

Abstract
To understand RNA, it is necessary to move beyond a descriptive categorization towards quantitative predictions of its molecular conformations and functional behavior. An incisive approach to understanding the function and folding of biological RNA systems involves characterizing small, simple components that are largely responsible for the behavior of complex systems including helix-junction-helix elements and tertiary motifs. State-of-the-art methods have permitted unprecedented insight into the conformational ensembles of these elements revealing, for example, that conformations of helix-junction-helix elements are confined to a small region of the ensemble, that this region is highly dependent on the junction’s topology, and that the correct alignment of tertiary motifs may be a rare conformation on the overall folding landscape. Further characterization of RNA components and continued development of experimental and computational methods with the goal of quantitatively predicting RNA folding and functional behavior will be critical to understanding biological RNA systems.
Science, almost by definition, is a search for order in Nature –simplifications, minimal descriptions, and categorizations of the world and universe around us. These descriptions are valuable when they summarize a considerable amount of information and when they attain the stature of a true model –i.e., one with the power to predict hitherto unseen phenomena. Our early molecular understanding of Nature in terms of the Central Dogma (Figure 1A) was thus highly satisfying [1]. It placed molecules into one of three classes based on type: DNA to store genetic information, RNA to carry the genetic information from DNA to proteins, and proteins to perform the functions encoded in the genetic information. This model thus predicted where the functions of newly discovered molecules would lie.
Figure 1.

From categories to molecules. (A) The Central Dogma of biology defined three categories and a unidirectional information flow (arrows) in which DNA stores the information, and RNA carries the information to proteins that then carry out all biological functions. (B) Updated view of the Central Dogma, in which information flow is not unidirectional and roles are not balkanized. In particular, RNA performs biological functions alone or in collaboration with proteins including translation, splicing, gene regulation, and chromosome maintenance. (C) Mechanism of protein targeting to the endoplasmic reticulum (ER) or plasma membrane by the signal recognition particle (SRP), which contains an RNA component (red) that serves as a scaffold for the SRP and as a means for molecular reorganization during the functional cycle [Adapted from 12]. SRP binds an N-terminal signal sequence (orange) in the nascent peptide and arrests elongation. The SRP–ribosome-nascent chain complex (SRP-RNC) then associates with the SRP-receptor (SR) in the membrane, followed by transfer of the RNC to the translocon and SRP dissociation. At the translocon, translation resumes, and the nascent peptide is translocated into the ER lumen or a membrane bilayer.
Nevertheless, while the Central Dogma was and is of great value, over time it was recognized to not have full predictive value –e.g., RNA is copied into DNA by reverse transcriptase; tRNA acts as a structured, functional adaptor to read the genetic code; RNA molecules carry out catalytic functions in chromosome maintenance, pre-mRNA splicing, protein targeting to membranes, and self-processing; and proteins form heritable structures (Figure 1B) [2–7]. Nature, driven by natural selection and oblivious to our classifications, used all of its molecular constituents to solve problems and to evolve new solutions that provide a selective advantage.
Consider why the discovery of catalytic RNA was greeted with widespread disbelief and enormous skepticism. Conventional wisdom held that “enzymes are (were) proteins.” But while it is true that there are only four RNA bases in contrast to 20 amino acids, no RNA pKa values near neutrality, and barriers to RNA packing due to backbone electrostatic repulsion and degrees of freedom, these properties do not mean that RNA cannot do catalysis. Here, again, we were limited by categorization: RNA is or is not a catalyst. However, if we resist our desire for neat and absolute categories, we can ask (or could have asked): “How good can RNA be as a catalyst?” And we can use (or could have used) our molecular and atomic understanding to assess this question in a manner unbiased by our prior categorizations.
Indeed, before the discovery of catalytic RNA, several researchers had suggested that the ability of RNA to form complex shapes could lead to it acting as a catalyst [8–10]. The discovery of catalytic RNA is less surprising when one considers the problem in terms of molecules instead of categories and realizes that other types of biomolecules can be catalysts. For example, lipids, forming micelles, can enhance reactions between hydrophobic groups within the micelle, between hydrophobic groups incorporated into micelles, and between charged groups attracted to an oppositely charged micelle surface [11]. By applying the guiding principle of natural selection and the properties of molecules, we can better understand biology and go beyond the limitations stemming from our desire to categorize. Here, we apply this perspective to RNA folding and function.
In the past decade, there has been an explosion of information about and understanding of RNA-mediated processes. The ribosome and the signal recognition particle (SRP) are two well-characterized systems that involve functional RNAs [12,13]. Figure 1C outlines the functional cycle of the SRP in which the RNA of the SRP plays a role in global rearrangement of the complex. This role suggests that dynamics, or conformational transitions, are required for this and other RNA-mediated processes. So how do we move beyond describing such processes to understanding them, with understanding ultimately defined by the ability to quantitatively predict the structural, conformational, and functional behavior of RNA systems? Such ability would be the bedrock for a true understanding, at the molecular level, of how biology works and the foundation for an ability to predict the effects of mutations, interventions and an ability to engineer new behaviors for applications in health and technology.
There have been several efforts to model the ribosome and its function using computational approaches [14,15]. While these represent extraordinary achievements, the size and complexity of the ribosome render it difficult to assess and develop these computational models via a series of quantitative predictions and subsequent experimental tests. A distinct approach, founded on the apparent greater modularity of RNA components (relative to the high interconnectivity of the structural components of proteins), is to begin with smaller RNA components that can be characterized at the atomic level and ultimately to assemble the behavior of these components to make atomic-level predictions about larger RNA systems. From such a thorough understanding, cellular factors and features that perturb the fundamental dynamics and thermodynamics of RNAs and RNA/protein complexes can be identified and their contributions dissected and understood.
Given that there is already a well-developed and reasonably predictive understanding of RNA secondary structure via nearest-neighbor, or “Turner,” rules [16–18], we focus here on helix-junction-helices (HJHs) and tertiary motifs as ubiquitous components of RNA structure and emphasize the need to address the ensemble nature of both of these elements to understand RNA behavior and function.
RNA Helix-Junction-Helix (HJH) Elements
Folding to a functional state requires an enormous loss in conformational entropy, and much of this entropy loss is accomplished for RNA by the formation of stable secondary structure elements. Once secondary structure is formed, helices are rigid, to a first approximation, and the primary source of conformational entropy lies in the junctions that connect the helices. Thus, we need to understand the behavior of these junctions to understand (and predict) RNA folding and to assess and understand their roles in functional conformational transitions.
Pioneering work on RNA junctions by Lilley, following his classic studies of the DNA Holliday junction, indicated that RNA junctions prefer particular geometries that then favor specific folded structures [19–21]. There are now several examples of junctions known to favor particular bent conformations [22–23]. However, the ensemble nature of HJHs, which is a manifestation of the free energy landscape, has only recently been investigated. As alluded to above, the longer term aim is to stitch together energy landscapes from the components of functional RNAs (and RNA-protein complexes) to quantitatively predict and understand the kinetic and thermodynamic properties of complex systems.
Chu et al. approached the problem of predicting HJH ensembles conceptually, showing that different types of connecting junctions lead to different energy landscapes for the attached helices (Figure 2A,2B) [24•]. In particular, they modeled a single-stranded junction connection and a double-stranded junction connection by molecular dynamics using short PEG tethers for the junctions to ensure accurate modeling. The helices were represented by cylinders to isolate the features of the resulting helix-helix energy landscape that are determined by the topology of the connections between the helices. Differences in the resulting landscapes (Figure 2A,2B) –i.e., the spatial positions of the helices emanating from the junctions– demonstrated the importance of the topology of the physical connection, which was further highlighted by considering the probabilities of forming tertiary structures.
Figure 2.

Predictions of the behavior of isolated junctions within RNA. [Reprinted from 24•.] (A) An idealized single-stranded PEG junction construct (left) and its predicted conformational landscape (right). Green/red sticks (with green distal and red proximal to the junction to aid visualization) represent the different orientations of one helix relative to the reference helix (gray) in the landscape. (B) An idealized double-stranded PEG junction construct (left) and its predicted conformational landscape (right) visualized as described in (A). (C) Prediction of the fraction folded for different positions of a tertiary contact, based on simulations of the allowed conformational space in (A) and (B). The left panel shows the positions of different pairs of tertiary contacts (diamonds, circles, or triangles) relative to the position of the single-stranded (A) and double-stranded (B) PEG junctions. The panel on the right shows the predicted folded fraction for different positions of the tertiary contact in the two junction contexts at 1 M monovalent salt. When the tertiary contacts are at the triangle position, the estimated fraction folded is less than 0.01%.
As described below, RNA tertiary structure is often enforced by motifs embedded in helices. Using simplified models for motifs to avoid other complicating factors and unknowns, Chu et al. placed tertiary motif partners at specific locations on the PEG-tethered helices and predicted large differences in folding free energies (Figure 2C). Since this work predicts that the junction region determines the orientation of individual helices even with simplified PEG junctions, resolving the structural ensembles of real HJH motifs is critical for predicting the folding behavior of complex RNA structures.
The next obvious question is what does the ensemble look like for a real RNA HJH? Al-Hashimi and colleagues addressed this question through extensive studies of the biologically important TAR HJH element (Figure 3A). They used NMR residual dipolar coupling (RDC) measurements that have the advantage—for this application—of providing dynamic information without discriminating between motional timescales over the range of picoseconds to milliseconds [25•]. Thus, information about the ensemble of TAR structures can be obtained rather than simply an averaged structure or information about the dynamics experienced over a narrow time window determined by the particular experimental method.
Figure 3.

Conformational ensembles of RNA helix-junction-helix motifs (HJHs). (A) The secondary structure of HIV-1 TAR. Wild-type TAR has a six-nucleotide hairpin loop that for the majority of studies is replaced with the more stable UUCG tetraloop. (B) The Euler angles of a HJH, including the twist angle about the two helices (αh, γh) and the interhelical bend angle (βh), describe the interhelical orientation. The Euler angle distribution of an NMR RDC-selected conformational ensemble of TAR reveals the presence of large amplitude bending and twisting motions. The population of specific conformations (squares) is indicated by color, with increasing probability from blue to red. Seven ligand-bound conformations of TAR from crystal structures (black circles) reside within the ligand-free ensemble [Reprinted from 26• with permission. Copyright 2013 American Chemical Society]. (C) The interhelical orientations of HJHs demonstrate topological confinement, illustrated by the Euler angle distribution map. PDB-derived HJHs, defined by the number of single-stranded residues in each strand of the junction (SXSY, with the subscripts denoting the number of nucleotides present on each side of the bulge; X ≥ Y; left panel). The HJHs exhibit changes in the distribution of interhelical orientations dependent on the junction topology (colored by Y-family) [From 31• •. Reprinted with permission from AAAS].
The preferred TAR conformations were determined and shown to represent a rather small region of the sterically allowed space for the TAR junction. Nevertheless, an ensemble was required to accurately represent the data (Figure 3B) [26•]. Binding of ligands to TAR RNA was shown to favor conformers within a region of the overall energy landscape for free TAR (Figure 3B) [26•–28], emphasizing the need for ensemble-based molecular descriptions to understand binding events. The probability of forming the subset of ligand-bound conformers in free TAR and the atomic-level properties of the binding event will determine the probability of following so-called tertiary capture or induced fit binding mechanisms [28]. We suggest that a rigorous ensemble view of a system such as TAR and its ligands, in the form of a free energy landscape, can ultimately lead to a quantitative and predictive understanding of the association process, an outcome that is not accessible from simply labeling pathways as tertiary capture or induced fit.
An important next question is to what extent does the specific sequence of the junction matter, and, as highlighted in the conceptual study of Chu et al., to what extent does the topology of the junction matter? “Matter” here can be defined as the effect on the ensemble of helix-helix positions in space emanating from the junctions and the free energy landscape describing the three Euler angles and three Cartesian coordinates that determine the positions of the helices with respect to one another (i.e., the interhelical distances and orientations) [29• •–31• •]. A summary of crystal structures of HJHs collated based on the number of single-stranded nucleotides on each side of the junction suggests that the preferred helix positions are highly dependent on the topology of the junction (Figure 3C) [31• •].
It remains to be determined how sensitive helix-helix positions are to particular sequences within junctions of specified length, e.g., whether most sequences behave the same with rare outliers such as the kink-turn [23] or if there is a wide range of landscapes that are highly sequence-idiosyncratic. The degree to which flexibility within the helices themselves affects the positioning of distal tertiary motifs and whether the sequences of Watson-Crick base pairs have significant effects also remain to be determined. Importantly, the helix-helix conformational landscape is highly sensitive to electrostatic screening [24•,32], which in turn depends on the ionic constituents of the solution, but popular models do not yet adequately treat the underlying electrostatics, as discussed elsewhere [33,34].
How will these major outstanding questions be answered? NMR RDC approaches have proven powerful for probing rotational degrees of freedom, and, in principle, complementary information can be obtained from X-ray scattering interference (XSI), an approach developed by Harbury and colleagues that employs two site-specifically attached Au-nanocrystals and provides an instantaneous distance-probability distribution via the scattering interference pattern of these nanocrystals [35•,36•,37• •, Shi et al. in press]. Given the large number of sequence combinations in need of exploration, developing information-rich, high-throughput biophysical assays is a key challenge. As highlighted in the next section, techniques are also needed to reveal rare or “hidden” conformations; NMR relaxation dispersion methods can detect discrete rare conformations [38• •,39• •], but approaches that look more generally at the ensemble of sparsely sampled conformations are also needed. Ultimately physics-based computational approaches have the promise to provide complete atomic-level descriptions of conformations, their exchange rates and paths, and their abundances across the full range of solution ion conditions. Realizing this goal will likely require considerable patience and methodical cycling through models, non-trivial predictions, and explicit quantitative tests [33], and infrastructure and support for such long-term initiatives will be required [40•].
RNA Tertiary Motifs
An apparent simplifying feature of RNA structure, relative to proteins, is the presence of reoccurring tertiary motifs [41,42]. Unlike protein three-dimensional structures with near uniform dense packing, RNA three-dimensional structures appear to have only occasional areas of direct and extended contact. These limited and distinct tertiary connections lead to the view that dissecting RNA tertiary folding and conformational transitions into quantifiable elements may be more tractable than the parallel goal for proteins where the driving force for folding is distributed throughout the packed structure.
In this section we address the ensemble properties of RNA tertiary motifs first from the standpoint of the components that come together to form the tertiary contact (i.e., bound or folded state). The conformational landscape and interconversion of the free states determines the probability of forming the tertiary motif (kinetics) and directly influences its stability (thermodynamics) via the number of accessible states in the unfolded (or partially folded) ensemble.
Tertiary contacts form when one region of a RNA “binds” to another, and this behavior has parallels to ligand and protein binding by RNA. Early NMR structural studies from Williamson revealed RNA conformational change accompanying ligand binding, and it was suggested that this might be a general property of RNA molecules [43,44]. More recently, it was discovered that Nature has exploited conformational changes coupled to ligand binding to control gene expression via riboswitches, providing another example of Nature’s ability to operate outside the predictions of the Central Dogma, with RNA rather than proteins acting as control sensors [45].
RNA folding generally appears to be a slow process [46,47], and different origins may be at play for different RNAs or under different conditions. The factors potentially responsible for slow folding include the HJH conformational landscape, as introduced above, tertiary structure motif formation, rearrangements of ion atmosphere ions and ion binding, and the making and breaking of nonnative secondary and tertiary structures. As noted above, a full and predictive understanding will require isolation and dissection of each of these underlying factors in simpler systems, and here we describe results that provide insight into a ubiquitous RNA tertiary motif, the tetraloop/tetraloop receptor (TL/TLR) (Figure 4A).
Figure 4.
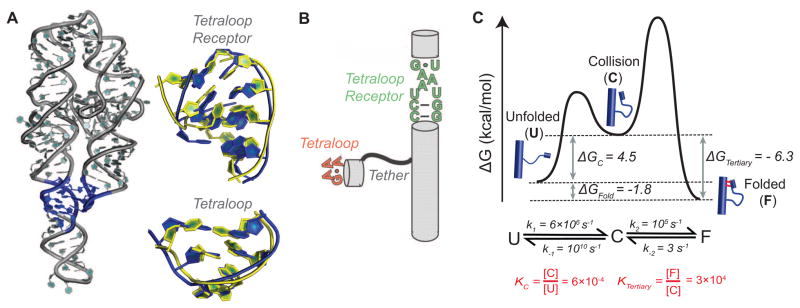
Dynamics and tetraloop-tetraloop receptor (TL/TLR) tertiary motif assembly. (A) The crystal structure of the P4–P6 domain of the Tetrahymena ribozyme with its TL/TLR highlighted in blue (PDB: 1GID [48]; left panel) and an overlay of the free (yellow, PDB: 1TLR [49]) and bound (blue, PDB: 1GID [48]) forms of the TLR (top) and the free (yellow, PDB: 1ZIF [50]) and bound (blue, PDB: 1GID [48]) forms of the TL (bottom) (right panel). (B) TL/TLR constructs used in single molecule FRET experiments [adapted from 53]. (C) Predicted free energy reaction profile for the formation of a tertiary contact based on functional measurements made on inter- and intramolecular tertiary contacts [51,52], as described in the text.
Comparison of X-ray crystal structures of TL/TLR tertiary motifs with the NMR structures of the free tetraloop and free tetraloop receptor structures reveals substantial rearrangement (Figure 4A) [48–50]. However, this structural knowledge alone does not tell us the pathways and energetics of motif formation –is there substantial or miniscule populations of the folded states are present in the unfolded ensembles, are there small or large barriers to rearrangement between these states, and can an initial non-native complex form and rearrange to the final state? We do not yet have answers to these questions, but literature results can be combined to provide quantitative estimates of and some rough boundary conditions for the association process.
Qin et al. studied the formation of a TL/TLR motif from intermolecular TL and TLR components via changes in relaxation of an attached EPR tag and determined a dissociation constant of Kd = 0.4 mM (at 125 mM Mg2+) [51]. Nesbitt, Pardi, and colleagues used smFRET to study the formation of a TL/TLR connected intramolecularly via flexible U7, A7, or U14 tethers (Figure 4B; [52]). Given the flexibility of this tether it is reasonable to assume that the rate constants for dissociation of intermolecular and intramolecular TL/TLRs are the same, and a value of kundock = 3 s−1 was measured in the smFRET studies under conditions similar to those used in the intermolecular experiments (at 0–10 mM Mg2+). Returning to the intermolecular process, an equilibrium dissociation constant can be represented in terms of association and dissociation rate constants: Kd = kdissoc/kassoc. With the observed Kd and the assumption that kdissoc =~kundock, then kassoc, the second order rate constant for TL/TLR formation, can be calculated: . While the rate and equilibrium constants used to estimate the value of kassoc were not obtained under the same conditions, it is likely that the value above provides a reasonable order-of-magnitude estimate.
We can compare this kassoc value with the value expected for diffusional collision –i.e., the rate constant expected if the molecular components are preformed such that essentially every collision results in a productive binding event. However, diffusion is several orders of magnitude faster than TL/TLR association (kdiff = ~109 M−1 s−1), indicating that roughly only 1 out of every 105 collisions leads to TL/TLR formation (Figure 4C). Thus, there is a substantial barrier for tertiary structure formation subsequent to collision.
We can now use the above estimates to help describe intramolecular tertiary contact formation for the intramolecular construct studied by Nesbitt, Pardi and colleagues [42,52,53] as a transition from an unfolded (U) to an folded ensemble (F) via an intermediate collisional ensemble (C) in which the tertiary elements are roughly aligned following an intramolecular collision but with one or both tertiary element in a nonproductive conformation (Figure 4B, 4C). Since our earlier calculations estimate that the TL/TLR is formed once every 105 collisions, the rate constant for transitioning from C to U (k−1) is approximately 105 times greater than that for the transition from C to F (k2 = 10−5k−1) (Figure 4C). As k−1 is much larger than k2, k−2 represents the rate-limiting step for dissociation and . A rate constant k−1 for escape from C, the reverse of diffusional collision, can be crudely estimated to be around 1010 s−1. Thus, k−1 is ~1010 s−1 and k2 (=k2 = 10−5k−1) is ~105 s−1. This leaves only k1 to calculate, and its value can be obtained from the values for the other three rate constants and the overall equilibrium constant of Kfold = 19 [52], according to the equation: , which gives k1 = 6 × 106 s−1. The equilibrium, crudely estimated, for the collision complex KC = k1/k−1 (= 6 x 106 s−1/1010 s−1) = ~6 x 10−4. Thus, only a very small fraction of either or both the TL and TLR exists at equilibrium in the productive form.
These calculations illustrate an approach to begin to break down and help describe and understand RNA folding processes and conformational transitions. Future challenges include determining how or whether these barriers change with solution conditions, how electrostatics contribute to the barrier of bringing together RNA elements, what the association rate constants and probabilities for other RNA tertiary motifs are, and what the atomic-level reasons for slow binding and rearrangements that occur prior to, during, and subsequent to binding are. In addition, it is likely that many of the aligned states that must form prior to tertiary motif formation (Figure 4C) are rare and below the threshold of detection in NMR RDC and XSI studies so that new approaches to find and quantitate these rarely visited sectors of HJH conformational landscapes will be needed.
Conclusion
The rapidly growing number of RNA systems and RNA-based functions creates the temptation to simplify through categorization. We suggest that, ultimately, a predictive understanding of RNA systems and other complex macromolecular assemblages will require understanding at the molecular and atomic level. Although the size and complexity of RNA systems present a formidable challenge, molecular understanding with predictive capabilities may be attainable by studying simpler systems, in parallel to biological and physical studies of complex RNAs, with the perspective that the energy landscapes of component parts can be used to predict the energy landscapes of larger, more complex RNAs and RNA/protein assemblies. These simple systems, in particular HJHs and tertiary motifs, along with recent methodological breakthroughs, allow us to begin to observe and understand the conformational ensembles of RNA and to progress toward this ambitious goal.
Highlights.
RNA folding studies are poised to move from descriptive to predictive
Prediction of RNA kinetics and thermodynamics requires energy landscape determination
RNAs can be considered collections of helices connected by junctions
Junctions determine relative orientations of helices and embedded tertiary motifs
New experimental methods determine energy landscapes of helix-junction-helices
Energy landscapes of components may be used to predict landscapes of complex RNAs
Quantitative molecular descriptions will provide deep understanding of RNA in biology
Acknowledgments
We thank Steve Bonilla, Xuesong Shi, and Shan Yang for helpful discussions. We thank members of the Herschlag laboratory for comments and suggestions. Research from our lab is supported by National Institutes of Health Grant P01 GM066275. G.S. was supported in part by an Office of Technology Licensing Stanford Graduate Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Benjamin E. Allred, Email: beallred@stanford.edu.
Seshadri Gowrishankar, Email: seshg@stanford.edu.
References
- 1.Crick FH. On protein synthesis. Symp Soc Exp Biol. 1958;12:138–163. [PubMed] [Google Scholar]
- 2.Crick F. Central dogma of molecular biology. Nature. 1970;227:561–563. doi: 10.1038/227561a0. [DOI] [PubMed] [Google Scholar]
- 3.Temin HM, Mizutani S. RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature. 1970;226:1211–1213. doi: 10.1038/2261211a0. [DOI] [PubMed] [Google Scholar]
- 4.Autexier C, Lue NF. The structure and function of telomerase reverse transcriptase. Annu Rev Biochem. 2006;75:493–517. doi: 10.1146/annurev.biochem.75.103004.142412. [DOI] [PubMed] [Google Scholar]
- 5.Fresco JR, Adams A, Ascione R, Henley D, Lindahl T. Tertiary structure in transfer ribonucleic acids. Cold Spring Harb Symp Quant. 1966;31:527–537. doi: 10.1101/sqb.1966.031.01.068. [DOI] [PubMed] [Google Scholar]
- 6.Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157:77–94. doi: 10.1016/j.cell.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 7.Kraus A, Groveman BR, Caughey B. Prions and the potential transmissibility of protein misfolding diseases. Annu Rev Microbiol. 2013;67:543–564. doi: 10.1146/annurev-micro-092412-155735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woese C. The Genetic Code: The Molecular Basis for Genetic Expression. Harper and Row; 1967. [Google Scholar]
- 9.Crick FHC. The origin of the genetic code. J Mol Biol. 1968;38:367–379. doi: 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- 10.Orgel LE. Evolution of genetic apparatus. J Mol Biol. 1968;38:381–393. doi: 10.1016/0022-2836(68)90393-8. [DOI] [PubMed] [Google Scholar]
- 11.Dwars T, Paetzold E, Oehme G. Reactions in micellar systems. Angew Chem Int Ed. 2005;44:7174–7199. doi: 10.1002/anie.200501365. [DOI] [PubMed] [Google Scholar]
- 12.Akopian D, Shen K, Zhang X, Shan S. Signal recognition particle: an essential protein-targeting machine. Annu Rev Biochem. 2013;82:693–721. doi: 10.1146/annurev-biochem-072711-164732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Voorhees RM, Ramakrishnan V. Structural basis of the translational elongation cycle. Annu Rev Biochem. 2013;82:203–236. doi: 10.1146/annurev-biochem-113009-092313. [DOI] [PubMed] [Google Scholar]
- 14.Sanbonmatsu KY. Computational studies of molecular machines: the ribosome. Curr Opin Struct Biol. 2012;22:168–174. doi: 10.1016/j.sbi.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitra K, Frank J. Ribosome dynamics: insights from atomic structure modeling into cryo-electron microscopy maps. Annu Rev Biophys Biomol Struct. 2006;35:299–317. doi: 10.1146/annurev.biophys.35.040405.101950. [DOI] [PubMed] [Google Scholar]
- 16.Freier SM, Kierzek R, Jaeger JA, Sugimoto N, Caruthers MH, Neilson T, Turner DH. Improved free-energy parameters for predictions of RNA duplex stability. Proc Natl Acad Sci USA. 1986;83:9373–9377. doi: 10.1073/pnas.83.24.9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sugimoto N, Kierzek R, Turner DH. Sequence dependence for the energetics of terminal mismatches in ribooligonucleotides. Biochemistry. 1987;26:4559–4562. doi: 10.1021/bi00388a059. [DOI] [PubMed] [Google Scholar]
- 18.Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 19.Murchie AIH, Clegg RM, von Krtzing E, Duckett DR, Diekmann S, Lilley DM. Fluorescence energy transfer shows that the four-way DNA junction is a right-handed cross of antiparallel molecules. Nature. 1989;341:763–766. doi: 10.1038/341763a0. [DOI] [PubMed] [Google Scholar]
- 20.Battacharyya A, Murchie AI, Lilley DM. RNA bulges and the helical periodicity of double-stranded RNA. Nature. 1990;343:484–487. doi: 10.1038/343484a0. [DOI] [PubMed] [Google Scholar]
- 21.Duckett DR, Murchie AI, Lilley DM. The global folding of four-way helical junctions in RNA, including that in U1 snRNA. Cell. 1995;83:1027–1036. doi: 10.1016/0092-8674(95)90218-x. [DOI] [PubMed] [Google Scholar]
- 22.Lilley DM. Structures of helical junctions in nucleic acids. Q Rev Biophys. 2000;33:109–159. doi: 10.1017/s0033583500003590. [DOI] [PubMed] [Google Scholar]
- 23.Lilley DMJ. The structure and folding of kink turns in RNA. WIREs RNA. 2012;3 :797–805. doi: 10.1002/wrna.1136. [DOI] [PubMed] [Google Scholar]
- 24•.Chu VB, Lipfert J, Bai Y, Pande VS, Doniach S, Herschlag D. Do conformational biases of simple helical junctions influence RNA folding stability and specificity? RNA. 2009;15:2195–2205. doi: 10.1261/rna.1747509. The work provided a conceptual basis for considering helix-junction-helix (HJH) motifs as fundamental elements in RNA folding, and the notion that their conformational ensemble properties determine the probability of tertiary structure formation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25•.Bothe JR, Nikolova EN, Eichhorn CD, Chugh J, Hansen AL, Al-Hashimi HM. Characterizing RNA dynamics at atomic resolution using solution-state NMR spectroscopy. Nature Methods. 2011;8:919–931. doi: 10.1038/nmeth.1735. The authors review NMR techniques including RDCs and relaxation dispersion that are used to characterize RNA dynamics over a broad range of timescales. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26•.Salmon L, Bascom G, Andricioaei I, Al-Hashimi HM. A general method for constructing atomic-resolution RNA ensembles using NMR residual dipolar couplings: the basis for interhelical motions revealed. J Am Chem Soc. 2013;135:5457–5466. doi: 10.1021/ja400920w. The authors use NMR RDC and MD simulation to construct and test an atomic resolution ensemble of TAR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pitt SW, Majumdar A, Serganov A, Patel DJ, Al-Hashimi HM. Argininamide binding arrests global motions in HIV-1 TAR RNA: comparison with Mg2+ -induced conformational stabilization. J Mol Biol. 2004;338:7–16. doi: 10.1016/j.jmb.2004.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Q, Stelzer AC, Fisher CK, Al-Hashimi HM. Visualizing spatially correlated dynamics that directs RNA conformational transitions. Nature. 2007;450:1263–1267. doi: 10.1038/nature06389. [DOI] [PubMed] [Google Scholar]
- 29••.Mustoe AM, Bailor MH, Teixeira RM, Brooks CL, III, Al-Hashimi HM. New insights into the fundamental role of topological constraints as a determinant of two-way junction conformation. Nucl Acids Res. 2012;40:892–904. doi: 10.1093/nar/gkr751. The authors present a general description of how junction topology provides a strong constraint for the relative spatial positions of helices. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bailor MH, Mustoe AM, Brooks CL, III, Al-Hashimi HM. 3D maps of RNA interhelical junctions. Nature Protocols. 2011;6:1536–1545. doi: 10.1038/nprot.2011.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31••.Bailor MH, Sun X, Al-Hashimi HM. Topology links RNA secondary structure with global conformation, dynamics, and adaption. Science. 2010;327:202–206. doi: 10.1126/science.1181085. The authors construct a map of inter-helical orientations of two-way RNA helix-junction-helix motifs allowed by steric and connectivity constraints across the junction. The map reveals that allowed RNA inter-helical orientations are highly confined, making up only ~10% of all theoretically possible inter-helical orientations. [DOI] [PubMed] [Google Scholar]
- 32.Bai Y, Chu VB, Lipfert J, Pande VS, Herschlag D, Doniach S. Critical assessment of nucleic acid electrostatics via experimental and computational investigation of an unfolded state ensemble. J Am Chem Soc. 2008;130:12334–12341. doi: 10.1021/ja800854u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lipfert J, Doniach S, Das R, Herschlag D. Understanding nucleic acid-ion interactions. Annu Rev Biochem. 2014;83:813–841. doi: 10.1146/annurev-biochem-060409-092720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chu VB, Bai Y, Lipfert J, Herschlag D, Doniach S. A repulsive field: advances in the electrostatics of the ion atmosphere. Curr Opin Chem Biol. 2008;12 :619–625. doi: 10.1016/j.cbpa.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35•.Mathew-Fenn RS, Das R, Harbury PB. Remeasuring the double helix. Science. 2008;322:446–449. doi: 10.1126/science.1158881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36•.Shi X, Herschlag D, Harbury PB. Structural ensemble and microscopic elasticity of freely diffusing DNA by direct measurement of fluctuations. Proc Natl Acad Sci USA. 2013;110:E1444–E1451. doi: 10.1073/pnas.1218830110. In this paper and reference 35 Harbury and colleagues introduce a new approach to accurately measure distances within nucleic acid structures in solution. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37••.Shi X, Beauchamp KA, Harbury PB, Herschlag D. From a structural average to the conformational ensemble of a DNA bulge. Proc Natl Acad Sci USA. 2014;111:E1473–E1480. doi: 10.1073/pnas.1317032111. The authors apply X-ray interferometry to estimate the conformational ensemble of a macromolecule, a DNA bulge. Testable atomic-level models were generated by combine the measured ensemble with molecular dynamics simulations. This work paves the way for further investigation of RNA ensembles by X-ray interferometry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38••.Lee J, Dethoff EA, Al-Hashimi HM. Invisible RNA state dynamically couples distant motifs. Proc Natl Acad Sci USA. 2014;111:9485–9490. doi: 10.1073/pnas.1407969111. The authors use NMR relaxation dispersion to detect a transient state that is involved in remodeling the structure of TAR between the apical loop and bulge. The results suggest a mechanism for long-range communications for complex processes such as RNA cooperative folding and ribonucleoprotein assembly. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39••.Dethoff EA, Petzold K, Chugh J, Casiano-Negroni A, Al-Hashimi HM. Visualizing transient low-populated structures of RNA. Nature. 2012;491:724–728. doi: 10.1038/nature11498. The authors characterize an ‘invisible’ RNA transient state existing in low abundance and short duration using NMR relaxation dispersion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40•.Cruz JA, Blanchet MF, Boniecki M, Bujnicki JM, Chen SJ, Cao S, Das R, Ding F, Dokholyan NV, Flores SC, Huang L, Lavender CA, Lisi V, Major F, Mikolajczak K, Patel DJ, Philips A, Puton T, Santalucia J, Sijenyi F, Hermann T, Rother K, Rother M, Serganov A, Skorupski M, Soltysinski T, Sripakdeevong P, Tuszynska I, Weeks KM, Waldsich C, Wildauer M, Leontis NB, Westhof E. RNA-Puzzles: a CASP-like evaluation of RNA three-dimensional structure prediction. RNA. 2012;18:610–625. doi: 10.1261/rna.031054.111. Making blind predictions from computational models provides the only truly unbiased means to test these models. This group has taken this important step for RNA structural prediction. A next critical step will be blind predictions of RNA folding kinetics and thermodynamics and community-based evaluations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jaeger L, Michel F, Westhof E. Involvement of a GNRA tetraloop in long-range RNA tertiary interactions. J Mol Biol. 1994;236:1271–1276. doi: 10.1016/0022-2836(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 42.Fiore JL, Nesbitt DJ. An RNA folding motif: GNRA tetraloop-receptor interactions. Q Rev Biophys. 2013;46:223–264. doi: 10.1017/S0033583513000048. [DOI] [PubMed] [Google Scholar]
- 43.Battiste JL, Tan R, Frankel AD, Williamson JR. Binding of an HIV Rev peptide to Rev responsive element RNA induces formation of purine-purine base pairs. Biochemistry. 1994;33:2741–2747. doi: 10.1021/bi00176a001. [DOI] [PubMed] [Google Scholar]
- 44.Puglisi JD, Tan R, Calnan BJ, Frankel AD, Williamson JR. Conformation of the TAR RNA-arginine complex by NMR spectroscopy. Science. 1992;257:76–80. doi: 10.1126/science.1621097. [DOI] [PubMed] [Google Scholar]
- 45.Winkler WC, Breaker RR. Regulation of bacterial gene expression by riboswitches. Annu Rev Microbiol. 2005;59:487–517. doi: 10.1146/annurev.micro.59.030804.121336. [DOI] [PubMed] [Google Scholar]
- 46.Zarrinkar PP, Williamson JR. Kinetic intermediates in RNA folding. Science. 1994;265:918–924. doi: 10.1126/science.8052848. [DOI] [PubMed] [Google Scholar]
- 47.Zarrinkar PP, Wang J, Williamson JR. Slow folding kinetics of RNase P RNA. RNA. 1996;2:564–573. [PMC free article] [PubMed] [Google Scholar]
- 48.Cate JH, Gooding AR, Podell E, Zhou K, Golden BL, Kundrot CE, Cech TR, Doudna JA. Crystal structure of a group I ribozyme domain: principles of RNA packing. Science. 1996;273:1678–1685. doi: 10.1126/science.273.5282.1678. [DOI] [PubMed] [Google Scholar]
- 49.Butcher SE, Dieckmann T, Feigon J. Solution structure of a GAAA tetraloop receptor RNA. EMBO J. 1997;16:7490–7499. doi: 10.1093/emboj/16.24.7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jucker FM, Heus HA, Yip PF, Moors EHM, Pardi A. A network of heterogenous hydrogen bonds in GNRA tetraloops. J Mol Biol. 1996;264:968–980. doi: 10.1006/jmbi.1996.0690. [DOI] [PubMed] [Google Scholar]
- 51.Qin PZ, Butcher SE, Feigon J, Hubbell WL. Quantitative analysis of the isolated GAAA tetraloop/receptor interaction in solution: a site-directed spin labeling study. Biochemistry. 2001;40:6929–6936. doi: 10.1021/bi010294g. [DOI] [PubMed] [Google Scholar]
- 52.Hodak JH, Downey CD, Fiore JL, Pardi A, Nesbitt DJ. Docking kinetics and equilibrium of a GAAA tetraloop-receptor motif probed by single-molecule FRET. Proc Natl Acad Sci USA. 2005;102:10505–10510. doi: 10.1073/pnas.0408645102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fiore JL, Holmstrom ED, Nesbitt DJ. Entropic origin of Mg2+-facilitated RNA folding. Proc Natl Acad Sci USA. 2012;109:2902–2907. doi: 10.1073/pnas.1114859109. [DOI] [PMC free article] [PubMed] [Google Scholar]