

Summary
AMP-activated protein kinase (AMPK), whose activity is a critical determinant of cell vitality, serves a fundamental role in integrating extracellular and intracellular nutrient information into signals that regulate various metabolic processes. Despite the importance of AMPK, its specific roles within the different intracellular spaces remain unresolved, largely due to the lack of real-time, organelle-specific AMPK activity probes. Here, we present a series of molecular tools that allows for the measurement of AMPK activity at the different subcellular localizations and that allows for the rapid induction of AMPK inhibition. We discovered that AMPKα1, not AMPKα2, was the subunit that preferentially conferred spatial specificity to AMPK, and that inhibition of AMPK activity at the mitochondria was sufficient for triggering cytosolic ATP increase. These findings suggest that genetically encoded molecular probes represent a powerful approach for revealing the basic principles of the spatiotemporal nature of AMPK regulation.
Introduction
Cell vitality and growth are critically dependent on nutrients such as glucose, fatty acids, and amino acids (Marshall, 2006). 5′ Adenosine monophosphate-activated protein kinase (AMPK) is an evolutionary conserved serine/threonine protein kinase which integrates nutrient signaling pathways to organize energy homeostasis (Hardie et al., 2012). Mammalian AMPK is a heterotrimeric complex that comprises a catalytic α subunit (α1 and α2), a regulatory β (β1 and β2) and γ (γ1, γ2, and γ3) subunits, each of which is encoded by a different gene (Shackelford and Shaw, 2009). Multiple physiological factors are known to activate AMPK through intracellular metabolic perturbations, most of which increase intracellular AMP/ATP and ADP/ATP ratios and result in allosteric binding of either AMP or ADP to an AMPK γ subunit (Xiao et al., 2011, Oakhill et al., 2011). This conformational change of an AMPK complex facilitates the subsequent phosphorylation of Thr172 in the α subunit by upstream kinases, such as LKB1 (Woods et al., 2003). The same phosphorylation could occur through a different upstream molecule such as calcium ions that activate CaMKKβ (Hawley et al., 2005; Hurley et al., 2005; Woods et al., 2005).
Befitting its role in maintaining energy homeostasis, AMPK switches on catabolic pathways and concomitantly switches off anabolic pathways (Hardie, 2007; Kahn et al., 2005). In addition, recent studies have revealed diverse roles for AMPK in regulating various cellular functions, including cell growth, autophagy, neuronal polarity, and gene transcription (Mihaylova and Shaw, 2011). However, the mechanism by which AMPK assures execution of different downstream functions remains to be determined. One solution for a signaling molecule to achieve multi-tasking within the differential cellular space is compartmentalization. Compartmentalization of the AMPK response could occur at three levels: regulators, effectors and AMPK itself. Indeed, an AMPK complex has differential localization dynamics. Specifically, AMPK shuttles between the nucleus and cytosol in response to various physiological inputs (Kodiha et al., 2007; Suzuki et al., 2007; Warden et al., 2001). Furthermore, several studies demonstrated that activated AMPK could accumulate within specific compartments, including endomembrane structures (Mitchelhill et al., 1997; Oakhill et al., 2010), several mitotic components (Vazquez-martin et al., 2009), cytosol and nucleus (Tsou et al., 2011), and the basal bodies of primary cilia (Boehlke et al., 2010). Such compartmentalization is known to be one of the properties of multi-task molecules, as exemplified by an oncogene product, Ras (Bivona and Philips, 2003). There are also several dozens of AMPK substrates that reside at distinct locations in cells and tissues (Hardie et al., 2012).
Despite the accumulating observations of the spatially defined AMPK regulation, the most conclusive evidence for the AMPK compartmentalization requires methods that enable both monitoring and perturbing AMPK activities at distinct subcellular locations. Genetically encodable Förster resonance energy transfer (FRET)-based reporters visualize dynamic posttranslational modifications such as phosphorylation of lipids and proteins in living cells, without the need to lyse cells for biochemical characterization (Antal and Newton, 2013; Zhang and Allen, 2007). One example of these phosphorylation reporters is an AMPK biosensor, with which AMPK activity in either cytoplasmic or nuclear compartments was successfully visualized (Tsou et al., 2011). However, it is still imperative to systematically monitor AMPK activity at subcellular compartments such as endoplasmic reticulum (ER), mitochondria, Golgi apparatus and lysosomes. Here, we report an improved AMPK biosensor that can selectively profile the kinase dynamics at each subcellular location of living cells. Gaining insights into causality of the observations on dynamic signal regulation inevitably requires target perturbation with high spatiotemporal precision. A common pharmacological inhibitor for AMPK like Compound C can only generate global AMPK inhibition. We thus developed another set of genetically encoded molecular tools based on a rationally designed AMPK inhibitor peptide (AIP), enabling the first-ever perturbation of AMPK activity exclusively at individual organelles on a timescale of minutes. With a complete panel of the molecular sensors and actuators, we herein provide direct evidence of subcellularly compartmentalized AMPK signaling.
Results
Design principle of biosensors that illuminate subcellular compartment-specific AMPK activity
We recently improved a previously reported AMPK biosensor (Tsou et al., 2011) by replacing fluorescent proteins. This new AMPK reporter named ABKAR for AMPK/BRSK activity reporter features a two-fold larger dynamic range compared to the original one (author's note: a manuscript of ABKAR development is under revision elsewhere, and is attached to the present manuscript). We also thoroughly characterized the binding profile and revealed that, in addition to LKB1 and CaMKKβ, brain specific kinases (BRSK1/2) can also phosphorylate ABKAR. We thus used non-neuronal cells lacking expression of BRSK1/2 in the present study to focus on the AMPK signaling. A salient feature of genetically encoded FRET reporters is that they can be targeted to predefined locations by appending the corresponding subcellular localization signals. Therefore, we applied this approach to probe AMPK regulation in different subcellular compartments. To measure AMPK activity at specific subcellular compartments, we fused it to well-established signal sequences for the cytosolic surface of the following organelles (Figure 1A): plasma membrane (PM), Golgi apparatus (Golgi), endoplasmic reticulum (ER), mitochondria (Mito), and lysosome (Lyso) as well as cytosol (Cyto) and nucleus (Nuc). Those biosensors were collectively referred to as organelle-specific ABKAR (osABKAR) (Figure 1B and Table S1, available online). We ensured their specific localization by expressing these biosensors together with the appropriate organelle markers in mouse embryonic fibroblasts (MEFs) (Figure S1). We confirmed that none of the osABKAR constructs had adverse effects on organelle morphology (Figure S1).
Figure 1. FRET response of osABKAR in differential subcellular compartment.
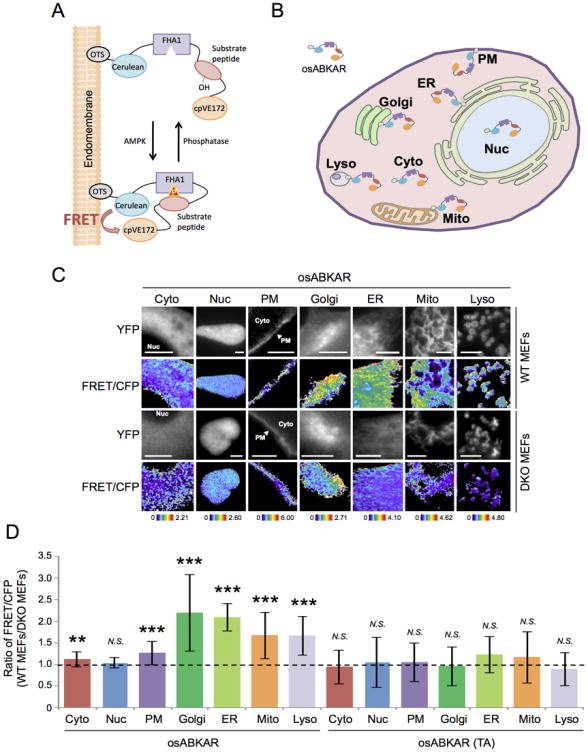
(A) Schematic diagram of osABKAR. OsABKAR is composed of organelle-targeting sequence (OTS), CFP variant; Cerulean, FHA1 phospho-amino acid binding domain, AMPK substrate motif, and YFP variant, cpV E172.
(B) The series of osABKAR. OsABKAR includes plasma membrane-specific ABKAR (PM-ABKAR), Golgi apparatus-specific ABKAR (Golgi-ABKAR), endoplasmic reticulum-specific ABKAR (ER-ABKAR), mitochondria-specific ABKAR (Mito-ABKAR), lysosome-specific ABKAR (Lyso-ABKAR), cytoplasm-specific ABKAR (Cyto-ABKAR), and nucleus-specific ABKAR (Nuc-ABKAR).
(C) Representative FRET image of osABKAR. Indicated osABKAR was transfected into either WT MEFs or DKO MEFs. Upper panels: representative YFP images. Lower panels: representative pseudocolor images of FRET/CFP ratio show the FRET response. Scale bar, 10 μm.
(D) AMPK activity at each subcellular compartment. WT MEFs and DKO MEFs were transiently transfected with either Cyto-ABKAR (n = 50 in WT MEFs, n = 37 in DKO MEFs), Nuc-ABKAR (n = 49 in WT MEFs, n = 47 in DKO MEFs), PM-ABKAR (n = 41 in WT MEFs, n = 28 in DKO MEFs), Golgi-ABKAR (n = 45 in WT MEFs, n = 31 in DKO MEFs), ER-ABKAR (n = 39 in WT MEFs, n = 33 in DKO MEFs), Mito-ABKAR (n = 47 in WT MEFs, n = 32 in DKO MEFs), Lyso-ABKAR (n = 47 in WT MEFs, n = 35 in DKO MEFs), Cyto-ABKAR (TA) (n = 10 in WT MEFs, n = 12 in DKO MEFs), Nuc-ABKAR (TA) (n = 68 in WT MEFs, n = 59 in DKO MEFs), PM-ABKAR (TA) (n = 29 in WT MEFs, n = 30 in DKO MEFs), Golgi-ABKAR (TA) (n = 28 in WT MEFs, n = 26 in DKO MEFs), ER-ABKAR (TA) (n = 17 in WT MEFs, n = 26 in DKO MEFs), Mito-ABKAR (TA) (n = 20 in WT MEFs, n = 23 in DKO MEFs), or Lyso-ABKAR (TA) (n = 57 in WT MEFs, n = 21 in DKO MEFs). Subsequently FRET signal in WT MEFs to DKO MEFs ratio was calculated from three to four independent experiments. Data are presented as mean ± standard deviation. *p < 0.05, **p < 0.01. ***p < 0.001. N.S., statistically nonsignificant.
Visualization of subcellular compartment-specific AMPK activity
To determine whether these AMPK biosensors changed FRET signal in an AMPK-dependent manner, we transfected osABKAR into either cells derived from AMPK catalytic α subunit-null MEFs (DKO MEFs) (Laderoute et al., 2006) or genetically matched wild-type MEFs (WT MEFs). Cyto-ABKAR, a cytosol-specific AMPK biosensor, indicated that the FRET signal in the cytosol was 1.11 ± 0.17-fold higher than that in DKO MEFs (n = 50 in WT MEFs; n = 37 in DKO MEFs), whereas in the nucleus there was no significant difference in the FRET signal between WT MEFs and DKO MEFs according to the nuclear-specific AMPK biosensor, Nuc-ABKAR (1.04 ± 0.12-fold; n = 49 in WT MEFs; n = 47 in DKO MEFs) (Figure 1C and 1D).
To examine AMPK activity in other subcellular locations, we measured the FRET signal at the plasma membrane, Golgi apparatus, mitochondria, ER, and lysosome using the corresponding osABKAR in WT and DKO MEFs (Figure 1C). Notably, the FRET signal in each endomembrane compartment in WT MEFs was considerably higher than that in DKO MEFs (plasma membrane: 1.26 ± 0.27-fold; n = 41 in WT MEFs; n = 28 in DKO MEFs, Golgi apparatus: 2.19 ± 0.88-fold; n = 45 in WT MEFs; n = 31 in DKO MEFs, ER: 2.08 ± 0.32-fold; n = 39 in WT MEFs; n = 33 in DKO MEFs, mitochondria: 1.67 ± 0.54-fold; n = 47 in WT MEFs; n = 32 in DKO MEFs, lysosome: 1.66 ± 0.45-fold; n = 47 in WT MEFs; n = 35 in DKO MEFs) (Figure 1D). We observed generally little to no correlation between the FRET signal of osABKAR biosensors and their expression level in the range we tested (-0.52 < r < 0.28) (Data not shown). For these FRET measurements, we used an objective lens that was maximally corrected for chromatic aberration to avoid optical artifacts potentially generated at thin membrane structures. We also compared the FRET signals between the WT and DKO MEFs to cancel out any possible variability generated from different signal sequences tagged to individual osABKAR. Interestingly, the AMPK activity exhibited heterogeneity even at each organelle (Figure 1C), which may reflect potential regulation of AMPK activity even within the same compartment. Despite our careful selection, we acknowledge possibilities of imaging artifacts derived from chromatic aberration may exist. More sophisticated microscopy together with a series of experiments would be needed to extend the breadth of exploration of the regulation of AMPK activity at individual organelles. By measuring the ABKAR FRET values in the presence of AMPK dominant mutants, we confirmed that the ABKAR sensors indeed respond to AMPK activity (see Supplementary text).
To test whether the observed FRET signals originate from the phosphorylation at a threonine residue within the AMPK substrate motif, we made a series of osABKAR (TA) biosensors whose threonine residue in the motif was replaced with alanine. As with osABKAR, osABKAR (TA) specifically localized to the intended compartments (Figure S1). However, unlike osABKAR, osABKAR (TA) did not exhibit different FRET values between WT and DKO MEFs (Figure 1D), verifying that the observed compartment specific ABKAR signals indeed reflect AMPK activity, rather than non-specific physical artifacts.
Effect of glucose availability on AMPK activity at membrane compartments
To address if and how the high AMPK activity at the membranes alters based on the availability of nutrients, we examined both localization and phosphorylation of endogenous AMPK at the cytosolic and membrane compartments with or without 2-deoxyglucose (2-DG), an AMPK activator by inhibiting glycolysis (Figure S2A). In agreement with previous studies (Mitchelhill et al., 1997; Oakhill et al., 2010), our fractionation assay indicated that AMPK levels in the membrane compartment were approximately 6-fold higher than the levels in the cytosolic compartment in the absence of 2-DG (Figure S2B and S2C). Although we did not observe detectable relocalization of AMPK upon 2-DG addition (Figure S2B and S2C), we did observe significantly more phosphorylated AMPK in the membranes than in the cytosol (Figure S2B and S2D). These results suggest that AMPK activity is maintained at a high level in the membrane compartment by concentrating the enzymes at the membranes, a feature not much affected by glucose availability.
Measurement of subcellular AMPK activity in response to metabolic inhibition
We next examined whether osABKAR was able to report endogenous AMPK activity at the subcellular compartment level in response to metabolic perturbation. To address this question, WT MEFs were cultured under a glucose-deprived condition that obstructs the ATP synthesizing processes, which results in increased AMPK activity (Figure S2A). In the cytosolic compartment, glucose starvation resulted in an increase in the FRET signal within thirty minutes (1.70 ± 0.61-fold; n = 35) and sustained the signal over four hours (Figure 2A). On the other hand, glucose starvation did not cause an increase in the FRET signal in the nuclear compartment even after four hours (0.95 ± 0.19-fold; n = 44) (Figure 2B). Probing outside the nuclear compartment, we found that glucose starvation gave a distinctive response within thirty minutes at the plasma membrane (1.12 ± 0.23-fold; n = 30) (Figure 2C), Golgi apparatus (1.22 ± 0.36-fold; n = 22) (Figure 2D), and ER (1.13 ± 0.24-fold; n = 37) (Figure 2E). In contrast, FRET signal at mitochondria did not change in thirty minutes (1.03 ± 0.25-fold; n = 32) nor at the two-hour time point (1.07 ± 0.23-fold; n = 29), but significantly increased after four hours (1.34 ± 0.44-fold; n = 25) (Figure 2F). Similar to the mitochondria, the FRET signal at the lysosomal compartment also did not change within thirty minutes (1.05 ± 0.29-fold; n = 26), but increased gradually after two hours (1.20 ± 0.23-fold; n = 30) (Figure 2G).
Figure 2. AMPK activity dynamics at each subcellular compartment in response to metabolic perturbation.
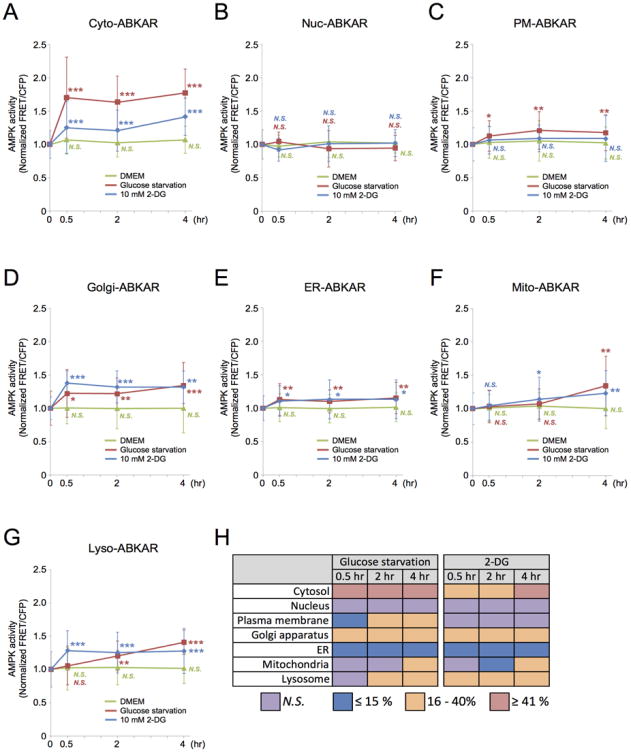
(A-G) WT MEFs transiently transfected with indicated osABKAR were incubated under metabolically perturbed conditions: glucose starvation or 10 mM 2-DG treatment. The FRET/CFP ratio was measured at each indicated time point. Measured FRET/CFP ratio was normalized to the cells incubated with DMEM (time point: 0 hour). Quantification was performed on three independent experiments. All data are presented as mean ± standard deviation.
(A) AMPK activity dynamics in cytosol. WT MEFs expressing Cyto-ABKAR were analyzed at each time point: 0 hour: n = 63; DMEM 0.5 hour: n = 42; DMEM 2 hours: n = 40; DMEM 4 hours: n = 40, glucose starvation 0.5 hour: n = 35; glucose starvation 2 hours: n = 37; glucose starvation 4 hours: n = 31, 2-DG 0.5 hour: n = 38; 2-DG 2 hours: n = 39; 2-DG 4 hours: n = 37.
(B) AMPK activity dynamics in nucleus. WT MEFs expressing Nuc-ABKAR were analyzed at each time point: 0 hour: n = 65; DMEM 0.5 hour: n = 50; DMEM 2 hours: n = 40; DMEM 4 hours: n = 41, glucose starvation 0.5 hour: n = 34; glucose starvation 2 hours: n = 38; glucose starvation 4 hours: n = 44, 2-DG 0.5 hour: n = 44; 2-DG 2 hours: n = 43; 2-DG 4 hours: n = 41.
(C) AMPK activity dynamics at plasma membrane. WT MEFs expressing PM-ABKAR were analyzed at each time point: 0 hour: n = 49; DMEM 0.5 hour: n = 31; DMEM 2 hours: n = 36; DMEM 4 hours: n = 32, glucose starvation 0.5 hour: n = 30; glucose starvation 2 hours: n = 34; glucose starvation 4 hours: n = 27, 2-DG 0.5 hour: n = 25; 2-DG 2 hours: n = 32; 2-DG 4 hours: n = 34.
(D) AMPK activity dynamics at Golgi apparatus. WT MEFs expressing Golgi-ABKAR were analyzed at each time point: 0 hour: n = 38; DMEM 0.5 hour: n = 29; DMEM 2 hours: n = 30; DMEM 4 hours: n = 30, glucose starvation 0.5 hour: n = 22; glucose starvation 2 hours: n = 24; glucose starvation 4 hours: n = 23, 2-DG 0.5 hour: n = 24; 2-DG 2 hours: n = 27; 2-DG 4 hours: n = 28.
(E) AMPK activity dynamics at ER. WT MEFs expressing ER-ABKAR were analyzed at each time point: 0 hour: n = 47; DMEM 0.5 hour: n = 30; DMEM 2 hours: n = 30; DMEM 4 hours: n = 41, glucose starvation 0.5 hour: n = 37; glucose starvation 2 hours: n = 41; glucose starvation 4 hours: n = 29, 2-DG 0.5 hour: n = 37; 2-DG 2 hours: n = 39; 2-DG 4 hours: n = 39.
(F) AMPK activity dynamics at mitochondria. WT MEFs expressing Mito-ABKAR were analyzed at each time point: 0 hour: n = 33; DMEM 0.5 hour: n = 31; DMEM 2 hours: n = 25; DMEM 4 hours: n = 38, glucose starvation 0.5 hour: n = 32; glucose starvation 2 hours: n = 29; glucose starvation 4 hours: n = 25, 2-DG 0.5 hour: n = 33; 2-DG 2 hours: n = 37; 2-DG 4 hours: n = 37.
(G) AMPK activity dynamics at lysosome. WT MEFs expressing Lyso-ABKAR were analyzed at each time point: 0 hour: n = 39; DMEM 0.5 hour: n = 29; DMEM 2 hours: n = 26; DMEM 4 hours: n = 29, glucose starvation 0.5 hour: n = 32; glucose starvation 2 hours: n = 30; glucose starvation 4 hours: n = 30, 2-DG 0.5 hour: n = 32; 2-DG 2 hours: n = 27; 2-DG 4 hours: n = 30.
(H) Differential AMPK activity at each subcellular compartment in response to metabolic perturbation inputs. All analyzed data in Figure 2 were summarized.
*p < 0.05, **p < 0.01. ***p < 0.001. N.S., statistically nonsignificant.
We then looked into the effects of another metabolic perturbation input, 2-DG. Western blot analysis revealed that AMPK activity in cells treated with 10 mM 2-DG or cultured with glucose withdrawn conditions were almost identical as indicated by the phosphorylation level of acetyl-CoA carboxylase (ACC), one of the downstream effectors of AMPK (Figure S3A, B). In stark contrast, 2-DG treatment did not alter FRET signal in nucleic compartment over four hours (1.02 ± 0.2-fold; n = 41) (Figure 2B). Similarly, FRET signal at the plasma membrane did not change after four hours of 2-DG treatment (1.09 ± 0.34-fold; n = 34) (Figure 2C). At the Golgi apparatus, ER, and lysosome, 2-DG stimulation resulted in an immediate response of 1.38 ± 0.18-fold (n =24), 1.11 ± 0.22-fold (n = 39), and 1.28 ± 0.3-fold (n = 29) within thirty minutes, respectively, and the FRET signals were maintained at subsequent time points (Figure 2D, E, and G). Mito-ABKAR showed a robust and sustained response of 1.14 ± 0.33-fold (n = 37) and 1.23 ± 0.34-fold (n = 37) to 2-DG after two and four hours, respectively (Figure 2F). As expected, the FRET signal in each compartment was rather stable when WT MEFs were cultured in nutrient abundant conditions (i.e., DMEM conditions in Figure 2). Additionally, we ascertained that the FRET signal in each compartment did not change in DKO MEFs cultured with nutrient-rich or metabolic perturbation conditions during the experiments (Figure S3C-I). Furthermore, we confirmed that neither 2-DG treatment nor glucose starvation resulted in changes in FRET signal in WT MEFs expressing Cyto-ABKAR (TA) (Figure 3A). We also measured intracellular pH under these conditions to find little to no pH change after 30 minutes (Figure 3B) as well as during the 30 minutes (Figure 3C, D). Collectively, the observation of the distinct FRET changes at different organelles indicates that AMPK activity under distinctive metabolic impediments is spatiotemporally diverse and osABKAR was a faithful indicator of these real-time events in each compartment by responding to AMPK-mediated phosphorylation (Figure 2H).
Figure 3. Glucose starvation and 2-DG treatment does not affect intracellular pH level.
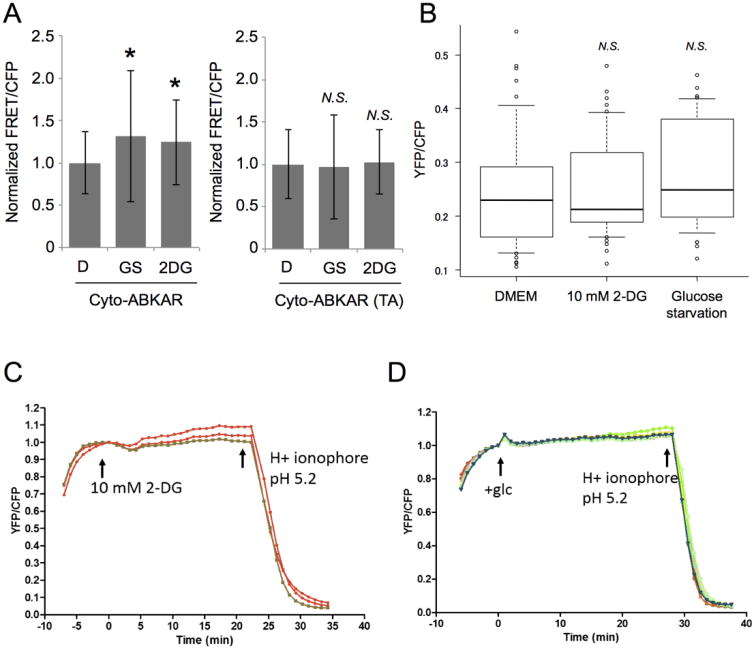
(A) WT MEFs were transiently transfected with either Cyto-ABKAR or Cyto-ABKAR (TA), and then AMPK activity was subsequently measured in cytosol under indicated condition after thirty minutes. D: DMEM. GS: glucose starvation. 2DG: 10 mM 2-DG. WT MEFs expressing either Cyto-ABKAR in DMEM (n = 37), in glucose-starved DMEM (n = 46), and in the presence of 2-DG (n = 48) or Cyto-ABKAR (TA) in DMEM (n = 28), in glucose-starved DMEM (n = 8), and in the presence of 2-DG (n = 19) were analyzed. Measured FRET/CFP ratio was normalized to the cells incubated with DMEM. Quantification was performed on three independent experiments. Data are presented as mean ± standard deviation.
(B) Intracellular pH level under same condition in Figure 3A (DMEM: n = 46, 2-DG: n = 42, and Glucose starvation: n = 39) was measured by pH biosensor in WT MEFs.
(C) Intracellular pH level in WT MEFs was monitored by time-lapse imaging. 10 mM 2-DG and H+ ionophore were added at indicated time point. n = 3.
(D) Intracellular pH level in WT MEFs was monitored by time-lapse imaging. Fresh DMEM containing glucose and H+ ionophore were added at indicated time point. n = 9.
*p < 0.05, N.S., statistically nonsignificant.
Differential kinase activity of the AMPK a subunit in each subcellular compartment
Different phenotypes have been observed between α1 and α2 knockout mice (Viollet B, 2009), making us speculate the difference may arise from distinct roles of these AMPK subunits and may underlie the observed spatially unique AMPK activity. We therefore set out to determine whether AMPK α1 and α2 subunits were able to reconstitute AMPK activity at each subcellular compartment in DKO MEFs. We first examined whether mChF-fused AMPK α1 and α2 subunits maintain kinase activity. We observed that both mChF-AMPK α1 and α2 subunits were diffusive in cells (Figure S4A). ABKAR showed that 2-DG stimulation resulted in 1.62 ± 0.24-fold increase of FRET signal in WT MEFs expressing mChF (n = 10) while no 2-DG stimulated response was observed in DKO MEFs expressing mChF (0.91 ± 0.25-fold, n = 9) (Figure S4B and S4C). However, the response of the biosensor in the cytosol of DKO MEFs expressing either mChF-AMPK α1 or α2 subunit was similar to 2-DG stimulation as the WT MEFs (α1: 1.41 ± 0.34-fold, n = 11, α2: 1.67 ± 0.52-fold, n = 9)(Figure S4B and S4C), revealing that reconstituted mChF-AMPK α1 and α2 subunits were functional. Interestingly, the expression level of mChF-AMPK α1 and mChF-AMPK α2 did not seem to affect the results within the range of protein expression we tested (Figure S4D). We then transiently transfected either mChF-AMPK α1 or α2 in either WT MEFs or DKO MEFs, and measured FRET signals in each compartment where basal AMPK activity in WT MEFs was higher than that in DKO MEFs (Figure 1D). Both AMPK α1 and α2 subunits recovered FRET signal to almost equivalent degree in the cytosolic compartment as judged by Cyto-ABKAR (Figure 4A). As was the case with the cytosol, FRET signals in ER and mitochondria in DKO MEFs were recovered by the expression of AMPK α subunits to the same degree (Figure 4D and 4E). Interestingly, reconstitution of AMPK α1, but not the α2 subunit recovered the FRET signal at the plasma membrane (Figure 4B). The functional difference of AMPK α subunits was also observed in the Golgi apparatus and lysosome, where the FRET signal was increased significantly more by the AMPK α1 subunit compared to the α2 subunit (Figure 4C and 4F). There was not any appreciable difference in the expression level of the AMPK α1 and α2 subunit in each experiment (Data not shown). Taken together, these results indicate that the FRET signal reported by osABKAR in each subcellular compartment was dynamically changeable in an AMPK α subunit-dependent manner. In addition, each of the AMPK α1 and α2 subunits apparently has a distinctive territory despite their overlapping functions.
Figure 4. Activity difference of AMPK a subunits at each subcellular compartment.
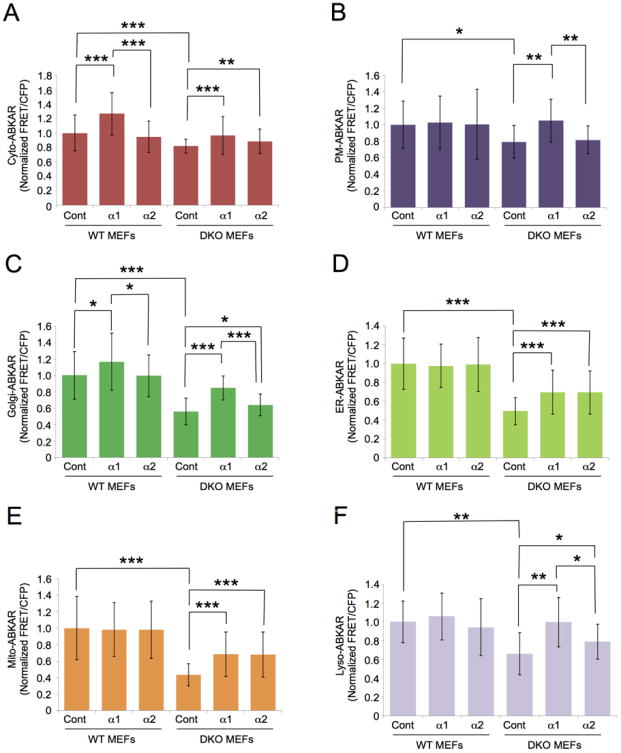
(A-F) Indicated osABKAR was co-transfected with mChF (Cont), mChF-AMPK α1 subunit (α1), or mChF-AMPK α2 subunit (α2) into either WT MEFs or DKO MEFs, then FRET/CFP ratio was measured under nutrient surplus condition. Measured FRET/CFP ratio was normalized to the WT MEFs expressing mChF (WT MEFs-Cont). Quantification was performed on three independent experiments. All data are presented as mean ± standard deviation.
(A) Reconstituted AMPK activity in cytosol. AMPK activity in cytosol in cells expressing Cyto-ABKAR and either mChF (WT cont: n = 56, DKO cont: n = 34), mChF-AMPK α1 (WT α1: n = 38, DKO α1: n = 45), or mChF-AMPK α2 (WT α2: n = 57, DKO α2: n = 49) were analyzed.
(B) Reconstituted AMPK activity at plasma membrane. AMPK activity at plasma membrane in cells expressing PM-ABKAR and either mChF (WT cont: n = 29, DKO cont: n = 16), mChF-AMPK α1 (WT α1: n = 20, DKO α1: n = 25), or mChF-AMPK α2 (WT α2: n = 33, DKO α2: n = 16) were analyzed.
(C) Reconstituted AMPK activity at Golgi apparatus. AMPK activity at Golgi apparatus in cells expressing Golgi-ABKAR and either mChF (WT cont: n = 33, DKO cont: n = 27), mChF-AMPK α1 (WT α1: n = 33, DKO α1: n = 31), or mChF-AMPK α2 (WT α2: n = 34, DKO α2: n = 31) were analyzed.
(D) Reconstituted AMPK activity at ER. AMPK activity at ER in cells expressing ER-ABKAR and either mChF (WT cont: n = 48, DKO cont: n = 36), mChF-AMPK α1 (WT α1: n = 55, DKO α1: n = 47), or mChF-AMPK α2 (WT α2: n = 43, DKO α2: n = 33) were analyzed.
(E) Reconstituted AMPK activity at mitochondria. AMPK activity at mitochondria in cells expressing Mito-ABKAR and either mChF (WT cont: n = 56, DKO cont: n = 36), mChF-AMPK α1 (WT α1: n = 46, DKO α1: n = 37), or mChF-AMPK α2 (WT α2: n = 46, DKO α2: n = 38) were analyzed.
(F) Reconstituted AMPK activity at lysosome. AMPK activity at lysosome in cells expressing Lyso-ABKAR and either mChF (WT cont: n = 52, DKO cont: n = 28), mChF-AMPK α1 (WT α1: n = 44, DKO α1: n = 36), or mChF-AMPK α2 (WT α2: n = 37, DKO α2: n = 24) were analyzed. *p < 0.05, **p < 0.01. ***p < 0.001.
Development of a genetically encoded AMPK inhibitor peptide
Determining causality of the observed spatiotemporally dynamic AMPK regulation necessitates organelle specific perturbation of AMPK activity. A small molecule, Compound C (6-[4-(2-Piperidin-1-ylethoxy) phenyl]-3-pyridin-4-ylpyrazolo [1,5-a]pyrimidine), is currently the only known cell-permeable agent that can inhibit AMPK in an ATP-competitive manner. Though useful as a pan-AMPK inhibitor, Compound C cannot suppress AMPK activity at specific subcellular compartments due to its high diffusivity. Therefore, we developed a new genetically encoded AMPK inhibitor peptide, termed AIP, which allows for inhibition of AMPK activity at specific subcellular locations. The amino acid sequence of AIP is similar to the substrate peptide used in ABKAR (Figure 5A), and contains an AMPK recognition motif with a phosphorylation site. To examine whether AIP is able to inhibit AMPK activity, we performed an in vitro AMPK activity assay, whereby a known AMPK substrate peptide (HMRSAMSGLHLVKRR) was used as a glutathione-S-transferase (GST) conjugate (GST-SAMS). As a result, AIP suppressed AMPK-mediated phosphorylation of GST-SAMS peptide competitively in a concentration-dependent manner with an IC50 value of 76 μM (Figure 5B and 5C). We next examined the inhibitory effect of AIP (TA) peptide, whose phosphorylation site was replaced with Ala residue (Figure 5A). In vitro AMPK activity assay showed that the inhibitory effect of AIP (TA) was lower than that of AIP (IC50 = 277 μM vs. 76 μM), but efficiently suppressed AMPK activity at the high concentration (Figure 5B and 5C). We confirmed that AIP, but not AIP (TA) peptide, was phosphorylated by AMPK (Figure S5A), implying that while AIP inhibits AMPK in a substrate-competitive manner, AIP (TA) works in a pseudosubstrate-competitive manner. The inhibitory effect of AIP was robust and reasonable, but not as strong as that of a known AMPK inhibitor, Compound C, whose IC50 was 3 μM when measured in the same in vitro AMPK assay (Fig. S5B). Next, we compared inhibitory effects on AMPK activity generated by AIP and Compound C. As a result, we observed that the inhibitory effect of mChF-fused AIP (mChF-AIP) and 40 μM Compound C on AMPK activity at Golgi apparatus, for example, was nearly equivalent in WT MEFs (21.4 ± 10.1% decrease, n = 11, vs. 21.1 ± 11.2% decrease, n = 9, respectively) as reported by Golgi-ABKAR (Figure S5C). AMPK activity at Golgi apparatus was synergistically inhibited in WT MEFs expressing mChF-AIP in the presence of Compound C (34.1 ± 13.3% decrease, n = 8) (Figure S5C).
Figure 5. Development of genetically encoded AMPK inhibitor peptide.
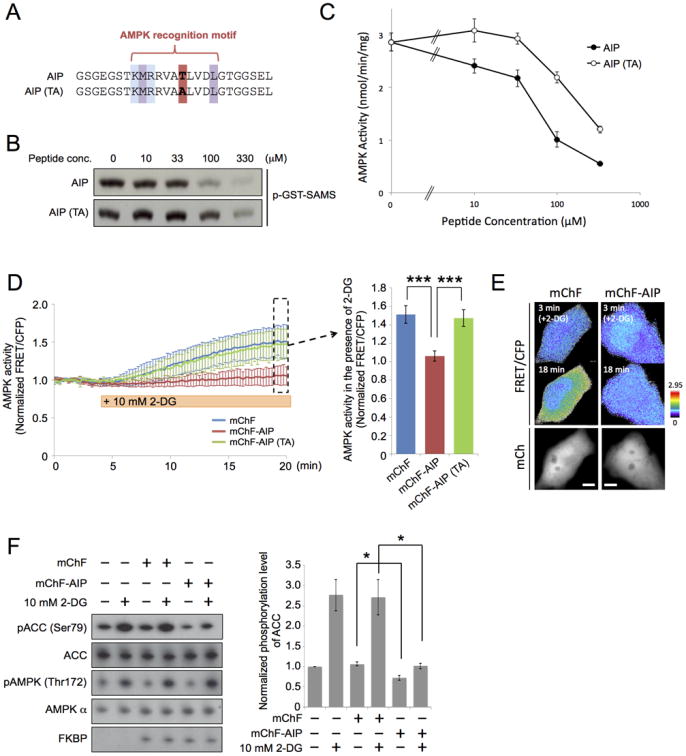
(A) The amino acid sequence alignment of AIP and AIP (TA).
(B) Inhibitory effect of AIP as judged by in vitro AMPK activity assay. Purified activated AMPK and GST-SAMS was incubated with indicated concentration of either AIP or AIP (TA). Representative autoradiography image of phosphorylated GST-SAMS is shown.
(C) Data of in vitro AMPK activity assay performed in Figure 5B are presented as mean ± standard deviation. Quantification was performed on three independent experiments.
(D) Inhibitory effect of AIP as judged by ABKAR. Cos7 cells expressing ABKAR and either mChF (n = 15), mChF-AIP (n = 10), or mChF-AIP (TA) (n = 11) were treated with 10 mM 2-DG. Left: mean of normalized FRET/CFP ratio ± standard deviation. Right: mean of last 5 frames (surrounded by a dotted line) ± standard deviation. ***p < 0.001. 15 sec/frame.
(E) Representative pseudocolor image of FRET/CFP ratio in cells expressing either mChF or mChF-AIP are shown.
(F) Inhibitory effect of AIP according to western blot assay. WT MEFs transiently transfected with either mChF or mChF-AIP were treated with or without 2-DG. Subsequently western blot was performed to determine the phosphorylation level of ACC and AMPK. Left: representative western blot. Right: Quantified phosphorylation level of ACC under indicated condition. Phosphorylation level of ACC was normalized to the non-treated cells expressing mChF. Data are presented as mean ± standard error from three independent experiments. *p < 0.05.
To examine whether AIP suppresses AMPK signaling in the presence of 2-DG, we performed time-lapse imaging with Cos7 cells expressing ABKAR. Stimulation with 2-DG in cells expressing mChF resulted in a 51.2 ± 9.4% response after fifteen minutes (n = 15) (Figure 5D and 5E). Notably, cells expressing mChF-AIP did not respond to 2-DG treatment (6.2 ± 5.6%, n = 10), indicating that mChF-AIP competitively attenuated AMPK activity in the presence of 2-DG (Figure 5D and 5E). On the other hand, mChF-AIP (TA) could not suppress 2-DG induced AMPK activation as judged by ABKAR (47.4 ± 8.9%, n = 11) (Figure 5D). It is of note here that the expression level of mChF-AIP was similar to mChF and mChF-AIP (TA) (Data not shown). To exclude the possibility that the direct interaction of phosphorylated mChF-AIP with FHA domain in ABKAR is the cause of FRET signal reduction, WT MEFs were transfected with phospho-mimic AIP peptide: mChF-AIP (TD) and mChF-AIP (TE), whose phosphorylation site was replaced with Asp and Glu, respectively (Figure S6A). All constructs showed a diffuse localization pattern in the cytosol when expressed in cells (Figure S6B). Among these three peptides, mChF-AIP was the only one that suppressed AMPK activity in WT MEFs as judged by ABKAR (Figure S6C and S6D). To further characterize the inhibitory effect of AIP, we transiently expressed mChF-AIP in WT MEFs, and then checked the phosphorylation status of endogenous ACC. mChF-AIP expression efficiently suppressed 2-DG-induced AMPK activation as detected by the phosphorylation level of ACC (Figure 5F). As an additional note, basal phosphorylation of ACC was also suppressed to some extent. These results suggest that mChF-AIP suppresses phosphorylation activity of AMPK in living cells.
Perturbation of AMPK signaling at specific subcellular compartments
Next, we assessed the functionality of AIP as an AMPK inhibitor. Defective AMPK signaling leads to an increase of intracellular ATP level in a mTORC1-dependent or -independent manner (Faubert et al., 2013). In agreement with this, we also observed that the concentration of intracellular ATP in DKO MEFs was 1.18 ± 0.28-fold higher than that in WT MEFs as judged by the ATP biosensor (Imamura et al., 2009) (Figure 6A). To examine whether AIP is able to mimic this phenotype, we expressed mChF, mChF-AIP, or mChF-AIP (TA) in cells. As a result, the mChF-AIP expression increased intracellular ATP in WT MEFs (1.23 ± 0.29-fold, n = 29) compared to the cells expressing mChF (Figure 6B). The expression of mChF-AIP (TA) did not alter intracellular ATP in WT MEFs (1.05 ± 0.24-fold, n = 20) (Figure 6B). We also confirmed that expression of mChF-AIP did not change intracellular ATP in DKO MEFs as reported by ATP biosensor (Figure 6B).
Figure 6. Manipulation of intracellular ATP level by subcellular compartment-specific AMPK signaling perturbation.
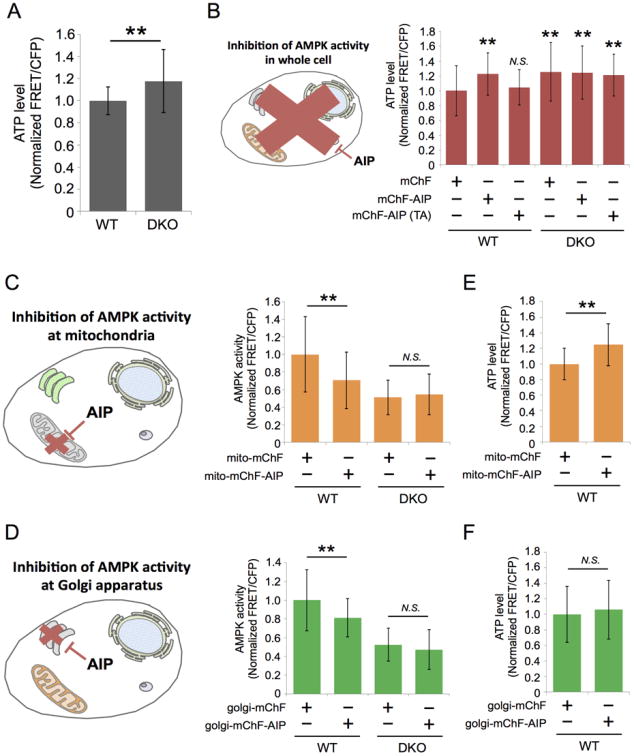
(A) ATP biosensor was transiently transfected in WT MEFs (n = 40) and DKO MEFs (n = 34). 48 hours after transfection, intracellular ATP level was measured from three independent experiments. Analyzed FRET/CFP ratio was normalized to the data obtained from WT MEFs. Quantification was performed on three independent experiments. Data are presented as mean ± standard deviation. **p < 0.01
(B) The effect of AIP on intracellular ATP level. Cytosolic ATP level in cells expressing either mChF (WT: n = 27, DKO: n = 40), mChF-AIP (WT: n = 29, DKO: n = 40), or mChF-AIP (TA) (WT: n = 20, DKO: n = 36) was measured under nutrient rich conditions. ATP level was normalized to that of WT MEFs expressing mChF. Quantification was performed on three independent experiments. Data are presented as mean ± standard deviation.
(C) The effect of mito-mChF-AIP on AMPK activity at mitochondria. WT MEFs or DKO MEFs were transiently transfected with either mito-mChF (WT: n = 40, DKO: n = 27) or mito-mChF-AIP (WT: n = 34, DKO: n = 20), then AMPK activity was measured at the mitochondria under nutrient surplus condition. Quantification was performed on three independent experiments. Data are presented as mean ± standard deviation.
(D) The effect of Golgi-mChF-AIP on AMPK activity at Golgi apparatus. WT MEFs or DKO MEFs were transiently transfected with either Golgi-mChF (WT: n = 64, DKO: n = 40) or Golgi-mChF-AIP (WT: n = 43, DKO: n = 29), then AMPK activity was measured at Golgi apparatus under nutrient surplus condition. Quantification was performed on three independent experiments. Data are presented as mean ± standard deviation.
(E) The effect of mito-mChF-AIP on intracellular ATP level. WT MEFs were transiently transfected with either mito-mChF (n = 29) or mito-mChF-AIP (n = 30), then ATP level was measured in cytosol by ATP biosensor. Quantification was performed on three independent experiments. Data are presented as mean ± standard deviation.
(F) The effect of Golgi-mChF-AIP on intracellular ATP level. WT MEFs were transiently transfected with either Golgi-mChF (n = 63) or Golgi-mChF-AIP (n = 52), then ATP level was measured in cytosol by ATP biosensor. Quantification was performed on three independent experiments. Data are presented as mean ± standard deviation.
**p < 0.01. N.S., statistically nonsignificant.
We then tested whether spatially specific perturbation of AMPK signaling yields this phenotype. Since mitochondria play a crucial role in ATP synthesis, we hypothesized that perturbation of AMPK signaling at the mitochondria affects intracellular ATP levels. Therefore we developed mitochondria-specific AIP (mito-mChF-AIP) (Figure S7A). Likewise, we designed Golgi apparatus-specific AIP (golgi-mChF-AIP) as a control for mito-mChF-AIP (Figure S7A). We ensured their localization in WT MEFs using the appropriate organelle markers (Figure S7B). To examine the inhibitory effect of these organelle-specific AIPs, we transiently expressed these constructs in WT MEFs with the appropriate osABKAR. Mito-ABKAR and Golgi-ABKAR showed that mito-mChF-AIP and Golgi-mChF-AIP suppressed AMPK activity at the mitochondria (29.4 ± 32% decrease in WT MEFs) (Figure 6C) and Golgi apparatus (18.7 ± 20.4% decrease in WT MEFs) (Figure 6D), respectively. These organelle-specific AIPs did not affect overall AMPK activity as judged by cytosolic ABKAR, indicating that the inhibitory effect of mito-mChF-AIP and Golgi-mChF-AIP was confined to mitochondria and Golgi apparatus, respectively (Figure S7C and S7D). We next examined the effect of the subcellular compartment-specific perturbation of AMPK signaling on the intracellular ATP level. WT MEFs expressing mito-mChF-AIP exhibited a 1.25 ± 0.39-fold increase of intracellular ATP as shown using the ATP biosensor (Figures 6E). On the other hand, WT MEFs expressing Golgi-mChF-AIP did not show any change in intracellular ATP levels (Golgi-mChF-AIP: 1.06 ± 0.37-fold) (Figure 6F). Mito-mChF-AIP and Golgi-mChF-AIP expression did not affect intracellular ATP levels in DKO MEFs (Figure S7E and S7F). Taken together, these results suggested that spatio-specific perturbation of AMPK signaling could both qualitatively and quantitatively reproduce the phenotype that we observed by overall AMPK inhibition, implying that AMPK inhibition at mitochondria is primarily responsible for the ATP homeostasis in cells.
Spatiotemporal regulation of AMPK activity at specific subcellular compartments
Chemically inducible dimerization (CID) system is a powerful tool to rapidly manipulate spatiotemporal information of target molecules in cells (DeRose et al., 2013). We therefore examined whether AIP is able to regulate AMPK activity in differential subcellular compartments on a timescale of seconds by employing the CID system. Here, a chemical dimerizer like rapamycin induces striking concentration of diffusive FKBP-fused protein such as mChF-AIP exclusively to one of the organelle surfaces within seconds (Figure 7A). In the following experiments, we transiently transfected mChF, mChF-AIP, or mChF-AIP (TA) in HEK293 epithelial cells together with a mitochondria-anchored FRB (Tom20-CFP-FRB), then monitored AMPK activity at mitochondria using mito-ABKAR before and after rapamycin addition. While the expression of mChF-AIP slightly suppressed basal AMPK activity at mitochondria, neither mChF nor mChF-AIP (TA) exhibited this inhibition (Figure 7B). As expected, mChF translocation to mitochondria did not affect AMPK activity at the mitochondria (6.2 ± 7.8% decrease; n = 39, thirty minutes) (Figure 7C and 7D). However translocation of mChF-AIP and mChF-AIP (TA) to mitochondria resulted in significant decrease of AMPK activity at the mitochondria (16.5 ± 9.8% decrease, n = 35 and 20.4 ± 23.3% decrease, n = 34, respectively) (Figure 7C and 7D). It is notable that AIP (TA) indicated very effective suppression of AMPK activity despite its little to no effect on the kinase prior to its translocation to the mitochondria. Conversely, wild type AIP requires precautions in this usage in cells due to the background effect. These results suggest that AIP (TA) is a powerful tool to rapidly and inducibly perturb AMPK signaling at a specific subcellular compartment. As these rapidly inducible experiments reproduced the earlier result obtained with “constitutive” inhibition of AMPK using mito-mChF-AIP, we may exclude the possibility that the Mito-ABKAR FRET signal decreased due to AMPK down-regulation triggered by the chronic accumulation of an AMPK substrate (i.e., AIP) at mitochondria, rather than the direct competition for AIP.
Figure 7. Spatiotemporal regulation of AMPK activity by CID system.
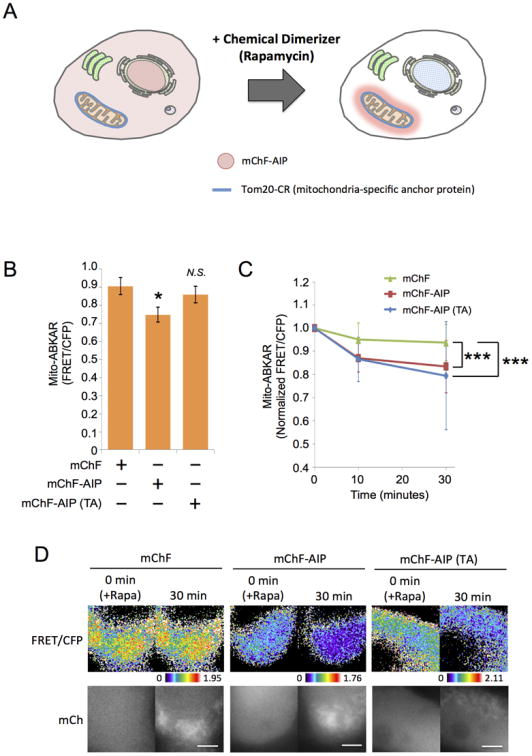
(A) Schematic diagram of CID system. Since cells express anchor protein, Tom20-CFP-FRB (Tom20-CR), diffusive FKBP-fused peptide (mChF-AIP) (left) was trapped at mitochondria in the presence of rapamycin (right).
(B) The effect of AIP and AIP (TA) on AMPK activity at mitochondria. HEK293 cells were transiently transfected with mito-ABKAR, Tom20-CR as anchor protein, and either mChF (n = 39), mChF-AIP (n = 35), or mChF-AIP (TA) (n = 34). Subsequently, AMPK activity at mitochondria was monitored before adding rapamycin. *p < 0.05. N.S., statistically nonsignificant.
(C) Inhibitory effect of sequestered AIP at mitochondria on AMPK activity. AMPK activity at mitochondria in HEK293 cells described in Figure 7B was monitored before (0 minute) and after adding rapamycin at each time points. AMPK activity at each time point was normalized to that in 0 minute. Data are presented as normalized mean ± standard deviation from two independent experiments. ***p < 0.001.
(D) Representative image of Figure 7C were shown. Upper panel: FRET/CFP. Lower panel: mCherry image. Scale bar, 10 μm.
Discussion
Fluorescent biosensors that can monitor signaling events in living cells have become an indispensable tool for gaining deeper insights into spatiotemporally dynamic signal transduction. Here, we designed, characterized and implemented a series of AMPK biosensors specifically targeted to individual organelles (osABKAR) to measure AMPK activity at designated subcellular regions of interest. In the present study, we demonstrated the unique osABKAR FRET signals distinctly changing in an AMPK-dependent manner at each subcellular location, revealing that AMPK activity at the membranes was higher than that in the cytosol under both basal and nutrient-rich conditions. Given that AMPK is able to associate with endomembrane (Mitchelhill et al., 1997; Oakhill et al., 2010), at least partially explains why AMPK activity is basally higher in membrane-bound organelles. In apparent contrast, Golgi-specific Brefeldin A resistance factor 1, which localizes to Golgi apparatus, is barely phosphorylated by AMPK even under nutrient-rich conditions (Miyamoto et al., 2008; Morohashi et al., 2010), as its phosphorylation level increases in response to ATP depletion (Miyamoto et al., 2008) or during mitosis (Mao et al., 2013; Morohashi et al., 2010). This preliminary phosphorylation of AMPK substrates at the organelle membranes might facilitate execution of AMPK-mediated acute responses to transient changes in environmental factors.
The glycolysis pathway plays an essential role in ATP synthesis. Both glucose starvation and 2-DG treatment cause metabolic perturbation at the first step of glycolysis, followed by a decrease in the intracellular ATP level. Since ATP is an essential molecule for virtually all cellular processes, the intracellular ATP level is tightly controlled by AMPK (Hardie, 2011). We found that a different input signal, glucose starvation or 2-DG simulation, leads to distinct patterns of signal propagation to increase AMPK activity in a subcellular location-dependent manner. The molecular mechanism of this difference induced by these seemingly similar metabolic perturbations is not clear, but one speculation would be that the perturbations result in different metabolite profiles (Sandulache et al., 2011), which may be in turn translated into AMPK activity at different subcellular locations. Alternatively, LKB1 may involve input-dependent AMPK activation at each organelle in response to these input stimuli. Interestingly, in agreement with a previous report (Tsou et al., 2011), glucose starvation and 2-DG treatment did not increase AMPK activity in the nucleus even under prolonged treatment. Given that other pharmacological AMPK activators are able to increase AMPK activity at specific subcellular locations, including nucleus (Kodiha et al., 2011; Tsou et al., 2011), additional input signals could be required to promote AMPK activation in the nucleus under the metabolically perturbed conditions we examined. Taken together, we speculate that AMPK continuously processes a multitude of input signals to make appropriate decisions among which are changes in gene expression, enzymatic activity, and rewiring of their signaling networks. Future studies may reveal the interplay between the spatio-temporally dynamic AMPK activity and its downstream cell functions.
One protein complex could play multiple roles by changing the number and/or members of its modular constituents. AMPK is a heterotrimeric complex, composed of α, β, and γ subunits (Shackelford and Shaw, 2009). Given the multiple subfamily members for each AMPK subunit, 12 different heterotrimeric enzymes are theoretically possible. However, the mechanism of functional distinctions among these AMPK complexes remains unknown. Here we showed that α subunits can rescue AMPK activity at individual organelles of DKO MEF cells, but the recovery efficiency varied depending on the type of α subunits as well as the compartments. This finding implies, among others, that α1- and α2-containing-AMPK complexes may be differentially regulated, and may thus regulate separate downstream effectors. While this notion does not contradict the observation of different phenotypes between α1 and α2 knockout mice (Viollet B, 2009), we observed little to no accumulation of the AMPK α subunit at organelle surfaces, contrary to our prediction. Future studies are therefore necessary to resolve the molecular mechanism underlying these observations.
It was mechanistically insightful to learn that, while both AIP and AIP (TA) peptide were able to perturb AMPK signaling when accumulated at mitochondria, only AIP (TA) exhibited a negligible background inhibition prior to accumulation. The difference appears to be logically explained by the shifted dose response curves obtained from the in vitro kinase assay (Fig. 5C). This speculation further suggests that wild type AIP could readily become a useful tool after optimizing its expression level in cells. In theory, a concurrent usage of AIP and AIP (TA) should allow us to manipulate AMPK activity at two different subcellular locations. While assessment of off-target inhibition by AIP should be performed, our results collectively suggest that genetically encoded AIP is a valuable tool to inhibit AMPK specifically at an intended subcellular location. Future studies may enable the precise control of cellular functions through the manipulation of AMPK activity at specific subcellular locations.
In conclusion, we have designed and characterized a genetically encoded organelle-specific FRET based biosensor, osABKAR, providing a real-time readout of AMPK activity exclusively at intended subcellular locations in a native context. Furthermore, we developed a genetically encoded competitive AMPK inhibitor, AIP, which enables perturbation of AMPK signaling at specific cellular locations within minutes. A pair of these molecular sensors and actuators offers a platform to disentangle compartmentalized AMPK signaling under both physiological and pathological conditions. Not only the knowledge obtained with these molecular tools, but also the tools themselves to harness the dynamic AMPK activity, may eventually lead to novel insights into treatment and/or cure of cancers and diabetes where AMPK regulation has gone awry.
Experimental Procedures
Detailed experimental procedures are provided in the Supplemental Experimental Procedures.
Plasmid Construction
Organelle-specific ABKAR was generated by fusing organelle-targeting signal with ABKAR. Details regarding organelle specific ABKAR and other constructs used here are provided in the Supplemental Experimental Procedures. All constructs were verified by sequence after subcloning.
Live-cell Imaging
Cells plated on cover glass were subjected to the live-cell imaging. For imaging under nutrient-rich condition, DMEM medium containing 25 mM HEPES, 25 mM glucose, and 2 mM glutamine (Gibco) was used. For imaging with cells under glucose starvation and 2-DG treatment, phenol red-free glucose withdrawal medium (Gibco) and 10 mM 2-DG containing DMEM medium containing 25 mM HEPES, 25 mM glucose, and 2 mM glutamine were used. For Compound C treatment, cells expressing AMPK biosensor were treated with 40 μM Compound C for 30 minutes before imaging. Fluorescent images were obtained using epifluorescence microscopy. Details regarding microscope setting are provided in the Supplemental Experimental Procedures. For time-lapse image, fluorescence images were taken every 15 seconds for 20 minutes. Images were normally collected for 5 minutes before addition of 2-DG, and then were collected for 15 minutes after addition of the drug. FRET between CFP and YFP was normalized to the mean of the reading for the first five time points. Mean was calculated from three to four independent experiments. Details regarding measurement of intracellular pH are provided in the Supplemental Experimental Procedures.
Statistical analysis
Statistical analysis was done with an unpaired two-tailed Student's t-test. F-test was used to determine whether variances were equal or not.
Supplementary Material
Acknowledgments
We are grateful to Yusuke Kageyama, Qing Li, and Xin Zhou for assistance and insightful suggestions on the experiments. We also thank Shiva Razavi and Atsuo Sasaki for critical comments on the manuscript. This work was supported by the National Institute of Health (NIH) (GM092930, DK102910, CA103175 and DK089502 to TI, and DK073368 and CA174423 to JZ), a Grant-in-aid for Scientific Research (26440056 to HT) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and the Japan Science and Technology Agency (10216 to TI). T.M. is a recipient of a fellowship from the Japanese Society for the Promotion of Science.
Footnotes
Accession numbers: AMPK α1: AAC52355
AMPK α2: AAA85033
HIV1-Rev: AGO06240.1
SV40 large T-antigen: CBL95275
Tyrosine-protein kinase Lyn: XP_004313862
Nitric oxide synthase, endothelial: XP_004702531.1
Cytochrome P450 2C1: AAA31436
A kinase anchor protein 1: AAC27100
LAMP1: NP_005552
Tom20: NP_055580
Supplemental Information: Supplemental Information including experimental procedures, text, seven figures, and one table can be found with this article online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antal CE, Newton AC. Spatiotemporal dynamics of phosphorylation in lipid second messenger signaling. Mol Cell Proteomics. 2013;12:3498–3508. doi: 10.1074/mcp.R113.029819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bivona TG, Philips MR. Ras pathway signaling on endomembranes. Curr Opin Cell Biol. 2003;15:136–142. doi: 10.1016/s0955-0674(03)00016-4. [DOI] [PubMed] [Google Scholar]
- Boehlke C, Kotsis F, Patel V, Braeg S, Voelker H, Bredt S, Beyer T, Janusch H, Hamann C, Gödel M, et al. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat Publ Gr. 2010;12:1115–1122. doi: 10.1038/ncb2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRose R, Miyamoto T, Inoue T. Manipulating signaling at will: chemically-inducible dimerization (CID) techniques resolve problems in cell biology. Pflugers Arch. 2013;465:409–417. doi: 10.1007/s00424-012-1208-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17:113–124. doi: 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–1908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Ross Fa, Hawley Sa. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley Sa, Pan Da, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Hurley RL, Anderson Ka, Franzone JM, Kemp BE, Means AR, Witters La. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–29066. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- Imamura H, Huynh KP, Togawa H, Saito K, Iino R, Kato-yamada Y. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based. Proc Natl Acad Sci USA. 2009;106:15651–15656. doi: 10.1073/pnas.0904764106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Kodiha M, Rassi JG, Brown CM, Stochaj U. Localization of AMP kinase is regulated by stress, cell density, and signaling through the MEK 3 ERK1 / 2 pathway. Am J Physiol Cell Physiol. 2007:1427–1436. doi: 10.1152/ajpcell.00176.2007. [DOI] [PubMed] [Google Scholar]
- Kodiha M, Ho-wo-cheong D, Stochaj U. Pharmacological AMP-kinase activators have compartment-specific effects on cell physiology. Am J Physiol Cell Physiol. 2011:1307–1315. doi: 10.1152/ajpcell.00309.2011. [DOI] [PubMed] [Google Scholar]
- Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, Foretz M, Viollet B. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol. 2006;26:5336–5347. doi: 10.1128/MCB.00166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao L, Li N, Guo Y, Xu X, Gao L, Xu Y, Zhou L, Liu W. AMPK phosphorylates GBF1 for mitotic Golgi disassembly. J Cell Sci. 2013;126:1498–1505. doi: 10.1242/jcs.121954. [DOI] [PubMed] [Google Scholar]
- Marshall S. Role of insulin, adipocyte hormones, and nutrient-sensing pathways in regulating fuel metabolism and energy homeostasis: a nutritional perspective of diabetes, obesity, and cancer. Sci STKE. 2006;2006:re7. doi: 10.1126/stke.3462006re7. [DOI] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchelhill KI, Michell BJ, House CM, Stapleton D, Dyck J, Gamble J, Ullrich C, Witters La, Kemp BE. Posttranslational Modifications of the 5′-AMP-activated Protein Kinase 1 Subunit. J Biol Chem. 1997;272:24475–24479. doi: 10.1074/jbc.272.39.24475. [DOI] [PubMed] [Google Scholar]
- Miyamoto T, Oshiro N, Yoshino K, Nakashima A, Eguchi S, Takahashi M, Ono Y, Kikkawa U, Yonezawa K. AMP-activated protein kinase phosphorylates Golgi-specific brefeldin A resistance factor 1 at Thr1337 to induce disassembly of Golgi apparatus. J Biol Chem. 2008;283:4430–4438. doi: 10.1074/jbc.M708296200. [DOI] [PubMed] [Google Scholar]
- Morohashi Y, Balklava Z, Ball M, Hughes H, Lowe M. Phosphorylation and membrane dissociation of the ARF exchange factor GBF1 in mitosis. Biochem J. 2010;427:401–412. doi: 10.1042/BJ20091681. [DOI] [PubMed] [Google Scholar]
- Oakhill JS, Chen ZP, Scott JW, Steel R, Castelli La, Ling N, Macaulay SL, Kemp BE. β-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK) Proc Natl Acad Sci U S A. 2010;107:19237–19241. doi: 10.1073/pnas.1009705107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakhill JS, Steel R, Chen ZP, Scott JW, Steel R, Ling N, Tam S, Kemp BE. AMPK Is a Direct Adenylate Charge-Regulated Protein Kinase. Science. 2011;332:1433–1435. doi: 10.1126/science.1200094. [DOI] [PubMed] [Google Scholar]
- Sandulache VC, Ow TJ, Pickering CR, Frederick MJ, Zhou G, Fokt I, Davis-Malesevich M, Priebe W, Myers JN. Glucose, not glutamine, is the dominant energy source required for proliferation and survival of head and neck squamous carcinoma cells. Cancer. 2011;117:2926–2938. doi: 10.1002/cncr.25868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Okamoto S, Lee S, Saito K, Shiuchi T, Minokoshi Y. Leptin stimulates fatty acid oxidation and peroxisome proliferator-activated receptor alpha gene expression in mouse C2C12 myoblasts by changing the subcellular localization of the alpha2 form of AMP-activated protein kinase. Mol Cell Biol. 2007;27:4317–4327. doi: 10.1128/MCB.02222-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou P, Zheng B, Hsu CH, Sasaki AT, Cantley LC. A fluorescent reporter of AMPK activity and cellular energy stress. Cell Metab. 2011;13:476–486. doi: 10.1016/j.cmet.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-martin A, Oliveras-ferraros C, Menendez JA. The active form of the metabolic sensor. Cell Cycle. 2009;8:2385–2398. doi: 10.4161/cc.8.15.9082. [DOI] [PubMed] [Google Scholar]
- Viollet B. AMPK: Lessons from transgenic and knockout animals. Front Biosci. 2009;14:19–44. doi: 10.2741/3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warden SM, Richardson C, Jr, OD J, Stapleton D, Kemp BE, Witters LA. Post-translational modifications of the beta-1 subunit of AMPK affect enzyme activity and cellular localization. Biochem J. 2001;283:275–283. doi: 10.1042/0264-6021:3540275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LGD, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 Is the Upstream Kinase in the AMP-Activated Protein Kinase Cascade. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker Pa, Eccleston JF, Haire LF, et al. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472:230–233. doi: 10.1038/nature09932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Allen MD. FRET-based biosensors for protein kinases: illuminating the kinome. Mol Biosyst. 2007;3:759–765. doi: 10.1039/b706628g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.