

Background: Abnormal NIK expression and activation trigger liver injury.
Results: CHIP bound to NIK and promoted NIK ubiquitination/degradation; liver-specific overexpression of NIK triggered fatal liver injury; and coexpression of CHIP reversed NIK detrimental effects.
Conclusion: CHIP negatively regulates NIK and protects against NIK-induced liver injury.
Significance: This study sheds light on a novel regulation of the NIK pathways by CHIP.
Keywords: Inflammation, Liver Injury, NF-kappa B (NF-KB), Protein Degradation, Ubiquitylation (Ubiquitination)
Abstract
Ser/Thr kinase NIK (NF-κB-inducing kinase) mediates the activation of the noncanonical NF-κB2 pathway, and it plays an important role in regulating immune cell development and liver homeostasis. NIK levels are extremely low in quiescent cells due to ubiquitin/proteasome-mediated degradation, and cytokines stimulate NIK activation through increasing NIK stability; however, regulation of NIK stability is not fully understood. Here we identified CHIP (carboxyl terminus of HSC70-interacting protein) as a new negative regulator of NIK. CHIP contains three N-terminal tetratricopeptide repeats (TPRs), a middle dimerization domain, and a C-terminal U-box. The U-box domain contains ubiquitin E3 ligase activity that promotes ubiquitination of CHIP-bound partners. We observed that CHIP bound to NIK via its TPR domain. In both HEK293 and primary hepatocytes, overexpression of CHIP markedly decreased NIK levels at least in part through increasing ubiquitination and degradation of NIK. Accordingly, CHIP suppressed NIK-induced activation of the noncanonical NF-κB2 pathway. CHIP also bound to TRAF3, and CHIP and TRAF3 acted coordinately to efficiently promote NIK degradation. The TPR but not the U-box domain was required for CHIP to promote NIK degradation. In mice, hepatocyte-specific overexpression of NIK resulted in liver inflammation and injury, leading to death, and liver-specific expression of CHIP reversed the detrimental effects of hepatic NIK. Our data suggest that CHIP/TRAF3/NIK interactions recruit NIK to E3 ligase complexes for ubiquitination and degradation, thus maintaining NIK at low levels. Defects in CHIP regulation of NIK may result in aberrant NIK activation in the liver, contributing to live injury, inflammation, and disease.
Introduction
NIK,4 also called MAP3K14, is an essential Ser/Thr kinase for the development and function of the immune system in both mice and humans (1–5). Systemic deletion of NIK results in severe immunodeficiency, leading to premature death in mice (1, 2, 6). A homozygous loss-of-function NIK mutation is associated with primary immunodeficiency disorders (5). In addition to regulating immune cells, NIK also regulates live metabolism and liver integrality (7, 8). NIK expression in the liver is higher in mice with obesity (7). High levels of hepatic NIK increase the hyperglycemic response to counterregulatory hormones, contributing to hyperglycemia and glucose intolerance in obesity (7). Abnormal activation of hepatic NIK also promotes hepatocytes to secret proinflammatory mediators that induce liver inflammation and liver destruction, leading to death in mice (8). A patient with a homozygous NIK mutation (expressing a kinase-dead P565R mutant) died at 3 years of age, with deterioration of liver function (5). Therefore, NIK levels appear to be maintained within a narrow range, and alterations in NIK levels (either above or below this range) are likely to cause impairment in cellular function and/or survival.
NIK is widely expressed in most tissues (9). Multiple cytokines as well as cellular stress and injury have been reported to activate NIK (10). NIK phosphorylates and activates IκB kinase α (IKKα), which in turn phosphorylates NF-κB2 precursor p100 (10). Phosphorylation triggers proteolytic cleavages of p100, generating a mature, transcriptionally active form of p52 NF-κB2 (10). NIK activation depends on an increase in its stability and protein levels (10). In quiescent cells, NIK is undetectable due to rapid degradation mediated by the ubiquitin/proteasome system (11, 12). cIAP1 and cIAP2 have been described as two important ubiquitin E3 ligases for NIK (13, 14). TRAF3 binds to NIK and recruits NIK to cIAP1/2 for ubiquitination and degradation (15, 16). In one model, cytokine stimulation causes TRAF3 degradation, thus uncoupling NIK from cIAP1/2, so newly synthesized NIK accumulates and becomes activated (15, 16).
In a screen for NIK-interacting proteins, we identified heat shock cognate 71-kDa protein (HSC70). HSC70 is a ubiquitously expressed chaperone that facilitates folding and maturation of numerous nascent proteins (17). HSC70 carries out folding reactions through cooperation with cochaperones, including CHIP (17). CHIP, also known as Stub1, is a ubiquitously expressed cochaperone (∼34.5 kDa). It contains three tetratricopeptide repeats (TPRs) at the N terminus, a coiled-coil region in the middle, and a U-box, a ring finger-like domain, at the C terminus (18, 19). The TPR domain mediates interactions of CHIP with HSC70 and other members of the HSP family (e.g. HSP70 and HSP90) (18, 20). The middle coiled-coil region mediates homodimerization of CHIP (19). The U-box domain contains intrinsic ubiquitin E3 ligase activity and mediates ubiquitination and degradation of HSC70 as well as misfolded, nonnative client proteins, providing a protein quality control mechanism (20–22). In addition to binding to HSP family members, CHIP also binds to several Thr/Ser kinases, including mixed lineage kinase 3 (MLK3), serum- and glucocorticoid-regulated kinase-1 (SGK-1), LKB1, and Akt; it promotes ubiquitination and degradation of these kinases via its intrinsic E3 ligase activity (23–26).
In this study, we demonstrate that CHIP binds via its TPR domain to NIK and promotes NIK degradation, leading to suppression of the noncanonical NF-κB2 pathway. CHIP-induced degradation of NIK is independent of CHIP intrinsic E3 ligase activity. CHIP binds to TRAF3, and CHIP and TRAF3 act coordinately to promote NIK degradation. Hepatocyte-specific overexpression of NIK triggers liver inflammation and injury, leading to death in mice; simultaneous overexpression of CHIP in the liver completely reverses NIK-induced death. These observations indicate that CHIP is a novel negative regulator of NIK.
EXPERIMENTAL PROCEDURES
Animal Experiments
Animal experiments were conducted following the protocols approved by the University Committee on the Use and Care of Animals (UCUCA). STOP-NIK mice (C57BL/6 background) were described previously (7, 27). Mice were housed on a 12-h light-dark cycle in the Unit for Laboratory Animal Medicine at the University of Michigan, and fed ad libitum a normal chow diet (9% fat; TestDiet, St. Louis, MO). STOP-NIK male mice (7–8 weeks) were coinfected with albumin-cre (1 × 1011 viral particles per mouse) and CHIP (1 × 1011 viral particles per mouse) adenoviruses, or albumin-cre (1 × 1011 viral particles per mouse) and GFP (1 × 1011 viral particles per mouse) adenoviruses, via tail vein injection as described previously (8). Blood samples were collected from tail veins, and blood glucose levels were determined using glucometers (Bayer Corp., Pittsburgh, PA). Plasma alanine aminotransferase (ALT) activity and bilirubin levels were measured using an ALT reagent set and a total bilirubin reagent set (Pointe Scientific Inc., Canton, MI), respectively.
Generation and Purification of shRNA Adenoviruses
CHIP shRNA sequences were: 5′-AAAUCCAUAUCUGCCAUGUAU-3′. shCHIP DNA oligonucleotides were inserted at 3′ to the H1 promoter in pAdeasy vectors. shCHIP pAdeasy vectors were linearized and transfected into Q293A cells to generate shCHIP adenoviruses. Adenoviruses were amplified in Q293A cells and purified using CsCl.
Cell Culture, Transient Transfection, and Adenoviral Infection
HEK293 cells were grown at 37 °C in 5% CO2 in DMEM supplemented with 25 mm glucose, 100 units ml−1 penicillin, 100 units ml−1 streptomycin, and 8% calf serum. For transient transfection, cells were split 16–20 h before transfection. Expression plasmids were mixed with polyethylenimine (Sigma) and introduced into cells. The total amount of plasmids was maintained constant by adding empty vectors. Primary hepatocytes were prepared from adult mice by liver perfusion with type II collagenase (Worthington Biochemical) as described previously (28). Hepatocytes were grown on collagen-coated plates in William's medium E (Sigma) supplemented with 2% FBS, 100 units ml−1 penicillin, and 100 μg ml−1 streptomycin, and infected with adenoviruses as described previously (28).
Luciferase Assays
NF-κB luciferase reporter plasmids were transfected into HEK293 cells. Cell extracts were prepared 48 h after transfection and subjected to luciferase activity assays using a kit (Promega, Madison, WI) following manufacturer instructions.
Immunoprecipitation and Immunoblotting
Cells or tissues were homogenized in an L-RIPA lysis buffer (50 mm Tris, pH 7.5, 1% Nonidet P-40, 150 mm NaCl, 2 mm EGTA, 1 mm Na3VO4, 100 mm NaF, 10 mm Na4P2O7, 1 mm benzamidine, 10 μg ml−1 aprotinin, 10 μg ml−1 leupeptin, 1 mm phenylmethylsulfonyl fluoride). For denatured immunoprecipitation, cells were homogenized in a denature buffer (50 mm Tris, pH 6.8; 1% SDS; 5 mm DTT), incubated at 95 °C for 10 min, and then diluted 10 times with L-RIPA buffer prior to immunoprecipitation. Cell extracts were incubated with primary antibodies at 4 °C for 2 h and then with protein A-agarose beads (RepliGen Corp., Waltham, MA) or protein G-Sepharose beads (GE Healthcare) for an additional hour at 4 °C. The immunocomplexes were washed three times with washing buffer (50 mm Tris, pH 7.5; 1% Nonidet P-40; 150 mm NaCl; 2 mm EGTA) and boiled at 95 °C for 5 min in loading buffer (50 mm Tris-HCl, pH 6.8; 2% SDS, 2% β-mercaptoethanol; 10% glycerol; 0.005% bromphenol blue). Protein was separated by SDS-PAGE, immunoblotted with the indicated antibodies, and visualized using the Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE) or ECL (Amersham Biosciences).
Reactive Oxygen Species Assays
Liver samples were homogenized in L-RIPA buffer. Liver extracts were incubated with dichlorofluorescein diacetate fluorescent (final concentration: 5 μm) probes (Sigma, D6883) at 37 °C for 1 h. Dichlorofluorescein diacetate fluorescent fluorescence was measured using a BioTek Synergy 2 multi-mode microplate reader (485 nm excitation and 527 nm emission).
Quantitative Real Time PCR (qPCR)
Total RNAs were extracted using TRIzol reagent (Invitrogen Life Technologies). The first-strand cDNAs were synthesized using random primers (Life Technologies, 48190-011) and M-MLV reverse transcriptase (Promega). Relative mRNA abundance of different genes was measured using SYBR Green PCR master mix (Life Technologies, 4367659) and Mx3000P real time PCR system (Stratagene, LA Jolla, CA). qPCR Primers were: MCP-1 forward, 5′-ACTGAAGCCAGCTCTCTCTTCCTC-3′, reverse, 5′-TTCCTTCTTGGGGTCAGCACAGAC-3′; inducible NOS (INOS) forward, 5′-CAGGGCCACCTCTACATTTG-3′, reverse, 5′-TGCCCCATAGGAAAAGACTG-3′; IL6 forward, 5′-AGCCAGAGTCCTTCAGA-3′, reverse, 5′-GGTCCTTAGCCACTCCT-3′; IL10 forward, 5′-CTGGACAACATACTGCTAACCG-3′, reverse, 5′-GGGCATCACTTCTACCAGGTAA-3′; TNFα forward, 5′-CATCTTCTCAAAATTCGAGTGACAA-3′, reverse, 5′-TGGGAGTAGACAAGGTACAACCC-3′; CXCL5 forward, 5′-TGCATTCCGCTTAGCTTTCT-3′, reverse, 5′-CAGAAGGAGGTCTGTCTGGA-3′; NIK forward, 5′-TCTCTGGAGGAACAGGAACAA-3′, reverse, 5′-GCCATTGAGAGACTGGATCTG-3′; 36B4 forward, 5′-AAGCGCGTCCTGGCATTGTCT-3′, reverse, 5′-CCGCAGGGGCAGCAGTGGT-3′; 18 S forward, 5′-CGCTTCCTTACCTGGTTGAT-3′, reverse, 5′-GAGCGACCAAAGGAACCATA-3′.
Statistical Analysis
Data were presented as means ± S.E. Differences between groups were analyzed by Student's t tests. p < 0.05 was considered statistically significant.
RESULTS
CHIP Binds via Its TPR Domain to NIK
To purify NIK-binding proteins, FLAG-tagged NIK was overexpressed in mouse primary hepatocytes and immunoprecipitated with anti-FLAG antibodies. NIK-bound proteins were identified using LTQ-XL mass spectrometers and proteomic analysis. HSC70 was a NIK-associated protein. To verify HSC70-NIK interaction, HSC70 (with an N-terminal Myc tag) and NIK (with an N-terminal FLAG tag) were transiently overexpressed in HEK293 cells. Myc-HSC70 was immunoprecipitated with antibody to Myc and immunoblotted with antibody to FLAG or Myc. Reciprocally, FLAG-NIK was immunoprecipitated with antibody to FLAG and immunoblotted with antibody to Myc or FLAG. In both assays, NIK was coimmunoprecipitated with HSC70 (Fig. 1, A and B).
FIGURE 1.

CHIP binds via its TPR domain to NIK. HEK293 cell extracts were prepared 48 h after transfection. A and B, Myc-HSC70 was transiently coexpressed with FLAG-NIK, immunoprecipitated (IP) with antibody to Myc, and immunoblotted with antibodies to FLAG or Myc (A). Reciprocally, FLAG-NIK was immunoprecipitated with antibody to FLAG and immunoblotted with antibodies to Myc or FLAG (B). C and D, FLAG-NIK was coexpressed with CHIP or Myc-CHIP. FLAG-NIK was immunoprecipitated with antibody to FLAG and immunoblotted with antibodies to CHIP or FLAG (C). Myc-CHIP was immunoprecipitated with antibody to Myc and immunoblotted with antibodies to FLAG or Myc (D). Cell extracts were immunoblotted with antibodies to CHIP, FLAG, or Myc. E, FLAG-NIK(KA) was coexpressed with Myc-CHIP, immunoprecipitated with antibody to FLAG, and immunoblotted with antibodies to Myc or FLAG. F, FLAG-NIK was coexpressed with CHIP, CHIP(H260Q), or CHIP(K30A), immunoprecipitated with antibody to FLAG, and immunoblotted with antibodies to CHIP or FLAG. Cell extracts were also immunoblotted with antibodies to FLAG, Myc, or CHIP.
We examined interactions between NIK and HSC70 cochaperone CHIP. FLAG-NIK was transiently coexpressed with CHIP in HEK293 cells and immunoprecipitated with antibody to FLAG. CHIP was detected in NIK immunoprecipitates (Fig. 1C). For reciprocal coimmunoprecipitation, we prepared expression vectors encoding CHIP with an N-terminal Myc tag. Myc-CHIP was transiently coexpressed with FLAG-NIK, immunoprecipitated with antibody to Myc, and immunoblotted with antibody to FLAG. FLAG-NIK was coimmunoprecipitated with Myc-CHIP (Fig. 1D).
To determine whether NIK kinase activity is required for NIK-CHIP interaction, FLAG-NIK(KA) was coexpressed with Myc-CHIP in HEK293 cells. NIK(KA) is catalytically inactive due to lack of the ATP-binding site (7). FLAG-NIK(KA) was immunoprecipitated with antibody to FLAG and immunoblotted with antibody to Myc. CHIP was similarly coimmunoprecipitated with NIK(KA) (Fig. 1E), indicating that the catalytic activity of NIK is dispensable for NIK-CHIP interaction.
To determine whether the TPR and the U-box domains are involved in CHIP-NIK interaction, we generated CHIP(K30A) and CHIP(H260Q). CHIP(K30A) has a defective TPR domain and is unable to bind to HSP70 and HSP90 (29). CHIP(H260Q) has a defective U-box domain and lacks intrinsic E3 ligase activity (30). FLAG-NIK was coexpressed with CHIP, CHIP(H260Q), or CHIP(K30A), immunoprecipitated with antibody to FLAG, and immunoblotted with antibody to CHIP. NIK was coimmunoprecipitated with CHIP and CHIP(H260Q) but not CHIP(K30A) (Fig. 1F). Antibody to CHIP detected two forms of CHIP, whereas antibody to Myc detected one form. Together, these data indicate that NIK binds to both HSC70 and CHIP. The TPR domain, but not the U-box domain of CHIP, mediates CHIP-NIK interaction.
CHIP Promotes NIK Degradation
To determine whether CHIP regulates NIK levels, FLAG-NIK was coexpressed with CHIP in HEK293 cells, and cell extracts were immunoblotted with antibodies to FLAG, CHIP, or the p85 regulatory subunit of the PI 3-kinase (a loading control). CHIP dose-dependently decreased NIK protein levels (Fig. 2A). To determine whether CHIP increases NIK degradation, FLAG-NIK was transiently coexpressed with Myc-CHIP, and protein synthesis was blocked by cycloheximide 24 h after transfection. Cell extracts were prepared 0, 2, 4, 6, and 8 h after cycloheximide treatments and immunoblotted with antibodies to FLAG, Myc, or tubulin. CHIP markedly accelerated NIK degradation rates (Fig. 2B, upper panels). NIK half-life was reduced from ∼4 h in the absence to ∼1.8 h in the presence of coexpression of CHIP (Fig. 2B, lower panels).
FIGURE 2.

CHIP decreases NIK stability. A, FLAG-NIK plasmids were cotransfected with increasing amounts of CHIP plasmids into HEK293 cells. Cell extracts were prepared 48 h after transfection and immunoblotted with antibodies to FLAG, CHIP, or p85. B, FLAG-NIK was coexpressed with or without Myc-CHIP in HEK293 cells. Cells were treated with cycloheximide (100 μm for 0, 2, 4, 6, or 8 h) 24 h after transfection. Cell extracts were immunoblotted with antibodies to FLAG, Myc, or tubulin. NIK levels were quantified by densitometry and normalized to tubulin levels. Con: n = 3, CHIP: n = 3. C, FLAG-NIK or FLAG-NIK(KA) was coexpressed with or without Myc-CHIP in HEK293 cells. Cell extracts were prepared 48 h after transfection and immunoblotted with antibodies to FLAG, CHIP, or p85. Con, control; CHX, cycloheximide. D, mouse primary hepatocytes were coinfected with NIK and CHIP or GFP (control) adenoviruses. Cell extracts were prepared 24 h after infection and immunoblotted with antibodies to FLAG, CHIP, or tubulin. E, mouse primary hepatocytes were infected with scramble or shCHIP adenoviruses. Cell extracts were prepared 48 h after infection and immunoblotted with antibodies to CHIP or tubulin. Values are presented as means ± S.E. *, p < 0.05.
To determine whether the catalytic activity of NIK is required for CHIP-mediated degradation of NIK, kinase-inactive FLAG-NIK(KA) was coexpressed with CHIP and immunoblotted with antibody to FLAG. Like NIK, NIK(KA) was also down-regulated by CHIP (Fig. 2C). Therefore, NIK kinase activity is not required for either CHIP-NIK interactions or CHIP-mediated destruction of NIK.
To determine whether CHIP promotes NIK degradation in hepatocytes, NIK or CHIP was introduced into mouse primary hepatocytes via FLAG-NIK or Myc-CHIP adenoviral infection. Cell extracts were prepared 24 h after infection and immunoblotted with antibodies to FLAG, CHIP, or tubulin. CHIP markedly decreased NIK protein levels in primary hepatocytes (Fig. 2D). Thus, CHIP promotes NIK degradation in multiple cell types.
To determine whether endogenous CHIP regulates NIK stability, we prepared shCHIP and scramble (control) adenoviruses. Mouse primary hepatocytes were coinfected with NIK and shCHIP (or scramble) adenoviruses, and cell extracts were prepared 48 h after infection and immunoblotted with antibodies to NIK, CHIP, or tubulin. Infection with shCHIP adenoviral vectors reduced the levels of endogenous CHIP as expected; shCHIP-mediated down-regulation of CHIP markedly increased NIK levels (Fig. 2E). These results suggest that endogenous CHIP is a physiological negative regulator of NIK.
The Ubiquitin/Proteasome System Mediates CHIP-induced Degradation of NIK
To determine whether the ubiquitin/proteasome system mediates CHIP-induced NIK degradation, FLAG-NIK was coexpressed in mouse primary hepatocytes with or without Myc-CHIP using adenovirus-mediated gene delivery methods. Hepatocytes were treated with MG132, a 26 S proteasome inhibitor, 20 h after adenoviral infection. Cell extracts were prepared 4 h after MG132 treatment and immunoblotted with antibodies to FLAG, CHIP, or tubulin. In the absence of MG132, CHIP markedly decreased NIK levels (Fig. 3A, lane 3 versus lane 1). MG132-mediated inhibition of proteasomes greatly inhibited CHIP-induced reduction of NIK (Fig. 3A, lane 4 versus lanes 1 and 3).
FIGURE 3.

CHIP promotes NIK degradation via the ubiquitin/proteasome system. A, mouse primary hepatocytes were infected with FLAG-NIK and Myc-CHIP adenoviruses and treated with MG132 (20 μm) 16–20 h after infection. Cell extracts were prepared 4 h after MG132 treatment and immunoblotted with antibodies to FLAG, CHIP, or tubulin. B, FLAG-NIK plasmids were cotransfected into HEK293 cells with CHIP plasmids in the presence of HA-ubiquitin expression. Sixteen hours after transfection, cells were treated with MG132 (20 μm) for 4 h. Cell extracts were immunoprecipitated (IP) with antibody to FLAG under denatured conditions and immunoblotted with antibodies to HA or NIK. Cell extracts were immunoblotted with antibodies to NIK or CHIP. C, mouse primary hepatocytes were infected with FLAG-NIK and Myc-CHIP adenoviruses and treated with MG132. NIK was immunoprecipitated with antibody to FLAG under denatured conditions and immunoblotted with antibodies to ubiquitin (Ub) or FLAG. Lysates were immunoblotted with antibody to CHIP. D, NIK was transiently coexpressed with CHIP in HEK293 cells. Cells were treated with chloroquine (30 μm) 36 h after transfection. Cell extracts were prepared 6 h after chloroquine treatment and immunoblotted with antibodies to FLAG, CHIP, or p85. E, CHIP was transiently expressed in HEK293 cells. Cell extracts were prepared 36 h after transfection and immunoblotted with antibodies to LC3, CHIP, or p85.
To determine whether CHIP promotes ubiquitination of NIK, FLAG-NIK was coexpressed with CHIP and HA-ubiquitin in HEK293 cells. Cells were treated with MG132 to block degradation of ubiquitinated NIK (Ub-NIK). FLAG-NIK was immunoprecipitated with antibody to FLAG under denatured conditions and immunoblotted with antibodies to HA or NIK. We detected modest levels of basal Ub-NIK, and overexpression of CHIP markedly increased Ub-NIK levels (Fig. 3B). To determine whether CHIP stimulates ubiquitination of NIK in hepatocytes, mouse primary hepatocytes were infected with NIK and CHIP (or GFP) adenoviruses and treated with MG132 20 h after infection. Cell extracts were prepared 4 h after MG132 treatment, immunoprecipitated with antibody to FLAG, and immunoblotted with antibodies to ubiquitin or FLAG. Overexpression of CHIP increased the amount of Ub-NIK in hepatocytes (Fig. 3C). Together, these results suggest that CHIP promotes NIK degradation through the ubiquitin/proteasome system.
To determine whether autophagy is involved in CHIP-induced destruction of NIK, cells were treated with chloroquine, a lysosome inhibitor, to block autophagy. Inhibition of autophagy did not attenuate CHIP-induced reduction of NIK levels (Fig. 3D). Additionally, overexpression of CHIP did not alter the levels of LC3-II, an autophagy marker (Fig. 3E). Thus, autophagy is unlikely to mediate CHIP-promoted NIK degradation.
The Intrinsic E3 Activity of CHIP Is Not Required for CHIP to Promote NIK Degradation
To determine the contributions of the TPR and the U-box domains to CHIP-induced degradation of NIK, FLAG-NIK was transiently coexpressed with CHIP, CHIP(H260Q), or CHIP(K30A). Cell extracts were prepared 48 h after transfection and immunoblotted with antibodies to FLAG, CHIP, or p85. CHIP decreased NIK levels in a dose-dependent manner (Fig. 4A, top three panels); surprisingly, CHIP(H260Q), a E3 ligase-inactive mutant, also dose-dependently decreased NIK levels (Fig. 4A, middle three panels). These results indicate that CHIP does not function as a ubiquitin E3 ligase for NIK. In contrast, CHIP(K30A), which contains the intact U-box domain but lacks the functional TPR domain, was unable to promote NIK degradation (Fig. 4A, bottom three panels). We quantified NIK protein levels and observed that CHIP and CHIP(H260Q), but not CHIP(K30A), dose-dependently promoted NIK degradation (Fig. 4B). These results suggest that TPR domain-mediated CHIP-NIK interaction, but not the E3 ligase activity of the U-box domain, is required for CHIP to promote NIK degradation.
FIGURE 4.

The TPR but not the U-box domain is required for CHIP to promote NIK degradation. A and B, NIK was transiently coexpressed with CHIP, CHIP(H260Q), or CHIP(K30A) in HEK293 cells. Cell extracts were prepared 48 h after transfection and immunoblotted with antibodies to FLAG, CHIP, or p85 (A). NIK protein levels were quantified and normalized to p85 levels (B). n = 3. a.u., arbitrary units. C, FLAG-NIK was coexpressed with Myc-CHIP and Myc-HSC70 in HEK293 cells. Cell extracts were prepared 48 h after transfection and immunoblotted with antibodies to FLAG, Myc, or tubulin. CHIP and HSC70 mass was ∼34.5 and ∼71 kDa, respectively. Values are presented as means ± S.E. *, p < 0.05.
HSC70 binds to both NIK and CHIP, so we examined the effect of HSC70 on the ability of CHIP to reduce NIK stability. Myc-HSC70 was transiently coexpressed with FLAG-NIK and Myc-CHIP in HEK293 cells, and cell extracts were prepared 48 h after transfection and immunoblotted with antibodies to FLAG, Myc, or tubulin. CHIP markedly decreased NIK levels, and coexpression of HSC70 attenuated the ability of CHIP to reduce NIK levels (Fig. 4C).
CHIP and TRAF3 Act Coordinately to Promote NIK Degradation
Like CHIP, TRAF3 binds to NIK and promotes NIK degradation (10). To compare destructive capability between CHIP and TRAF3, FLAG-NIK was coexpressed with Myc-CHIP, TRAF3, or both. Cell extracts were prepared 48 h after transfection and immunoblotted with antibodies to FLAG, Myc, or TRAF3 (Fig. 5A). NIK levels were quantified by densitometry (Fig. 5B). CHIP was more potent than TRAF3 to reduce NIK levels, and a combination of both CHIP and TRAF3 decreased NIK to the lowest levels (Fig. 5, A and B).
FIGURE 5.

CHIP and TRAF3 act coordinately to promote NIK degradation. A and B, FLAG-NIK was coexpressed with Myc-CHIP and TRAF3 in HEK293 cells. Cell extracts were prepared 48 h after transfection and immunoblotted with antibodies to FLAG, Myc, or TRAF3 (A). NIK levels were quantified by densitometry and normalized to tubulin levels (B). n = 3. n.s.: nonspecific bands. C, TRAF3 was cotransfected with Myc-CHIP or Myc-CHIP(K30A) in HEK293 cells, and cell extracts were prepared 48 h after transfection. CHIP was immunoprecipitated (IP) with antibody to Myc and immunoblotted with antibodies to TRAF3 or Myc. Cell extracts were immunoblotted with antibodies to TRAF3 or Myc. D, FLAG-NIK was coexpressed with Myc-CHIP or Myc-CHIP(K30A) in the presence or absence of overexpression of TRAF3 in HEK293 cells. Cell extracts were prepared 48 h after transfection and immunoblotted with antibodies to FLAG, Myc, TRAF3, or tubulin. Values are presented as means ± S.E. *, p < 0.05.
To determine whether CHIP binds to TRAF3, Myc-CHIP or Myc-CHIP(K30A) was coexpressed with TRAF3, immunoprecipitated with antibody to Myc, and immunoblotted with antibodies to TRAF3 or Myc. TRAF3 was coimmunoprecipitated with CHIP but not CHIP(K30A) (Fig. 5C). The residual levels of TRAF3 in the absence of CHIP (Fig. 5C, lane 1) or presence of CHIP(K30A) (Fig. 5C, lane 3) may result from nonspecific binding of TRAF3 to Myc antibody-coupled beads. CHIP did not alter (or slightly reduced) TRAF3 levels (Fig. 5C). These data indicate that the TPR domain of CHIP is required not only for CHIP-NIK, but also for CHIP-TRAF3, interactions.
To determine whether the TPR domain is required for CHIP to enhance TRAF3-induced degradation of NIK, FLAG-NIK and TRAF3 were coexpressed with Myc-CHIP or Myc-CHIP(K30A) in HEK293 cells, and cell extracts were prepared 48 h after transfection. Under these conditions, CHIP alone modestly reduced NIK levels (Fig. 5D, lanes 1 and 2), and coexpression of TRAF3 with CHIP reduced NIK levels to extremely low levels (Fig. 5D, lane 3). CHIP(K30A) had dramatically reduced capability to enhance TRAF3-induced NIK reduction (Fig. 5D, lane 4). These findings suggest that the TPR domain-mediated CHIP-TRAF3 interaction is required for the additive/synergistic action of TRAF3 and CHIP to promote NIK degradation.
CHIP Suppresses NIK Activation of the Noncanonical NF-κB2 Pathway
To examine the effect of CHIP on signaling events downstream of NIK, NF-κB2 precursors (p100) were coexpressed with FLAG-NIK in the presence or absence of Myc-CHIP. Cell extracts were prepared 48 h after transfection and immunoblotted with antibodies to NF-κB2, FLAG, or CHIP. Overexpression of NIK activated the noncanonical NF-κB2 pathway as revealed by production of the mature form of p52 NF-κB2 (Fig. 6A, lanes 2 and 3 versus lane 1). Coexpression of CHIP substantially reduced the levels of both NIK and p52 NF-κB2 (Fig. 6A, lanes 5 and 6 versus lanes 2 and 3).
FIGURE 6.
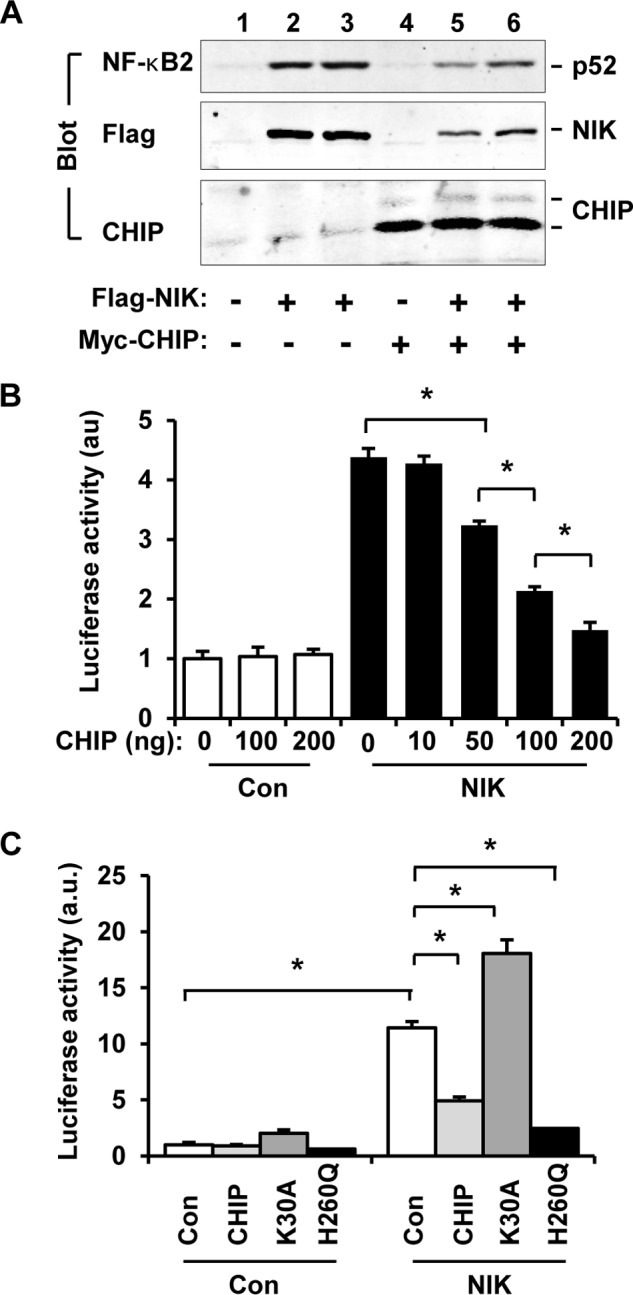
CHIP attenuates NIK-stimulated activation of the noncanonical NF-κB2 pathway. A, FLAG-NIK and p100 was coexpressed with or without Myc-CHIP in HEK293 cells. Cell extracts were prepared 48 h after transfection and immunoblotted with antibodies to NF-κB2, FLAG, or CHIP. B, NF-κB luciferase reporter, CHIP, and NIK plasmids were cotransfected into HEK293 cells. Luciferase activity was measured 48 h after transfection. Con, control. C, NF-κB luciferase reporter plasmids were cotransfected with CHIP, CHIP(K30A), or CHIP(H260Q) plasmids into HEK293 in the presence or absence of NIK overexpression. Luciferase activity was measured 48 h after transfection. Values are presented as means ± S.E. *, p < 0.05.
We then measured NF-κB2 activity using NF-κB luciferase reporter assays. NF-κB luciferase reporter plasmids were transiently cotransfected into HEK293 cells with NIK plasmids in the presence or absence of coexpression of CHIP, and luciferase activity was measured 48 h after transfection. NF-κB luciferase reporters are expected to be activated by endogenous NF-κB. NIK markedly increased the activity of NF-κB luciferase reporters, and CHIP dose-dependently suppressed NIK-induced activation of NF-κB luciferase reporters (Fig. 6B). In the absence of NIK, CHIP alone did not alter basal NF-κB luciferase reporter activity. These data suggest that CHIP suppresses activation of endogenous NF-κB2 by NIK.
To determine whether the TPR and the U-box domains mediate CHIP suppression of endogenous NF-κB2, NF-κB luciferase reporter plasmids were cotransfected into HEK293 cells with CHIP, CHIP(K30A), or CHIP(H260Q) plasmids in the presence or absence of coexpression of NIK. NIK increased NF-κB luciferase reporter activity, and CHIP suppressed NIK action (Fig. 6C). Like CHIP, CHIP(H260Q) also dramatically suppressed NIK-stimulated activation of the NF-κB luciferase reporters (Fig. 6C). These data indicate that the U-box E3 ligase activity is not required for CHIP suppression of the noncanonical NF-κB2 pathway. In contrast to CHIP and CHIP(H260Q), CHIP(K30A), which was unable to bind to NIK and to reduce NIK levels, further increased NF-κB luciferase reporter activity at both basal and NIK-stimulated conditions (Fig. 6C). Together, these results suggest that CHIP suppresses the noncanonical NF-κB2 pathway through its TPR domain-mediated interaction with NIK and subsequent degradation of NIK.
CHIP Suppresses NIK-triggered Fatal Liver Inflammation and Injury
To determine whether CHIP down-regulates NIK in animals, we obtained STOP-NIK mice. In these mice, a STOP-NIK cassette was knocked in the Rosa26 allele, and Cre-mediated excision of the STOP sequences is able to reactivate the NIK transgene (27). We purified albumin-cre adenoviruses that express Cre under the control of the murine albumin promoter. STOP-NIK mice were infected with albumin-cre adenoviruses via tail vein injection. We previously reported that under these conditions, Cre is expressed only in the hepatocytes of the infected mice, resulting in hepatocyte-specific activation of the NIK transgene (8). In agreement, liver NIK mRNA levels were much higher in the albumin-cre group than in the empty albumin adenoviral vector group (control) (Fig. 7A). In separate groups, livers were harvested 7 days after infection and then used for measuring the levels of NF-κB2 and CHIP by immunoblotting with antibodies to NF-κB2, or CHIP. Overexpression of CHIP decreased the levels of the mature form of p52 NF-κB2 in the liver (Fig. 7B), presumably due to CHIP-mediated degradation of NIK.
FIGURE 7.

CHIP protects against NIK-induced liver inflammation and fibrosis. A, STOP-NIK mice (7–8 weeks) were infected with albumin-cre or empty albumin adenoviral vectors via tail vein injection. Liver NIK mRNA levels were measured 18 days after infection by qPCR and normalized to 18 S levels. n = 6. Con, control. B, STOP-NIK mice were coinfected with Cre and CHIP or Cre and GFP adenoviruses. Liver extracts were prepared 7 days after infection and immunoblotted with antibodies to NF-κB2, CHIP, or tubulin. C–I, STOP-NIK male mice (7–8 weeks) were coinfected with Cre and CHIP or Cre and GFP adenoviruses. Cre;GFP: n = 7, Cre;CHIP: n = 6. C, survival curves. D, blood ALT activity and bilirubin and glucose levels. E, body weight 6 days after infection. F, liver weight (normalized to body weight (BW)) 6 days after infection. G, H&E staining of liver paraffin sections (5 μm) (6 days after infection). H, liver mRNA abundance was measured 6 days after infection by qPCR and normalized to 36B4 levels. a.u., arbitrary units; INOS, inducible NOS. I, liver reactive oxygen species (ROS) levels (normalized to liver weight) were measured 6 days after infection. Values are presented as means ± S.E. *, p < 0.05.
We previously reported that hepatocyte-specific activation of the NIK transgene, triggered by albumin-cre adenoviral infection of STOP-NIK mice, results in liver injury and failure, leading to death (8). To determine whether CHIP attenuates the detrimental effect of NIK in the liver, STOP-NIK mice were infected with albumin-cre together with CHIP or GFP (control) adenoviruses via tail vein injection. Hepatocyte-specific activation of the NIK transgene caused death of STOP-NIK mice within 2 weeks after infection with albumin-cre adenoviruses, and coexpression of CHIP in the liver completely rescued NIK-induced death (Fig. 7C). These data demonstrate that CHIP antagonizes the death effects of abnormally high levels of hepatic NIK in animals, presumably by increasing NIK degradation.
Hepatocyte-specific activation of the NIK transgene caused liver injury, as revealed by increased blood ALT activity, increased bilirubin levels, and decreased blood glucose levels, in STOP-NIK mice infected with albumin-cre adenoviruses (Fig. 7D). Co-expression of CHIP significantly reduced blood ALT activity and bilirubin levels and increased blood glucose levels in STOP-NIK mice infected with albumin-cre and CHIP adenoviruses (Fig. 7D). We previously reported that hepatocyte-specific activation of the NIK transgene decreases body weight and increases liver weight, liver inflammation, and liver oxidative stress in STOP-NIK mice (8). Coexpression of CHIP largely attenuated these harmful effects of aberrant hepatic NIK in STOP-NIK mice infected with albumin-cre and CHIP adenoviruses, as revealed by significantly increased body weight (Fig. 7E), decreased hepatomegaly (Fig. 7F), decreased immune cell infiltration into the liver (Fig. 7G), decreased expression of proinflammatory genes (Fig. 7H), and decreased levels of reactive oxygen species (Fig. 7I). In obesity, a modest elevation of liver NIK enhances the hyperglycemic response to glucagon, thus increasing hepatic glucose production (7). In contrast, extremely higher levels of NIK, as in the current model, decrease hepatic glucose production by inducing hepatocyte injury and death (8). Together, these results demonstrate that CHIP counteracts the majority of the deteriorating effects of aberrant activation of hepatic NIK.
DISCUSSION
We provide multiple lines of evidence showing that CHIP is a novel negative regulator of NIK. First, CHIP binds via its TPR domain to NIK. Second, CHIP dose-dependently decreases NIK protein levels and markedly reduces NIK stability and half-life. CHIP-NIK interaction is required for CHIP-induced NIK destruction. Third, CHIP markedly suppresses NIK activation of the noncanonical NF-κB2 pathway. Forth, overexpression of CHIP in the liver completely reverses death induced by hepatocyte-specific overexpression of NIK and markedly attenuates the multiple detrimental effects of NIK.
CHIP promotes NIK degradation at least in part by increasing ubiquitination of NIK and subsequent degradation of NIK by the 26 S proteasome system. We observed that CHIP promoted ubiquitination of NIK. Proteasome inhibitors attenuated the ability of CHIP to decrease NIK levels. CHIP functions as a ubiquitin E3 ligase for multiple substrates, including membrane proteins (e.g. CFTR, CYP3A4, EGFR, and ErbB2), cytoplasmic proteins (e.g. MLK3, Akt, SGK-1, and Pten), and nuclear proteins (e.g. FOXO1, Smad3, myocardin, and inducible cAMP early repressor); its intrinsic E3 ligase activity in the U-box is required for CHIP to promote ubiquitination and degradation of these proteins (21, 24–26, 29, 31–37). Surprisingly, the intrinsic E3 ligase activity is not involved in mediating CHIP-induced NIK degradation. CHIP(H260), an E3 ligase-inactive mutant, promoted NIK destruction and suppressed activation of the noncanonical NF-κB2 pathway to a similar degree as CHIP did. In contrast, the TPR domain, which mediated CHIP-NIK interaction, was required for CHIP to decrease NIK levels and suppress the activation of the noncanonical NF-κB2 pathway. These findings raise the possibility that CHIP-NIK interaction may recruit NIK to an E3 ligase complex that ubiquitinates NIK. Alternatively, CHIP may function as a scaffold to coordinate dynamic interactions between NIK and its E3 ligase(s), thus promoting NIK ubiquitination. In agreement with this idea, CHIP binds to other E3 ligases, including Parkin (38). However, our data do not exclude the possibility that CHIP may decrease NIK levels by an additional mechanism.
CHIP appears to act in coordination with TRAF3 to efficiently promote ubiquitination and degradation of NIK. CHIP bound via its TPR domain to TRAF3. CHIP or TRAF3 alone promoted degradation of NIK to relatively modest levels; together, they induced NIK destruction to the lowest levels. CHIP(K30A) bound to neither NIK nor TRAF3, and was also unable to enhance TRAF3-induced degradation of NIK, indicating that CHIP-NIK and/or CHIP-TRAF3 complex formation is critical for NIK degradation. The middle coiled-coil regions mediate dimerization of CHIP (19). A CHIP dimer has two TPR domains. One may bind to NIK, whereas the other may bind to TRAF3, leading to formation of a TRAF3-CHIP-NIK complex. The formation of this tertiary complex may facilitate ubiquitination and degradation of NIK. Furthermore, HSC70 decreased the ability of CHIP to decrease NIK levels, providing an additional route through which external and/or internal signals regulate activation of NIK and the noncanonical NF-κB2 pathway.
The liver is an essential metabolic organ. Liver injury is the leading cause of mortality and morbidity. Hepatic NIK is aberrantly activated in multiple liver disease, including alcoholic liver disease, drug-induced liver disease, and primary biliary cirrhosis (8). In mice, hepatocyte-specific overexpression of NIK is sufficient to cause severe liver inflammation and injury, leading to death within a few weeks (8). We observed that liver-specific overexpression of CHIP completely reversed hepatocyte NIK-induced death and largely corrected the abnormalities caused by hepatocyte-specific overexpression of NIK (e.g. loss of body weight, hypoglycemia, liver inflammation, liver oxidative stress, and hepatomegaly). These findings raise the possibility that CHIP may act a “brake” to suppress abnormal activation of NIK in hepatocytes, thus protecting against liver injury and liver disease.
In conclusion, we have identified CHIP as a previously unknown negative regulator of NIK. CHIP binds to NIK and promotes degradation of NIK independently of its intrinsic E3 ligase activity. Liver-specific expression of CHIP reverses the detrimental effects of abnormal activation of hepatocyte NIK. Our data suggest that hepatic CHIP protects against liver injury in certain pathological conditions and serve as a potential therapeutic target for the treatment of liver diseases.
Acknowledgments
We thank Drs. Liang Sheng, Lin Jiang, and Zheng Chen and Nish Patel for assistance and discussion. We thank Dr. Klaus Rajewsky (Immune Disease Institute, Harvard Medical School, Boston, MA) for providing STOP-NIK mice, and we thank Drs. Patterson (University of North Carolina, Chapel Hill) and Yoichi Osawa (University of Michigan) for providing CHIP and HSC70 cDNAs. This work utilized the cores supported by the Michigan Diabetes Research and Training Center (National Institutes of Health Grant DK020572), Michigan Metabolomics and Obesity Center (National Institutes of Health Grant DK089503), the University of Michigan Comprehensive Cancer Center (National Institutes of Health Grant CA46592), the University of Michigan Nathan Shock Center (National Institutes of Health Grant P30AG013283), and the University of Michigan Gut Peptide Research Center (National Institutes of Health Grant DK34933).
This study was supported by National Institutes of Health Grants DK091591 and DK094014 (to L. R.). This work was also supported by grants from the China Scholarship Council (CSC) (to B. J.) and the National Beef and Yak Industrial Technology System (Grant CARS-38) and the China National 863 Program (Grant 2013AA102505) (to L. Z.).
- NIK
- NF-κB-inducing kinase
- CHIP
- carboxyl terminus of HSC70-interacting protein
- TPR
- tetratricopeptide repeats
- HSP
- heat shock protein
- ALT
- alanine aminotransferase
- qPCR
- quantitative real time PCR
- Ub-NIK
- ubiquitinated NIK.
REFERENCES
- 1. Yin L., Wu L., Wesche H., Arthur C. D., White J. M., Goeddel D. V., Schreiber R. D. (2001) Defective lymphotoxin-β receptor-induced NF-κB transcriptional activity in NIK-deficient mice. Science 291, 2162–2165 [DOI] [PubMed] [Google Scholar]
- 2. Aya K., Alhawagri M., Hagen-Stapleton A., Kitaura H., Kanagawa O., Novack D. V. (2005) NF-κB-inducing kinase controls lymphocyte and osteoclast activities in inflammatory arthritis. J. Clin. Invest. 115, 1848–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miyawaki S., Nakamura Y., Suzuka H., Koba M., Yasumizu R., Ikehara S., Shibata Y. (1994) A new mutation, aly, that induces a generalized lack of lymph nodes accompanied by immunodeficiency in mice. Eur. J. Immunol. 24, 429–434 [DOI] [PubMed] [Google Scholar]
- 4. Koike R., Nishimura T., Yasumizu R., Tanaka H., Hataba Y., Hataba Y., Watanabe T., Miyawaki S., Miyasaka M. (1996) The splenic marginal zone is absent in alymphoplastic aly mutant mice. Eur. J. Immunol. 26, 669–675 [DOI] [PubMed] [Google Scholar]
- 5. Willmann K. L., Klaver S., Doğu F., Santos-Valente E., Garncarz W., Bilic I., Mace E., Salzer E., Conde C. D., Sic H., Májek P., Banerjee P. P., Vladimer G. I., Haskoloğlu S., Bolkent M. G., Küpesiz A., Condino-Neto A., Colinge J., Superti-Furga G., Pickl W. F., van Zelm M. C., Eibel H., Orange J. S., Ikincioğulları A., Boztuğ K. (2014) Biallelic loss-of-function mutation in NIK causes a primary immunodeficiency with multifaceted aberrant lymphoid immunity. Nat. Commun. 5, 5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Häcker H., Chi L., Rehg J. E., Redecke V. (2012) NIK prevents the development of hypereosinophilic syndrome-like disease in mice independent of IKKα activation. J. Immunol. 188, 4602–4610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sheng L., Zhou Y., Chen Z., Ren D., Cho K. W., Jiang L., Shen H., Sasaki Y., Rui L. (2012) NF-κB-inducing kinase (NIK) promotes hyperglycemia and glucose intolerance in obesity by augmenting glucagon action. Nat. Med. 18, 943–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shen H., Sheng L., Chen Z., Jiang L., Su H., Yin L., Omary M. B., Rui L. (2014) Mouse hepatocyte overexpression of NF-κB-inducing kinase (NIK) triggers fatal macrophage-dependent liver injury and fibrosis. Hepatology 60, 2065–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fagarasan S., Shinkura R., Kamata T., Nogaki F., Ikuta K., Tashiro K., Honjo T. (2000) Alymphoplasia (aly)-type nuclear factor κB-inducing kinase (NIK) causes defects in secondary lymphoid tissue chemokine receptor signaling and homing of peritoneal cells to the gut-associated lymphatic tissue system. J. Exp. Med. 191, 1477–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sun S. C. (2012) The noncanonical NF-κB pathway. Immunol. Rev. 246, 125–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saitoh Y., Yamamoto N., Dewan M. Z., Sugimoto H., Martinez Bruyn V. J., Iwasaki Y., Matsubara K., Qi X., Saitoh T., Imoto I., Inazawa J., Utsunomiya A., Watanabe T., Masuda T., Yamamoto N., Yamaoka S. (2008) Overexpressed NF-κB-inducing kinase contributes to the tumorigenesis of adult T-cell leukemia and Hodgkin Reed-Sternberg cells. Blood 111, 5118–5129 [DOI] [PubMed] [Google Scholar]
- 12. Liao G., Zhang M., Harhaj E. W., Sun S. C. (2004) Regulation of the NF-κB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J. Biol. Chem. 279, 26243–26250 [DOI] [PubMed] [Google Scholar]
- 13. Varfolomeev E., Blankenship J. W., Wayson S. M., Fedorova A. V., Kayagaki N., Garg P., Zobel K., Dynek J. N., Elliott L. O., Wallweber H. J., Flygare J. A., Fairbrother W. J., Deshayes K., Dixit V. M., Vucic D. (2007) IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 131, 669–681 [DOI] [PubMed] [Google Scholar]
- 14. Vince J. E., Wong W. W., Khan N., Feltham R., Chau D., Ahmed A. U., Benetatos C. A., Chunduru S. K., Condon S. M., McKinlay M., Brink R., Leverkus M., Tergaonkar V., Schneider P., Callus B. A., Koentgen F., Vaux D. L., Silke J. (2007) IAP antagonists target cIAP1 to induce TNFα-dependent apoptosis. Cell 131, 682–693 [DOI] [PubMed] [Google Scholar]
- 15. Vallabhapurapu S., Matsuzawa A., Zhang W., Tseng P. H., Keats J. J., Wang H., Vignali D. A., Bergsagel P. L., Karin M. (2008) Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nat. Immunol. 9, 1364–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zarnegar B. J., Wang Y., Mahoney D. J., Dempsey P. W., Cheung H. H., He J., Shiba T., Yang X., Yeh W. C., Mak T. W., Korneluk R. G., Cheng G. (2008) Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 9, 1371–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stricher F., Macri C., Ruff M., Muller S. (2013) HSPA8/HSC70 chaperone protein: structure, function, and chemical targeting. Autophagy 9, 1937–1954 [DOI] [PubMed] [Google Scholar]
- 18. Ballinger C. A., Connell P., Wu Y., Hu Z., Thompson L. J., Yin L. Y., Patterson C. (1999) Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol. 19, 4535–4545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nikolay R., Wiederkehr T., Rist W., Kramer G., Mayer M. P., Bukau B. (2004) Dimerization of the human E3 ligase CHIP via a coiled-coil domain is essential for its activity. J. Biol. Chem. 279, 2673–2678 [DOI] [PubMed] [Google Scholar]
- 20. Connell P., Ballinger C. A., Jiang J., Wu Y., Thompson L. J., Höhfeld J., Patterson C. (2001) The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 3, 93–96 [DOI] [PubMed] [Google Scholar]
- 21. Meacham G. C., Patterson C., Zhang W., Younger J. M., Cyr D. M. (2001) The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 3, 100–105 [DOI] [PubMed] [Google Scholar]
- 22. Jiang J., Ballinger C. A., Wu Y., Dai Q., Cyr D. M., Höhfeld J., Patterson C. (2001) CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J. Biol. Chem. 276, 42938–42944 [DOI] [PubMed] [Google Scholar]
- 23. Blessing N. A., Brockman A. L., Chadee D. N. (2014) The E3 ligase CHIP mediates ubiquitination and degradation of mixed-lineage kinase 3. Mol. Cell. Biol. 34, 3132–3143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Belova L., Sharma S., Brickley D. R., Nicolarsen J. R., Patterson C., Conzen S. D. (2006) Ubiquitin-proteasome degradation of serum- and glucocorticoid-regulated kinase-1 (SGK-1) is mediated by the chaperone-dependent E3 ligase CHIP. Biochem. J. 400, 235–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gaude H., Aznar N., Delay A., Bres A., Buchet-Poyau K., Caillat C., Vigouroux A., Rogon C., Woods A., Vanacker J. M., Höhfeld J., Perret C., Meyer P., Billaud M., Forcet C. (2012) Molecular chaperone complexes with antagonizing activities regulate stability and activity of the tumor suppressor LKB1. Oncogene 31, 1582–1591 [DOI] [PubMed] [Google Scholar]
- 26. Su C. H., Wang C. Y., Lan K. H., Li C. P., Chao Y., Lin H. C., Lee S. D., Lee W. P. (2011) Akt phosphorylation at Thr308 and Ser473 is required for CHIP-mediated ubiquitination of the kinase. Cell. Signal. 23, 1824–1830 [DOI] [PubMed] [Google Scholar]
- 27. Sasaki Y., Calado D. P., Derudder E., Zhang B., Shimizu Y., Mackay F., Nishikawa S., Rajewsky K., Schmidt-Supprian M. (2008) NIK overexpression amplifies, whereas ablation of its TRAF3-binding domain replaces BAFF:BAFF-R-mediated survival signals in B cells. Proc. Natl. Acad. Sci. U.S.A. 105, 10883–10888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhou Y., Jiang L., Rui L. (2009) Identification of MUP1 as a regulator for glucose and lipid metabolism in mice. J. Biol. Chem. 284, 11152–11159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu W., Marcu M., Yuan X., Mimnaugh E., Patterson C., Neckers L. (2002) Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc. Natl. Acad. Sci. U.S.A. 99, 12847–12852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hatakeyama S., Yada M., Matsumoto M., Ishida N., Nakayama K. I. (2001) U box proteins as a new family of ubiquitin-protein ligases. J. Biol. Chem. 276, 33111–33120 [DOI] [PubMed] [Google Scholar]
- 31. Wang T., Yang J., Xu J., Li J., Cao Z., Zhou L., You L., Shu H., Lu Z., Li H., Li M., Zhang T., Zhao Y. (2014) CHIP is a novel tumor suppressor in pancreatic cancer through targeting EGFR. Oncotarget 5, 1969–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ahmed S. F., Deb S., Paul I., Chatterjee A., Mandal T., Chatterjee U., Ghosh M. K. (2012) The chaperone-assisted E3 ligase C terminus of Hsc70-interacting protein (CHIP) targets PTEN for proteasomal degradation. J. Biol. Chem. 287, 15996–16006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li F., Xie P., Fan Y., Zhang H., Zheng L., Gu D., Patterson C., Li H. (2009) C terminus of Hsc70-interacting protein promotes smooth muscle cell proliferation and survival through ubiquitin-mediated degradation of FoxO1. J. Biol. Chem. 284, 20090–20098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xie P., Fan Y., Zhang H., Zhang Y., She M., Gu D., Patterson C., Li H. (2009) CHIP represses myocardin-induced smooth muscle cell differentiation via ubiquitin-mediated proteasomal degradation. Mol. Cell. Biol. 29, 2398–2408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shang Y., Xu X., Duan X., Guo J., Wang Y., Ren F., He D., Chang Z. (2014) Hsp70 and Hsp90 oppositely regulate TGF-β signaling through CHIP/Stub1. Biochem. Biophys. Res. Commun. 446, 387–392 [DOI] [PubMed] [Google Scholar]
- 36. Woo C. H., Le N. T., Shishido T., Chang E., Lee H., Heo K. S., Mickelsen D. M., Lu Y., McClain C., Spangenberg T., Yan C., Molina C. A., Yang J., Patterson C., Abe J. (2010) Novel role of C terminus of Hsc70-interacting protein (CHIP) ubiquitin ligase on inhibiting cardiac apoptosis and dysfunction via regulating ERK5-mediated degradation of inducible cAMP early repressor. FASEB J. 24, 4917–4928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang Y., Guan S., Acharya P., Liu Y., Thirumaran R. K., Brandman R., Schuetz E. G., Burlingame A. L., Correia M. A. (2012) Multisite phosphorylation of human liver cytochrome P450 3A4 enhances its gp78- and CHIP-mediated ubiquitination: a pivotal role of its Ser-478 residue in the gp78-catalyzed reaction. Mol. Cell. Proteomics 11, M111.010132, 10.1074/mcp.M111.010132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Imai Y., Soda M., Hatakeyama S., Akagi T., Hashikawa T., Nakayama K. I., Takahashi R. (2002) CHIP is associated with Parkin, a gene responsible for familial Parkinson's disease, and enhances its ubiquitin ligase activity. Mol. Cell 10, 55–67 [DOI] [PubMed] [Google Scholar]