

Abstract
The injury inflammatory response mediated by the innate immune system is an important contributor to neurodegeneration in the central nervous system (CNS) and retina. A major branch of the innate immune system is regulated by the Toll-like receptors (TLRs), which are receptors for endogenous damage associated molecules released from injured cells as well as pathogen-derived molecules, and interleukin-1 receptors (IL-1R), which are activated by IL-1α, IL-1β and IL-18 cytokines. TLRs and IL-1R are expressed on immune and non-immune cell types and act as first responders to cell damage, which results in tissue repair, or inflammation and apoptosis. Both TLR and IL-1R require the adaptor protein myeloid differentiation primary response gene 88 (MyD88) for signaling. Although inflammation is implicated in neuronal death in the retina, the role of MyD88-dependent TLR and IL-1R signaling in retinal degeneration is unknown. Therefore, the purpose of this study was to investigate the role of MyD88-mediated signaling in neuronal degeneration in the retinal degeneration 1 (rd1) mouse model, which exhibits a phenotype of rapid photoreceptor death and inflammation. To generate rd1 mice lacking the MyD88 gene, rd1 were bred with MyD88 knockout mice (MyD88-/-) for several generations to produce rd1/MyD88+/+ and rd1/MyD88-/- genotypes. Chemokine mRNA expression levels were analyzed by qRT-PCR, and recruitment of activated microglia was quantified by immunodetection of the IBA-1 protein. Retinal outer nuclear layer cell counts were performed to quantify photoreceptor degeneration, and retinal function was assessed using electroretinograms (ERG). Our results revealed that retinal expression of Ccl2, Ccl4, Ccl7 and Cxcl10 was reduced by 2 to 8-fold in rd1/MyD88-/- mice compared with rd1/MyD88+/+ mice (p<0.05), which coincided with attenuated microglial activation, higher numbers of photoreceptors and higher retina responses to photopic and scotopic stimuli. At later ages, rd1/MyD88-/- had reduced chemokine expression and higher photopic responses but no change in microglial recruitment compared with rd1 mice with functional MyD88. In conclusion, lack of MyD88-mediated signaling increased photoreceptor survival and retina function in rd1 mice, which implicates MyD88-mediated innate immunity pathways as an important pathogenic factor during retinal degeneration.
Keywords: TLR signaling, IL-1R, MyD88, photoreceptor, retinal degeneration, chemokines, microglia, retina, innate immunity
Introduction
Activation of the innate immune system is implicated in the pathogenesis of numerous neurodegenerative conditions in the brain (Glass et al., 2010) and retina (Donoso et al., 2006). Toll-like receptors (TLRs) and interleukin-1 receptor (IL-1R) form a superfamily of innate immunity receptors that require the adaptor protein MyD88 for signaling. TLRs recognize exogenous pathogen-associated proteins, such as lipopolysaccharide, and are also the major receptors for endogenous host-derived danger-associated proteins, such as HMGB1 and heat shock proteins (Mathur et al., 2011). There are 10 TLRs, and all except TLR3 and a minor portion of TLR4 signal through MyD88 (Takeda and Akira, 2004). IL-1R is activated by the IL-1 pro-inflammatory family of molecules, including IL-1β and IL-18.
Upon ligand binding to TLR and IL-1R, MyD88 is recruited to the receptors and in turn recruits the kinases IRAK-1 and IRAK-4, which leads to activation of NF-κB, AP-1, and subsequent induction of cytokines, including IL-6 and TNF-α (Elner et al., 2005; Kumar et al., 2004). During non-pathogen injury such as oxidative stress, TLR and IL-1R are believed to be activated by molecules released from injured cells, and induce MyD88-dependent intracellular signaling cascades that stimulate inflammation and promote further neuronal death (Caso et al., 2007; Kaczorowski et al., 2008; Lehnardt et al., 2003; Marsh et al., 2009; Okun et al., 2009; Tang et al., 2008). Therefore, characterizing how MyD88-mediated signaling influences neuronal viability is important for understanding the role of innate immunity and inflammation in the onset and rate of degeneration.
The retina is a light-sensitive specialized neuroepithelial tissue composed of three layers of functionally distinct neurons and glia that act together to generate vision. Because of its accessibility and low complexity, the retina is often used to study neuroinflammatory events that also occur elsewhere in the CNS. Chemokines and microglia are implicated in photoreceptor death in inherited retinal degeneration (Zeng et al., 2005), but the question of whether the prominent danger signal responders, the TLRs and IL-1R, influence these factors, and more importantly, disease outcome, has not been elucidated. Whereas the contribution of MyD88-independent TLR3 and complement to retinal degeneration have been described (Hageman et al., 2005; Khandhadia et al.; Klein et al., 2008; Yu et al., 2012), the role of MyD88-dependent TLR and IL-1R signaling in inherited photoreceptor degeneration is not known. Therefore, the overall understanding of inflammatory pathways during retinal disease remains incomplete.
Several lines of evidence suggest a pathogenic role for TLR and IL-1R receptors in the retina. TLRs are expressed throughout the retina, and induction of TLR expression was demonstrated in retinal degenerations in mouse and age-related macular degeneration (AMD) patients (Kohno et al., 2013; Kokkinopoulos et al., 2005; Maloney et al., 2010; Shiose et al., 2011; Zhu et al., 2013). An active role for TLR signaling via MyD88 in photoreceptor death has been shown in several recent studies using acute injury. TLR4 signaling in microglia contributed to light-induced photoreceptor degeneration (Kohno et al., 2013), and TLR4 activation caused photoreceptor death during oxidative stress in vitro (Yi et al., 2012) and mitochondrial oxidative stress damage in vivo (Ko et al., 2011). Additionally, TLR2 signaling enhanced choroidal neovascularization and promoted the development of diabetic retinopathy (Fujimoto et al., 2010). Inhibiting TLR2/4 increased retinal endothelial cell survival (Tang et al., 2013) and mice lacking TLR4 show improved survival of retinal ganglion cells from axotomy-induced death (Kilic et al., 2008) and have smaller infarct sizes in CNS injury models (Caso et al., 2007; Tang et al., 2007), suggesting that MyD88 functions as a common regulator of cell death. A role for IL-1R in regulating photoreceptor death has also been demonstrated by nature of the involvement of its proinflammatory ligands, cytokines IL-1β and IL-18, in retinal degeneration (Anderson et al., 2013; Tarallo et al., 2012). The lipofuscin component A2E stimulated IL-1β in retinal pigment epithelium (RPE) (Anderson et al., 2013), indicating a potential link between drusen and IL-1R signaling. In AMD retinas, IL-1β and IL-18 secretion was induced by the NLRP3 inflammasome, resulting in detrimental effects in dry AMD (Tarallo et al., 2012) and protective effects in neovascular AMD (Doyle et al., 2012). These studies suggest that TLR and IL-1R are associated with retinal disease, but their precise contribution to degeneration, especially in an inherited model of photoreceptor death, has not been investigated.
In this study, we tested the involvement of TLR and IL-1R signaling in retinal degeneration using the retinal degeneration 1 (rd1) mouse model. The rd1 mouse has a spontaneously-derived mutation in the rod photoreceptor-expressed cGMP phosphodiesterase β-subunit (PDE6β) gene, which also causes the human retinal degeneration retinitis pigmentosa (Chang et al., 2002; Huang et al., 1995). The PDE6β mutation in rd1 mice leads to aggressive retinal degeneration with photoreceptor apoptosis beginning at post-natal day (P)10, and causes rapidly declining photoreceptor layer thickness (Bowes et al., 1990). The pattern of chemokine expression and microglial recruitment in rd1 has been well characterized, thus making it a suitable model for this study. We used a genetic approach to block TLR and IL-1R signaling in rd1 by removing MyD88, and demonstrated that innate immunity mediates the inflammatory response in early stages of retinal degeneration, with reduced chemokine expression and microglial recruitment in mice lacking MyD88. Furthermore, the dampened initial immune response is associated with increased photoreceptor survival and retina function. These findings reveal new insights into the contribution of TLR and IL-1R to retinal degeneration, and identify MyD88 as a potential target for delaying retinal disease progression.
Materials and Methods
Animals
All procedures involving mice were performed in accordance with the ARVO Statement for the Use of Animals in Ophthalmology and Vision Research and were approved by the Institutional Animal Care and Use Committee at the University of Miami. To block TLR and IL-1R signaling pathways, we bred rd1 mice with global MyD88 knock-out mice (MyD88-/-), which have a constitutive deletion of MyD88 exon 3 and show reduced innate immune responses to TLR ligands and IL-1 and IL-18 (Hou et al., 2008). The rd1 mice (C3H/HeOuJ, The Jackson Laboratory (Bar Harbor, ME, USA), Stock number 000635), are homozygous for the retinal degeneration 1 mutation (Pde6βrd1) (Bowes et al., 1990; Gibson et al., 2013) and were bred with MyD88-/- mice (B6.129P2(SJL)-MyD88tm1.1Defr/J, Stock number 009088, The Jackson Laboratory) (Hou et al., 2008) to produce breeders that were homozygous for the rd1 mutation and heterozygous for the MyD88 genotype (MyD88+/-). The breeders were backcrossed for 4 generations and were used to produce litters containing the rd1/MyD88+/+ and rd1/MyD88-/- genotypes. All mice were housed in a 12-hour light/dark cycle and were kept at equivalent distances from the overhead light.
Electroretinograms (ERGs)
ERGs were performed as previously described (Patel and Hackam, 2014). After 4 hours of dark adaptation, mice were anesthetized by intraperitoneal injection of a solution containing 100 mg/Kg body weight ketamine (Vedco, Inc., St. Joseph, MO) and 10 mg/Kg xylazine solution (AnaSed, Akron, Lake Forest, IL) and the pupils were dilated with 10% phenylepherine hydrochloride (Akorn, Lake Forest, IL). The mice were placed on a small stage and wrapped in a modified heating blanket attached to a constant running water bath to maintain body temperature. A reference electrode was placed subcutaneously at the head and a ground electrode was inserted into the tail. Silver wire electrodes shaped into a large loop were placed on both corneas, and the stage was placed into a Ganzfeld light-emitting chamber (Li et al., 2010). Single flash stimuli were produced using the UTAS system controlled by EM for Windows software (LKC Technologies, Gaithersburg, MD). Scotopic stimuli were flashes of white light ranging from 1 to 100 cds/m2 on a white background light, and photopic stimuli were flashes of green light on low intensity green background light.
Immunohistochemistry and outer nuclear layer cell counts
Enucleated eyes were fixed with cold 4% paraformaldehyde in PBS and processed through increasing sucrose concentrations from 5% to 20%, then embedded in optimal cutting temperature compound and frozen. Embedded globes were sectioned into 8 μm thick sections and mounted onto Superfrost micro slides (VWR International, Radnor, PA) (Yi et al., 2012). To detect activated microglia, the slides were first blocked with 20% goat serum then incubated with primary antibody rabbit anti-IBA-1 (Wako, Osaka, Japan) overnight at 4°C, washed with PBS and then incubated with secondary anti-rabbit antibody conjugated with Alexa Fluor-488 (Invitrogen, Carlsbad, CA). The retinas were washed and counterstained with 4′,6-diamidino-2-phenylindole (DAPI), and then viewed using a fluorescent microscope (Zeiss Axiovert 200). Control slides omitting the primary antibody were viewed using the same microscopy and exposure settings. Microglia counts were performed by counting IBA-1 positive microglia with an ameboid shape from the outer plexiform layer (OPL) to the RPE, and expressed as density of activated microglial cells within a specific area of outer retina. Microglia were counted in the entire section in 3 adjacent retinal sections, in the equivalent regions of the retina in each animal.
For photoreceptor nuclei counts, columns of nuclei were counted at the periphery, near the ora serrata, and centrally, near the optic nerve region, to account for the recognized central-peripheral temporal gradient of degeneration (Carter-Dawson et al., 1978). Ten columns of DAPI-positive nuclei in the outer nuclear layer (ONL) were counted at each site centrally on either side of the optic nerve, and at each side of the cornea peripherally, adjacent to the ora serrata, while viewed at a 20× magnification. Three adjacent retinal sections were counted in each region, and nuclear column counts averaged for central and peripheral regions, for a total of 60 columns per region per eye.
RNA isolation and Quantitative PCR (qPCR)
Total mRNA was isolated from dissected retinas using Trizol reagent (Invitrogen), as described (Silva et al., 2010). One microgram of RNA was reverse-transcribed into cDNA using Thermoscript RT (Invitrogen), and the cDNA samples were used for qPCR in the Mastercycler ep realplex (Eppendorf, Biotech, Hamburg, Germany) using SYBRGreen (Biorad, Hercules, CA). Primer sequences specific to the genes of interest were selected to span at least one intron (Table 1). Amplification of each sample was performed in triplicate for each gene, and the expression levels were normalized to the housekeeping gene Acid Ribosomal Phosphoprotein (ARP) using the delta-delta Ct method (Hackam et al., 2004). The expression of each gene was then normalized to expression levels in wild type adult mouse retina, except for TNF-α, which was not detected in wild type retina, and was instead normalized to TNF-α expression levels in the murine microglial cell line BV2.
Table 1. Primers used for real-time PCR.
| Gene | Primers: Oligonucleotide sequence 5′-3′ | |
|---|---|---|
| ARP | Forward | CGACCTGGAAGTCCAACTAC |
| Reverse | ATCTGCTGCATCTGCTTG | |
| Ccl2 | Forward | AGGTCCCTGTCATGCTTCTG |
| Reverse | ATTTGGTTCCGATCCAGGTT | |
| Ccl3 | Forward | GCAACCAAGTCTTCTCAGCG |
| Reverse | TGGAATCTTCCGGCTGTAGG | |
| Ccl4 | Forward | TGTGCAAACCTAACCCCGAG |
| Reverse | ATACCACAGCTGGCTTGGAG | |
| Ccl5 | Forward | AGCAGCAAGTGCTCCAATCT |
| Reverse | ATTTCTTGGGTTTGCTGTGC | |
| Ccl7 | Forward | CCATGAGGATCTCTGCCACG |
| Reverse | ACCCACTTCTGATGGGCTTC | |
| Cxcl10 | Forward | GCTGCAACTGCATCCATATC |
| Reverse | CACTGGGTAAAGGGGAGTGA | |
| TNF-α | Forward | TCTTCTCATTCCTGCTTGTGG |
| Reverse | CACTTGGTGGTTTGCTACGA | |
Statistical Analysis
A two-tailed Student's t-test was used where two groups were compared, and ANOVA was used with post-hoc Tukey-Kramer analysis where multiple groups were compared. Null hypothesis was rejected at p<0.05.
Results
Loss of MyD88-mediated signaling reduced chemokine mRNA expression during retinal degeneration
Pro-inflammatory chemokines are upregulated during retinal degeneration and are thought to contribute to photoreceptor apoptosis (Guo et al., 2012; Kohno et al., 2014; Rutar et al., 2012). To investigate whether removing MyD88-mediated innate immunity signaling pathways blocks expression of these potentially destructive molecules in the presence of a mutation, we compared rd1 mice with a genetic deletion of MyD88 (rd1/MyD88-/-) with their littermates that had intact MyD88 (rd1/MyD88+/+). The rd1 mouse is an aggressive model of inherited retinal degeneration, with rod photoreceptor death beginning at P10, reaching a peak of degeneration at P16 (Zeng et al., 2005), and resulting in almost complete loss of rods by P17 (Carter-Dawson et al., 1978). A secondary slow progressive cone photoreceptor degeneration follows, in a mechanism that remains poorly understood. The inflammation associated with inherited retinal degeneration occurs prior to and during this period of elevated neuronal death (Gupta et al., 2003; Roque et al., 1996; Yoshida et al., 2013; Zeiss and Johnson, 2004). Therefore, we sought to identify whether MyD88 had a role during the peak of degeneration in the early time-points of degeneration.
QPCR analysis was performed on retinal lysates during the early stage of photoreceptor death at P12, at P14, and during the peak of degeneration at P16. As shown in Fig. 1, expression of Ccl2 (MCP-1), Ccl4 (MIP-1β), Ccl7 (MCP-3) and Cxcl10 mRNA peaked at P12 in the rd1MyD88+/+ mouse and reduced thereafter, which is consistent with previous studies in rd1 that showed maximal chemokine expression at P12, reducing subsequently to baseline levels at P18 (Zeng et al., 2005). Notably, at the same time-point, expression of these chemokines was 2-8 fold lower in the rd1/MyD88-/- compared with rd1/MyD88+/+ (p<0.05; Fig. 1A-D), suggesting a dampened inflammatory response. In contrast, Ccl3 (MIP-1α) expression increased from P12 to P14 in both rd1/MyD88+/+ and rd1/MyD88-/- (p<0.05 for both rd1/MyD88+/+ and rd1/MyD88-/-; Fig. 1E), indicating that the delayed and reduced levels of the other chemokine mRNAs were not a global defect in transcription in the rd1/MyD88-/- mice. Furthermore, Ccl5 expression remained constant across the ages in both genotypes, but mRNA transcripts in rd1/MyD88-/- were consistently 2-fold lower than rd1/MyD88+/+ at all three time-points (p<0.05; Fig. 1F). These results indicate that reduced MyD88-dependent TLR and IL-1R signaling in rd1/MyD88-/- leads to reduction in mRNA expression of specific chemokines associated with retinal degeneration.
Fig. 1.
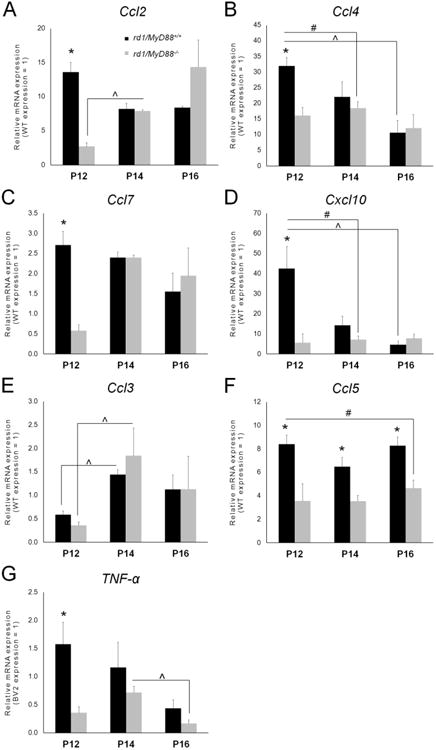
Reduced pro-Inflammatory chemokine and TNF-α transcript expression in the rd1 mouse in the absence of MyD88-mediated signaling. (A-F) Expression of Ccl2 (MCP-1), Ccl4 (MIP-1β), Ccl7 (MCP-3), Cxcl10, Ccl3 (MIP-1α), and Ccl5 (RANTES) in rd1/MyD88+/+ and rd1/MyD88-/- mice at postnatal day 12 (P12), P14 and P16, as measured by qPCR. Reduced transcript levels and a shift in the peak expression from P12 to P14 was observed in MyD88-/- compared with MyD88+/+, for Ccl4, Ccl5, Ccl7, and Cxcl10 (n=3-5, p<0.05). In contrast, Ccl5 expression level remains relatively constant as retinal degeneration progresses with a consistent 2-fold reduction in the rd1/MyD88-/- mouse (n=3-5, p<0.05), whereas Ccl3 expression was not different between the two genotypes. (G) Retinal expression of TNF-α is reduced by 4.4 fold in the rd1/MyD88-/- compared with rd1/MyD88+/+ at P12 (n=3 each, p<0.05). TNF-α expression was normalized to ARP and was expressed relative to expression in the BV2 microglial cell line due to lack of expression in WT retina. Mean ± SEM are shown. * p<0.05, comparison between genotypes within a given time-point p<0.05; (x0005E) p<0.05, comparison among ages of the same genotype; # p<0.05, comparison of peak expression between rd1/MyD88+/+ and rd1/MyD88-/-across all time-points. Samples were compared with the house-keeping gene acid ribosomal phosphoprotein (ARP). All samples were normalized to that of a wild type adult mouse retina, except for TNF-α, which was not detected in wild type retina, and was instead compared with the murine microglial cell line BV2.
Loss of MyD88-mediated signaling reduced TNF-α mRNA expression during retinal degeneration
The cytokine TNF-α exacerbates photoreceptor death in retinal degenerations (de Kozak et al., 1997; Nakazawa et al., 2011) and is produced at highest quantities during early degeneration at P12 in the rd1 mouse, returning to baseline by P18 (Zeng et al., 2005). We previously demonstrated that TNF-α induced photoreceptor death after TLR4-mediated MyD88 signaling in primary retina dissociated cultures (Yi et al., 2012). To determine the contribution of MyD88 to the expression of TNF-α during retinal degeneration, we examined TNF-α mRNA levels in the rd1/MyD88-/- and rd1/MyD88+/+ mice at each time-point. As was the case with chemokines, TNF-α expression was maximal at P12 in the rd1/MyD88+/+ and reduced thereafter. In contrast, there was a delay in peak expression to P14 in the rd1/MyD88-/- (Fig. 1G). Also, TNF-α expression at P12 was 4.4-fold lower in the rd1/MyD88-/- compared with rd1/MyD88+/+ (p<0.05).
Loss of MyD88 attenuates activated microglial recruitment only during early degeneration
Microglia are recruited to the outer retina during retinal degeneration where they are thought to exacerbate photoreceptor loss through the release of additional chemokines and toxic proteins (Ng and Streilein, 2001; Zeiss and Johnson, 2004; Zhang et al., 2005). Because TLRs are implicated in microglia activation and subsequent chemokine production (Esen and Kielian, 2006; Jack et al., 2005), and because MyD88-mediated signaling is implicated in microglia recruitment into injured tissue in the CNS (Lim et al., 2011), we next investigated whether MyD88-mediated signaling was involved in microglia recruitment to the ONL in the rd1 retina. Resting microglia reside in the ganglion cell layer (GCL) and inner retina in non-degenerating retina, and are induced to migrate towards the outer retina in conditions of retinal pathology (Zeiss and Johnson, 2004). Therefore, we quantified IBA-1-positive ameboid (activated) microglia in the region from the outer plexiform layer (OPL) to the RPE (Fig. 2A).
Fig. 2.
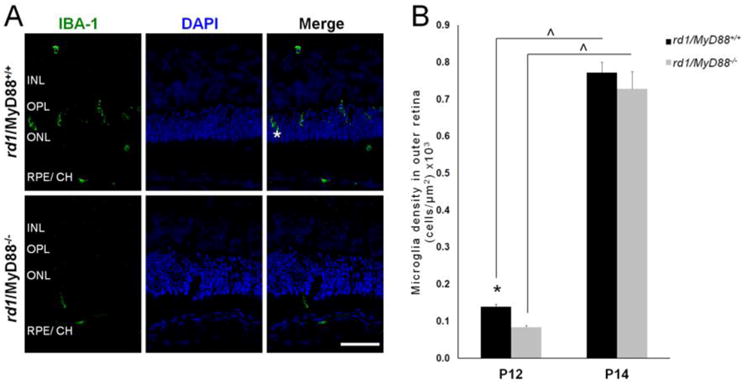
Activated microglia recruitment to the outer retina is reduced at P12 but not at P14 in the rd1/MyD88-/- mouse. (A) Retinal sections from P12 rd1/MyD88+/+ and rd1/MyD88-/- mice immunostained for the activated microglia marker IBA-1 (asterisk), alongside DAPI and merge images. INL= Inner Nuclear Layer, OPL= Outer Plexiform Layer, ONL= Outer Nuclear Layer, RPE/ CH= Retinal Pigment Epithelium/Choroid. Scale bar: 50μm. (B) Microglia cell density in outer retina (from OPL to RPE). Numbers of activated microglia were counted and expressed as number of cells per area (μm2) of outer retina ×103. At P12, there is a 40% reduction in activated microglia in the outer retina of rd1/MyD88-/-compared with rd1/MyD88+/+ mice (n=3 each, p<0.05). Microglia density increases in both mice from P12 to P14, at which point outer retinal microglia numbers are similar in both rd1/MyD88+/+ and rd1/MyD88-/- (n=4, 3 respectively). Mean ± SEM are shown. * p<0.05, comparison among genotypes within a given time-point; (x0005E) p<0.05, comparison between ages of the same genotype.
The density of microglia in the outer retina increased with disease progression from P12 to P14 in both rd1/MyD88+/+ and rd1/MyD88-/- (p<0.01; Fig. 2B), consistent with previous findings (Zeng et al., 2005). However, at P12, mice lacking MyD88 had a 40% reduction in microglial density compared with rd1/MyD88+/+ mice (p<0.05). The difference in microglia density between the two genotypes diminished dramatically at P14. The reduced microglial recruitment at P12 but not at P14 in the rd1/MyD88-/- mice suggests that MyD88-mediated signaling may be involved in early microglial recruitment, but as the disease progresses other signaling pathways may play a more prominent role. Loss of MyD88 leads to reduced expression of chemotractant chemokines at P14, therefore one would expect a subsequent attenuation in recruited microglia, which does not occur, suggesting other factors may be involved in the recruitment of these cells.
Loss of MyD88-mediated signaling increased photoreceptor survival during retinal degeneration
To determine whether MyD88-mediated signaling and associated reduction in pro-inflammatory cytokine mRNA expression and microglia influences photoreceptor viability, we next quantified photoreceptor survival and function. Counting columns of photoreceptor (rod and cone) nuclei in the ONL is an established accurate marker of photoreceptor survival (Michon et al., 1991). As retinal degeneration progressed from P12 to P16, the number of photoreceptor nuclei declined by 5.4-fold in the rd1/MyD88+/+ (p<0.0001; Fig. 3B) and by 3.9-fold in rd1/MyD88-/- mice (p<0.0001). However, the rate of photoreceptor loss that occurred between P12 to P14 differed from rate of cell loss between P14 to P16, because while the rate of cell loss was 2.26-fold greater in rd1/MyD88+/+ between P12 to P14, the rate of cell loss in rd1/MyD88-/- caught up to rd1/MyD88+/+ between P14 to P16 and was equivalent. Although photoreceptor nuclei do diminish in the rd1/MyD88-/- mouse, the numbers of remaining nuclei are higher in rd1/MyD88-/- compared with rd1/MyD88+/+, with the ONL cell count 1.43-fold higher at P16 (p<0.05) in the rd1/MyD88-/- mouse compared to rd1/MyD88+/+. Therefore, eliminating MyD88-mediated signaling in the rd1 mouse results in a moderate but significant protective effect, and dampens the severity of photoreceptor loss during early retinal degeneration.
Fig. 3.
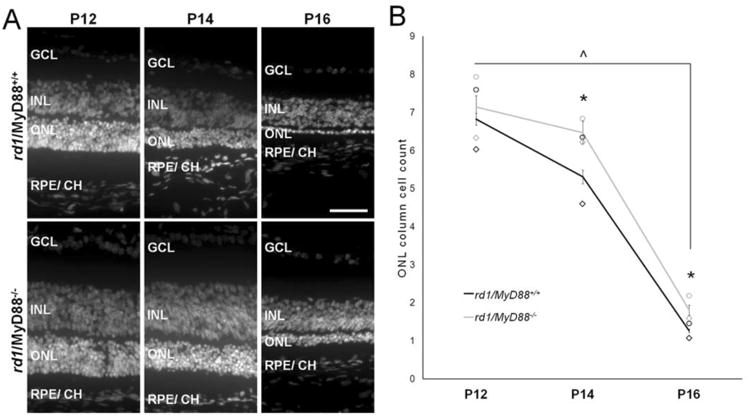
Fewer photoreceptors are lost during retinal degeneration in the rd1/MyD88-/- mouse. (A) Representative IHC sections with nuclear DAPI stain of P12 to P16 rd1/MyD88+/+ and rd1/MyD88-/-retinas revealing increased cells in the outer nuclear layer in the rd1/MyD88-/- across all ages. GCL= Ganglion Cell layer, INL= Inner Nuclear Layer, ONL= Outer Nuclear Layer, RPE/ CH= Retinal Pigment Epithelium/ Choroid. Scale bar: 50μm. (B) Average ONL column counts at P12, P14 and P16 in rd1/MyD88+/+ and rd1/MyD88-/- including averages for central (◊) and peripheral (o) counts. ONL cell number decreases in both genotypes with age, with a 4.9-fold decline from P12 to P16 in the rd1/MyD88+/+ (P12 n=6, P16 n=3, p<0.0001) and a 3.3-fold decline in rd1/MyD88-/- (P12, P16 n=3, p<0.0001). The amount of photoreceptor death was lower at P14 and P16 in the rd1/MyD88-/- mice compared with rd1/MyD88+/+, with 22% more nuclei remaining at P14 (rd1/MyD88+/+ n=4, rd1/MyD88-/-n= 3, p<0.05) and 43% more nuclei at P16 (rd1/MyD88+/+ n=3, rd1/MyD88-/- n= 3, p<0.05) in the rd1/MyD88-/- mice compared to rd1/MyD88+/+. Mean ± SEM are shown. * p<0.05, comparison between genotypes within a given time-point; ^ p<0.05, comparison among ages of the same genotype.
Retinal function is higher in the rd1/MyD88-/- mouse during degeneration
Retinal recordings in response to flashes of light, which are measured by ERG, were used to compare retinal function between the rd1/MyD88-/- and rd1/MyD88+/+ mice. In ERGs, light-stimulated a-waves represent photoreceptor hyperpolarization, while the b-waves represent photoreceptor to bipolar cell synaptic connections. The amplitudes of these waves are proportional to the number of surviving photoreceptors and are a reliable measure of retinal function (Dalke et al., 2004). Additionally, the time it takes the waves to reach their peak, known as the implicit time, is prolonged in retinal disease (Jae et al., 2013) and therefore is often used as a second measure of disease progression in rd1 mice. We used b-waves to characterize the retina response in the two genotypes because the photoreceptor a-waves were variable and close to background in the rapidly degenerating rd1 mouse retina. Analysis of ERG b-waves indicated that loss of MyD88 signaling resulted in improvement of scotopic retina response (rod-driven) at P12, with a maximal 1.46-fold increase in b-wave amplitude, and faster time-to-peak (implicit time) by 18.6 milliseconds compared with rd1/MyD88+/+ (p<0.05) (Fig. 4A). The increased scotopic b-wave amplitudes in rd1/MyD88-/- were not maintained at later time points, likely due to the aggressive nature of the rod degeneration in the rd1 mouse (Fig. 4B, 4C). In contrast, significant improvements of photopic retina responses (cone-driven) b-wave amplitudes were demonstrated in rd1/MyD88-/- mice at all time points (p<0.05) (Fig. 5A-C). To rule out the possibility that MyD88 loss itself affects the function of the retina, comparisons of adult non-degenerating C57Bl/6J mice with adult MyD88-/- mice indicated no difference in ERG responses (Fig. 6A-B). Therefore, loss of MyD88 signaling in rd1 improved retina responses to both scotopic and photopic stimuli.
Fig. 4.
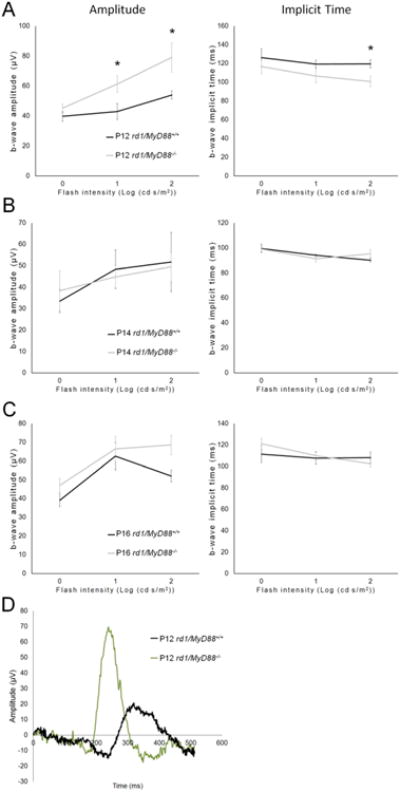
The scotopic retina response is improved at P12 in rd1 mice lacking MyD88. Graphs (A-C) show the retina response to scotopic (rod-driven) light stimuli at different flash intensities. Absence of MyD88-mediated signaling in rd1 resulted in improved retina function compared with rd1/MyD88+/+ at P12 only (rd1/MyD88+/+ n=6-7, rd1/MyD88-/- n=5). Representative ERG waveforms for each genotype at P12 are shown in panel D. Mean ± SEM are shown. * p<0.05
Fig. 5.
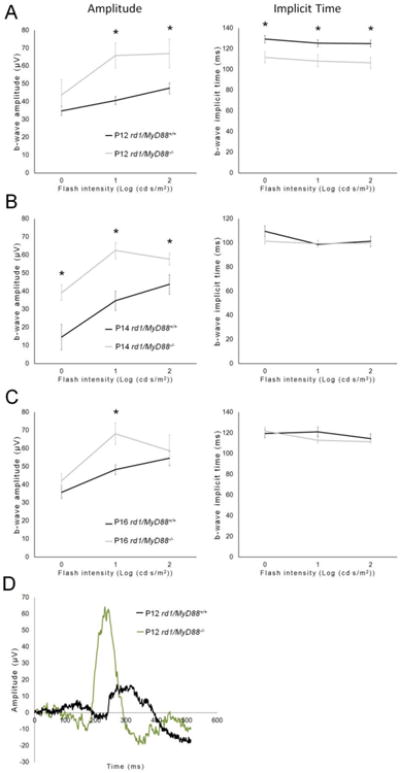
The photopic retina response is improved during retinal degeneration in rd1/MyD88-/- mice. Graphs (A-C) show the retina response to photopic (cone-driven) stimuli at different flash intensities. An improvement in retina function was demonstrated in rd1/MyD88-/- at all three time-points compared with rd1/MyD88+/+ (rd1/MyD88+/+ n=4-7, rd1/MyD88-/- n=4-10). Representative ERG waveforms for each genotype at P12 are shown in panel D. Mean ± SEM are shown. * p<0.05
Fig. 6.
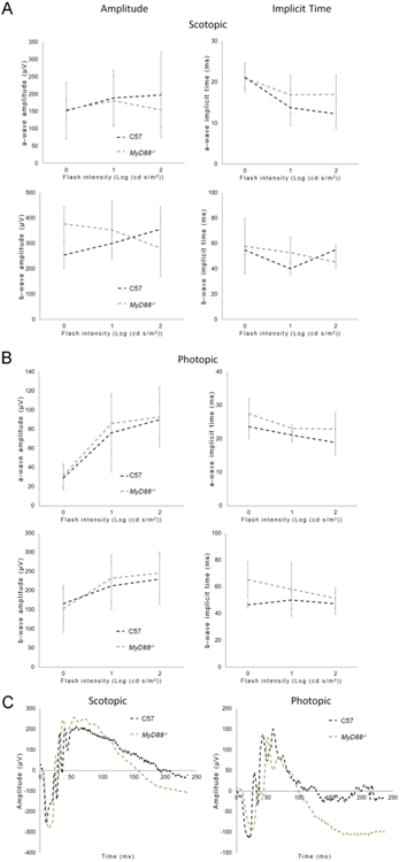
MyD88 loss in the absence of the rd1 mutation does not affect retina function. Average ERG retinal response (amplitude) to (A) scotopic and (B) photopic stimuli in adult non-degenerating C57Bl/6J mice and adult MyD88-/- mice show equivalent retina responses between the genotypes (C57 n=5, MyD88-/- n=4). (C) Representative ERG waveforms are shown. Mean ± SD are shown.
Discussion
The purpose of this study was to investigate whether MyD88-mediated signaling contributes to retinal degeneration. The immune response is known to affect disease severity, but it was unknown whether the absence of MyD88-mediated signaling, downstream of TLRs and IL-1R, would change inflammatory markers of degeneration and the extent of photoreceptor loss. This investigation is novel in that by blocking the majority of TLR and IL-1R signaling during degeneration using a genetic knockout of MyD88, we eliminated a crucial component of the innate immune response to neurodegeneration in the retina. We discovered attenuated retinal microglial recruitment during early degeneration and reduced pro-inflammatory chemokine transcript expression. Furthermore, loss of MyD88-mediated signaling resulted in increased photoreceptors, improved early scotopic retina function and improved photopic retina function. Our evidence thus suggests involvement of MyD88-mediated signaling in the inflammation-associated exacerbation of retinal degeneration during early disease.
All TLRs signal through MyD88, with the exception of TLR3 and a portion of TLR4, and are expressed throughout the retina (Kindzelskii et al., 2004). TLR4 has been localized to rod photoreceptors in wild-type and degenerating mice (Yi et al., 2012) and the expression of TLR2-9 confirmed in microglia (Jack et al., 2005), and TLR1-10 in RPE (Kindzelskii et al., 2004; Kumar et al., 2004). During non-pathogen injury such as ischemia and oxidative stress, as exhibited in retinal degeneration, TLRs and IL-1R are believed to be activated by molecules released from injured neurons early in the disease process, and induce intracellular signaling cascades that cause further photoreceptor death. TLRs are activated by molecules collectively known as damage-associated molecular pattern molecules (DAMPs) that are released from injured cells, such as Hsp70 (Vabulas et al., 2002) and HMGB1 (Lotze and Tracey, 2005; Qiu et al., 2010), and signature molecules in degenerating retinas, including oxidized lipids (Chavez-Sanchez et al., 2010; Higgins et al., 2003) and drusen components (Higgins et al., 2003; Kaarniranta and Salminen, 2009). Indeed, photoreceptor outer segments co-incubated with microglia caused their activation and subsequent pro-inflammatory chemokine production in vivo (Kohno et al., 2013). IL-1R is activated by cytokines induced during inflammation. Our findings indicate that blocking the TLR and IL-1R response to injury-associated molecules reduces but does not eliminate subsequent retinal degeneration. Our findings of reduced chemokine expression, increased photoreceptor numbers and improved retina function are supported by a recent study by Kohno et al. By knocking out a single upstream component of MyD88 signaling, the TLR4 receptor, in a light-induced retinal degeneration mouse model, Kohno et al. demonstrated reduced chemokine production and microglial activation and increased rod and cone photoreceptor numbers (Kohno et al., 2013).
IL-1R, which also functions through MyD88, is activated by the pro-inflammatory cytokines IL-1β and IL-18 and contributes to retinal degeneration (Anderson et al., 2013; Tarallo et al., 2012). IL-1β and IL-18 are cleaved from their pro-forms by another major class of innate immune receptors, the NLRP3, an important danger signal receptor that responds to signature proteins of macular degeneration as ligands, including lipofuscin component A2E (Anderson et al., 2013), drusen and drusen component complement C1Q (Doyle et al., 2012), and Alu RNA (Tarallo et al., 2012). In addition to TLR and IL-1R, MyD88 regulates a viral-response IFN-γR1 pathway independent of TLRs (Sun and Ding, 2006). However, this pathway is unlikely to be relevant in the rd1 retinal degeneration because viral infection is absent. MyD88-independent inflammatory pathways, including NF-κB and NLRP3 inflammasome activation are associated with the pathogenesis of retinal degenerations. MyD88 regulates complement and the inflammasome and it is possible that there is a compensatory change in these pathways in rd1/MyD88-/- mice. Furthermore, blocking MyD88 leaves non-MyD88 TLR pathways active (TLR3/TRIF, some TLR4 signaling), as well as other DAMP receptors (e.g. RAGE), which may contribute to retinal degeneration in the mice, and may account for the fact that we observed only a delay in degeneration instead of halting degeneration entirely.
Photoreceptor degeneration is preceded by, or accompanied by, pro-inflammatory C-C motif chemokine (Ccl) induction. Increased retinal Ccl2, Ccl3, Ccl4 and Ccl12 were detected in mouse models of AMD and light-induced retinal degeneration (Kohno et al., 2014) and increased Ccl2-Ccl5 and Ccl7 expression were observed prior to and during photoreceptor loss in the rd1 mouse (Zeng et al., 2005). Additionally, in retinal degeneration 10 (rd10) mice that genetically lacked CCR2, there was improved retinal function and structure (Guo et al., 2012), suggesting that pro-inflammatory chemokines are active participants in the pathology of retinal degeneration. Expression of Ccl2, Ccl4, Ccl5, Ccl7 and Cxcl10 and TNF-α transcripts were reduced in rd1/MyD88-/- compared with rd1/MyD88+/+ mice, and maximal expression levels attained in the rd1/MyD88-/- mice did not reach those of rd1/MyD88+/+ at the time-points analyzed. However, the fact that chemokines are induced at all in the rd1/MyD88-/- mouse also implicates MyD88-independent pathways. Certainly, administration of TLR4 ligand LPS to MyD88-deficient cells induced several cytokines such as Ccl3, Ccl4, and TNF-α (Covert et al., 2005). Additionally, TLR-mediated activation of transcription factor NF-κB is biphasic and occurs first via the MyD88-dependent followed by the MyD88-independent TRIF pathway (Covert et al., 2005; Yamamoto et al., 2003), therefore highlighting a potential mechanism for the delayed peak chemokine expression exhibited in the rd1/MyD88-/- mouse.
TNF-α is known to be cytotoxic to neurons (Neniskyte et al., 2014) and exacerbates photoreceptor death in retinal degeneration (de Kozak et al., 1997; Nakazawa et al., 2011). TNF-α produced by primary retinal microglia isolated from the RCS rat retinal degeneration model was toxic to photoreceptors in vitro (de Kozak et al., 1997). In fact, microglia and indeed photoreceptors themselves have been shown to produce TNF-α in response to stimulation by each other in vivo, via microglia stimulated by degenerating photoreceptors (Zeng et al., 2005), and photoreceptors co-incubated with activated microglia (Yang et al., 2007). As in previous studies (Zeiss and Johnson, 2004), TNF-α expression in the rd1/MyD88+/+ retina peaked at P12 prior to maximal photoreceptor death and reduced thereafter, whereas expression in the rd1/MyD88-/- mouse at the same time-point was 4.4-fold lower, and remained 2- to 3-fold lower compared with rd1/MyD88+/+ at further time-points. Therefore, reduced TNF-α expression as a result of absent MyD88-mediated signaling may contribute to functional and cellular photoreceptor protection in rd1/MyD88-/-.
Chemokines released secondary to tissue injury are chemotractants for inflammatory cells, most prominently, the microglia. These cells serve not only as phagocytes clearing cellular debris, but also as neurotoxic substance secretors in the CNS (Boje and Arora, 1992) and retina (de Kozak et al., 1997; Roque et al., 1999; Zeng et al., 2005), provoking further neuronal loss in both settings. Maximal expression of the chemotractant chemokines precedes microglial recruitment to the outer retina during retinal degeneration (Zeng et al., 2005), a phenomenon also exhibited in our rd1/MyD88+/+ mice. However, although reduced chemokine expression and microglial recruitment was observed at P12 in rd1/MyD88-/-, this reduction did not translate to attenuated microglial recruitment at P14. While MyD88-mediated signaling influences chemokine expression during retinal degeneration, it does not affect the microglial recruitment that follows. An explanation for normal microglial recruitment at P14 lies in the possibility that despite the 2- to 5-fold decline in chemokine expression in rd1/MyD88-/- at P12, a minimal cytokine threshold required for microglial recruitment and activation was reached nonetheless. In addition, microglial activation secondary to TLR4 ligand LPS in MyD88-deficient mice was demonstrated by Esen et al. (Esen and Kielian, 2006), raising the possibility that MyD88-independent factors and alternative molecules may also play a role in their recruitment.
Chemokine expression and microglial recruitment precede maximal photoreceptor loss (Zeng et al., 2005), and our trends support this, with the biggest drop of photoreceptor nuclei occurring between P14 and P16. In addition, the rate of photoreceptor nuclei loss between P14 to P16 in rd1/MyD88-/- caught up to that of rd1/MyD88+/+ mice, possibly a reflection of the effect of higher outer retinal microglial recruitment at P14 in rd1/MyD88-/-. By knocking out MyD88-mediated signaling, we have managed to dampen the initial inflammatory response to dying photoreceptors, preserving photoreceptors and conveying not only a protective effect on scotopic function of the retina during early degeneration, but interestingly, also improved photopic function, which represents the vulnerable bystanders, the cone photoreceptors. The rd1 mutation affects rod photoreceptors primarily, however cone photoreceptor loss also follows, and although the exact mechanism is unknown, a suggested cause is noxious stimuli produced by the inflammatory response mounted against degenerating rod photoreceptors (Zeng et al., 2005). The improved scotopic function in rd1/MyD88-/- does not extend past P12 perhaps because the rod photoreceptors that initiate the scotopic response eventually succumb to the ongoing pathology resulting from the aggressive rod PDE6β gene mutation. Because cone photoreceptor loss is known to occur after P18 in the rd1 mouse (Carter-Dawson et al., 1978), the improvement of photopic retina function, which is primarily mediated by cones, in rd1/MyD88-/- thus appears to occur prior to photoreceptor loss. Furthermore, photopic b-wave amplitudes do not reduce between P12 to P16 in the rd1/MyD88+/+ mouse, suggesting no functional deterioration with age. It is also possible that the delayed immune response may preserve the function of the inner retina only, and the outer retina (the photoreceptors) is only preserved at the structural and not functional level.
In addition to microglia and photoreceptors, Muller glia and RPE also express TLRs and signal via MyD88 (Jack et al., 2005; Kindzelskii et al., 2004; Kumar et al., 2004; Yi et al., 2012). Future avenues for exploration would investigate how MyD88 inhibition in each cell type, particularly RPE, would affect their role in the recognition of endogenous danger signals and the subsequent initiation of inflammation in retinal degeneration. In addition, TLR-independent MyD88-mediated signaling in the RPE plays a role in pathogenesis of retinal degeneration (Kerur et al., 2013; Tarallo et al., 2012). Alu RNA accumulation in RPE is implicated in dry AMD and stimulated TLR-independent NF-κB activation and subsequent NLRP3 inflammasome activation, which lead to IL-18 maturation and subsequent release. IL-18-induced IL-1R MyD88-mediated signaling in the RPE resulted in RPE loss and subsequent photoreceptor death (Kerur et al., 2013). Interestingly, TLR4 signaling regulates complement (Song, 2012), and activates the NLRP3 inflammasome in RGCs after injury (Qi et al., 2014), indicating interactions among MyD88-dependent and -independent pathways. Additional studies would indicate whether MyD88-dependent and -independent pathways cross-talk in the RPE to regulate retinal degeneration.
In this study, we used the rd1 retinal degeneration mouse model because the inflammatory response has been well characterized in the literature. Rd1 is an aggressive degeneration model with rapid photoreceptor loss that begins prior to retinal maturation (Gibson et al., 2013), and was chosen because its inflammatory profile and rate of degeneration is well established. Future avenues will investigate whether protection from lack of MyD88 would also be observed in models of less severe inherited retinal degeneration. Because pathways that regulate photoreceptor death in inherited retinal degenerations are often also implicated in the pathogenesis of AMD, these findings provide insight into potential therapeutic strategies for dry AMD.
Conclusion
In summary, knocking out innate immunity MyD88-mediated signaling in an aggressive inherited retinal degeneration mouse model dampens the initial immune response, which results in a protective delay in loss of the scotopic retina response, and improvement in photopic retina function. This study highlights the destructive role of TLR/IL-1R MyD88 mediated inflammatory pathways in neurodegenerative tissue injury.
Highlights.
Knocking out MyD88-dependent TLR/IL-1R signaling in retinal degeneration reduces retinal proinflammatory chemokine and TNF-α mRNA expression
Absence of MyD88-mediated signaling leads to attenuated microglial recruitment during early degeneration but not at later time-points.
Dampened immune response in the absence of MyD88 leads to an improvement in retina function.
Therefore, TLR/IL-1R MyD88 mediated inflammatory pathways play a destructive role in neurodegenerative tissue injury in the retina.
Acknowledgments
We would like to thank BaoXiang Li and Hany Azcuy for technical support, William J. Feuer for helpful discussions and statistical analyses, as well as Drs. Dmitiry V. Ivanov and Ali M. Saeed for reagents. This work was supported by the Karl Kirchgessner Foundation and a Research to Prevent Blindness Ernest & Elizabeth Althouse Special Scholar Award. Institutional support to BPEI was received from a Research to Prevent Blindness Unrestricted Grant and NEI Center Core Grant P30 EY014801
List of Abbreviations
- AMD
age-related macular degeneration
- Ccl
chemokine (C-C motif) ligand
- Cxcl10
chemokine (C-X-C motif) ligand 10
- HMGB1
High-mobility group protein B1
- Hsp70
70 kilodalton heat shock protein
- IFN-γ
interferon- γ
- IL-1R
interleukin-1 receptor
- IRAK
Interleukin-1 receptor-associated kinase
- MAPK
mitogen-activated protein kinase
- MyD88
myeloid differentiation primary response gene 88
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NLRP3
NOD-like receptor family, pyrin domain containing 3
- PDE6β
phosphodiesterase β-subunit
- RPE
retinal pigment epithelium
- TLR
Toll-like receptor
- TNF-α
tumor necrosis factor-α
- rd1
retinal degeneration 1.
Footnotes
Competing Interests: The authors declare they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson OA, et al. A2E induces IL-1ss production in retinal pigment epithelial cells via the NLRP3 inflammasome. PLoS One. 2013;8:e67263. doi: 10.1371/journal.pone.0067263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boje KM, Arora PK. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992;587:250–6. doi: 10.1016/0006-8993(92)91004-x. [DOI] [PubMed] [Google Scholar]
- Bowes C, et al. Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature. 1990;347:677–80. doi: 10.1038/347677a0. [DOI] [PubMed] [Google Scholar]
- Carter-Dawson LD, et al. Differential effect of the rd mutation on rods and cones in the mouse retina. Invest Ophthalmol Vis Sci. 1978;17:489–98. [PubMed] [Google Scholar]
- Caso JR, et al. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599–608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- Chang B, et al. Retinal degeneration mutants in the mouse. Vision Res. 2002;42:517–25. doi: 10.1016/s0042-6989(01)00146-8. [DOI] [PubMed] [Google Scholar]
- Chavez-Sanchez L, et al. The activation of CD14, TLR4, and TLR2 by mmLDL induces IL-1beta, IL-6, and IL-10 secretion in human monocytes and macrophages. Lipids Health Dis. 2010;9:117. doi: 10.1186/1476-511X-9-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covert MW, et al. Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science. 2005;309:1854–7. doi: 10.1126/science.1112304. [DOI] [PubMed] [Google Scholar]
- Dalke C, et al. Electroretinography as a screening method for mutations causing retinal dysfunction in mice. Invest Ophthalmol Vis Sci. 2004;45:601–9. doi: 10.1167/iovs.03-0561. [DOI] [PubMed] [Google Scholar]
- de Kozak Y, et al. Tumor necrosis factor and nitric oxide production by resident retinal glial cells from rats presenting hereditary retinal degeneration. Ocul Immunol Inflamm. 1997;5:85–94. doi: 10.3109/09273949709085056. [DOI] [PubMed] [Google Scholar]
- Donoso LA, et al. The role of inflammation in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2006;51:137–52. doi: 10.1016/j.survophthal.2005.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle SL, et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med. 2012;18:791–8. doi: 10.1038/nm.2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elner SG, et al. TLR4 mediates human retinal pigment epithelial endotoxin binding and cytokine expression. Trans Am Ophthalmol Soc. 2005;103:126–35. discussion 135-7. [PMC free article] [PubMed] [Google Scholar]
- Esen N, Kielian T. Central role for MyD88 in the responses of microglia to pathogen-associated molecular patterns. J Immunol. 2006;176:6802–11. doi: 10.4049/jimmunol.176.11.6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto T, et al. Choroidal Neovascularization Enhanced by Chlamydia pneumoniae via Toll-like Receptor 2 in the Retinal Pigment Epithelium. Invest Ophthalmol Vis Sci. 2010;51:4694–702. doi: 10.1167/iovs.09-4464. [DOI] [PubMed] [Google Scholar]
- Gibson R, et al. Functional and neurochemical development in the normal and degenerating mouse retina. J Comp Neurol. 2013;521:1251–67. doi: 10.1002/cne.23284. [DOI] [PubMed] [Google Scholar]
- Glass CK, et al. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–34. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, et al. Knockout of ccr2 alleviates photoreceptor cell death in a model of retinitis pigmentosa. Exp Eye Res. 2012;104:39–47. doi: 10.1016/j.exer.2012.08.013. [DOI] [PubMed] [Google Scholar]
- Gupta N, et al. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp Eye Res. 2003;76:463–71. doi: 10.1016/s0014-4835(02)00332-9. [DOI] [PubMed] [Google Scholar]
- Hackam AS, et al. Identification of gene expression changes associated with the progression of retinal degeneration in the rd1 mouse. Invest Ophthalmol Vis Sci. 2004;45:2929–42. doi: 10.1167/iovs.03-1184. [DOI] [PubMed] [Google Scholar]
- Hageman GS, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102:7227–32. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins GT, et al. Induction of angiogenic cytokine expression in cultured RPE by ingestion of oxidized photoreceptor outer segments. Invest Ophthalmol Vis Sci. 2003;44:1775–82. doi: 10.1167/iovs.02-0742. [DOI] [PubMed] [Google Scholar]
- Hou B, et al. Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity. 2008;29:272–82. doi: 10.1016/j.immuni.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SH, et al. Autosomal recessive retinitis pigmentosa caused by mutations in the alpha subunit of rod cGMP phosphodiesterase. Nat Genet. 1995;11:468–71. doi: 10.1038/ng1295-468. [DOI] [PubMed] [Google Scholar]
- Jack CS, et al. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175:4320–30. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- Jae SA, et al. Electrophysiological and Histologic Evaluation of the Time Course of Retinal Degeneration in the rd10 Mouse Model of Retinitis Pigmentosa. Korean J Physiol Pharmacol. 2013;17:229–35. doi: 10.4196/kjpp.2013.17.3.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaarniranta K, Salminen A. Age-related macular degeneration: activation of innate immunity system via pattern recognition receptors. J Mol Med. 2009;87:117–23. doi: 10.1007/s00109-008-0418-z. [DOI] [PubMed] [Google Scholar]
- Kaczorowski DJ, et al. Early events in the recognition of danger signals after tissue injury. J Leukoc Biol. 2008;83:546–52. doi: 10.1189/jlb.0607374. [DOI] [PubMed] [Google Scholar]
- Kerur N, et al. TLR-independent and P2X7-dependent signaling mediate Alu RNA-induced NLRP3 inflammasome activation in geographic atrophy. Invest Ophthalmol Vis Sci. 2013;54:7395–401. doi: 10.1167/iovs.13-12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandhadia S, et al. Age-related macular degeneration and the complement system. Immunobiology. 217:127–46. doi: 10.1016/j.imbio.2011.07.019. [DOI] [PubMed] [Google Scholar]
- Kilic U, et al. TLR-4 deficiency protects against focal cerebral ischemia and axotomy-induced neurodegeneration. Neurobiol Dis. 2008;31:33–40. doi: 10.1016/j.nbd.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Kindzelskii AL, et al. Toll-like receptor 4 (TLR4) of retinal pigment epithelial cells participates in transmembrane signaling in response to photoreceptor outer segments. J Gen Physiol. 2004;124:139–49. doi: 10.1085/jgp.200409062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, et al. Inflammation, complement factor h, and age-related macular degeneration: the Multi-ethnic. Study of Atherosclerosis. Ophthalmology. 2008;115:1742–9. doi: 10.1016/j.ophtha.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko MK, et al. The role of TLR4 activation in photoreceptor mitochondrial oxidative stress. Invest Ophthalmol Vis Sci. 2011;52:5824–35. doi: 10.1167/iovs.10-6357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno H, et al. Photoreceptor proteins initiate microglial activation via Toll-like receptor 4 in retinal degeneration mediated by all-trans-retinal. J Biol Chem. 2013;288:15326–41. doi: 10.1074/jbc.M112.448712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno H, et al. CCL3 production by microglial cells modulates disease severity in murine models of retinal degeneration. J Immunol. 2014;192:3816–27. doi: 10.4049/jimmunol.1301738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokkinopoulos I, et al. Toll-like receptor mRNA expression patterns in human dendritic cells and monocytes. Mol Immunol. 2005;42:957–68. doi: 10.1016/j.molimm.2004.09.037. [DOI] [PubMed] [Google Scholar]
- Kumar MV, et al. Innate immunity in the retina: Toll-like receptor (TLR) signaling in human retinal pigment epithelial cells. J Neuroimmunol. 2004;153:7–15. doi: 10.1016/j.jneuroim.2004.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnardt S, et al. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A. 2003;100:8514–9. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, et al. CNTF induces regeneration of cone outer segments in a rat model of retinal degeneration. PLoS One. 2010;5:e9495. doi: 10.1371/journal.pone.0009495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JE, et al. MyD88 deficiency ameliorates beta-amyloidosis in an animal model of Alzheimer's disease. Am J Pathol. 2011;179:1095–103. doi: 10.1016/j.ajpath.2011.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–42. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- Maloney SC, et al. Choroidal neovascular membranes express toll-like receptor 3. Ophthalmic Res. 2010;44:237–41. doi: 10.1159/000313989. [DOI] [PubMed] [Google Scholar]
- Marsh BJ, et al. Toll-like receptor signaling in endogenous neuroprotection and stroke. Neuroscience. 2009;158:1007–20. doi: 10.1016/j.neuroscience.2008.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur S, et al. Extracellular heat shock protein 70 induces cardiomyocyte inflammation and contractile dysfunction via TLR2. Circ J. 2011;75:2445–52. doi: 10.1253/circj.cj-11-0194. [DOI] [PubMed] [Google Scholar]
- Michon JJ, et al. A comparative study of methods of photoreceptor morphometry. Invest Ophthalmol Vis Sci. 1991;32:280–4. [PubMed] [Google Scholar]
- Nakazawa T, et al. Tumor necrosis factor-alpha mediates photoreceptor death in a rodent model of retinal detachment. Invest Ophthalmol Vis Sci. 2011;52:1384–91. doi: 10.1167/iovs.10-6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neniskyte U, et al. Tumour necrosis factor alpha-induced neuronal loss is mediated by microglial phagocytosis. FEBS Lett. 2014 doi: 10.1016/j.febslet.2014.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng TF, Streilein JW. Light-induced migration of retinal microglia into the subretinal space. Invest Ophthalmol Vis Sci. 2001;42:3301–10. [PubMed] [Google Scholar]
- Okun E, et al. Toll-like receptors in neurodegeneration. Brain Res Rev. 2009;59:278–92. doi: 10.1016/j.brainresrev.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AK, Hackam AS. A novel protective role for the innate immunity Toll-Like Receptor 3 (TLR3) in the retina via Stat3. Mol Cell Neurosci. 2014;63C:38–48. doi: 10.1016/j.mcn.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, et al. Retinal ischemia/reperfusion injury is mediated by Toll-like receptor 4 activation of NLRP3 inflammasomes. Invest Ophthalmol Vis Sci. 2014 doi: 10.1167/iovs.14-14380. [DOI] [PubMed] [Google Scholar]
- Qiu J, et al. High-mobility group box 1 promotes metalloproteinase-9 upregulation through Toll-like receptor 4 after cerebral ischemia. Stroke. 2010;41:2077–82. doi: 10.1161/STROKEAHA.110.590463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roque RS, et al. Microglial cells invade the outer retina as photoreceptors degenerate in Royal College of Surgeons rats. Invest Ophthalmol Vis Sci. 1996;37:196–203. [PubMed] [Google Scholar]
- Roque RS, et al. Retina-derived microglial cells induce photoreceptor cell death in vitro. Brain Res. 1999;836:110–9. doi: 10.1016/s0006-8993(99)01625-x. [DOI] [PubMed] [Google Scholar]
- Rutar M, et al. Small interfering RNA-mediated suppression of Ccl2 in Muller cells attenuates microglial recruitment and photoreceptor death following retinal degeneration. J Neuroinflammation. 2012;9:221. doi: 10.1186/1742-2094-9-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiose S, et al. Toll-like receptor 3 is required for development of retinopathy caused by impaired all-trans-retinal clearance in mice. J Biol Chem. 2011;286:15543–55. doi: 10.1074/jbc.M111.228551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AK, et al. Lithium chloride regulates the proliferation of stem-like cells in retinoblastoma cell lines: a potential role for the canonical Wnt signaling pathway. Mol Vis. 2010;16:36–45. [PMC free article] [PubMed] [Google Scholar]
- Song WC. Crosstalk between complement and toll-like receptors. Toxicol Pathol. 2012;40:174–82. doi: 10.1177/0192623311428478. [DOI] [PubMed] [Google Scholar]
- Sun D, Ding A. MyD88-mediated stabilization of interferon-gamma-induced cytokine and chemokine m. RNA Nat Immunol. 2006;7:375–81. doi: 10.1038/ni1308. [DOI] [PubMed] [Google Scholar]
- Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Tang J, et al. MyD88-dependent pathways in leukocytes affect the retina in diabetes. PLoS One. 2013;8:e68871. doi: 10.1371/journal.pone.0068871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SC, et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A. 2007;104:13798–803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SC, et al. Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid beta-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Exp Neurol. 2008;213:114–21. doi: 10.1016/j.expneurol.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarallo V, et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell. 2012;149:847–59. doi: 10.1016/j.cell.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabulas RM, et al. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem. 2002;277:15107–12. doi: 10.1074/jbc.M111204200. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–3. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- Yang LP, et al. A possible mechanism of microglia-photoreceptor crosstalk. Mol Vis. 2007;13:2048–57. [PubMed] [Google Scholar]
- Yi H, et al. Novel role for the innate immune receptor Toll-like receptor 4 (TLR4) in the regulation of the Wnt signaling pathway and photoreceptor apoptosis. PLoS One. 2012;7:e36560. doi: 10.1371/journal.pone.0036560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida N, et al. Laboratory evidence of sustained chronic inflammatory reaction in retinitis pigmentosa. Ophthalmology. 2013;120:e5–12. doi: 10.1016/j.ophtha.2012.07.008. [DOI] [PubMed] [Google Scholar]
- Yu M, et al. A novel role of complement in retinal degeneration. Invest Ophthalmol Vis Sci. 2012;53:7684–92. doi: 10.1167/iovs.12-10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeiss CJ, Johnson EA. Proliferation of microglia, but not photoreceptors, in the outer nuclear layer of the rd-1 mouse. Invest Ophthalmol Vis Sci. 2004;45:971–6. doi: 10.1167/iovs.03-0301. [DOI] [PubMed] [Google Scholar]
- Zeng HY, et al. Identification of sequential events and factors associated with microglial activation, migration, and cytotoxicity in retinal degeneration in rd mice. Invest Ophthalmol Vis Sci. 2005;46:2992–9. doi: 10.1167/iovs.05-0118. [DOI] [PubMed] [Google Scholar]
- Zhang C, et al. Activation of microglia and chemokines in light-induced retinal degeneration. Mol Vis. 2005;11:887–95. [PubMed] [Google Scholar]
- Zhu Y, et al. Increase in peripheral blood mononuclear cell Toll-like receptor 2/3 expression and reactivity to their ligands in a cohort of patients with wet age-related macular degeneration. Mol Vis. 2013;19:1826–33. [PMC free article] [PubMed] [Google Scholar]