

Abstract
Artemisinins are the corner stone of anti-malarial drugs1. Emergence and spread of resistance to them2–4 raises risk of wiping out recent gains achieved in reducing world-wide malaria burden and threatens future malaria control and elimination on a global level. Genome wide association studies (GWAS) have revealed parasite genetic loci associated with artemisinin resistance5–10. However, there is no consensus on biochemical targets of artemisinin. Whether and how these targets interact with genes identified by GWAS, remains unknown. Here we provide biochemical and cellular evidence that artemisinins are potent inhibitors of Plasmodium falciparum phosphatidylinositol-3-kinase (PfPI3K), revealing an unexpected mechanism of action. In resistant clinical strains, increased PfPI3K was associated with the C580Y mutation in P. falciparum Kelch13 (PfKelch13), a primary marker of artemisinin resistance. Polyubiquitination of PfPI3K and its binding to PfKelch13 were reduced by PfKelch13 mutation, which limited proteolysis of PfPI3K and thus increased levels of the kinase as well as its lipid product phosphatidylinositol 3-phosphate (PI3P). We find PI3P levels to be predictive of artemisinin resistance in both clinical and engineered laboratory parasites as well as across non-isogenic strains. Elevated PI3P induced artemisinin resistance in absence of PfKelch13 mutations, but remained responsive to regulation by PfKelch13. Evidence is presented for PI3P-dependent signaling, where transgenic expression of an additional kinase confers resistance. Together these data present PI3P as the key mediator of artemisinin resistance and the sole PfPI3K as an important target for malaria elimination.
Our prior work identified an important role for PI3P in protein export from the P. falciparum endoplasmic reticulum (ER) to the erythrocyte, at the early ‘ring’ stage of blood-infection11. Consequently, a secretory reporter that binds PI3P remains in the ring ER, but in absence of PI3P, undergoes default secretion to the parasitophorous vacuole (PV). This yielded a cell-based screen for drugs that inhibit PI3P production (Fig. 1a). We were particularly interested in artemisinins because clinical resistance to them develops at the early ring stage3. Low nanomolar concentrations of dihydroartemisinin (DHA), the active form of all artemisinins block production of PI3P (Fig. 1a). This effect is fast acting (within 30 min), reversed by washing out the drug and without effect on subsequent parasite growth (Extended Data Fig. 1a). Wortmannin or LY294002, active against the sole parasite PfPI3K12,13, but not the inactive LY303511 blocked PI3P production. Artemisinin and artesunate were also inhibitory (Extended Data Fig. 1b, c), but deoxyartemisinin, anti-folates and aminoquinolines had no effect (Fig. 1a and Extended Data Fig. 1b–e). Biochemical analyses confirmed that DHA reduced mass PI3P levels (and drug washout restored PI3P levels; Fig. 1b). Quantitative inhibition of immunopurified PfPI3K was achieved by 4 nM DHA but not by deoxyartemisinin (Fig. 1c). DHA at 10 μM failed to significantly inhibit 46 mammalian kinases, including its closest human orthologue VPS34 (a class III kinase; Fig. 1d, Extended Data Table 1) strongly supporting that DHA is not a promiscuous kinase inhibitor.
Figure 1. DHA targets PfPI3K.

a, SS-EEA1WT-mCherry detects ring PI3P in punctate (ER) domains11. Mutant SS-EEA1R1374A-mCherry secretes to the PV (second row; 11). 4 nM DHA redistributes SS-EEA1WT-mCherry to the PV. Washout restores ER-PI3P. 4 nM deoxyartemisinin, no effect. Blue, nucleus; scale, 5 μm; P, parasite; E, Erythrocyte. Mean (SD) with three experimental replicates with image analysis from 400 optical sections. b-d, Effects of DHA on (b) PI3P mass (c) immunopurified PfPI3K (raw data in Supplementary Data 2) and (d) mammalian PI3Kinases. Mean from three experimental replicates (each with triplicate data points). For (b), SD < 3; (c) upper graph, SD < 1.5; lower graph SD < 5; (d) SD < 0.5. e, Overlay of the model of PfPI3K (cyan) and human class III PI3K VPS34 (grey, pdb code 3IHY) with active site marked (asterisk). f, DHA in PfPI3K model (cyan) binding site. g, Surface representation of f. Additional details in Extended Data Figs. 1–3.
Extended Data Table 1.
Percentage inhibition of mammalian kinases by 10 μM DHA.
| Kinase | % Inh | Kinase | % Inh | Kinase | % Inh | Kinase | % Inh |
|---|---|---|---|---|---|---|---|
| ABL1 T3151 | −2 | FGFR3 K650E | −2 | LCK | 1 | PRKACA (PKA) | 2 |
| ALK | −1 | FGFR4 | 4 | MAP3K8 (COT) | 3 | PRKCA (PKC alpha) | −2 |
| AKT1 (PKB alpha) | 3 | FRAP1 (mTOR) | 3 | MAPK1 (ERK2) | 2 | PRKCQ (PKC theta) | −3 |
| AURKA (Aurora A) | 3 | GSK3B (GSK3 beta) | 0 | MAPK14 (p38 alpha) | 1 | RET | 3 |
| AXL | −1 | IGF1R | 2 | MAPKAPK2 | 5 | ROCK2 | 1 |
| BTK | −1 | INSR | 4 | MET (cMet) | 2 | SYK | −2 |
| CDK2/cyclin A | 1 | JAK1 | 0 | PDGFRA V561D | −1 | TYK2 | 5 |
| EGFR (ErbB1) | −1 | JAK2 | 1 | PDK1 Direct | −1 | ZAP70 | 1 |
| EPHA4 | 2 | JAK3 | −4 | PI4KB (PI4K beta) | −13 | ||
| EPHB4 | 1 | KDR (VEGFR2) | 0 | PKN1 (PRK1) | 0 | ||
| ERBB2 (HER2) | −4 | KIT | −1 | PKN2 (PRK2) | −3 | ||
SD <0.5.
In the absence of a crystal structure of PfPI3K, a computational model can provide structural hypotheses to help understand experimental results. We therefore used homology modeling of PfPI3K and 20 ns molecular dynamics (MD) simulations14 to study the binding of DHA and several structural analogues (Fig. 1e–g, Supplementary Data 1, Extended Data Fig.2 and Extended Data Fig. 3a). The model suggests polar contacts of DHA with the D1889 hydroxyl and the Y1915 lactol oxygen of PfPI3K at the binding site (Fig. 1f) as well as an excellent shape complementarity of DHA with PfPI3K at its hydrophobic region (Fig. 1g). This is in agreement with the rapid inhibition of PfPI3K by DHA at nanomolar concentrations. MD simulation also rationalize the experimentally observed lack of inhibition of VPS34, mammalian class I or class II PI3Kinases by DHA (Extended Data Fig. 3b–d). Artesunate (at high 10 μM concentrations) has been reported to inhibit activities of the PI3K-AKT pathway in mammalian systems15–17. However, whether this was due to direct inhibition of PI3K or AKT, the DHA metabolite or other indirect effects (such as high concentration that far exceed inhibitory concentrations for malaria parasites) was not studied.
Together the data in Fig. 1 suggest that DHA specifically targets PfPI3K. Yet GWAS studies suggest that >1000 genes (including PfPI3K) show clinical resistance to artemisinins6; moreover PfPI3K polymorphisms are not detected in all resistant strains. Rather, there is selective pressure in regions of Chromosome 135,9 and in particular pfkelch137,8,10 but the mechanism is unknown. The mammalian orthologue of PfKelch13 functions is a substrate adapter for an E3 ubiquitin ligase18. The putative substrate binding domain of PfKelch13 shows characteristic ‘Kelch’ propeller domains7,18 (Fig. 2a) mutations in which associate with artemisinin resistance7,8,10. We hypothesized that mutations may decrease affinity for a protein substrate, thereby increasing its steady state levels by reducing ubiquitination and (proteosomal) degradation.
Figure 2. Proteostatic regulation of PfPI3K by PfKelch13.

a, PfKelch13 ‘Kelch’ propeller domains. b, PfPI3K, PfKelch13 polymorphisms, PfPI3K levels in clinical (ANL) resistant (2, 4, and 9) and sensitive (1, 7) strains and laboratory strain 3D7; loading control PfFKBP. ANL-7, contaminated with resistance mutation R539T. c–d, Western blots show PfPI3K in NF54-PfKelch13C580Y and NF54-PfKelch13WT,; 3D7-PfKelch13C580Y-HA and 3D7-PfKelch13WT-HA; loading controls, BiP, PfFKBP. e, Panels: (Top left) PfKelch13 (arrow) binding to PfPI3K is reduced by PfKelch13C580Y. In the PfKelch13WT background, IP PfPI3K displays K48-ubiquitination (top center, arrow), atypical poly-ubiquitination (lower left, *) and degradation (lower right), all reduced by PfKelch13C580Y. K63-ubiquitination was not observed (top right). IP Lysate input PfFKBP. f, PfKelch13 but not PfKelch13C580Y binds PfPI3K (arrow). Loading control, PfFKBP. Molecular weights, in kDa. In b-f, data are representative of three experimental replicates. g, A model for Fig. 2. Additional details in Extended Data Fig. 4. Raw data for b-f, in Supplementary Data 2.
We therefore queried whether changes in PfPI3K levels were associated with PfKelch13 mutations in clinical and engineered laboratory strains. Analysis of clinical isolates from Cambodia19 (also see Methods) suggested ~ 1.5 to 2 fold increase in PfPI3K levels in resistant versus sensitive strains (Fig. 2b). One sensitive clinical strain (ANL-7) contained the resistance mutation R539T: genome sequencing revealed it was not clonal suggesting contamination with a resistant parasite strain (data not shown). The PfKelch13 mutation C580Y was not present in clinical sensitive strains but detected in two of three resistant strains. C580Y was also recognized to be the most prevalent mutation in resistant populations7,8 and thus investigated further in two distinct laboratory strains. We utilized parasites with chromosomally inserted PfKelch13C580Y in the P. falciparum NF5420 (Extended Data Fig. 4a). Additionally, we expressed a HA-tagged form of PfKelch13C580Y in a second strain 3D7 (Extended Data Fig. 4b, Extended Data Table 2). Both mutated strains showed 2 to 3-fold increase in levels of PfPI3K relative to their PfKelch13WT counterparts (Fig. 2c, d) without changes in levels of PfKelch13 (Extended Data Fig. 4a, c).
Extended Data Table 2.
Primers used for cloning.
| Primer | Sequence(5′–3′) |
|---|---|
| PfKelch13AvrIIF | AGATCTCCTAGGATGGAAGGAGAAAAAGTAAAAACAAAAGCAAATAG |
| PfKelch13HAXhoIR | GGATCCCTCGAGTTAAGCATAATCTGGAACATCATATGGATAAGCAGCTGCTATATTTGCTATTAAAACGGAGTGACCAAATCTGG |
| C580Y reverse | TTATTATCAAAAGCAACATACATAGCTGATGATCTAGG |
| C580Y forward | CCTAGATCATCAGCTATGTATGTTGCTTTTGATAATAA |
| HsVps34AvrIIF | GGATCCCCTAGGATGGGGGAAGCAGAGAAGTTTCACTACATC |
| HsVps34mycXhoIR | GGATCCCTCGAGTTATAAATCTTCTTCAGATATTAATTTTTGTTCAGCTGCTGCTTTTCTCCAGTACTGGGCAAACTTGTGAATC |
| Vps34–742AAA745F | GGAGTTGGAGCTGCAGCACTGGATAACCTTTTGCTAACAAAAACAGGCAAACTC |
| Vps34–742AAA745R | GTTATCCAGTGCTGCAGCTCCAACTCCAAGTATATAGGTGATCACGCAATATCC |
| AKTAvrII_F | GTCCTAGGATGAATCATATAAACTTTTTCAAATG |
| AKTBgIII_R | GTAAGATCTTTTTTGTTGACCTGAAAATTCATAATAG |
| Kelch13_F | ATGGAAGGAGAAAAAGTAAAAACAAAAG |
| Kelch13_R | TATATTTGCTATTAAAACGGAGTGACC |
| Kelch13-1 | ATGGAAGGAGAAAAAGTAAAAAC |
| Kelch13-2 | GATATGAGTGTATTAGATTCGAAC |
| Kelch13-3 | GATACTTATGAAAAGAAAATTATTG |
| Kelch13-4 | GAAAAAGAAAAATATTATCAAGAA |
| Kelch13-5 | CAACAATGCTGGCGTATGTGTACACC |
| Kelch13-6 | GATAATAAAATTTATGTCATTGGTGG |
| PfPI3K-I682T-F | CCTCATAGTACGAATAATACAAATCTCCAATCC |
| PfPI3K-I682T-R | ATATATTAATTCTTTATTAAAATTAAGGAAATTCAAAATCATGTATCC |
| PfPI3K-Q431K-F | AACCTTTTCATATGATAACAATCCAAACAATATC |
| PfPI3K-Q431K-R | TTTTCTTTTTATATGTCTTATAATTGTAACGACTGG |
| PfPI3K-Y1330C-F | CCAAGAATAAAACAGACAGAATACAAAATAAAACC |
| PfPI3K-Y1330C-F | CTCATAGGAGTATCCAGCGAATTTTCC |
Further analysis revealed that immunopurified (IP) PfPI3K was bound to PfKelch13WT and this was reduced by the C580Y mutation (Fig. 2e). K48-linked and atypical ubiquitination, both signals for proteosomal degradation were prominent in PfPI3K and concomitantly reduced by PfKelch13C580Y. K63-linked ubiquitination21 was not detected suggesting ubiquitination drives degradation rather than a change in cellular localization and endocytic trafficking (and PfPI3K continued to be cytoplasmically distributed as seen in absence of mutation; Extended Data Fig. 1f). Fragmentation detected in PfPI3K was diminished in presence of the C580Y mutation. The data in Fig. 2f suggested that cleavage of kinase began even before it was fully released from PfKelch13WT and confirmed that lack of effect of PfKelch13C580Y was due to its failure to bind PfPI3K. Notably, Kelch adapters for E3 ligases are substrate specific and therefore our data well support the model in Fig. 2g. Since PfPI3K is an early ring-stage target of artemisinins, elevation of the kinase and/or its products may provide a mechanism of artemisinin resistance at this stage. The data in Fig. 2c, suggested that in an isogenic background, PfKelch13 mutation linked to resistance may increase levels of PfPI3K. Nonetheless equal amounts of PfPI3K across two distinct non-isogenic strains may not show equivalent activity for a variety of reasons, including different levels of precursor lipid substrate pools. We therefore examined whether PfPI3K activity, as measured by PI3P production provided a quantitative estimation of resistance across non-isogenic strains. PI3P can only be produced by (the sole) PfPI3K12. PI3,4P2 and PI3,4,5P3 (derived from PI3P) are both detected as minor fractions of the total cellular PI3P and under 1% in rings12 the stage of clinical artemisinin resistance. Thus ring parasite PI3P levels directly reflect the activity of PfPI3K. As shown in Fig. 3a, there was linear correlation between the levels of PI3P and resistance (as measured by Ring-stage Survival Assays or RSA22; Extended Data Fig. 5a) in both clinical and genetically engineered laboratory strains.
Figure 3. Elevated PI3P and artemisinin-resistance in presence and absence of PfKelch13 mutations.

a, RSA vs. PI3P levels in clinical (triangle) and laboratory (circle) strains. 1–4: sensitive strains ANL-1, 3D7, NF54-PfKelch13WT, 3D7-PfKelch13WT-HA with RSA ≤1. * ANL-7 contaminated with (R539T) resistant parasites show intermediate PI3P/RSA levels. 3D7-PfKelch13C580Y-HA contains a chromosomal wild type copy of PfKelch13 and thus shows lower PI3P and RSA (49 and 9) than NF54-PfKelch13C580Y (59 and ~ 13). b, 3D7 expressing transgenic myc-tagged VPS34 (left) or VPS34-catalytically dead mutant (right); loading control PfFKBP; raw data in Supplementary Data 2. Molecular weights, in kDa. Data are representative of three experimental replicates. c, PI3P levels in indicated strains. d, PI3P, RSA in VPS34 strains respond to PfKelch13. 1–4, sensitive strains ANL-1, PfVPS34-MUTANT-myc, SS-EEA1R1374A-mCherry and 3D7. In a, c, d, Mean from three experimental replicates (each in triplicate). (a) RSA, SD < 0.5; (c) PI3P, SD < 8 (shown by error bars). e, a model for PI3P-induced artemisinin resistance. Further details in Extended Data Fig. 5.
To elevate PI3P in absence of PfKelch13, we transgenically expressed human VPS34 (Fig. 3b and Extended Data Fig. 5b–d). Human VPS34 synthesizes only PI3P and accordingly in the absence of PfKelch13 mutation increased ring PI3P and resistance in two independent 3D7 transgenic lines (PfVPS34-myc1 and PfVPS34-myc2); but a catalytically inactive VPS34 or a control reporter had no effect (Fig. 3c, d and Extended Data Fig. 5b–e). Introduction of PfKelch13WT into VPS34 transgenic parasites reduced PI3P and RSA while the PfKelch13C580Y mutation reciprocally increased cellular PI3P and RSA levels (Fig. 3c–d and Extended Data Fig. 5b–f). The best fit line in Fig. 3d is closely comparable to that in Fig. 3a, suggesting the VPS34 transgenic lines remained responsive to PfKelch13. These data prove that elevated PI3P levels confer artemisinin resistance (Fig. 3e). Notably, a ~2.5 fold increase in PI3P induced greater than one order of magnitude change in resistance suggesting that since PI3P is a signaling lipid, small changes in its levels may induce downstream activation of large amplitudes of resistance (Fig. 3e).
We next queried whether additional cellular components could also influence PI3P and RSA levels. PI3K-AKT is a primary signal transduction pathway in eukaryotes but poorly understood in malaria parasites. In addition to PI3P, PfPI3K can also synthesize PI3,4,5P312 which in most cells is needed for activation of AKT23. P. falciparum has an orthologue of AKT (PfAKT/PF3D7_1246900; Extended Data Fig. 6a). However PfAKT appears different from its mammalian counterparts because it lacks a PH domain and a conserved Ser473. Rather unexpectedly we found that DHA blocks cellular PfAKT activity (Fig. 4a) but did not inhibit purified PfAKT (Fig. 4b). Since it targets PfPI3K (Fig. 1), we reasoned that in parasites DHA may block PfAKT through inhibition of PfPI3K. Although PfAKT lacks a PI3,4,5P3-binding PH domain, it may function through a calmodulin-binding PH domain protein24 since PfAKT contains a calcium/calmodulin activator domain. As indicated earlier, low levels of PI3,4,5P3 are made by PfPI3K12 (and in this regard, PfPI3K is also different from its closest homologue VPS34 which produces solely PI3P). Transgenic elevation of PfAKT (Extended Data Fig. 6b) induced a ~1.8 fold elevation of PI3P (presumably stimulated by feedback mechanisms) and RSA of 6.5, which is comparable to the resistance level seen in one of the clinically derived lines (ANL-4; Fig. 4c).
Figure 4. PI3P-mediated resistance linked to GWAS.

a–b Effect of DHA on a, cellular and b, immunopurified, PfAKT. c, Expression of PfAKT-GFP in Pf 3D7. Loading controls, PfFKBP, erythrocyte Band 3. Molecular weights in kDa. RSA, PI3P, as indicated. In a-c, mean of three experimental replicates (each with triplicate data points). AKT activity and RSA SD < 0.5; PI3P SD < 5. In b-c: western blots are representative of three experimental replicates; raw data in Supplementary Data 2. d, PI3P-mediated resistance pathways. Bold script: Lipid (PI3P) or protein components from this study and GWAS; Plain: GWAS only 6,7. Solid lines, established relationships; broken/dotted lines, partial functional validation; broad grey arrows, in silico prediction. Additional details in Extended Data Fig. 6.
Evidence that PfAKT is a major downstream effector of PfPI3K is still lacking. Nonetheless, the data suggest that secondary genes may influence levels of PfPI3K/PI3P product perhaps explaining why AKT (and other kinases) are also under artemisinin selective pressure6. Differences between the pools of available substrates (phosphatidylinositol; PI, ATP) extent of ubiquitination (and additional unknown regulation) of PfPI3K may also be influenced by complex lipid, biosynthetic, transport and signaling networks (Fig. 4d) that vary across genetic backgrounds. Therefore absolute levels of PfPI3K across non-isogenic strains may not provide an accurate measure of PfPI3K activity/PI3P product and artemisinin resistance. Genes involved in lipid uptake25 are also implicated in resistance6 and serum lipids are known to be essential for intracellular blood stage parasite growth26. This may explain the wide range of RSA values displayed by resistant, clinical strains and why two resistant strains like ANL-2 and ANL-4 bear the same mutation C580Y and equivalent levels of PfPI3K, but different amounts of PI3P product and thus different levels of resistance.
DHA has many other targets in the later trophozoite parasite stage27–29, but PfPI3K is the only known target in early ring stages: clinical resistance to artemisinins develops in early rings (and in absence of hemoglobin digestion associated with either late rings or trophozoites). Two most prevalent mutations (C580Y and R539T) associate with elevation of PI3P. All resistance mutations of PfKelch13 are limited to the Kelch propeller domain, suggesting they too regulate PfPI3K activity to contribute to differences in RSA. The molecular identities of other putative PfKelch13 targets and the ubiquitin ligase activity of the complex have yet to be investigated. PI3P may influence host remodeling, and functions of the apicoplast and food vacuole11–13 as well as cell survival through redox, transcriptional and DNA repair pathways (Fig. 4d, grey arrows), all of which have also been implicated in artemisinin resistance5,6,9,30. Sustained, selective targeting of the PfPI3kinase and/or its regulation will be critical to developing new therapies that allow eliminating artemisinin resistance and rendering effective malaria control.
Online Content Methods, along with any additional Extended Data display items and Source Data, are available in the online version of the paper; references unique to these sections appear only in the online paper.
METHODS
Cloning and generation of constructs for P. falciparum transfection
All constructs used for generating transgenic P. falciparum 3D7 parasites were assembled in different plasmids and finally cloned into pA150 or pA15611. This resulted in the expression of the proteins under the calmodulin (cam) promoter. The generation of constructs pA156 (SS-EEA1WT-mCherry) and pA156 (SS-EEA1WT-mCherry) have been described earlier11.
For the generation of constructs pA150 (PfKelch13WT-HA), pfkelch13 (PF3D7_1343700) was amplified from P. falciparum genomic DNA using PfKelch13AvrIIF and PfKelch13HAXhoIR; digested with AvrII-XhoI and cloned into corresponding sites of pA150. The construct pA150 (PfKelch13C580Y-HA) was generated using overlapping PCR strategy. Briefly, the products of PCR1 (using PfKelch13AvrIIF and C580Yreverse) and PCR2 (using C580Yforward and PfKelch13HAXhoIR) reactions were used as template for the overlapping PCR3 to generate full length pfkelch13 (with residues conferring C580Y mutation in the protein sequence), which was subsequently digested with AvrII-XhoI and cloned into pA150 to generate pA150 (PfKelch13C580Y-HA).
The plasmid containing human vps34 (Cat# SC118487) was purchased from Origene Inc. (Rockville, MD, USA). This was used as template to PCR amplify human vps34 using HsVps34AvrIIF and HsVps34mycXhoIR that was subsequently digested and cloned to the AvrII-XhoI site of either pA156 (with bsd resistance cassette) or pA150 (with hdhfr resistance cassette) to generate pA150 (VPS34-myc1) and pA150 (VPS34-myc2), respectively. The construct pA150 (VPS34MUTANT-myc1) was generated using overlapping PCR strategy. Briefly, the products of PCR1 (using HsVps34AvrIIF and Vps34-742AAA745R) and PCR2 (using Vps34-742AAA745F and HsVps34mycXhoIR) reactions were used as template for the overlapping PCR3 to generate full length vps34mutant-myc1 (with residues 742DRH745 changed to AAA in the protein sequence), which was subsequently digested with AvrII-XhoI and cloned into pA150 to generate pA150 (VPS34MUTANT-myc1).
The construct pA150 (PfAKT-GFP) was generated as follows. Briefly, the full length pfakt was amplified from P. falciparum genomic DNA using AKTAvrII_F and AKTBglII_R primers and cloned into AvrII-BglII site of pA150 (SSGFP)11. This resulted in an in-frame fusion with gfp at the 5′ end.
Single amino acid mutation replacing cysteine at position 580 with tyrosine (C580Y) in PfKelch13 (PF3D7_1343700) was generated using CRISPR-cas9 technology as described elsewhere20.
Sequence analysis of pfkelch13 and pfpi3k polymorphisms in the clinical strains
Clinical strains were first propagated in tissue culture and genomic DNA isolated using Quick-gDNA® Blood MiniPrep kit from ZYMO Research following manufacturer’s instructions.
For the sequence analysis for polymorphism in pfkelch13, either the full length pfkelch13, the full-length gene was amplified using Kelch13_F and Kelch13_R primers, or specific regions were amplified using Kelch13-1, Kelch13-2, Kelch13-3, Kelch13-4, Kelch13-5 and Kelch13-6 primers and analyzed.
To check for pfpi3k polymorphism in clinical strains, specific regions of pfpi3k were amplified using PfPI3K-I682T-F/ PfPI3K-I682T-R, PfPI3K-Q431K-F/ PfPI3K-Q431K-R, PfPI3K-Y1330C-F/ PfPI3K-Y1330C-F primer pairs and sequenced.
Generation of anti-peptide antibodies
All anti-peptide antibodies were custom-generated by Thermo Scientific Inc. (Rockford, IL, USA). Antibodies against P. falciparum PI3K (PfPI3K, PF3D7_0515300) were generated in guinea pigs and rabbits against the C-terminal peptides CVDKLHEWALNWK and CVLKVQEKFRLDLNDE, respectively. Antibodies were also custom-generated in rat, against the peptide RKRFDEERLRFLQEIDKI and corresponding to the amino acids 299–316 of PfKelch13 (PF3D7_1343700). Antibodies to BiP (PF3D7_0917900) were generated against the peptide DYFIKMFKKKNNIDLRTDKR corresponding to amino acids 261–280. For the generation of rabbit antibodies to PfAKT (RAC-beta serine/threonine protein kinase, PF3D7_1246900), amino acids corresponding to 229–246 (KKDKKIINLKKYNNAHRD) were selected. Antibodies were also custom-generated in rats, against the peptide REIVGDNTIEKKTEKALRE corresponding to amino acids 90–108 of PfExp-2 (PF3D7_1471100).
All guinea pig antibody generations involved collection of control sera on day 0 followed by immunization on day 1 with 0.1 mg of Keyhole Limpet Hemocyanin (KLH)-conjugated peptide with complete Freund’s adjuvant (CFA) subcutaneously at 4 sites. Booster immunizations were performed on days 21 and 42 with 50 μg of KLH-peptide with incomplete Freund’s adjuvant subcutaneously. Test bleeds were checked by western blotting with uninfected and infected erythrocytes and corresponding animals were finally boosted again on day 62 followed by exsanguination bleed and peptide-affinity purification of the antibodies.
For antibody generation in rabbits, control sera was collected on day 0 and animals were immunized on day 1 with 500 μg of KLH-conjugated peptides subcutaneously at 10 sites. Animals were again boosted on days 14 and 28 with 250 μg antigen and sera collected on day 35 to check the reactivity. Finally, after a booster immunization of selected animals on day 56/58, production bleed was collected on day 72 and processed for peptide-affinity purification of the antibodies.
The reactivity of all custom-generated antibodies were confirmed by western blots using uninfected and infected red cells and used either for immunofluorescence, immunoprecipitation assays or western blot analyses.
Parasite culture, transfection and live cell imaging
P. falciparum 3D7 parasites were propagated in A+ human erythrocytes (bought from Biochemed Services, Winchester VA, USA: protocol approved by the University of Notre Dame) in RPMI supplemented with 0.5% Albumax II, 0.2 mM Hypoxanthine, 11 mM Glucose, 0.17% NaHCO3, 10 μg/ml gentamycin and maintained at 37°C in 5% C02 in a humidified incubator. Synchronous P. falciparum 3D7 parasites in culture were transfected with indicated plasmids by standard procedures as described earlier11. Forty-eight hours after transfection, the cultures were selected either with 2.5 nM WR99210 (Jacobus Pharmaceuticals; for transfections involving pA150) or 1 μg/ml Blasticidin hydrochloride until stable cells lines were obtained. Culture-adapted, clinical field strains of P. falciparum as well as NF54 parasites expressing PfKelch13WT and PfKelch13C580Y (20) were propagated in the same media at 37°C in 90% N2, 5% 02 and 5% C02 in a humidified incubator. Cultures were monitored daily by Giemsa staining of methanol-fixed smears and fed as necessary. Parasitemia were usually maintained below 12% for healthy culture growth.
Patient sample collection and short term cultures
After hospital admission and informed consent, 10 ml of whole P. falciparum-infected blood was drawn from adults at multiple field sites in western Cambodia. White blood cells were removed over a CF11 column. The infected red blood cells were subjected to short term in vitro culture in RPMI1640 containing 20% human serum or Albumax II (0.5%) for a maximum of eight weeks. This study was approved by the Oxford Tropical-medicine Research Ethical Committee (OXTREC) as well as the Ministry of Health in Cambodia; the trial was registered under NCT00493363. The study was also approved by the University of Notre Dame.
Treatments with PI3K inhibitors, wortmannin, LY294002, the inactive ortholog LY303511 and artemisinins
Highly synchronized parasites at schizont stages from P. falciparum 3D7 (transgenically expressing secretory SS-EEA1WT-mCherry) were purified using percoll density gradients. Purified schizonts were then allowed to invade fresh batch of red blood cells for 6 h. The resulting intracellular rings were exposed to mock treatment (0.1% DMSO) or the following compounds: wortmannin, LY294002, inactive LY294002 analog LY303511 (from Selleckchem), artemisinin, artesunate or dihydroartemisinin (DHA) (from Sigma-Aldrich) at the indicated concentrations in DMS0 (0.1%). Drug treatments were carried out as indicated for 30 min or 4 h. Cells were subsequently processed for live cell imaging, immunofluorescence assay (IFA) and western blotting. Drugs were removed by washing thrice with serum-free RPMI, and infected erythrocytes were imaged after 1 h. In addition, where indicated parasite growth in culture was monitored 24 h later in Giemsa smears.
Imaging of live and cells by fluorescence microscopy and quantitative analyses
For live cell imaging, parasites were processed as described previously31. In both live and fixed cells, the parasite nuclei were stained with Hoechst 33342 and cells were imaged with a 100X, NA 1.4 objective on an Olympus IX inverted fluorescence microscope with a temperature controlled stage and a Photometrix cooled CCD camera (CH350/LCCD) driven by DeltaVision software from Applied Precision Inc. (Seattle, WA).
Quantitative analysis for the fraction of perinuclear SS-EEA1WT-mCherry fluorescence was undertaken using DeltaVision software. Briefly, consecutive 0.2 micron optical z-sections per cell were observed for either perinuclear/ER-like fluorescence pattern or peripheral/PV-like pattern under control or drug-treated condition and represented graphically. 400 optical sections were quantified per treatment (10 in-focus optical sections of the parasite per infected cell). While perinuclear/ER fluorescence was an indication of no observable effect on the PI3P level, peripheral/PV fluorescence was an indication of a reduction in PI3P level on drug treatment.
Immunoprecipitation of PfPI3K and PI3kinase assay
PfPI3K (PF3D7_0515300) was immunoprecipitated from parasite stocks previously frozen at −80°C. Proteins were extracted 1 h at 4°C using extraction/lysis buffer containing 10 mM Tris. HCl, pH 7.5, 100 mM NaCl, 5 mM EDTA, 1% Triton X-100, 100 μM sodium orthovanadate, 20 μM sodium fluoride, 20 μM β-glycerophosphate, and 1×protease inhibitor cocktail, Roche Diagnostics; 10% glycerol). After removal of insoluble debris, the extract was incubated with guinea pig anti-PfPI3K for 12 h at 4°C. Protein A agarose beads (pre-equilibrated in lysis buffer) was then added and additionally incubated for 6 h. Immune complexes bound to beads were extensively washed with extraction buffer. PfPI3K bound beads were either processed for western blotting or kinase activity assay. For western blotting beads were directly boiled in reducing SDS-PAGE sample buffer, resolved by SDS-PAGE and probed with rabbit anti-PfPI3K. Kinase assay of PfPI3K in immunoprecipitated beads was measured using Class III PI3-Kinase kit from Echelon Biosciences (K-3000) following manufacturer’s instructions. Mean (SD) from three experimental replicates, each containing duplicate data points are shown.
Mammalian kinase assays
The kinase activity of mammalian kinases was performed by Life Technologies (Grand Island, NY, USA) in the absence or presence of 10 μM DHA, through SelectScreen® Biochemical Kinase Profiling Service, and expressed as percentage inhibition.
Immunolocalization by indirect immunofluorescence assays (IFA)
Indirect immunofluorescence assays (IFAs) were performed on non-transfected P. falciparum 3D7 or transgenic parasites fixed with glutaraldehyde/paraformaldehyde as described previously32 using the following antibody concentrations-rabbit anti-PfPI3K (custom-made, 20 μg/ml), guinea pig anti-Exp-2 (custom-made, 20 μg/ml) and mouse anti-myc antibodies from Abcam (Cambridge, MA, USA). The appropriate FITC-or TRITC labeled secondary IgG antibodies (ICN Biochemicals) were used at 1:200 dilution. Parasite nuclei were stained with 5 mg/ml Hoechst 33342 (Molecular Probes) and slides were mounted with DABCO.
Immunoprecipitation and western blotting
PfPI3K was immunopurified from P. falciparum 3D7 transgenic parasites (3D7-PfKelch13WT-HA and 3D7-PfKelch13C580Y-HA) using guinea pig anti-PfPI3K as described above. Immunoprecipitations using Protein A agarose beads were used as negative control. Western blots were subsequently performed using custom-generated rabbit anti-PfPI3K, rat anti-PfKelch13 antibodies, commercial rabbit anti-polyubiquitin (MBL-PW8810), K48-linkage specific polyubiquitin (Cell Signaling Technology-4289S), K63-linkage specific polyubiquitin (Cell Signaling Technology-5621S) or mouse monoclonal anti-HA tag antibody from (Abcam-ab18181) antibodies.
To measure the relative amount of PfPI3K expression in P. falciparum 3D7 and clinical isolates, 4 × 106 infected erythrocytes permeabilized with 0.01% saponin followed by extensive washing with PBS to remove hemoglobin. Cells were the solubilized in Laemmli’s sample buffer, resolved by SDS-PAGE and western blotting performed using guinea-pig anti-PfPI3K, followed by HRP-conjugated donkey-anti guinea pig antibodies from Jackson ImmunoResearch (West Grove, PA, USA). Blots were developed using chemiluminescence assay kit from Thermo Scientific (Rockford, IL, USA). Antibodies to the parasite protein PfFKBP served as a loading control. The intensities of PfPI3K and PfFKBP signals were quantitated by densitometry and expressed as a ratio.
The relative amount of PfPI3K expression in the transgenic parasites NF54-PfKelch13WT, NF54-PfKelch13C580Y, 3D7-PfKelch13WT-HA and 3D7-PfKelch13C580Y-HA was measured by resolving the saponin-permeabilized pellet from 2 × 107 parasites by SDS-PAGE and western blotting using guinea pig anti-PfPI3K followed by HRP-conjugated donkey-anti guinea pig antibodies and chemiluminescence detection. Antibodies to parasite proteins BiP and PfFKBP were used as loading controls. Antibodies to BiP were custom generated, while antibodies to PfFKBP were kindly provided by Nirbhay Kumar. The intensities of PfPI3K and PfFKBP signals were quantitated by densitometry and expressed as a ratio.
Transgenic P. falciparum 3D7 parasites, either expressing myc-tagged VPS34 (VPS34-myc1 and VPS34-myc2), those expressing PfKelch13WT-HA/PfKelch13C580Y-HA, as well as those expressing PfKelch13WT-HA/PfKelch13C580Y-HA in VPS34-myc1 background were also checked for the expression by western blotting. Primary antibody either involved mouse anti-myc antibodies from Abcam (Cambridge, MA, USA) or rabbit anti-HA antibodies (Thermo Fisher Scientific, Grand Island, NY, USA), followed by HRP-conjugated secondary antibody from Bio-Rad (Hercules, CA, USA). Blots were developed using chemiluminescence detection kit. Antibodies to parasite PfFKBP or BiP were used as loading controls.
Expression of SS-EEA1R1374A-mCherry was detected using Living Colors polyclonal rabbit anti-DsRed2 (Clontech, Mountain View, CA, USA).
Expression of PfAKT-GFP in transgenic parasites was confirmed by western blotting of uninfected erythrocytes, non-transfected 3D7 parasites and transgenic (PfAKT-GFP) parasites, using rabbit anti-GFP antibodies from Thermo Fisher Scientific (Grand Island, NY, USA) and HRP-conjugated secondary goat anti-rabbit antibodies from Bio-Rad (Hercules, CA, USA). Antibodies to parasite PfFKBP and human Band 3 were used as loading controls.
Model building, docking and molecular dynamics (MD) simulations
The homology model of P. falciparum PI3K was build based on the structure of the class III PI3 kinase from Drosophila (pdb code 2X6F)33, using the multiple threading alignment in I-TASSER34,35 using additional information for the definition of the active site from the structure of the human class III PI3K (pdb code 3IHY).
The known PI3K inhibitor, LY29400236 was docked to the model using GlideXP37 to generate the starting structure for model refinement. The initial structure for the PI3K-dihydroartemisinin (DHA) complex was generated by removing the LY294002 ligand from the refined structure and docking DHA to this model using GlideXP.
This model of PfPI3K then served as the initial structure for 20ns MD simulations. The model in these simulation consisted of the protein, surrounded by a periodic box of TIP3P38 water molecules that extended 10 Å from the protein. Na+ counter ions were placed by the LEaP module of AMBER1239 to neutralize the system. Ionizable residues were set to their normal ionization states at pH 7.
The MD simulations were carried out using the PMEMD module of AMBER1239. The protein was modeled using the ff03.r1 version of the AMBER force field while the DHA ligand was represented by the GAFF force field40,41. Atom-centered partial charges were generated based on B3LYP/6-31G* optimized geometries using RESP42,43 as implemented in the antechamber module of AMBER12. A time step of 2 fs, combined with the SHAKE algorithm44 to constrain all bonds involving hydrogen atoms, was chosen. A non-bonded cutoff of 10.0 Å was used, and the non-bonded pair list was updated every 25 time steps.
The temperature (300K) and pressure (1 atm) of the NPT ensemble were controlled by Langevin dynamics and isotropic position scaling45 respectively. Long-range interactions were treated by the Particle-Mesh-Ewald (PME)46,47 methods with a grid spacing of ~1 Å and a fourth-order B-spline interpolation to compute the potential and forces in between grid points. The trajectories were analyzed using the PTRAJ module of AMBER12. The model of the PfPI3K-DHA complex was simulated analogously for a total time of 20 ns.
The same MD protocol was used for the study of the complexes of artelinic acid, artemisone, and deoxyartemisinin in the pfPI3K model as well as DHA in VPS34 and p110γ. In short, initial structures were generated by docking of the small molecules to the PfPI3K model, human VPS34 (pdb code 3IHY), and human p110γ (pdb code 4ANV). After manual inspection of the generated poses, the initial models were subjected to 20 ns MD simulations as described above.
Assay for measuring cellular and immunoprecipitated PfAKT activity
P. falciparum 3D7 early-ring stage parasites were synchronized by sorbitol treatment and exposed either to mock treatment: 0.1% DMSO (control/Mock) or 4 nM DHA for 3 h. An aliquot was washed using serum-free RPMI and returned to culture for 6 h (washout). AKT activity was measured in mock-treated, 4 nM DHA-treated and post-washed DHA treated cells (4 × 107) using the K-LISA Akt Activity Kit (#CBA019) from Calbiochem (Darmstadt, Germany) following manufacturer’s instructions. Briefly, phosphorylation of AKT biotinylated peptide substrate was detected with phosphoserine antibodies. Serial dilutions of active human Akt1/PKBα (Millipore) were used as standard and absorbance read at 450 nm on a plate reader. Mean (SD) from three experimental replicates, (each with triplicate data points) are shown.
Endogenous PfAKT was also immunopurified from P. falciparum 3D7 following saponin treatment (as described earlier) and the resulting parasite pellet extracted with extraction buffer (25 mM Tris.HCl pH 8.0, 150 mM NaCl, 1% (v/v) Triton X-100, 1% (w/v) Deoxycholate-Na, 2 mM EDTA containing complete protease inhibitor for 1 hour at 4°C. PfAKT was immunopurified using 10 μg of custom-generated antibodies (for 2 h at 4°C, under shaking condition) followed by Protein A agarose beads for 1 h. Protein A agarose beads incubated with parasite lysate in absence of antibodies served as negative control. Immunopurified PfAKT-beads and control beads were extensively washed in PBS, pH 7.4 and processed further for the measurement of activity in the absence or presence of 4 nM DHA as described previously. Immunoprecipitation was also confirmed by western blotting using anti-PfAKT antibodies.
Measurement of PI3P level in clinical and laboratory strains
Early ring stage parasites from P. falciparum 3D7 laboratory strains (non-transfected or transgenic) and clinical strains were tightly synchronized with sorbitol treatment and lyzed with saponin to remove hemoglobin. Resulting pellets (corresponding to 6 × 107 parasites) were washed with PBS. Lipids were extracted and PI3P level assessed (in triplicates) using Echelon PI3P Mass ELISA Kit (K-3300) following the manufacturer’s instructions.
PI3P level was also measured in 3D7 parasites under control condition, after 3 h treatment with 4 nM DHA, as well as after 6 h in culture following DHA washout. Mean (SD) from three experimental replicates, (each with triplicate data points) are shown.
Ring-stage survival assay (RSA)
In vitro RSA was assessed as described by elsewhere22. Briefly, tightly synchronized early ring parasites (0–3 h post invasion) were exposed to 700 nM DHA or 0.1% DMSO (control) for 6 h, washed thrice with serum-free RPMI and returned to culture for 66 h. Blood smears were prepared and stained with Giemsa. Each sample was done in duplicate and 10,000 erythrocytes were assessed independently by light microscope. Parasite survival rates (%RSA) were expressed as a percentage by comparing the number of viable parasites between the drug-treated and untreated control. Mean (SD) of RSA based on four replicated carried out by two independent laboratory personnel.
Extended Data
Extended Data Figure 1. Ring Parasite PI3P and PI3K: effect of inhibitors, and characterization of antibodies.

a, Rings at 6 h post-invasion, were either mock treated or exposed to 4 nM DHA for 4 h. An aliquot was washed in serum-free RPMI and cultured for 24 h. Parasitemia (ring and trophozoite stages) were determined by Giemsa staining and light microscopy. Mean (error bars SD < 2) from three experimental replicates are shown. b, Effect of artemisinins, deoxyartemisinin, PI3K inhibitors wortmannin and LY294002 (and its inactive orthologue LY303511) on PI3P level, as observed by redistribution of the PI3P reporter in transgenic 3D7 parasites expressing SS-EEA1WT-mCherry (red). Blue, parasite nucleus, P, parasite, E, erythrocyte; scale bar, 5 μm. Fluorescence and phase-merged images are as indicated. c, Quantitative analysis of the data in b. Mean (error bars SD < 21) from three experimental replicates are shown. d, Effect of anti-folates and chloroquine on PI3P production, as observed by redistribution of the PI3P reporter in transgenic 3D7 parasites expressing SS-EEA1WT-mCherry (red). Blue, parasite nucleus, P, parasite, E, erythrocyte; scale bar, 5 μm. Fluorescence and phase-merged images are as indicated. e, Quantitative analysis of the data in d. Mean (error bars SD < 27) from three experimental replicates are shown. As indicated in Fig. 1, the PI3P reporter SS-EEA1WT-mCherry (red) detects PI3P in punctate, perinuclear ER domains in 6 h rings (top panel; 11) or reduced PI3P when the reporter diffuses to the PV (periphery). f, Schematic showing structural features of PfPI3K. Custom antibodies were generated in rabbits and guinea pigs against the indicated peptides. Western blots indicate that both antibodies specifically recognize a ~255-kDa protein in infected (IE) but not uninfected (U) erythrocytes and are representative of 10 experimental replicates; raw data in Supplementary Data 2. Indirect immunofluorescence assays using rabbit (top panel) and guinea pig (bottom panel) antibodies confirm the PfPI3K (green) is localized in the parasite. Exp-2 (red) marks the parasite boundary. Blue, parasite nucleus, P, parasite, E, erythrocyte; scale bar, 5 μm. Fluorescence and phase-merged images are shown. Data are representative of 10 experimental replicates.
Extended Data Figure 2. Structural analyses of PfPI3K.

Sequence alignment of the putative catalytic domain of PfPI3K and its closest human orthologs human VPS34 and Drosophila VPS34. * indicates identity; dots indicate homology. D1889 and Y1915 residues are highlighted in yellow.
Extended Data Figure 3. Structural analyses of PfPI3K and human PI3K, with and without artemisinin derived compounds.

a Snapshots from 20 ns MD simulations and chemical structure of artelinic acid (top right), artemisone (bottom left) and deoxyartemisinin (bottom right) bound to PfPI3K. Snapshot from MD simulation of DHA bound to PfPI3K is shown (at the top left) for reference. Due to the lack of the hydroxyl group in artelinic acid and artemisone, no interactions involving D1889 and Y1915 were observed. Instead, the carboxylic group of artelinic acid forms a salt bridge with R1368 and the sulfone moiety of artemisone interacts with R1368. Deoxyartemisinin lacks hydrogen bond donors, so D1889 is not involved in protein-ligand interaction. Instead, the carbonyl oxygen of deoxyartemisinin forms a hydrogen bond with Y1915. Boxed inset shows the change in the overall shape due to the replacement of the endoperoxide with an ether bridge in deoxyartemisinin (green) and compared to DHA (red), thus leading to loss of the shape complementarity and reorientation in the binding site. As shown in Fig. 1 panels a, c, deoxyartemisinin failed to block PI3P production, in transgenic 3D7 parasites expressing SS-EEA1WT-mCherry (red) or purified PfPI3K. Thus, modeling studies explain a lack of effect of deoxyartemisinin. They would also suggest lack of effect of artelinic acid and artemisone compounds on PfPI3K. It should be noted the target of these compounds is unknown and it remains unclear whether they would share a ring stage target with DHA important for clinical resistance to artemisinins. b, Snapshots of MD simulation of DHA-human Vps34 complex. The initial coordinates of DHA…human Vps34 complex was obtained by the overlay of DHA…PfPI3K model (EDFig. 2b) with the human Vps34 crystal structure (PDB: 3IHY). The obtained complex was then subjected to 20 ns MD simulations. As can be seen, DHA does not interact with D644 and Y670, which correspond to D1889 and Y1915 in PfPI3K, respectively. Instead, the hydroxyl group of DHA now interacts with D761 (left). Snapshot at the right shows an overlay of DHA…human VPS34 complex with the PfPI3K model. In human VPS34 (gray), the loop that contains the D761 residue bends down to interact with DHA. In contrast, D2008 of PfPI3K (cyan), which is in the same position as D761 in human VPS34, has a different orientation which makes it unable to interact with DHA. c, Both D1889 and Y1915 in PfPI3K are conserved in human PI3K p110γ. 20 ns MD simulations of the DHA …p110γ complex (pdb code 4ANV), reveal that although the hydrogen bond between DHA and D841 still remains, the interaction between DHA and Y867 is lost. This suggests a weaker binding of DHA to p110γ compared to PfPI3K. The same argument can be made of p110δ based on sequence homology, but no crystal structure of human p110δ is available. d, Partial sequence alignment of PfPI3K, with human PI3K-C2α, PI3K-C2β, p110δ, p110γ and VPS34. The key residues of PfPI3K (D1889 and Y1915) that interact with DHA and similar positions in the human kinases are colored red. While D1889 in PfPI3K is conserved in both PI3K-C2α and PI3K-C2β, Y1915 in PfPI3K is replaced by F1172 and F1117 in PI3K-C2α and PI3K-C2β, respectively. Therefore, the hydrogen bond of the hydroxyl group in Y1915 suggested to be crucial in the computational analysis of PfPI3K cannot be formed with F. To the best of our knowledge, no crystal structures of PI3K-C2α and PI3K-C2β have been reported. In p110δ, p110γ and VPS34, a proline N-terminal to Y1915 is conserved. It is likely the reason for the different position and low flexibility of this loop observed in the MD simulations of p110δ, and VPS34.
Extended Data Figure 4. Transgenic P. falciparum expressing PfKelch13C580Y mutation in two different strains of P. falciparum using distinct approaches and markers.
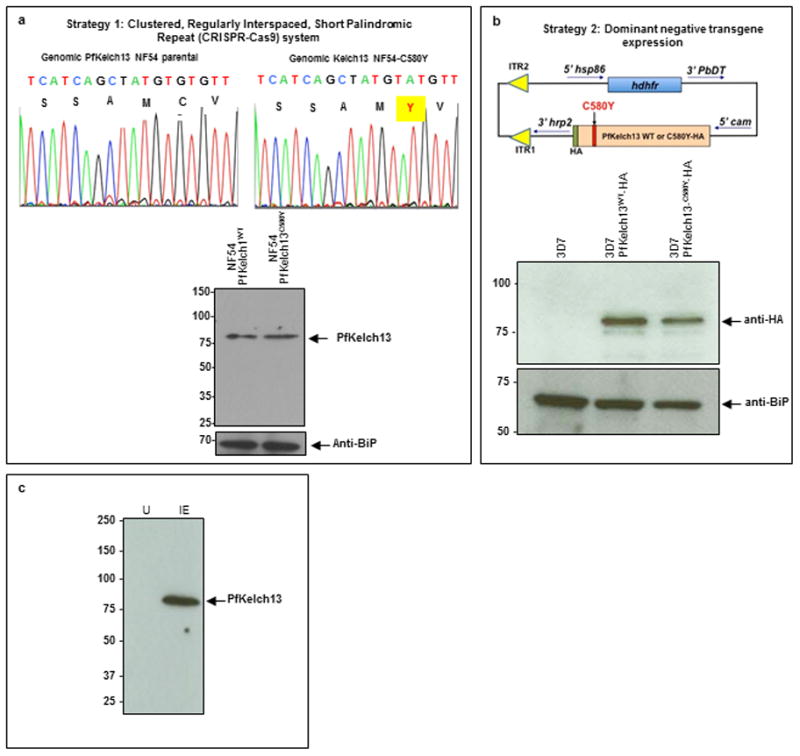
a, Strategy 1: CRISPR-Cas9 used to introduce a single point mutation in the P. falciparum NF54 genome as described elsewhere20. Both parent and mutant strains were sequenced to ensure that the only change detected was PfKelch13C580Y. Western blots using anti-PfKelch13 antibodies confirm that mutation does not change the level of PfKelch13 protein. b, Strategy 2 expresses a dominant negative PfKelch13C580Y and wild type PfKelch13WT tagged with HA, and driven by the constitutive cam promoter in P. falciparum 3D7. Western blots confirm that anti-HA antibodies detected tagged forms of PfKelch13 in transfected but not non-transfected 3D7. Comparable levels of wild type and mutant transgenes are expressed in resulting two strains of parasites (as shown). In a-b, BiP, an ER marker serves as a parasite loading control in western blots. Molecular weight standards (in kDa) are as indicated. c, Western blot indicate that antibodies to PfKelch13 specifically recognize a ~83-kDa protein in infected (IE) but not uninfected erythrocytes (U). Molecular weight standards (in kDa) are as indicated. In a-c: data are representative of three experimental replicates; raw data are in Supplementary Data 2.
Extended Data Figure 5. An in vitro ring-stage survival assay (RSA) level is associated with PI3P elevation in the presence and absence of Kelch mutations.

a, Schematic of the RSA assay22. b, Summary of RSA, PI3P, PfKelch13 mutation and PfPI3K polymorphisms in clinical and laboratory strains utilized in this study. Means are shown for three experimental replicates. RSA SD < 0.5; PI3P SD < 8 (as shown by error bars in Fig. 3c). c–d, Construction of 3D7 transgenic parasites expressing myc-tagged forms of human VPS34, the mammalian ortholog of PfPI3K. The resulting PfVPS34-myc1/2 express the vps34 transgene in ring stage parasites, as detected in indirect immunofluorescence assays (PfVPS34-MUTANT-myc data not shown in IFA). Exp-2 (red) marks the parasite boundary. Blue, parasite nucleus, P, parasite, E, erythrocyte; scale bar, 5 μm. Fluorescent and merged images are shown. Whole genome Illumina sequencing indicated VPS34-myc1 was inserted into PF3D7_1363900 and VPS34-myc2 is inserted in PF3D7_0718800. Both PF3D7_1363900 and PF3D7_0718800 encode for unknown function and neither has been reported in GWAS on artemisinin resistance. e, Construction of the strain 3D7-SS-EEA1R1374A-mCherry (used as transfection control) and western blot to confirm expression of SS-EEA1R1374A-mCherry (arrow). Molecular weight standards (in kDa) are as indicated. f, Construction of PfVPS34-myc1: PfKelch13WT-HA and PfVPS34-myc1: PfKelch13C580Y-HA parasites and western blots confirming expression of the transgenes. Raw data for western blots in e-f are shown in Supplementary Data 2. Molecular weight standards (in kDa) are as indicated. In d-f, data are representative of three experimental replicates.
Extended Data Figure 6. Evidence that the PI3K-AKT pathway is responsive to DHA.
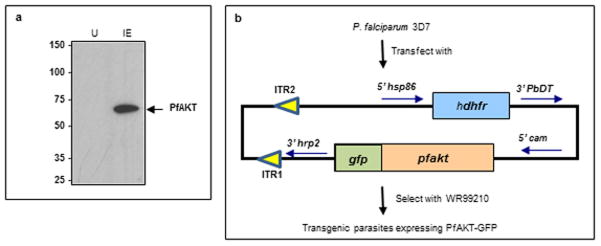
a, Western blot shows specificity of anti-PfAKT antibody that recognizes a band of expected size in infected (IE) but not uninfected (U) erythrocytes (1x 107 total cells loaded in each lane). Raw data is in Supplementary Data 2. Data are representative of three experimental replicates. b, Strategy and construct for generating transgenic 3D7 parasites expressing PfAKT-GFP.
Supplementary Material
3D coordinates of the PI3K model shown in Fig. 1e–g (as Excel file).
Raw data associated with Fig. 1–4 and Extended Data Fig. 1 and 4–6
Footnotes
Author contributions A.M., S.B., T.P., K.H., H.L., G.E., S.R., D. L.N., Y.F., O.W., M.P. and S.E. designed, performed and interpreted the experimental work. K.H. along with A.M., O.W. and S.B. wrote the manuscript. R.V.S, N.M. provided intellectual insight into aspects of this study. K.C., C.N., M.G., J.L.R. and A.M.D. provided reagents and intellectual input into study design. All authors commented on the manuscript.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
References
- 1.WHO. World malaria report 2012. http://www.who.int/malaria/publications/world_malaria_report_2012/en/
- 2.Noedl H, et al. Evidence of artemisinin-resistant malaria in western Cambodia. N Engl J Med. 2008;359:2619–2620. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- 3.Dondorp AM, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dondorp AM, et al. The threat of artemisinin-resistant malaria. N Engl J Med. 2011;365:1073–1075. doi: 10.1056/NEJMp1108322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheeseman IH, et al. A major genome region underlying artemisinin resistance in malaria. Science. 2012;336:79–82. doi: 10.1126/science.1215966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miotto O, et al. Multiple populations of artemisinin-resistant Plasmodium falciparum in Cambodia. Nature genetics. 2013;45:648–655. doi: 10.1038/ng.2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ariey F, et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014;505:50–55. doi: 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ashley EA, et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2014;371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takala-Harrison S, et al. Genetic loci associated with delayed clearance of Plasmodium falciparum following artemisinin treatment in Southeast Asia. Proc Natl Acad Sci U S A. 2013;110:240–245. doi: 10.1073/pnas.1211205110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takala-Harrison S, et al. Independent emergence of Plasmodium falciparum artemisinin resistance mutations in Southeast Asia. J Infect Dis. 2014 doi: 10.1093/infdis/jiu491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhattacharjee S, Stahelin RV, Speicher KD, Speicher DW, Haldar K. Endoplasmic reticulum PI(3)P lipid binding targets malaria proteins to the host cell. Cell. 2012;148:201–212. doi: 10.1016/j.cell.2011.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tawk L, et al. Phosphatidylinositol 3-phosphate, an essential lipid in Plasmodium, localizes to the food vacuole membrane and the apicoplast. Eukaryot Cell. 2010;9:1519–1530. doi: 10.1128/EC.00124-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaid A, Ranjan R, Smythe WA, Hoppe HC, Sharma P. PfPI3K, a phosphatidylinositol-3 kinase from Plasmodium falciparum, is exported to the host erythrocyte and is involved in hemoglobin trafficking. Blood. 2010;115:2500–2507. doi: 10.1182/blood-2009-08-238972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen Y, LJ, Estiu G, Isin B, Ahn YY, Lee DS, Barabási AL, Kapatral V, Wiest O, Oltvai ZN. Blueprint for antimicrobial hit discovery targeting metabolic networks. Proc Natl Acad Sci U S A. 2010;107:1082–1087. doi: 10.1073/pnas.0909181107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Efferth T, Marschall M, Wang X, Huong SM, Hauber I, Olbrich A, Kronschnabl M, Stamminger T, Huang ES. Antiviral activity of artesunate towards wild-type, recombinant, and ganciclovir-resistant human cytomegaloviruses. J Mol Med (Berl) 2002;80:233–242. doi: 10.1007/s00109-001-0300-8. [DOI] [PubMed] [Google Scholar]
- 16.Xu H, et al. Anti-malarial agent artesunate inhibits TNF-alpha-induced production of proinflammatory cytokines via inhibition of NF-kappaB and PI3 kinase/Akt signal pathway in human rheumatoid arthritis fibroblast-like synoviocytes. Rheumatology (Oxford, England) 2007;46:920–926. doi: 10.1093/rheumatology/kem014. [DOI] [PubMed] [Google Scholar]
- 17.Cheng C, et al. Anti-malarial drug artesunate attenuates experimental allergic asthma via inhibition of the phosphoinositide 3-kinase/Akt pathway. PLoS One. 2011;6:e20932. doi: 10.1371/journal.pone.0020932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang DD, LS, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chotivanich K, et al. Laboratory detection of artemisinin-resistant Plasmodium falciparum. Antimicrob Agents Chemother. 2014;58:3157–3161. doi: 10.1128/AAC.01924-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghorbal M, et al. Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat Biotechnol. 2014;32:819–821. doi: 10.1038/nbt.2925. [DOI] [PubMed] [Google Scholar]
- 21.Ikeda F, DI Atypical ubiquitin chains: new molecular signals. ‘Protein Modifications: Beyond the Usual Suspects’ review series. EMBO Rep. 2008;9:536–542. doi: 10.1038/embor.2008.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Witkowski B, et al. Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in Cambodia: in-vitro and ex-vivo drug-response studies. Lancet Infect Dis. 2013;13:1043–1049. doi: 10.1016/S1473-3099(13)70252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yano S, Tokumitsu H, Soderling TR. Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature. 1998;396:584–587. doi: 10.1038/25147. [DOI] [PubMed] [Google Scholar]
- 25.van Ooij C, W-MC, Ringel A, Cockcroft S, Haldar K, Blackman MJ. Identification of a Plasmodium falciparum phospholipid transfer protein. J Biol Chem. 2013;288:31971–31983. doi: 10.1074/jbc.M113.474189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mi-Ichi F, Kano S, Mitamura T. Oleic acid is indispensable for intraerythrocytic proliferation of Plasmodium falciparum. Parasitology. 2007;134:1671–1677. doi: 10.1017/S0031182007003137. [DOI] [PubMed] [Google Scholar]
- 27.Eckstein-Ludwig U, et al. Artemisinins target the SERCA of Plasmodium falciparum. Nature. 2003;424:957–961. doi: 10.1038/nature01813. [DOI] [PubMed] [Google Scholar]
- 28.Klonis N, et al. Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proc Natl Acad Sci U S A. 2011;108:11405–11410. doi: 10.1073/pnas.1104063108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng Q, Kyle DE, Gatton ML. Artemisinin resistance in Plasmodium falciparum: A process linked to dormancy? International journal for parasitology, drugs and drug resistance. 2012;2:249–255. doi: 10.1016/j.ijpddr.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Painter HJ, Campbell TL, Llinas M. The Apicomplexan AP2 family: integral factors regulating Plasmodium development. Mol Biochem Parasitol. 2011;176:1–7. doi: 10.1016/j.molbiopara.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Osborne AR, et al. The host targeting motif in exported Plasmodium proteins is cleaved in the parasite endoplasmic reticulum. Mol Biochem Parasitol. 2010;171:25–31. doi: 10.1016/j.molbiopara.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tonkin CJ, et al. Localization of organellar proteins in Plasmodium falciparum using a novel set of transfection vectors and a new immunofluorescence fixation method. Mol Biochem Parasitol. 137:13–21. doi: 10.1016/j.molbiopara.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 33.Miller S, et al. Shaping development of autophagy inhibitors with the structure of the lipid kinase Vps34. Science. 2010;327:1638–1642. doi: 10.1126/science.1184429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nature protocols. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- 37.Friesner RA, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 38.Jorgensen WL, Chandrasekhar J, Buckner JK, Madura JD. Computer simulations of organic reactions in solution. Ann N Y Acad Sci. 1986;482:198–209. doi: 10.1111/j.1749-6632.1986.tb20951.x. [DOI] [PubMed] [Google Scholar]
- 39.Case DA, Darden TA, Cheatham I, TE, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KMJRB, Hayik S, Roitberg A, Seabra G, Swails J, Gotz AW, Kolossvary I, Wong KF, Paesani F, Vanicek J, Wolf RM, Liu J, Wu X, Brozell S, Steinbrecher H, Gohlke H, Cai Q, Ye X, Wang J, Hsieh M-J, Cui G, Roe DR, Mathews DH, Seetin MG, Salomon-Ferrer R, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. AMBER 12. University of California; San Francisco: 2012. www.ambermd.org. [Google Scholar]
- 40.Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. Journal of molecular graphics & modelling. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 41.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. Journal of computational chemistry. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 42.Christopher I, Bayly PC, Cornell Wendy, Kollman Peter A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- 43.Fox T, Kollman PA. The application of different solvation and electrostatic models in molecular dynamics simulations of ubiquitin: how well is the X-ray structure “maintained”? Proteins. 1996;25:315–334. doi: 10.1002/(SICI)1097-0134(199607)25:3<315::AID-PROT4>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 44.Jean-Paul Ryckaert GC, Berendsen Herman JC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. Journal of Computational Physics. 1977;23:327–341. [Google Scholar]
- 45.Pastor RWB, Bernard R, Attila Szabo. An analysis of the accuracy of Langevin and molecular dynamics algorithms. Molecular Physics. 1988;65:1409–1419. [Google Scholar]
- 46.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 47.Petersen HG. Accuracy and efficiency of the particle-mesh-ewald method. J Chem Phys. 1995;103:3668–3679. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
3D coordinates of the PI3K model shown in Fig. 1e–g (as Excel file).
Raw data associated with Fig. 1–4 and Extended Data Fig. 1 and 4–6