] [Google Scholar]
- 96. Baillie J., Sahlender D. A., Sinclair J. H. (2003) Human cytomegalovirus infection inhibits tumor necrosis factor α (TNF-α) signaling by targeting the 55-kilodalton TNF-α receptor. J. Virol. 77, 7007–7016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Smith W., Tomasec P., Aicheler R., Loewendorf A., Nemčovičová I., Wang E. C., Stanton R. J., Macauley M., Norris P., Willen L., Ruckova E., Nomoto A., Schneider P., Hahn G., Zajonc D. M., Ware C. F., Wilkinson G. W., Benedict C. A. (2013) Human cytomegalovirus glycoprotein UL141 targets the TRAIL death receptors to thwart host innate antiviral defenses. Cell Host Microbe 13, 324–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Seirafian S., Prod'homme V., Sugrue D., Davies J., Fielding C., Tomasec P., Wilkinson G. W. (2014) Human cytomegalovirus suppresses Fas expression and function. J. Gen. Virol. 95, 933–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. DeFilippis V. R., Alvarado D., Sali T., Rothenburg S., Früh K. (2010) Human cytomegalovirus induces the interferon response via the DNA sensor ZBP1. J. Virol. 84, 585–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kaiser W. J., Upton J. W., Mocarski E. S. (2008) Receptor-interacting protein homotypic interaction motif-dependent control of NF-κB activation via the DNA-dependent activator of IFN regulatory factors. J. Immunol. 181, 6427–6434 [DOI] [PMC free article] [PubMed] [Google Scholar]