

Abstract
Immunomodulator drugs, of which thiopurines can be considered the backbone, are widely used in the treatment of inflammatory bowel disease. They have been shown to be highly effective and safe; however, a significant proportion of patients are deemed to have a poor response or suffer adverse reactions. Knowing how to monitor and optimize thiopurine therapy in these scenarios is crucial to effective management. We discuss the metabolism of thiopurines, the use of enzyme/metabolite testing to guide treatment, as well as strategies to circumvent toxicity and side effects, such as allopurinol coprescription. The indications, use in pregnancy, safety profile and duration of thiopurine therapy are also discussed.
Keywords: gastroenterology, inflammatory bowel disease, ulcerative colitis, Crohn’s disease, drugs, thiopurines, azathioprine
Introduction
Inflammatory bowel disease (IBD) comprises ulcerative colitis (UC) and Crohn’s disease (CD). The disease courses are characterized by periods of relapse and remission. Both IBDs are clinically and histologically distinct but also share many common genetic and clinical factors.
UC was first described in 1859 by Samuel Wilks. Features of UC are confluent mural inflammation which begins in the rectum and extends proximally with sparing of the terminal ileum and small bowel. Sulfasalazine was the first drug introduced to treat the disease in the 1940s, followed by steroid treatment in the 1950s.
Patients with terminal ileitis were first reported in the early 1900s. Burrill Bernard Crohn described a series of cases in 1932 and subsequently this form of IBD became known as CD. Its hallmark is discontinuous chronic granulomatous inflammation, which commonly involves the terminal ileum but can affect any part of the gastrointestinal tract. In contrast with UC, CD may feature transmural inflammation which can result in penetrating or structuring disease.
The treatment for both diseases involves immunosuppressant medication. Mesalazine preparations are used as first-line treatment for UC, with corticosteroid therapy used in the early treatment of UC and CD. Thiopurine therapy is usually introduced as steroid-sparing therapy in UC when a patient’s condition fails to respond to two courses of steroid therapy. In addition, certain clinical scenarios such as presentations of acute severe colitis treated with rescue therapy may require early thiopurine therapy to maintain remission. Thiopurines are generally used earlier in CD as they provide long-term disease course modification which steroid therapy does not provide. A recent prospective study concluded that early azathioprine (AZA) therapy (within 8 weeks of diagnosis) provided no benefit in sustaining steroid-free remission compared with placebo [Panés et al. 2013]. It did, however, show that AZA was more effective in preventing moderate–severe relapses. The main limitation of this study was the use of steroid-free remission as an endpoint. There is a wealth of evidence including a Cochrane Library analysis reporting the effectiveness of thiopurines in maintenance of remission in CD [Prefontaine et al. 2010]. This suggests that thiopurines provide long-term disease modification which steroids do not; however, steroid therapy has a role in early disease treatment as a bridge to maintenance thiopurine therapy. Surgery is usually indicated for patients in whom medical therapy has failed or is refractory.
AZA was first synthesized in 1957 by George Herbert Hitchings and Gertrude Elion. They hypothesized that the growth of rapidly dividing cells could be arrested with antimetabolites of nucleic acid bases [Elion, 1989]. Their work led to the discovery of thioguanine (TG, 2,6-diaminopurine) and mercaptopurine (MP). AZA, a derivative of MP, was subsequently produced to increase the bioavailability of MP. Collectively, these drugs belong to a group known as thiopurine analogues.
Despite being unlicensed for IBD, thiopurines are indicated in the treatment of both UC and CD, primarily as corticosteroid-sparing therapy and disease-modifying drugs. For patients with UC taking AZA, steroid-free, clinical and endoscopic remission has been described in 53% of patients compared with 21% receiving mesalazine therapy [Aridizzone et al. 2006]. In patients with CD, two Cochrane reviews have shown that AZA/MP is more efficacious than placebo in inducing and maintaining remission [Mowat et al. 2010]. More recently, a meta-analysis has shown that thiopurine use is highly effective with an associated 40% reduction of surgical resection in CD [Chatu et al. 2014].
Biologic drugs are the latest addition to the therapeutic armamentarium in managing IBD. Strong evidence advocating combination therapy in treating moderate to severe CD comes from the SONIC trial, which found biologic with concomitant thiopurine therapy more efficacious than either drug as monotherapy [Colombel et al. 2010]. This finding indicates yet another use for thiopurine drugs in the evolving treatment of IBD.
Metabolism of thiopurines
AZA and MP are prodrugs that undergo extensive metabolism via a complex enzymatic pathway. Figure 1 shows a simplified version of the pathway illustrating the important enzymes and metabolites [Blaker et al. 2012]. Initially, the majority of absorbed AZA is metabolized to MP; however, a small amount is metabolized to purine bases implicated with hypersensitivity reactions [McGovern et al. 2002]. MP is then metabolized via three different pathways. It may be oxidized by xanthine dehydrogenase and aldehyde oxidase to form thiouric acid which undergoes urinary excretion [Remy, 1963]. Alternatively, MP can be methylated by thiopurine-S-methyltransferase (TPMT) to form meth-ylmercaptopurine (MeMP). MP can also be metabolized by a group of enzymes known as the purine salvage pathway to produce the pharmacologically active metabolites, TG monophosphate, TG diphosphate and TG triphosphate. Collectively, these end metabolites are referred to as TG nucleotides (TGNs). MeMP may also be therapeutically efficacious, however it is widely accepted that TGNs are the primary mediators of therapeutic response.
Figure 1.
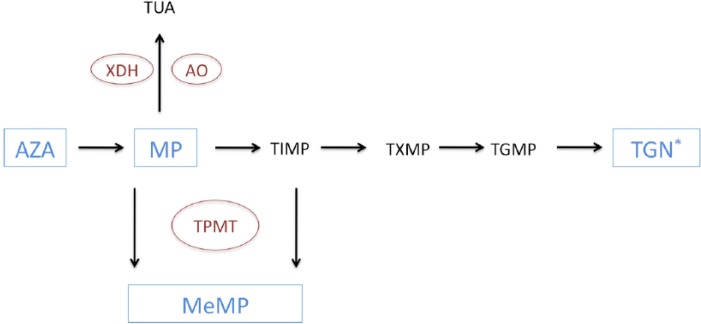
A simplified schematic of the thiopurine metabolic pathway. AO, aldehyde oxidase; AZA, azathioprine; MeMP, methylmercaptopurine; MP, mercaptopurine; TGMP, thioguanine monophosphate; TGN, thioguanine nucleotide; TIMP, thioinosine monophosphate; TPMT, thiopurine-S-methyltransferase; TUA, thiouric acid; XDH, xanthine dehydrogenase.
TGNs are incorporated into DNA, inhibiting its synthesis and leading to DNA strand breakage thus hindering cell proliferation [Aarbakke et al. 1997]. However, the main mechanism of immunomodulation is by inducing T-cell apoptosis by modulating cell (Rac1) signalling [Tiede et al. 2003].
Initiating and dosing thiopurine therapy
There is much inter-individual variation in the metabolism of thiopurines, both in their side effects and efficacy. This is due to differences in the amount of drug absorbed, idiosyncratic reactions, drug–drug reactions and polymorphic variation in thiopurine enzyme metabolism. The appropriate ideal dose for AZA is 2–2.5 mg/kg/day and for MP is 0.75–1.5 mg/kg/day [Mowat et al. 2010]. The dose of TG is not dependent on the patient’s weight and is usually prescribed at a dose of 20 mg/day.
The therapeutic onset after thiopurine initiation is delayed as it takes between 12 and 17 weeks for TGNs to be incorporated into DNA [Prefontaine et al. 2010]. This can have a great impact on clinical practice and acutely unwell patients often have to be bridged to thiopurine therapy with intermediary immunosuppressants such as corticosteroids.
MP is mainly metabolized by TPMT and has a pivotal role in determining the bioavailability of TGNs. TPMT activity has a trimodal phenotypic distribution according to allelic polymorphism. Within white populations, 0.3% of individuals have little or no activity, 12.4% have intermediate activity and 87.3% have normal activity.
Severe pancytopenia has been reported in patients with TPMT deficiency and it should therefore be measured prior to starting thiopurine therapy [Higgs et al. 2010]. It is worth noting that TPMT levels do not predict the majority of myelotoxicity cases and ongoing haematological monitoring is crucial [Gearry et al. 2003]. Reports indicate between 50% and 75% of thiopurine-related leucopenia occurs in patients with normal TPMT levels [Colombel et al. 2000; Ansari et al. 2002].
Prior to commencing thiopurine treatment, patients should be counselled on possible risks and side effects. As well as TPMT measurement, screening for opportunistic infections should be performed. Current European guidelines suggest serological screening for varicella zoster virus, hepatitis B virus, hepatitis C virus and human immunodeficiency virus [Rahier et al. 2009]. Screening for tuberculosis with a chest radiograph and interferon γ release assay should also be considered, particularly if risk factors are present. Young women should be encouraged to participate in national cervical cancer screening programs. Patients should also undergo vaccination prior to thiopurine initiation for the following diseases: varicella zoster virus, human papilloma virus, influenza, pneumococcus and hepatitis B [Rahier et al. 2009]. Naturally, live vaccines are contraindicated once immunomodulator therapy has begun.
Monitoring thiopurine therapy
When commencing thiopurine therapy, the full blood count (FBC) and liver function tests (LFTs) should be monitored every 2 weeks for the first 2 months followed by every 3 months for the duration of therapy. Pancreatitis and hepatotoxicity are uncommon, however the purpose of blood monitoring is primarily to detect thiopurine-induced leucopenia during treatment with approximately half of patients developing signs within 2 months and nearly two-thirds within 4 months of drug initiation [Colombel et al. 2000]. As a result, it is recommended that the FBC and LFTs be rechecked following any dose escalation. Patients developing flu-like illness, jaundice, abdominal pain and unexplained bleeding/bruising should report these signs to their physician immediately.
We recommend that measurement of red cell TGN levels from blood should be checked after 4 weeks of starting treatment with AZA/MP with the specific purpose of confirming adherence and guiding dose optimization. Subsequently, the levels should be checked again at 12–16 weeks once the TGN metabolites have reached a steady-state concentration. Although, it takes up to 4 months for the TGN tissue concentrations to plateau, early blood levels do appear to correlate with eventual tissue concentrations, enabling benefit in early blood level measurement. During maintenance treatment, TGN levels can be measured if patients experience a flare of their symptoms or if compliance or toxicity is suspected. TGN measurement is irrelevant for TG as the levels do not correlate with the dose. Figure 2 shows an algorithm illustrating when to measure TGN levels.
Figure 2.
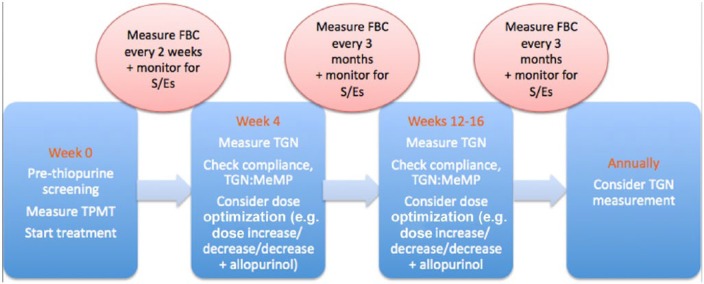
When to measure thioguanine nucleotides and blood test monitoring. FBC, full blood count; MeMP, methylmercaptopurine; S/Es, side effects; TGN, thioguanine nucleotide; TPMT, thiopurine-S-methyltransferase.
TGN levels have been shown to correlate well with clinical response. For IBD, TGN levels greater than 235 pmol/8 × 108 red blood cells (RBC) have been shown to maintain steroid-free remission in 65% of patients receiving thiopurines [Dubinsky et al. 2000]. Recently, a meta-analysis of nearly 2500 patients has supported this level as a therapeutic threshold [Moreau et al. 2013]. When the TGN level is less than 235 pmol/8 × 108 RBC, patients may still enter clinical remission but it is statistically unlikely [Dubinsky et al. 2000].
TGN levels greater than 450 pmol/8 × 108 RBC have been found to be associated with a greater occurrence of myelotoxicity and leucopenia [Dubinsky et al. 2000, 2002; Osterman et al. 2006].
TGN level measurement is particularly useful for monitoring how much of the drug is converted to the potentially toxic metabolite, MeMP. Raised levels of MeMP indicates that the patient preferentially metabolizes the thiopurine to MeMP as opposed to therapeutically active TGN. This phenomenon of skewed drug metabolism is known as thiopurine hypermethylation and occurs in approximately 18% of patients with normal TPMT levels.
Optimizing thiopurine therapy
Clinically relevant dosing strategies to optimize treatment are further discussed. Table 1 summarizes the interpretation of TGN levels in thiopurine maintenance treatment [Irving, 2014].
Table 1.
Interpretation of TGN levels in patients on maintenance AZA/MP therapy.
| TGN level | MeMP level | Interpretation | Action |
|---|---|---|---|
| Zero | Zero | Poor/variable compliance | Patient education (consider poor absorption: rare) |
| Low (<235 × 108 pmol/RBC) | Low | Subtherapeutic dosing | Increase dose and recheck levels |
| Low (<235 × 108 pmol/RBC) | High (MeMP:TGN > 11) | Thiopurine hypermethylator | Reduce drug dose to 25–33% + commence allopurinol and recheck levels |
| Therapeutic (>235 × 108 pmol/RBC) | Any (<5700 pmol/8 × 108) | Therapeutic or thiopurine/class resistance | Continue drug or change thiopurine/class if no response |
| High (>450 × 108 pmol/RBC) | High (>5700 pmol/8 × 108) | Supratherapeutic dosing | Reduce dose and recheck levels |
AZA, azathioprine; MeMP, methylmercaptopurine; MP, mercaptopurine; RBC, red blood cell; TGN, thioguanine nucleotide.
When commencing a thiopurine, it should be noted that the therapeutic benefit is usually only achieved after approximately 3 months. During this time, patients should be supported with alternative therapies (e.g. corticosteroids) until TGN levels reach a therapeutic steady state.
TPMT measurement allows for those patients with intermediary or absent levels of enzyme to be identified. Patients with intermediate TPMT activity should receive 50% of their thiopurine dose, whereas patients with low TPMT activity should avoid thiopurines, though they may tolerate 5–10% of their standard dose [Kaskas et al. 2003; Relling et al. 2011].
The commonest cause of an inadequate response to thiopurine treatment is underdosing [Seidman, 2003] or poor compliance. Measurement of TGN levels can detect when this occurs and dose incrementation or patient education usually achieves therapeutic levels [Gearry and Barclay, 2005]. A prospective study of steroid-dependent patients with IBD showed that dose optimization of AZA to achieve levels greater than 250 pmol/8 × 108 RBC was significantly associated with a higher rate of disease remission [Roblin et al. 2005]. This provides strong evidence for TGN level monitoring and dose optimization and our experience indicates that this usually correlates with clinical response. Rarely, TGN levels may be low due to poor absorption of the drug. The mechanism for this is not fully understood and switching to another class of drug may be necessary [Irving, 2014].
Thiopurine hypermethylation appears to occur in up to 20% of the population [Smith et al. 2012], where TGN levels are found to be low with conversely raised MeMP levels. An MeMP:TGN ratio of greater than 11 has been shown to greatly increase the risk of toxicity [Dubinsky et al. 2002; Smith et al. 2012]. Specifically, an MeMP level greater than 5700 pmol/8 × 108 RBC is associated with hepatotoxicity [Dubinsky et al. 2000]. Unfortunately, the initial thiopurine dose or TMPT level does not predict the risk of hypermethylation [Cuffari et al. 2001; Blaker et al. 2012]. Increasing the dose in patients who exhibit thiopurine shunting can increase the levels of MeMP with no rise in TGN levels [Cuffari et al. 2001]. Indeed, this practice can increase the risk of toxicity and highlights the importance of TGN/MeMP measurement. A clinically important strategy to circumvent this problem is to reduce the thiopurine dose to 25–33% and coprescribe the xanthine oxidase inhibitor, allopurinol (100 mg daily) [Sparrow et al. 2007; Leung et al. 2009; Smith et al. 2012]. The precise mechanism of this observation remains unknown but may be related to the finding that allopurinol increases thioxanthine levels which in turn inhibits TPMT activity [Blaker et al. 2013]. Recognition of thiopurine hypermethylation is clinically relevant and steroid-free remission at 1 year has been described in up to 73% of patients in a recent large series [Smith et al. 2012]. A crucial footnote is that failure to reduce the thiopurine dose in conjunction with allopurinol coprescription carries a great risk of myelotoxicity. Another benefit of low-dose AZA/MP and allopurinol can be to bypass certain thiopurine-related adverse effects, including gastrointestinal upset, flu-like symptoms, myalgia and hepatoxicity [Ansari et al. 2010].
Thiopurines are generally well tolerated, however approximately 9% of patients are thiopurine resistant and a further 15–28% develop an adverse drug reaction [Schwab et al. 2002]. The drug reaction may be related to drug metabolism, in which case TPMT and TGN measurement can guide treatment, but between 1% and 6.5% of patients experience idiosyncratic reactions [de Boer et al. 2007]. Most reactions (such as flu-like illness, arthralgia, rash, headache) seem to occur in the first 2–3 weeks of treatment and improve after a few weeks if the drug is continued. If symptoms persist, the patient can be informed that they stop rapidly upon drug withdrawal. Commencing therapy at a lower dose such as 50% of the ideal weight-based therapeutic dose may reduce the severity of an adverse reaction and allows for early identification and intervention. Once tolerated, the dose can then be increased with FBC, LFT and TGN monitoring guiding treatment. In addition, there is now robust evidence that subdividing the total ideal thiopurine dose into two smaller daily doses can be of benefit in reducing some side effects such as nausea in individuals who are preferential methylators [Shih et al. 2012; Pavlidis et al. 2014]. This simple intervention has also been shown to improve long-term remission rates [Shih et al. 2013].
AZA is usually the first line thiopurine to be prescribed. Intolerance due to side effects may lead to early discontinuation of the drug. In this instance, benefit may be derived from using a metabolic descendant such as MP or TG [Nagy et al. 2008; Amin et al. 2014]. This strategy has been shown to benefit certain patients who are intolerant of the initially trialled thiopurine. In patients who experience AZA/MP-induced pancreatitis, both further AZA/MP use is contra-indicated, however, patients have been shown to subsequently respond to and tolerate TG [Amin et al. 2015].
Risks and benefits in continuing or withdrawing thiopurine therapy
Generally, thiopurine therapy should be continued indefinitely once remission has been achieved. A randomized controlled trial of withdrawal of AZA therapy in patients with IBD found that 21% of patients who ceased AZA therapy experienced a relapse of their disease compared with 8% on ongoing therapy [Lémann et al. 2005]. The median duration of treatment and clinical remission was 5.7 and 5.3 years respectively. Logically, raised inflammatory markers (C-reactive protein) and anaemia were found to predict relapse. Further follow up of the cohort which ceased AZA found high rates of just over 50% relapse at 3 years with almost two-thirds of patients relapsing after 5 years [Treton et al. 2009].
Despite having a US Food and Drug Administration (FDA) category D rating (risk of adverse events to foetus), thiopurines are thought to be generally safe in pregnancy [Mowat et al. 2010]. The FDA rating is based on anecdotal evidence of high abortion rates; however, studies of AZA/MP in IBD exist, showing no increased risk of adverse events in pregnancy [Van Assche et al. 2010]. Patients who are in clinical remission at the time of conception are less likely to suffer flare of their disease during the course of their pregnancy. It is for this reason that it is generally advised that thiopurine therapy be continued in pregnancy. At present, there are few data to support that thiopurines are relatively contraindicated in breastfeeding. A recent small study has shown that there are negligible concentrations of thiopurine metabolites expressed in breast milk [Sau et al. 2007].
All immunomodulators carry a slightly increased risk of infection with a specifically increased susceptibility to viral infections seen with thiopurines [Toruner et al. 2008]. Fungal and mycobacterial infections are more associated with corticosteroid and biologic use respectively. In the case of an acute infection, the thiopurine can be withdrawn, the infection treated, and the thiopurine safely reintroduced once the infection has been cleared. It is important to note that there is a much greater risk of infection when immunomodulators are used in combination therapy [Toruner et al. 2008].
Postoperative patients do not appear to have an increased complication rate with thiopurine use and the drugs can be used safely in high-risk patients. Interestingly, preliminary data from the POCER trial suggest a reduction in postoperative endoscopic recurrence in patients treated with thiopurines or adalulimab [Kamm et al. 2014].
Thiopurine therapy has been shown to carry a slightly increased risk of malignancy, specifically lymphoproliferative disease. Studies indicate a four- to fivefold increased risk compared with the background population, although the absolute risk remains very low [Kandiel et al. 2005; Beaugerie et al. 2009]. A recent decision analysis study found that alternative therapy would be favoured if there were a tenfold risk in lymphoma [Vos et al. 2011]. The overall consensus is that the benefits of thiopurine therapy outweigh the risk of malignant lymphoma. Exposure to ultraviolet radiation, such as heavy sun exposure, has been shown to carry an increased risk of nonmelanomatous skin cancer. A retrospective study found that thiopurine exposure over 47 years was associated with an odds ratio of 5.0 for nonmelanomatous skin cancer. Caucasian descent carried an increased risk with an odds ratio of 12.4 [Setshedi et al. 2012]. More recently, a large retrospective analysis of thiopurine therapy both with and without anti-tumour necrosis factor (TNF) treatment has been published. Thiopurine monotherapy was found to be associated with an increased risk of malignancy compared with combination therapy with anti-TNF drugs. The rate of malignancy for patients aged more than 50 years receiving thiopurine therapy was 18.2%, significantly greater than the rate of 3.8% for patients aged less than 50 years. Thiopurine treatment duration of greater than 4 years was also shown to carry a greater risk for skin cancer and lymphoma [Beigel et al. 2014]. A large prospective observational study has recently shown that patients previously exposed to thiopurines have a sevenfold increased risk of developing a myeloproliferative disorder. Interestingly, patients receiving ongoing thiopurine therapy were not at greater risk [Lopez et al. 2014]; however, the results of this study should be considered when initiating and withdrawing therapy. Advancing age certainly seems to confer a greater risk of malignancy, particularly for patients over the age of 50 years. The risks and benefits should be carefully weighed up, with patients appropriately counselled and also considered for alternative therapy if appropriate. We currently practice annual outpatient screening of skin lesions for all patients receiving thiopurine therapy, with suspicious lesions referred for specialist dermatology assessment.
Historically, there has been concern regarding the risk of nodular regenerative hyperplasia (NRH) and the need for interval magnetic resonance imaging (MRI) screening. The cumulative incidence of NRH in patients with IBD treated with AZA is approximately 1% at 10 years. High doses of TG have been associated with frequencies of NRH of up to 62%; however, low-dose TG (20 mg once daily) appears safe with no cases of NRH seen [Musumba, 2013]. Until a prospective trial occurs reliably evaluating the risk of NRH with TG, our current practice is to survey for NRH with annual MRI scanning. The development of thrombocytopenia or raised liver enzymes detected by blood testing every 2 months may also be appropriate triggers to consider MRI screening.
There is no ideal duration of therapy, however 5 years would appear to be a pragmatic time to review the need for ongoing treatment based on previous study data. Consideration should be given to the long-term risks with thiopurine therapy, which may become increasingly important with advancing age [Beigel et al. 2014]. As always, the risks and benefits need to be carefully discussed with each individual patient. Thiopurine withdrawal has been shown to be associated with a greater risk of relapse [Wenzl et al. 2014]. Most recently, a retrospective study reported that thiopurine withdrawal in patients in sustained remission was associated with a 1-year moderate to severe relapse rate of 23% in CD and 12% in UC [Kennedy et al. 2014]. A raised C-reactive protein level prior to thiopurine withdrawal was found to be highly predictive of relapse, with rates also cumulatively increasing with time [Kennedy et al. 2014]. However, in those who decide to stop therapy and suffer a relapse, therapeutic response can be recaptured in almost all patients with recommencement of the thiopurine [Treton et al. 2009; Kennedy et al. 2014]. This differs somewhat from biologic therapy when a ‘drug-holiday’ can be associated with antidrug antibodies, an increased incidence of drug reactions and possibly a reduced chance of recapturing therapeutic response.
Summary
Thiopurine therapy should be personalized and tailored to the individual being treated. Thiopurine metabolism remains complex but is becoming increasingly understood. It is important to understand and identify reasons as to why a thiopurine is ineffective or not tolerated. Dose changes, switching to another thiopurine/drug, allopurinol coprescription and management of side effects are crucial to ensure thiopurines are used in the best way. In addition, the use of TPMT and TGN measurement is clinically relevant and can help optimize existing thiopurine treatment. Further studies specifically to document the benefit of TGN monitoring, allopurinol coprescription and TG efficacy and safety will aid in guiding treatment and ensure thiopurines are used to maximize response.
Footnotes
Conflict of interest statement: The authors declare no conflict of interest in preparing this article.
Funding: This research received no specific grant from any funding agency in the public, commercial, or not- for-profit sectors.
Contributor Information
Rishi M. Goel, Guy’s & St Thomas’ Hospitals – Gastroenterology, Westminster Bridge Road, London SE1 7EH, UK
Paul Blaker, Guy’s & St Thomas’ Hospitals – Gastroenterology, London, UK.
Alex Mentzer, Guy’s & St Thomas’ Hospitals – Gastroenterology, London, UK.
Steven C.M. Fong, Guy’s & St Thomas’ Hospitals – Gastroenterology, London, UK
Anthony M. Marinaki, Guy’s & St Thomas’ Hospitals - Clinical Chemistry, London, UK
Jeremy D. Sanderson, Guy’s & St Thomas’ Hospitals – Gastroenterology, London, UK
References
- Aarbakke J., Janka-Schaub G., Elion G. (1997) Thiopurine biology and pharmacology. Trends Pharmacol Sci 18: 3–7. [DOI] [PubMed] [Google Scholar]
- Amin J., Huang B., Yoon J., Shih D. (2015) Update 2014: advances to optimize 6-mercaptopurine and azathioprine to reduce toxicity and improve efficacy in the management of IBD. Inflamm Bowel Dis 21: 445–452. [DOI] [PubMed] [Google Scholar]
- Ansari A., Hassan C., Duley J., Marinaki A., Shobowale-Bakre E., Seed P., et al. (2002) Thiopurine methyltransferase activity and the use of azathioprine in inflammatory bowel disease. Aliment Pharmacol Ther 16: 1743–1750. [DOI] [PubMed] [Google Scholar]
- Ansari A., Patel N., Sanderson J., O’Donohue J., Duley J., Florin T. (2010) Low-dose azathioprine or mercaptopurine in combination with allopurinol can bypass many adverse reactions in patients with inflammatory bowel disease. Aliment Pharmacol Ther 31: 640–647. [DOI] [PubMed] [Google Scholar]
- Aridizzone S., Maconi G., Russo A., Imbesi V., Colombo E., Bianchi Porro G., et al. (2006) Randomised controlled trial of azathioprine and 5-aminosalicylic acid for treatment of steroid dependent ulcerative colitis. Gut 55: 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaugerie L., Brousse N., Bouvier A., Colombel J., Lemann M., Cosnes J., et al. (2009) Lymphoproliferative disorders in patients receiving thiopurines for inflammatory bowel disease: a prospective observational cohort study. Lancet 374: 1617–1625. [DOI] [PubMed] [Google Scholar]
- Beigel F., Steinborn A., Schnitzler F., Tillack C., Breiteneicher S., John J., et al. (2014) Risk of malignancies in patients with inflammatory bowel disease treated with thiopurines or anti-TNF alpha antibodies. Pharmacoepidemiol Drug Saf 23: 735–744. [DOI] [PubMed] [Google Scholar]
- Blaker P., Arenas-Hernandez M., Marinaki A., Sanderson J. (2012) The pharmacogenetic basis of individual variation in thiopurine metabolism. J Pers Med 9: 707–725. [DOI] [PubMed] [Google Scholar]
- Blaker P., Arenas-Hernandez M., Smith M., Shobowale-Bakre E., Fairbanks L., Irving P., et al. (2013) Mechanism of allopurinol induced TPMT inhibition. Biochem Pharmacol 86: 539–547. [DOI] [PubMed] [Google Scholar]
- Chatu S., Subramanian V., Saxena S., Pollok R. (2014) The role of thiopurines in reducing the need for surgical resection in Crohn’s disease: a systematic review and meta-analysis. Am J Gastroenterol 109: 23–34. [DOI] [PubMed] [Google Scholar]
- Colombel J., Ferrari N., Debuysere H., Marteau P., Gendre J., Bonaz B., et al. (2000) Genotypic analysis of thiopurine S-methyltransferase in patients with Crohn’s disease and severe myelosuppression during azathioprine therapy. Gastroenterology 118: 1025–1030. [DOI] [PubMed] [Google Scholar]
- Colombel J., Sandborn W., Reinisch W., Mantzaris G., Kornbluth A., Rachmilewitz D., et al. (2010) Infliximab, azathioprine, or combination therapy for Crohn’s disease. N Engl J Med 362: 1383–1395. [DOI] [PubMed] [Google Scholar]
- Cuffari C., Hunt S., Bayless T. (2001) Utilisation of erythrocyte 6-thioguanine metabolite levels to optimize azathioprine therapy in patients with inflammatory bowel disease. Gut 48: 642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer N., Zondervan P., Gilissen L., den Hartog G., Westerveld B., Derijks L., et al. (2007) Drug insight: pharmacology and toxicity of thiopurine therapy in patients with IBD. Nat Clin Gastroenterol Hepatol 4: 686–694. [DOI] [PubMed] [Google Scholar]
- Dubinsky M., Lamothe S., Yang H., Targan S., Sinnett F., Theoret Y., et al. (2000) Pharmacogenomics and metabolite measurement for 6-mercaptopurine therapy in inflammatory bowel disease. Gastroenterology 118: 705–713. [DOI] [PubMed] [Google Scholar]
- Dubinsky M., Yang H., Hassard P., Seidman E., Kam L., Abreu M., et al. (2002) 6-MP metabolite profiles provide a biochemical explanation for 6-MP resistance in patients with inflammatory bowel disease. Gastroenterology 122: 904–915. [DOI] [PubMed] [Google Scholar]
- Elion G. (1989) The purine path to chemotherapy. Science 244: 41–47. [DOI] [PubMed] [Google Scholar]
- Gearry R., Barclay M. (2005) Azathioprine and 6-mercaptopurine pharmacogenetics and metabolite monitoring in inflammatory bowel disease. J Gastroenterol Hepatol 20: 1149–1157. [DOI] [PubMed] [Google Scholar]
- Gearry R., Barclay M., Burt M., Collett J., Chapman B., Roberts R., et al. (2003) Thiopurine S-methyltransferase (TPMT) genotype does not predict adverse drug reactions to thiopurine drugs in patients with inflammatory bowel disease. Aliment Pharmacol Ther 18: 395–400. [DOI] [PubMed] [Google Scholar]
- Higgs J., Payne K., Roberts C., Newman W. (2010) Are patients with intermediate TPMT activity at increased risk of myelosuppression when taking thiopurine medications? Pharmacogenomics 11: 177–188. [DOI] [PubMed] [Google Scholar]
- Irving P. (2014) Are we using and monitoring thiopuines and biologics optimally? Digestive Diseases 32: 410–418. [DOI] [PubMed] [Google Scholar]
- Kamm M., De Cruz P., Wright E., Hamilton A., Ritchie K., Krejany E., et al. (2014) Optimising post-operative Crohn’s disease management: best drug therapy alone versus endoscopic monitoring, disease evolution, and faecal calprotectin monitoring. The POCER study. J Crohns Colitis 8(Suppl. 1): S13. [Google Scholar]
- Kandiel A., Fraser A., Korelitz B., Brensinger B., Lewis J. (2005) Increased risk of lymphoma among inflammatory bowel disease patients treated with azathioprine and 6-mercaptopurine. Gut 54: 1121–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaskas B., Louis E., Hindorf U., Schaeffeler E., Deflandre J., Graepler F., et al. (2003) Safe treatment of thiopurine S-methyltransferase deficient Cohn’s disease patients with azathioprine. Gut 52: 140–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy N., Kalla R., Warner B., Gambles C., Musy R., Reynolds S., et al. (2014) Thiopurine withdrawal during sustained clinical remission in inflammatory bowel disease: relapse and recapture rates, with predictive factors in 237 patients. Aliment Pharmacol Ther 40: 1313–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lémann M., Mary J., Colombel J., Duclos B., Soule J., Lerebours E., et al. (2005) A randomized, double-blind, controlled withdrawal trial in Crohn’s disease patients in long-term remission on azathioprine. Gastroenterology 128: 1812–1818. [DOI] [PubMed] [Google Scholar]
- Leung Y., Sparrow M., Schwartz M., Hanauer S. (2009) Long term efficacy and safety of allopurinol and azathioprine or 6-mercaptopurine in patients with inflammatory bowel disease. J Crohns Colitis 3: 162–167. [DOI] [PubMed] [Google Scholar]
- Lopez A., Mounier M., Bouvier A., Carrat F., Maynadie M., Beaugerie L., et al. (2014) Increased risk of acute myeloid leukemias and myelodysplastic syndromes in patients who received thiopurine treatment for inflammatory bowel disease. Clin Gastroenterol Hepatol 12: 1324–1329. [DOI] [PubMed] [Google Scholar]
- McGovern D., Travis S., Duley J., Shobowale-Bakre E., Dalton H. (2002) Azathioprine intolerance in patients with IBD may be imidazole-related and is independent of TPMT activity. Gastroenterology 122: 838–839. [DOI] [PubMed] [Google Scholar]
- Moreau A., Laporte S., Del Tedesco F., Rinaudo-Gaujous M., Phelip J., Paul S., et al. (2013) Association between thiopurines metabolites levels and clinical remission in IBD patients: an updated meta-analysis. Gastroenterology 144: S-92. [Google Scholar]
- Mowat C., Cole A., Windsor A., Ahmad T., Arnott I., Driscoll R., et al. (2010) Guidelines for the management of inflammatory bowel disease in adults. Gut 60: 571–607. [DOI] [PubMed] [Google Scholar]
- Musumba C. (2013) Review article: the association between nodular regenerative hyperplasia, inflammatory bowel disease and thiopurine therapy. Aliment Pharmacol Ther 38: 1025–1037. [DOI] [PubMed] [Google Scholar]
- Nagy F., Molnar T., Szepes Z., Farkas K., Nyari T., Lonovics J. (2008) Efficacy of 6-mercaptopurine treatment after azathioprine hypersensitivity in inflammatory bowel disease. World J Gastroenterol 14: 4342–4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterman M., Kundu R., Lichtenstein G., Lewis J. (2006) Association of 6-thioguanine nucleotide levels and inflammatory bowel activity: a meta-analysis. Gastroenterology 130: 1047–1053. [DOI] [PubMed] [Google Scholar]
- Panés J., López-Sanromán A., Bermejo F., García-Sánchez V., Esteve M., Torres Y., et al. (2013) Early azathioprine therapy is no more effective than placebo for newly diagnosed Crohn’s disease. Gastroenterology 145: 766–774. [DOI] [PubMed] [Google Scholar]
- Pavlidis P., Ansari A., Duley J., Oancea I., Florin T. (2014) Splitting a therapeutic dose of thioguanine May avoid liver toxicity and be an efficacious treatment for severe inflammatory bowel disease: a 2-center observational cohort study. Inflamm Bowel Dis 20: 2239–2246. [DOI] [PubMed] [Google Scholar]
- Prefontaine E., Macdonald J., Sutherland L. (2010) Azathioprine or 6-mercaptopurine for induction of remission in Crohn’s disease. Cochrane Database Syst Rev (6): CD000545. [DOI] [PubMed] [Google Scholar]
- Rahier J., Ben-Horin S., Chowers Y., Conlon C., De Munter P., D’Haens G., et al. (2009) European evidence-based consensus on the prevention, diagnosis and management of opportunistic infections in inflammatory bowel disease. J Crohns Colitis 3: 47–91. [DOI] [PubMed] [Google Scholar]
- Relling M., Gardner E., Sandborn W., Schmiegelow K., Pui C., Yee S., et al. (2011) Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 89: 387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy C. (1963) Metabolism of thiopyrimidines and thiopurines S-methylation with S-adenosylmethionine transmethylase and catabolism in mammalian tissues. J Biol Chem 238: 1078–1084. [PubMed] [Google Scholar]
- Roblin X., Serre-Debeauvais F., Phelip J., Faucheron J., Hardy G., Chartier A., et al. (2005) 6-tioguanine monitoring in steroid-dependent patients with inflammatory bowel diseases receiving azathioprine. Aliment Pharmacol Ther 21: 829–839. [DOI] [PubMed] [Google Scholar]
- Sau A., Clarke S., Bass J., Kaiser A., Marinaki A., Nelson-Piercy C. (2007) Azathioprine and breastfeeding: is it safe? BJOG 114: 498–501. [DOI] [PubMed] [Google Scholar]
- Schwab M., Schaffeler E., Marx C., Fischer C., Lang T., Behrens C., et al. (2002) Azathioprine therapy and adverse drug reactions in patients with inflammatory bowel disease: impact of thiopurine S-methyltransferase polypmorphism. Pharmacogenetics 12: 429–436. [DOI] [PubMed] [Google Scholar]
- Seidman E. (2003) Clinical use and practical application of TPMT enzyme and 6-mercaptopurine metabolite monitoring in IBD. Rev Gastroenterol Disord 3(Suppl. 1): S30–S38. [PubMed] [Google Scholar]
- Setshedi M., Epstein D., Winter T., Myer L., Watermeyer G., Hift R. (2012) Use of thiopurines in the treatment of inflammatory bowel disease is associated with an increased risk of non-melanoma skin cancer in an at-risk population: a cohort study. J Gastroenterol Hepatol 27: 385–389. [DOI] [PubMed] [Google Scholar]
- Shih D., Nguyen M., Zheng L., Ibanez P., Mei L., Kwan L., et al. (2012) Split-dose administration of thiopurine drugs: a novel and effective strategy for managing preferential 6-MMP metabolism. Aliment Pharmacol Ther 36: 449–458. [DOI] [PubMed] [Google Scholar]
- Shih D., Yoon J., Huang B., Karsan S., Melmed G., Ippoliti A., et al. (2013) Potential synergism between anti-TNF and thiopurine therapy: increased thiopurine metabolites by anti-TNF. Gastroenterology 144: S772. [Google Scholar]
- Smith M., Blaker P., Marinaki A., Anderson S., Irving P., Sanderson J. (2012) Optimising outcome on thiopurines in inflammatory bowel disease by co-prescription of allopurinol. J Crohns Colitis 6: 905–912. [DOI] [PubMed] [Google Scholar]
- Sparrow M., Hande S., Friedman S., Cao D., Hanauer S. (2007) Effect of allopurinol on clinical outcomes in inflammatory bowel disease nonresponders to azathioprine or 6-mercaptopurine. Clin Gastroenterol Hepatol 5: 209–214. [DOI] [PubMed] [Google Scholar]
- Tiede I., Fritz G., Strand S., Poppe D., Dvorsky R., Strand D., et al. (2003) CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J Clin Invest 111: 1133–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toruner M., Loftus E., Scott Harmsen W., Zinsmeister A., Orenstein R., Sandborn W., et al. (2008) Risk factors for opportunistic infections in patients with inflammatory bowel disease. Gastroenterology 134: 929–936. [DOI] [PubMed] [Google Scholar]
- Treton X., Bouhnik Y., Mary J., Colombel J., Duclos B., Soule J., et al. (2009) Azathioprine withdrawal in patients with Crohn’s disease maintained on prolonged remission: a high risk of relapse. Clin Gastroenterol Hepatol 7: 80–85. [DOI] [PubMed] [Google Scholar]
- Wenzl H., Primas C., Novacek G., Teml A., Offerlbauer-Ernst A., Högenauer C., et al. (2014) Withdrawal of long-term maintenance treatment with azathioprine tends to increase relapse risk in patients with Crohn’s disease. Dig Dis Sci 8 November (epub ahead of print). [DOI] [PubMed] [Google Scholar]
- Van Assche G., Dignass A., Reinisch W., Janneke van der Woude C., Sturm A., De Vos M., et al. (2010) The second European evidence-based Consensus on the diagnosis and management of Crohn’s disease: current management. J Crohns Colitis 4: 63–101. [DOI] [PubMed] [Google Scholar]
- Vos A., Bakkal N., Minnee R., Casparie M., de Jong D., Dijkstra G., et al. (2011) Risk of malignant lymphoma in patients with inflammatory bowel diseases: a Dutch nationwide study. Inflamm Bowel Dis 17: 1837–1845. [DOI] [PubMed] [Google Scholar]