

Abstract
Leishmania are protozoan pathogens of humans that exist as extracellular promastigotes in the gut of their sand fly vectors and as obligate intracellular amastigotes within phagolysosomes of infected macrophages. Between infectious blood meal feeds, sand flies take plant juice meals that contain sucrose and store these sugars in their crop. Such sugars are regurgitated into the sand fly anterior midgut where they impact the developing promastigote parasite population. In this report we showed that promastigotes of all Leishmania species secreted an invertase/sucrase enzyme during their growth in vitro. In contrast, neither L. donovani nor L. mexicana amastigotes possessed any detectable invertase activity. Importantly, no released/secreted invertase activity was detected in culture supernatants from either Trypanosoma brucei or Trypanosoma cruzi. Using HPLC, the L. donovani secretory invertase was isolated and subjected to amino acid sequencing. Subsequently, we used a molecular approach to identify the LdINV and LmexINV genes encoding the ~72 kDa invertases produced by these organisms. Interestingly, we identified high fidelity LdINV-like homologs in the genomes of all Leishmania sp. but none were present in either T. brucei or T. cruzi. Northern blot and RT-PCR analyses showed that these genes were developmentally/differentially expressed in promastigotes but not amastigotes of these parasites. Homologous transfection studies demonstrated that these genes in fact encoded the functional secretory invertases produced by these parasites. Cumulatively, our results suggest that these secretory enzymes play critical roles in the survival/growth/development and transmission of all Leishmania parasites within their sand fly vector hosts.
Keywords: Leishmania, Protozoan pathogen, Human parasite, Invertase, Secretory enzyme
Introduction
Leishmania sp. are a group of trypanosomatid protozoan pathogens which are transmitted to their mammalian hosts by the bite of infected female sand fly vectors. Collectively, in humans, these parasites cause over 2 million new cases of cutaneous, mucocutaneous, and fatal visceral diseases (i.e., leishmaniasis) per year worldwide [1]. All Leishmania parasites have a developmental life cycle that includes two major parasite developmental stages: (1) an extracellular, flagellated promastigote form that resides and multiply in the alimentary tract of their sand fly vector hosts and (2) an obligate intracellular, amastigote form which resides and multiplies within phagolysosomal compartments of infected mammalian macrophages [2]. Following their uptake in an infectious blood meal, Leishmania amastigotes transform within ~24–48 h into extracellular promastigotes within the sand fly vector midgut. These promastigotes subsequently rapidly divide and multiply, and undergo a complex series of transformations that result in the generation of mammalian-infective meta-cyclic promastigotes [3–6]. During this entire developmental process, members of the promastigote population continue to differentiate and migrate anteriorly in the sand fly gut culminating with the arrival of infectious metacyclics in the sand fly vector mouth parts. It is important to point out that all of these activities and developmental processes require an exogenous source of energy which the parasites must salvage/scavenge from the nutrients present within the insect vector midgut. To sustain themselves between their infectious blood feeds, female sand flies characteristically take plant juice meals including sucrose and other complex polysaccharides [7]. Following ingestion, such sugar meals are shunted and stored within the crop compartment of the insect’s gut. When needed, these sugar meals are regurgitated back into the sand fly anterior midgut where they would impact/interface with the anterior-migrating population of Leishmania parasites. To harness the energy present in these complex polysaccharide sugar meals (e.g., sucrose) the sand fly must hydrolyze them into smaller transportable monosaccharide units. To that end, it has been reported that sand fly guts are capable of synthesizing and secreting invertase-like activities which are capable of hydrolyzing sucrose into glucose and fructose [7]. Interestingly, Blum and Opperdoes reported that Leishmania donovani promastigotes also produced and released/secreted an extracellular soluble sucrase (i.e., invertase) enzyme activity during their growth in vitro [8]. Further these authors suggested that this enzyme may play “an important role in the nutrition and development of the promastigotes in the insect gut,…” [sic] [8]. Subsequently, these observations were verified by Gontijo et al. who reported that promastigotes of all New and Old World Leishmania species which they tested, also produced extracellular/released invertase activities [9]. In light of the published observations above, we initiated our studies toward identifying, characterizing, and episomally expressing in situ the gene(s) encoding these putative leish-manial released/secretory invertase-like enzymes.
Results
The first report of a released/secreted sucrase/invertase activity by any Leishmania sp. was reported from culture supernatants of L. donovani promastigotes (MHOM/SD/62/1S/CL2D) by Blum and Opperdoes [8]. Subsequently, these observations were confirmed and extended to a variety of promastigotes of Old and New World Leishmania sp. by Gontijo et al. [9]. These two reports served as the basis for the current report concerning the identification and characterization of the gene(s) encoding these potentially important parasite enzymes.
Identification of secreted/released invertase activities by in vitro grown Leishmania promastigotes
As indicated above, invertase-like activity was detected in culture supernatants of L. donovani promastigotes [8, 9]. To verify and extend those observations, concentrated culture supernatants from L. donovani (1SCL-2D) promastigotes were tested for invertase activity using our agarose-gel-triphenyltetrazolium chloride (TTC) reaction product capture assay as described below. Results of these qualitative assays demonstrated that this L. donovani promastigote isolate synthesized and released/secreted significant quantities of soluble invertase activity into its culture supernatant which accumulated during growth in vitro (Fig. 1a). No invertase activity was detected in similarly processed unused parasite growth medium controls (Fig. 1c). The gel shown in Fig. 1 is representative of the results obtained from numerous gel assays which were carried out in these experiments. Results of parasite growth experiments indicated that the accumulation of such soluble invertase activity in these parasite cell cultures was in relative proportion to total parasite cell number (i.e., parasite cell growth). It is important to point out that the “soluble” invertase activity measured in these parasite culture supernatants was not enhanced by the addition of non-ionic detergents (i.e., showed no detectable compartmentalized latency, data not shown). Results similar to those shown in Fig. 1a were also obtained from in vitro short-term release assays carried out with L. donovani (1SCL-2D) promastigotes (data not shown). Both concentrated (20×) and unconcentrated (1×) culture supernatants and aliquots of short-term release assays were also analyzed for their invertase activity using the glucose oxidase (GO) spectrophotometric assays described below. Results of a typical experiment obtained from unconcentrated (1×) culture supernatants from L. donovani promastigotes are shown in Fig. 1d. Approximately 20-fold higher levels of invertase activity were obtained in these assays when 20× concentrated parasite culture supernatants were used. Thus, in our agarose gel assays we determined that we needed at least 100 GO spectrophotometric units to readily detect invertase activity. Results of various assays demonstrated that ~1 × 107 L. donovani promastigotes produced ~100 units of invertase activity as determined by GO spectrophotometric assays. Overall, the results of our quantitative assays were in very good agreement with our qualitative results obtained in our agarose-based invertase assays. Having demonstrated that L. donovani 1SCL-2D promastigotes constitutively produced considerable quantities of invertase activity, we decided to assess whether any other Old World Leishmania strains and species might also produce similar invertase activities. To that end, concentrated culture supernatants were obtained from an Indian isolate of L. donovani (LRC-751); two strains of L. infantum, one from Spain (FVM10001JL) and one from Israel (LRC-639); one isolate from Israel of L. major (Friedlen) and one isolate from the Soviet Union of L. tropica (WR683). In addition, similar culture supernatants were obtained from a single Old World saurian strain of Leishmania, L. tarentolae (Jena 101), which does not cause disease in humans. Such culture supernatants were assayed using our agarose-gel-TTC reaction product capture assay. Interestingly, results of those assays showed that each of these Old World Leishmanias including L. tarentolae constitutively produced and released/secreted similar levels of invertase activity (data not shown) to those produced by the L. donovani (1SCL-2D) parasites. Similar results were also obtained with aliquots of in vitro short-term release assays from each of these Old World Leishmania species (data not shown). Results of spectrophotometric assays with samples derived from all of these Old World Leishmanias demonstrated that all of them produced comparable levels of invertase activity (i.e., ~107 promastigotes produce ~100 units of invertase activity/ml; data not shown). Taken together, results of these various assays demonstrated that all of the Old World Leishmanias tested appear to constitutively produce/release significant quantities of soluble invertase activity (i.e., non-latent, as indicated above). Having found this, it was of interest to determine whether any New World Leishmania species might also produce invertase activities. To test this hypothesis, concentrated culture supernatants obtained from L. mexicana (M379) promastigotes were tested in our agarose-gel-TTC reaction product capture assays. Results of those assays showed that L. mexicana (M379) promastigotes did in fact produce invertase activity but at qualitatively lesser amounts than that produced by similar numbers of L. donovani (1SCL-2D) promastigotes (c.f. Fig. 1a, b). No invertase activity was detected in similarly processed unused parasite growth medium controls (Fig. 1c). As above, the gel depicted in Fig. 1 is representative of one example of the results obtained from numerous gel assays which were carried out in these experiments. Results similar to those shown in Fig. 1b were also obtained from in vitro short-term release assays carried out with L. mexicana (M379) promastigotes (data not shown). Similarly, culture supernatants and aliquots of short-term release assays from L. mexicana (M379) promastigotes were also analyzed for their invertase activity using the GO spectrophotometric assay. Results of those quantitative assays indicated that ~1 × 107 L. mexicana (M379) promastigotes produced ~10 units of invertase activity as determined by GO spectrophotometric assays. Thus, the combined results of both our qualitative and quantitative assays indicated that L. mexicana (M379) did in fact produce invertase activity but at lower levels than that produced by any of the other Old World Leishmanias tested. These observations suggested that it would be of interest to assess whether any other New World species of Leishmania also produced invertase activity. To test this hypothesis, concentrated culture supernatants were obtained from in vitro grown promastigotes of a second L. mexicana isolate (BEL21, from a patient in Belize); an isolate of L. braziliensis (WR668, from a Brazilian patient); two isolates of L. amazonensis (PH8 and GML584, from a vector in Brazil and a patient in Panama, respectively); one isolate of L. pifanoi (WHOLL1, from a Venezuelan patient); two isolates of L. panamensis (WR470 and WHO594 isolated from several individual patients in Brazil); and an isolate of L. guyanensis (M4147, from a patient in Brazil). Such culture supernatants and aliquots of release assays obtained from each of these New World species were assayed for their invertase activity using our agarose-based-TTC capture method. Results of those assays showed that each of the New World Leishmanias tested constitutively produced and released/secreted similar levels of invertase activity (data not shown) to those produced by the L. mexicana (M379) promastigotes (re: Fig. 1b). Results of these qualitative assays were fully supported by those obtained from our GO spectrophotometric invertase assays (data not shown). Taken together, results of all of these assays demonstrated that each of the New World species tested did in fact produce soluble (non-latent) invertase activity, similar to each other, but none of them produced invertase at levels equivalent to that produced by any of the Old World Leishmania species tested above.
Fig. 1.

Detection of invertase activities produced by Leishmania promastigotes in vitro. a–c Agarose-gel assays of invertase activity. Aliquots of ×20 concentrated culture supernatants from L. donovani (1SCL-2D) and L. mexicana (M379) promastigotes and unused parasite culture medium were loaded to precut wells of an agarose gel containing 25 mM sucrose as substrate and allowed to diffuse into the gel for 4 h at 37 °C. Following incubation, the resulting hydrolysis product (fructose) was visualized in these plates using triphenylte-trazolium chloride (TTC) as a capture reagent. The resulting invertase activity is shown in these plates by the presence of a dark red/purple colored ring of TTC-captured product radiating from the sample wells. The gel shown here is one example of the results obtained from numerous gel assays which were carried out in these experiments. d Quantitation of invertase activity released/secreted by L. donovani promastigotes during their growth in vitro. Invertase activity produced by L. donovani promastigotes was measured using sucrose as substrate in a glucose oxidase spectrophotometric assay. Aliquots of culture supernatants were taken at various times during cell growth and assayed for invertase activity. The bars represent invertase activity expressed as μg of glucose produced by × number of cells/ml/h. The solid squares reflect the parasite cell number/ml prior to harvesting their culture supernatants. (Color figure online)
The cumulative results of our invertase assays for the Old and New World Leishmania sp. are summarized in Table 1A. While not tested in the current report, it is of interest to point out that similar sucrase/invertase activities were reported previously by Gontijo et al. for the non-Leishmania, monoxenic insect trypanosomatids (i.e., Crithidia fasciculata, Herpetomonas samuelpessoai, and Leptomonas seymouri) as well as a Phytomonas sp. (Table 1B). In contrast to the foregoing, despite numerous attempts, neither we nor Gontijo et al. were able to detect any invertase activity in the culture supernatants of either African or American Trypanosoma sp. (i.e., T. brucei and T. cruzi; Table 1B).
Table 1.
Detection of invertase enzyme activities and the presence of LdINV-like homologs in various kinetoplastid parasites
| Invertase activity | Possess LdINV-like homologs | |
|---|---|---|
| Production of invertase activity and the presence of LdINV-like homologs of various Old World and New World Leishmania species | ||
| Old World Leishmania sp. | ||
| L. donovani | +++ | ü |
| L. infantum | +++ | ü |
| L. major | +++ | ü |
| L. tropica | +++ | ü |
| L. tarentolae | +++ | ü |
| New World Leishmania sp. | ||
| L. mexicana | + | ü |
| L. braziliensis | + | ü |
| L. amazonensis | + | ü |
| L. pifanoi | + | ü |
| L. panamensis | + | ü |
| L. guyanensis | + | ü |
| Production of invertase activity and the presence of LdINV-like homologs in various non-Leishmania kinetoplastids | ||
| Non-Leishmania kinetoplastids | ||
| C. fasciculataa | + | ü |
| Phytomonas sp.a | + | ü |
| H. samuelpessoaia | + | NI |
| Leptomonas seymouria | + | NI |
| T. brucei | − | None |
| T. cruzi | − | None |
Invertase activity denoted as (+++) above reflects ≥100 units of invertase activity produced by 107 promastigotes/ml/h and (+) denotes ~10 units of invertase activity. The (ü) designates the presence of one or more LdINV-like homologs in this parasite’s genome
NI no LdINV-like homologs annotated in this partially sequenced genome, None there was no LdINV-like homologs annotated in this fully sequenced genome
Data from Gontijo et al.
Isolation of the L. donovani (1SCL-2D) invertase
Results of our qualitative and quantitative invertase assays indicated that the L. donovani (1SCL-2D) promastigotes, as virtually all other Old World species tested, produced significant quantities of invertase activity. Based on those observations it was deemed practical to attempt to isolate the Leishmania invertase synthesized and released/secreted by L. donovani (1SCL-2D) promastigotes. In preliminary experiments, we attempted to isolate invertase from concentrated culture supernatants of L. donovani (1SCL-2D) promastigotes using a variety of affinity-based chromatographic methods. Unfortunately, none of these methods proved successful because during the concentration steps, culture medium components tended to aggregate and precipitate out of solution (i.e., hemin aggregates/ppts). Subsequently, we found that this problem could be obviated by using samples obtained from short-term release assays (as described in “Experimental procedures” section below) using L. donovani (1SCL-2D) promastigotes. In that regard, dialyzed cell-free release assay supernatants obtained from L. donovani (1SCL-2D) promastigotes were loaded on to a Mono-Q column and subjected to fast protein liquid chromatography (FPLC). During fractionation, eluates were scanned at A220 nm and A280 nm. Aliquots of these samples were collected and subjected to SDS-PAGE and silver staining. An example of one such chromatographic separation obtained with these samples is shown in Fig. 2a. Results of such silver staining demonstrated that one of these fractions (INV) contained a single protein band with an apparent molecular mass of approximately 70 kDa (Fig. 2b). Subsequently, an aliquot of this selected fraction was separated by SDS-PAGE, blotted onto a polyvinylidene fluoride (PVDF) membrane and stained with Coomassie Blue. The resulting single Coomassie Blue-stained protein band was excised from such blots and subjected to N-terminal End Edman Amino Acid Sequencing.
Fig. 2.
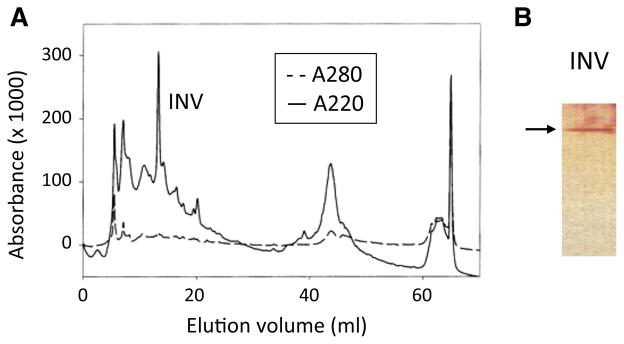
Isolation of the L. donovani (1SCL-2D) invertase using FPLC fractionation. Concentrated supernatants from L. donovani (1SCL-2D) promastigote release assays were subjected to FPLC on a Mono-Q column. Individual fractions from this column were collected, optically scanned and their absorbance at 220 and 280 nm were plotted (a). One of these fractions (INV) when subjected to SDS-PAGE and silver staining (b) contained only a single ~70 kDa protein band (arrow). (Color figure online)
Identification and characterization of the L. donovani INV gene
Results of N-terminal End Edman Amino Acid Sequencing of the 70 kDa LdINV protein revealed that it contained KDGVPYE amino acid sequence. This sequence was used to do a BLASTP search of GeneBank. That search revealed that the LdINV sequence fragment was common to numerous annotated invertases. Results of these BLASTP searches showed that many of these invertases contained a conserved consensus sequence of FYASKTFYD. Based on these two target sequences, oligonucleotide primers were designed and used in PCR reactions with L. donovani (1SCL-2D) genomic DNA as template. The resulting 858 bp PCR product was cloned into pCR®2.1-TOPO and the resulting plasmid LdINV-PCR858 was subjected to nucleotide sequencing. Results of such sequencing showed that LdINV-PCR858 had very high sequence identity (~96 %) with that of an L. major Friedlin gene sequence (gi|15027087|emb|AL389894.4|LMFLCHR4A) which was annotated as an invertase (β-D fructofuranosidase). While this L. major sequence was annotated as an invertase no evidence of its functional activity was provided. Subsequently, the LdINV-PCR858 plasmid was DIG labeled and used to screen an L. donovani (1SCL-2D) cosmid library. A positive cosmid clone (16.3) was isolated and sequenced in the region of the putative LdINV gene. Results of such sequencing demonstrated that the LdINV consisted of an open reading frame (ORF) of 1932 bp including a methionine start codon of ATG at the 5′ end and a stop codon of TAG at its 3′ end. As indicated in the “Experimental procedures” section below, the 5′ splice leader (SL) acceptor site of the LdINV gene was mapped using a Leishmania SL primer in an RT-PCR reaction. Results of the latter confirmed the authenticity of the methionine start codon of the LdINV ORF. Further, the insert nucleotide sequence of the LdINV-PCR858 plasmid was found to have 100 % identity to that contained within the LdINV ORF. Sequence analyses showed that the composition of the LdINV ORF was GC-rich, which is consistent with the overall GC content of the Leishmania genome [10].
Furthermore, such analyses showed that the LdINV ORF encodes a polypeptide of 644 amino acids with a calculated molecular mass of 72,290 Da and a pI of 5.18. The nucleotide sequence of the LdINV ORF was used in multiple sequence alignments of invertases from various organisms (e.g., yeast, plant, fungi, etc.). Analyses of the LdINV-deduced protein using the Pfam database indicated that it belongs to the glycohydrolase family 32 of proteins [11, 12]. Characteristic features of this family of hydrolases include an N-terminal five-bladed beta propeller and a C-terminal beta sandwich structure [12]. Each of these domains was mapped on to the LdINV-deduced protein sequence. Typical members of this family hydrolyse the glycosidic bond between two or more carbohydrates. One of the major sub-groups of the glyco hydro family 32 includes the invertases (i.e., protein/enzymes which can hydrolyze sucrose into glucose and fructose).
The hydrophobic, N-terminal, 28 amino acids of the LdINV-deduced protein constitutes a putative signal peptide based on the SignalP 3.0 algorithm [13]. Cleavage at this site, presumably in the endoplasmic reticulum (ER) of the parasite, would result in a mature protein with Lys27 as its N-terminal amino acid residue of the mature protein (Fig. 3). This hypothesis was fully supported by the results obtained of N-terminal sequencing of the ~70 kDa LdINV protein obtained from culture supernatants of FPLC fractionations (Fig. 3). Further the deduced amino acid sequence of the LdINV gene (KDGVPYE) is identical to that obtained from FPLC-isolated LdINV protein above (Fig. 3). Thus, cleavage at this site would result in a mature protein consisting of 618 amino acids with a calculated molecular mass of 69.6 kDa and a theoretical pI of 5.19. The LdINV-deduced protein was also analyzed using other structural algorithms. Those analyses indicated that this parasite enzyme lacked any apparent hydrophobic transmembrane domains or glycosylinositol phosphate anchor signature sequences [14]. Similarly, no KDEL or analogous ER retention sequences [15] or any other intracellular organelle specific-targeting sequences were identified in the LdINV-deduced protein. Based on its overall hydrophilicity, the presence of an N-terminal signal peptide and the absence of both membrane anchors and ER retention motifs suggest that the LdINV represents a soluble/released protein. These deduced structural features are in good agreement with our experimental observations, which demonstrated that the endogenous wild-type invertase was constitutively released/secreted by L. donovani parasites during their growth in vitro.
Fig. 3.

N-terminal end sequence alignment of LdINV and related Leishmania sp. homologs. The N-terminal end-deduced amino acid sequence of the LdINV gene (LdonINV) was compared to those of several Leishmania species present in the TriTryp genome database [L. infantum (LinfINV: LinJ.04.0300), L. major (LmajINV: LmjF.04.0310), L. tarentolae (LtarINV: LtaP04.0290), L. mexicana (LmexINV: LmxM.04.0310), and L. braziliensis (LbraINV: LbrM.04.0350)]. Met1 through Ala28 represents the putative signal peptide of the LdonINV. The arrowhead shown above the LdonINV sequence designates the putative signal peptidase cleavage site of this protein. The KDGVPYE gene-deduced amino acid sequence was verified by N-terminal end AA sequencing of the FPLC-isolated LdINV protein. The area highlighted in green designates the conserved 14 amino acid signature sequence characteristically found in all members of the glyco/hydrolase 32 subfamily of invertases. The yellow-highlighted aa denotes the critical aspartic acid residue within the active/hydrolytic site of this invertase family. The asterisks (*) designate the conservation of amino acid residues among all six Leishmania species; the periods (.) and colons (:) represent amino acids that are semi-conserved substitutions and conserved substitutions, respectively. The blue line denotes geographical separation between the Old World (above) and New World (below) Leishmania species. (Color figure online)
The LdINV-deduced protein was also analyzed for potential N- and O-linked glycosylation and phosphorylation sites using NetNGlyc, NetOGlyc, and NetPhos web-based tools. Results of such analyses indicated that the LdINV possessed six potential N-linked glycosylation sites at Asn120, Asn282, Asn396, Asn439, Asn480, and Asn628. These predictions are consistent with our earlier preliminary observations, which showed that the native, wild-type parasite (promastigote) released/secreted invertase, was a mannose-containing glycoprotein (i.e., was bound to concanavalin A beads, and such binding was inhibited with α-methylmannoside; data not shown). Results of NetOGlyc analyses failed to identify any potential O-linked glycosylation sites in the LdINV-deduced protein. In contrast, results of NetPhos analyses showed that LdINV contained at least 24 potential sites for phosphorylation by several different mechanisms (e.g., casein kinase II, protein kinase C, etc.). These include 8 potential phosphorylation sites on serines (i.e., Ser5, Ser219, Ser269, Ser423, Ser447, Ser476, Ser514, and Ser567), 15 on threonines (i.e., Thr59, Thr84, Thr92, Thr129, Thr253, Thr284, Thr299, Thr311, Thr326, Thr355, Thr386, Thr408, Thr502, Thr522, and Thr581), and one on tyrosines (i.e., Tyr634).
As indicated above, results of our bioinformatic searches indicated that invertases in general possessed a conserved block of 14 amino acids including a critical Asp residue near their N-terminal end which define their substrate binding and hydrolytic site [11]. It is important to point out that this conserved invertase signature sequence was readily apparent in the N-terminal end of the LdINV-deduced protein (Fig. 3).
Identification of LdINV homologs in other Leishmania parasites
Since L. donovani is a human pathogen it was of interest to ascertain whether an LdINV-like gene might also be present in the genomes of other related trypanosomatid parasites. In that regard, the LdINV sequence was used as our reference to examine the TriTryp/Leishmania genomes at www.tritrypdb.org. Results of such searches showed that LdINV homologs existed in the genomes of L. infantum (LinJ.04.0300), L. major (LmjF.04.0310), L. tarentolae (LtaP04.0290), L. mexicana (LmxM.04.0310), and L. braziliensis (LbrM.04.0350). To validate whether such LdINV homologs were actually present in these various Leishmania species they were subjected to PCR analyses. For these assays, forward and reverse primers based on the LdINV ORF were used in conjunction with gDNA as template from L. donovani (1S-CL2D and LRC-751), L. amazonensis, L. mexicana, L. tropica, L. major, L. infantum, L. braziliensis, and L. tarentolae. Results of these assays confirmed the presence of an 850 bp product which was amplified from each of these species indicating that they all possessed an LdINV homolog. The products of these PCR reactions were cloned and subjected to nucleotide sequencing. Results of such sequencing confirmed that each of these species possessed an LdINV homolog. All of the above data are in agreement with our experimental results which showed that the culture supernatants of each of these species possessed secreted/released invertase-like activities. Having identified these LdINV homologs, it was of interest to compare their amino acid sequences. This was done using the MAFFT alignment algorithm as referenced below. A portion of this alignment analysis is shown in Fig. 3. Further, results of those analyses showed that each of the four annotated genomes of Old World Leishmania (i.e., L. donovani, L. infantum, L. major, and L. tarentolae) showed an overall identity of 82 % between amino acid 1 and 60 of their sequences. This included a putative signal peptide sequence of approximately 28 amino acids (c.f. L. donovani aa 1–29; Fig. 3). Signal peptidase cleavage at this putative site for LdINV would result in a mature protein with KDGVPYE as an amino acid sequence at its N-terminal end. This prediction was verified by N-terminal end amino acid sequencing of the LdINV FPLC-purified protein. It is of interest to point out that cleavage at this site in each of the LdINV homologs would result in mature proteins with a conserved N-terminal 10 amino acid sequence [i.e., virtually identical with that of LdINV (KDGVPYEPIF)]. Of equal or greater significance, is the immediate downstream conservation of a 14 amino acid glyco/hydrolase 32 subfamily invertase signature sequence among all of the LdINV homologs (Fig. 3). It is also of significance to point out that the critical aspartic acid residue, which is the critical aa residue within the active/hydrolytic site of all invertases, is structurally conserved in all of the LdINV homologs (Fig. 3). In addition, this high level of amino acid sequence conservation among all of the LdINV homologs ensues downstream of this 14 aa “INV family signature block” for a total of 324 amino acids (Fig. 4). This 324 amino acid block represents an N-terminal end conserved domain among all of the invertase members of the glyco/hydrolase 32 protein family. The purported three-dimensional structure of this region has been characterized as a “five-bladed beta propeller” structure [12]. Also preserved downstream from this region in all of the LdINV homologs is a 71 aa sequence characteristically conserved among all invertases of the glyco/hydrolase 32 protein family (Fig. 4). This “invertase family” C-terminal domain has a purported three-dimensional structure composed of a sandwich of beta-pleated sheets (Fig. 4) [12]. Interspersed between and connecting these two invertase family signature domains is a 142 amino acid sequence (i.e., aa 363–505) which appears to be common to all of the LdINV homologs (Fig. 4). In addition, each of the LdINV homologs also possess a common C-terminal extension of 67 amino acids (i.e., aa 576–643) terminating with a non-polar aliphatic amino acid residue (e.g., Val643 for LdINV; Fig. 4). As indicated above, results of NetNGlyc analysis indicated that LdINV possessed six potential N-linked glycosylation sites at Asn120, Asn282, Asn396, Asn439, Asn480, and Asn628 (Fig. 4). Among these, only two N-linked glycosylation sites (Asn396 and Asn628) were present in all of the LdINV homologs (Fig. 4). Interestingly, these two N-linked glycosylation sites are restricted to the two amino acid domains that were unique to only the LdINV homologs. Of the other four potential N-linked glycosylation sites, Asn120 was present in all of the human LdINV homologs (Fig. 4). Similarly, Asn282 and Asn480 were present in all of the LdINV homologs except L. braziliensis (Fig. 4). The remaining potential N-linked glycosylation site at Asn439 was conserved in all of the Old World LdINV homologs and was positionally conserved near this amino acid in the New World LdINV homologs (i.e., −2 aa L. braziliensis, +6 aa L. mexicana). The identification of N-linked glycosylation sites in LdINV is consistent with our experimental results which showed that it is a released/secreted glycoprotein, as described above. It is conceivable that such N-linked glycosylation might be a common functional characteristic of each of the Leishmania secreted/released invertase activities which we demonstrated above.
Fig. 4.
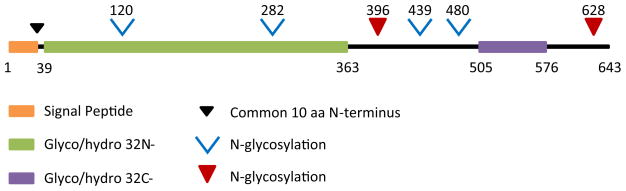
Diagrammatic representation of the features common to LdINV and its associated Leishmania homologs (c.f. Fig. 3). The orange bar represents a signal peptide present in all of the LdINV homologs. The black arrow delineates the unique 10 amino acid N-terminal sequence present in all the LdINV homologs. The green bar (glyco/hydro 32 N-; aa 39–aa 363) represents the 324 N-terminal amino acid sequence characteristic of all invertases in the glyco/hydrolase 32 protein family including its characteristic 14 amino acid active site (aa 39–aa 53). This domain has a purported 3-D structure characterized as a five-bladed beta propeller. The purple bar (glyco/hydro 32 C-; aa 505–aa 576) represents the 71 C-terminal amino acid sequence characteristic of all invertases in the glyco/hydrolase 32 protein family. This domain has a purported 3-D structure composed of a sandwich of beta- pleated sheets. The black bars (i.e., aa 363–aa 505 and aa 576–aa 643) represent aa sequences common/unique to all of the LdINV homologs. The red arrows represent N-linked glycosylation sites common to each of the LdINV homologs. The blue chevrons designate potential N-linked glycosylation sites in LdINV and most of its homologs. (Color figure online)
Taken together, the general conservation in protein structure among the LdINV homologs is in good agreement with our experimental results which showed that each of these Leishmania species actually released/secreted functional invertase activities. Further, this high level of conservation among LdINV homologs predicts that these secreted/released invertase enzymes must play important roles in the biology of all of these Leishmania parasites within their insect vectors.
As mentioned above, Gontijo et al. showed that various Leishmania sp. produced soluble invertase activities. Interestingly, they also showed that a variety of non-Leishmania, monoxenic insect trypanosomatids (i.e., C. fasciculata, H. samuelpessoai, and L. seymouri) also produced/released soluble sucrase/invertase activities. In addition these authors showed that the pathogenic plant trypanosomatid Phytomonas sp. also produced sucrase/invertase activity during their growth in vitro. In light of those observations, we searched various databases and found that multiple LdINV-like homologs were annotated (18 and 9, respectively) in the genomes of the non-Leishmania trypanosomatids C. fasciculata and Phytomonas sp. (i.e., HART-1 strain). These observations lend further support to the putative evolutionary relationships between Leishmania sp. and their insect trypanosomatid relatives. In contrast to the latter observations, it is important to point out that in the current report we were unable to detect any invertase activity, similar to the observations of Gontijo et al. in either African or American Trypanosoma sp. (i.e., T. brucei and T. cruzi). Congruent with these findings, despite multiple database searches, we were unable to identify any annotations for invertase in the genomes of any African or American Trypanosoma species. These data are compared and summarized in Table 1B.
Identification of an INV message during parasite development
Northern blot analyses were performed to evaluate the expression of LdINV mRNA during the parasite developmental life cycle. To that end, equivalent amounts of total RNA from both L. donovani promastigotes and axenic amastigotes were separated in agarose/formaldehyde gels (Fig. 5b), blotted onto nylon membranes, and hybridized with the LdINV-DIG858 probe. Results obtained from such blots showed that the LdINV probe specifically hybridized to two distinct messages, predominantly to one of approximately 4.4 kb and to a lesser extent to one greater than 7.5 kb, in both promastigote and axenic amastigote developmental forms (Fig. 5a). In that regard, the ~4.4 kb message is sufficiently large enough to encode the entire ~72 kDa L. donovani invertase protein, and the >7.5 kb message could represent its precursor. Alternatively, the >7.5 kb mRNA might reflect hybridization of the LdINV-DIG858 probe with some other structurally related genomic sequence(s). This remains a possibility due to the fact that during our genomic database searches for LdINV homologs we discovered a larger beta-fructofuranosidase-like gene (LdBPK_350650.1 which encodes ~120 kDa protein) that contained a partial homologous sequence of the LdINV gene. Interestingly, even though equivalent amounts of mRNA from each parasite developmental form were probed (Fig. 5b), the overall hybridization signals obtained in these blots appeared to be significantly more intense with RNA isolated from promastigotes (i.e., the insect vector stage of the parasite) than from axenic amastigotes (i.e., the developmental stage that produces disease in mammals). While these results indicated that the LdINV gene was transcribed throughout the parasite developmental life cycle, they also suggested that this gene’s expression might be developmentally regulated. The latter hypothesis is in full agreement with our experimental results which consistently showed that L. donovani promastigotes, but not axenic amastigotes, produced/secreted LdINV enzymatic activity (data not shown). Thus, our Northern results showed that LdINV mRNA was transcribed in both developmental forms but these results could not demonstrate that said message was actually translatable into protein. To investigate this further, it was of interest to determine whether the LdINV gene was indeed transcribed into mature mRNAs by both parasite developmental forms. To examine this, RT-PCR analyses were performed. It is important to point out that in trypanosomatid parasites all of their mature, translatable mRNAs are capped at the 5′ end with a conserved 39 nt, “splice leader” (SL) sequence [16]. Thus, for these experiments cDNAs were reverse transcribed from total RNA isolated from both L. donovani developmental forms. Aliquots of such cDNAs were used as template in PCR amplifications. For these reactions we used the 5′ forward primer corresponding to a portion of the SL sequence (SpliceFwd, see “Experimental procedures” section) and a Reverse primer corresponding to an internal sequence within the LdINV ORF (i.e., INV Rev, see “Experimental procedures” section). Results of these RT-PCR analyses showed that an approximately 500 bp product was amplified from both L. donovani promastigote and axenic amastigote developmental forms (data not shown). These results indicated that each of the parasite developmental forms did in fact produce a mature translatable LdINV mRNA from the LdINV gene. As anticipated from our Northern results, considerably more of the 500 bp RT-PCR product was detected in promastigotes than in axenic amastigote samples. Taken together with our Northern results, these RT-PCR results further suggest that LdINV protein expression is presumably post-transcriptionally regulated in these parasites.
Fig. 5.

Detection of mRNAs encoding invertase in both Leishmania sp. promastigote and axenic amastigote developmental forms. a Northern blot of total RNA isolated from L. donovani (Ld) promastigotes (Pro) and axenic amastigotes (AxAm) probed with the LdINV-DIG858 probe. Molecular mass markers in kb are shown at the left (arrows). b Ethidium bromide stained gel showing equivalent amounts of Pro and AxAm RNA were used to generate the Northern blot shown in a above. Molecular mass markers in kb are shown at the left (arrows). c Ethidium bromide-stained agarose gel showing the ~500 bp product (arrow) obtained from RT-PCR reactions using RNA isolated from L. mexicana (Lmex) promastigotes (Pro) and axenic amastigotes (AxAm). This image was inverted using Adobe Photoshop CS2 to accentuate the RT-PCR product obtained in these reactions
Having made these observations above for L. donovani (i.e., our archetypical Old World Leishmania causing visceral disease in humans); it was deemed of interest to determine whether this phenomenon also pertained to L. mexicana (i.e., our prototypical New World Leishmania causing human cutaneous disease). To test this hypothesis, RT-PCR analysis was carried out using total RNA isolated from both L. mexicana promastigotes and axenic amastigotes. These reactions were carried out using a SL primer and an internal LmexINV primer (RLmexINVRT) similar to those done with L. donovani above. Results of these assays showed that an approximately 500 bp product was amplified from both L. mexicana promastigote and axenic amastigote developmental forms (Fig. 5c). Thus, these RT-PCR results in conjunction with those from L. donovani above, strongly suggest that the expression of these parasite enzymes must be developmentally regulated, presumably involving post-transcriptional mechanisms. The latter hypothesis would account for the absence of detectable invertase activity in axenic amastigotes of either of these parasite species.
Reactivity of the anti-E. coli-recombinant LdINV antibody
As indicated, the LdINV ORF was used to generate a recombinant LdINV protein that was expressed in E. coli. Aliquots of purified E. coli-recombinant LdINV (rLdINV) were subjected to SDS-PAGE, blotted onto PVDF membranes and tested for invertase activity. Cumulative results of these assays demonstrated that this single ~72 kDa E. coli-recombinant protein possessed invertase enzymatic activity. Having shown that this recombinant protein actually possessed functional invertase activity, we used it to generate a rabbit anti-E. coli rLdINV antibody (α-rLdINV). Subsequently, Western blot analyses were done to determine whether the α-rLdINV antibody would recognize the endogenous, wild-type L. donovani invertase. To examine this, concentrated culture supernatants from wild-type L. donovani promastigotes and aliquots of purified E. coli rLdINV protein were separated in SDS-PAGE gels and transblotted onto PVDF membranes. Such membranes were probed with our rabbit α-rLdINV antibody or its control NRS. In such blots the α-rLdINV antibody reacted specifically with a single ~72 kDa protein present in the L. donovani culture supernatants (Fig. 6a). As anticipated, this antibody reacted very strongly with a major ~70 kDa protein present in the purified E. coli rLdINV preparations. Presumably the difference in molecular mass between the ~72 kDa LdINV and the ~70 kDa E. coli rLdINV reflects post-translational modifications (e.g., N-linked glycosylations and phosphorylations) to the parasite protein but not the bacterial protein. In addition, this α-rLdINV antibody also reacted with several other proteins of lower molecular mass in the purified E. coli rLdINV preparations. These bands presumably reflect potential protein degradations of the purified rLdINV. A similar conclusion pertains to the minor ~50 kDa protein observed in native LdINV preparations. In parallel, NRS blots showed no reactivity with the native LdINV or E. coli rLdINV samples (data not shown). Further, the α-rLdINV antibody showed no reactivity in control blots containing lysates of non-transfected E. coli or unused parasite culture medium (data not shown). Results of these Western blots demonstrated two significant points: (1) that our E. coli rLdINV expressed protein actually possessed functional invertase activity and (2) that the antibody we made against it specifically recognized the native LdINV enzyme produced/secreted by promastigotes during their growth in vitro. Taken together these observations indicate the LdINV gene that we identified actually encodes a secreted/released LdINV parasite enzyme. Results of our preliminary studies showed that the native released/secreted LdINV enzyme is a mannose-containing glycoprotein which was in agreement with our NetNGlyc predictions above. Presumably such N-linked glycosylations aid in secretion and potential stability of the parasite enzyme. In light of our observations with the E. coli rLdINV protein, such N-linked glycosylations are not necessary for invertase enzymatic activity per se.
Fig. 6.

Western blots demonstrating the reactivity of our rabbit antiE. coli-recombinant LdINV antibody. a Western blot showing the immunoreactivity of our rabbit anti-E. coli-recombinant LdINV antibody (α-rLdINV) with concentrated L. donovani culture supernatants (Ld Sup) and purified E. coli-recombinant LdINV protein (Ec rLdINV) separated in SDS-PAGE, blotted onto PVDF membranes, and probed with the α-rLdINV antibody. Protein standards in kDa are shown at the left (arrows). b Western blot demonstrating the immuno cross-reactivity of our rabbit α-rLdINV antibody with the native L. donovani (Ld) and L. mexicana (Lmex) released/secreted invertases. Culture supernatants (×1) from L. donovani and L. mexicana promastigotes and unused culture media controls were separated in SDS-PAGE, blotted onto PVDF membranes, and probed with the α-rLdINV antibody. Only a single ~72 kDa (arrow) band of INV protein was detected in culture supernatants from each of these parasite species but not present in their media controls
Results of our molecular searches above demonstrated that an LdINV homolog was present in L. mexicana. Further, we showed that this homolog had high structural identity with the LdINV-deduced protein and that L. mexicana promastigote developmental forms made mature mRNA for this gene product. Moreover, these observations are in good agreement with results of our qualitative invertase gel assays which showed that L. mexicana promastigotes produced/secreted functional invertase activity during their growth in vitro. In light of these observations, it was of importance to determine whether the antibody that we made against the rLdINV-expressed protein (α-rLdINV) would also recognize the native released/secreted invertase produced by L. mexicana promastigotes. To examine this, aliquots of 1× culture supernatants from both L. donovani and L. mexicana promastigotes and unused culture medium controls were separated by SDS-PAGE, transblotted onto PVDF membranes and probed with the α-rLdINV antibody (Fig. 6b). As anticipated, results of these assays showed that the α-rLdINV antibody reacted with only a single ~72 kDa protein present in the homologous L. donovani culture supernatants. More importantly, this antibody specifically reacted with only a single ~72 kDa protein present in the culture supernatants of L. mexicana promastigotes (Fig. 6b). The unused culture medium controls showed no activity with the α-rLdINV antibody. To extend our observations, cell-free culture supernatants from additional LdINV homologs [L. donovani (LRC-751), L. amazonensis, L. tropica, L. major, L. infantum, L. braziliensis, and L. tarentolae] promastigotes were tested in Western blots against the rabbit α-rLdINV antibody. Results of these assays showed that the α-rLdINV antibody reacted with only a single ~72 kDa protein present in the culture supernatants of each of these Leishmania species (data not shown). Taken together, these results in conjunction with our molecular observations strongly suggest that in addition to the native released/secreted L. donovani and L. mexicana invertase enzymes, each of the Old and New World species tested in this report all possess invertase enzymes with common structural features recognized by the α-rLdINV antibody.
Transfection of Leishmania promastigotes with epitope-tagged INV gene constructs
Having shown that the α-rLdINV antibody specifically recognized both the wild-type L. donovani and L. mexicana released/secreted invertases, it was deemed necessary to determine whether the LdINV and LmexINV genes in fact encoded functional invertase activities. To test this, a homologous expression system was devised. For these experiments, two separate chimeric constructs were generated: one contained the complete ORF of the cloned LdINV gene and the second contained the complete ORF of the cloned LmexINV gene. Each of these ORFs were fused, in-frame, at their 3′-end with a sequence encoding a 9-aa HA epitope of the influenza virus (all as described in the “Experimental procedures” section). Subsequent to ligation into the pKSNEO leishmanial expression vector, these constructs (designated subsequently as LdINV-HA and LmexINV-HA) were used to transfect L. donovani and L. mexicana promastigotes, respectively. Promastigotes of both species were transfected with the pKSNEO vector alone and these served as controls in all subsequent transfection experiments. Following drug selection, the growth kinetics of these transfected promastigotes were compared as described below. Results of those in vitro assays showed that promastigotes transfected with either the LdINV-HA or the LmexINV-HA chimeric constructs had very similar growth kinetics to their pKSNEO-transfected controls (data not shown). Furthermore, non-transfected (“wild type”) L. donovani and L. mexicana promastigotes, grown in complete medium lacking G418, displayed growth kinetics identical to those obtained with the transfectants above (data not shown). Taken together, our observations indicated that these episomal transfectants did not appear to alter the characteristic growth kinetics of the parental L. donovani or L. mexicana promastigote cell lines. Subsequently, such transfected promastigotes were analyzed for their expression of either the LdINV-HA or LmexINV-HA chimeric proteins using Western blots, immunoprecipitations (IPs), in situ enzyme activity gels, and spectrophotometric invertase enzyme assays.
Expression of the LdINV-HA and LmexINV-HA chimeric proteins in transfected parasites
Western blot analyses were performed to determine whether LdINV-HA and LmexINV-HA transfected promastigotes actually synthesized and released/secreted their respective chimeric protein during their growth in vitro. For these experiments, the transfected promastigotes were grown in chemically defined media, and their unconcentrated cell-free culture supernatants were subjected to IP as indicated below with either the α-rLdINV antibody or the rabbit α-HA antibody. The resulting immunoprecipitates were subjected to SDS-PAGE, transblotted onto PVDF membranes, and probed appropriately with either the respective α-rLdINV or α-HA antibodies. Western blots of samples immunoprecipitated with the α-HA antibody were probed with our α-rLdINV antibody. Results of such blots showed that the α-rLdINV antibody specifically reacted with only a single ~72 kDa LdINV-HA or LmexINV-HA-expressed protein in these α-HA immunoprecipitated samples (Fig. 7a). As anticipated, similar immunoprecipitates obtained from L. donovani pKSNEO and L. mexicana pKSNEO control transfectants showed no reactivity with our α-rLdINV antibody (Fig. 7a). Similarly, Western blots of culture supernatants immunoprecipitated with our α-rLdINV antibody were probed with the α-HA antibody. Results of such assays showed that the α-HA antibody specifically reacted only with an ~72 kDa LdINV-HA or LmexINV-HA-expressed protein in these immunoprecipitated samples (Fig. 7b). Immunoprecipitates obtained from L. donovani pKSNEO and L. mexicana pKSNEO control transfectants showed no reactivity with the α-HA antibody (Fig. 7b).
Fig. 7.

Expression of the LdINV-HA and LmexINV-HA chimeric proteins in transfected parasites. a Western blots of samples immunoprecipitated with the α-HA antibody and probed with our α-rLdINV antibody. Unconcentrated (×1) culture supernatants from LdINV-HA and LmexINV-HA-transfected promastigotes and their pKSNEO controls were immunoprecipitated with the α-HA antibody. Western blots of such immunoprecipitates were probed with our α-rLdINV antibody. The latter antibody only reacted with a single ~72 kDa (arrow) HA-abeled protein in the culture supernatants from LdINV-HA and LmexINV-HA promastigotes but not with their pKSNEO-transfected controls. b Western blots of samples immunoprecipitated with our α-rLdINV antibody and probed with the α-HA antibody. Unconcentrated (×1) culture supernatants from LdINV-HA and LmexINV-HA transfected promastigotes and their pKSNEO controls were immunoprecipitated with our α-rLdINV antibody. Western blots of such immunoprecipitates were probed with our αHA antibody. This α-HA antibody only reacted with a single ~72 kDa (arrow) protein in the culture supernatants from LdINV-HA and LmexINV-HA promastigotes but not with their pKSNEO-transfected controls
Taken together, results of these Western blot experiments demonstrated the following: (1) that the LdINV-HA and LmexINV-HA chimeric gene constructs were readily transcribed and translated into single ~72 kDa LdINV-HA and LmexINV-HA chimeric proteins in each of the transfected parasite species, respectively, (2) that these chimeric proteins were actually released/secreted by the transfected parasites during their growth in vitro, and (3) that the rabbit α-rLdINV antibody (i.e., made against the recombinantly expressed L. donovani invertase in E. coli) in fact recognized and immunoprecipitated both the LdINV-HA and the LmexINV-HA-expressed recombinant leishmanial invertases. Cumulatively, these observations showed that both the native parasite released/secreted invertases and their HA epitope-tagged episomally expressed recombinant proteins (i.e., encoded by the LdINV and LmexINV genes) possessed common structurally conserved antigenic epitopes.
Results of our IP and Western blot analyses above showed that both the Ld- and Lmex-INV-HA-transfected parasites synthesized and released/secreted their respective chimeric proteins. Therefore, it was relevant to determine whether these chimeric proteins in fact possessed functional invertase activity. For those assays, unconcentrated (1×) culture supernatants from LdINV-HA and LmexINV-HA transfected promastigotes and their pKSNEO controls were assayed for invertase activity in our qualitative agarose-gel-TTC reaction product capture assay. Results of those qualitative assays demonstrated that the LdINV-HA-transfected promastigotes synthesized and released/secreted significantly enhanced levels of invertase activity versus their pKSNEO controls (Fig. 8a, b). The latter observations were verified by results obtained with these culture supernatants in our GO spectrophotometric invertase assays (Fig. 8e). The enhanced levels of invertase activity produced by the LdINV-HA transfectants (i.e., ~10,000 units of invertase activity/107 parasites/ml) presumably reflect an additive contribution of both their wild-type activity and that of the expressed chimeric enzyme (c.f. Figs. 1d, 8e). In contrast, the activity observed with culture supernatants from pKSNEO control transfectants (i.e., ~100 units of invertase activity/107 parasites/ml) is equivalent to the endogenous levels of invertase activity produced and released by “wild-type” promastigotes (c.f. Figs. 1d, 8e). As anticipated, very similar results were obtained in these assays using culture supernatants from LmexINV-HA-transfected promastigotes and their pKSNEO controls (Fig. 8c, d). The gel shown in Fig. 8 is representative of the results obtained from numerous gel assays which were carried out in these experiments. Conclusions drawn based on our results from the L. donovani transfectants above similarly apply to those obtained with the L. mexicana transfectants. Results of those assays showed that the L. mexicana INV-HA transfectants did in fact produce and secrete the chimeric invertase activity but interestingly only at qualitatively lesser amounts than that produced by similar numbers of L. donovani transfectants (c.f. Fig. 8a, c). The latter observations mirror those which we obtained above with wild-type parasites from both of these species (c.f. Figs. 1, 8). Taken together, these observations further support the hypothesis which we stated above suggesting that the New World species produce invertase activity but at overall lower levels than that produced by Old World parasites. These observations also suggest that even though driven by episomal expression, synthesis and release/secretion of invertase by the New World L. mexicana parasites must involve some level of regulation.
Fig. 8.

Detection of invertase activity produced by INV-HA transfected promastigotes. Aliquots of unconcentrated (1×) culture supernatants from L. donovani LdINV-HA (a) and L. mexicana LmexINV-HA-(c) transfected promastigotes and their respective pKSNEO controls (b Ld Ctl and d Lmex Ctl, respectively) were loaded to precut wells of an agarose gel containing 25 mM sucrose as substrate and allowed to diffuse into the gel for 4 h at 37 °C. Following incubation, the resulting hydrolysis product (fructose) was visualized in these plates using triphenyltetrazolium chloride (TTC) as a capture reagent. The resulting invertase activity is shown in these plates by the presence of a dark red/purple-colored ring of TTC-captured product radiating from the sample wells. The gel shown here is one example of the results obtained from numerous gel assays which were carried out in these experiments. e Quantitation of invertase activity produced by LdINV-HA-transfected promastigotes and their pKSNEO controls. Aliquots of unconcentrated (1×) culture supernatants from LdINV-HA and pKSNEO control promastigotes (Ld Ctl) were assayed using sucrose as substrate in a glucose oxidase spectrophotometric assay. Aliquots of culture supernatants were taken and assayed for invertase activity at a cell density of ~107 parasites/ml. The invertase activity is shown as μg of glucose produced by 107 parasites/ml/hour. (Color figure online)
Discussion
Species of the protozoan parasite Leishmania cause over 2 million new cases of human cutaneous, mucocutaneous, and fatal visceral disease per year worldwide [1]. All Leishmania parasites undergo a digenetic life cycle which includes differentiation, development, and transmission between a sand fly vector and a mammalian host. Within these hosts, Leishmania parasites reside and multiply in highly restricted microenvironments (i.e., as extracellular, flagellated, promastigote forms within the alimentary tract of their sand fly vectors, and as obligate intracellular amastigote forms within the phagolysosomal compartments of infected macrophages) [2]. In this report we chose to use L. donovani (1S/CL2D) as an archetypical example of an Old World Leishmania which can cause fatal human visceral disease. Conversely, we chose to use L. mexicana (M379) in this study because it is an important protozoan pathogen of humans throughout Central and South America and that it is the major causative agent of human cutaneous leishmaniasis in the New World [17].
It is of relevance to point out that the focus of our laboratory concerns the identification and characterization of both cell surface membrane and released/secreted enzymes produced by these pathogenic Leishmania parasites. The rationale for our studies is to address how these parasites access essential nutrients from both their mammalian and insect vector hosts to facilitate their survival, multiplication, differentiation, and transmission throughout their developmental life cycle. With that in mind, we were motivated to initiate the current study based on the report of Blum and Opperdoes [8] which described for the first time, the identification of a released/secreted sucrase/invertase-like activity from L. donovani promastigotes. In that report, those investigators showed that a soluble sucrase/invertase-like enzyme activity was constitutively produced and released/secreted by L. donovani promastigotes (i.e., the 1S/CL2D strain) during their growth in vitro. Further in their discussion, Blum and Opperdoes concluded: “The sucrase that is secreted by promastigotes is likely to play an important role in the nutrition and development of the promastigotes in the insect gut,…” [sic]. Subsequently, the results of Blum and Opperdoes were verified and extended by Gontijo et al. [9]. In that report, those authors showed that promastigotes of a geographically distinct isolate of L. donovani from India (MHOM/80/DD8) also produced and released/secreted sucrase/invertase-like enzyme during their growth in vitro. Further, they showed that promastigotes of several other Old World Leishmanias also produced and released/secreted sucrase/invertase-like activities [i.e., L. aethiopica, L. infantum, L. major, L. (Sauroleishmania) tarentolae, and L. tropica]. Of equal or greater significance, Gontijo et al. demonstrated that such sucrase/invertase-like activities were also produced and released/secreted by promastigotes of at least six different New World Leishmanias (i.e., L. amazonensis, L. braziliensis, L. chagasi, L. guyanensis, L. mexicana, and L. panamensis). Cumulatively, the results of Gontijo et al. indicated that the production and release of sucrase(s)/invertase-like activities by promastigotes is a common biological characteristic of both Old and New World Leishmanias.
In light of those reports, we initiated our studies toward the identification, characterization and expression of the gene(s) encoding these putative leishmanial secretory/released invertase-like enzymes. In preliminary experiments, we devised an agarose-gel-TTC reaction product capture assay to qualitatively measure the invertase activity produced and released/secreted by L. donovani promastigotes in vitro. The invertase activities of such promastigote culture supernatants were also quantitated using the GO spectrophotometric assay. Results of these assays were in full agreement with the observations reported by Blum and Opperdoes concerning L. donovani. In subsequent experiments, we demonstrated that invertase activity was also produced and released/secreted by promastigotes of numerous different Old and New World species of Leishmania. Thus, our current results confirmed and extended the observations of Gontijo et al. and provide strong evidence showing that the synthesis and release/secretion of invertase-like activities is a common feature of promastigotes of all Leishmania species. It is of interest to note, that the results of our quantitative assays showed that promastigotes of each of the Old World Leishmania species tested typically produced at least 100 units of invertase activity per 1 × 107 cells. In contrast, promastigotes of each of the New World species tested produced only ~10 units of invertase activity per 1 × 107 cells. The dichotomy in invertase production observed between Old and New World Leishmania parasites presumably reflects their individual adaptation for growth and survival within the alimentary tracts of their geographically unique sand fly vector hosts.
As indicated above, the cumulative results of our qualitative and quantitative invertase assays demonstrated that promastigotes of virtually all of the Old World Leishmania species tested produced significant quantities of invertase activity. In that regard, experiments were devised to isolate the released/secretory invertase produced by L. donovani (1SCL-2D) promastigotes. Following Mono-Q FPLC fractionation and SDS-PAGE separation, a single ~70 kDa L. donovani putative invertase (LdINV) was isolated. Subsequently N-terminal end Edman Amino Acid Sequencing of this ~70 kDa LdINV protein revealed that it contained an amino acid sequence KDGVPYE. Results of BLASTP searches of GeneBank revealed that the LdINV sequence fragment was common to numerous annotated invertases. Further, many of these invertases also contained a downstream conserved consensus sequence of FYASKTFYD. Oligonucleotide primers of these two target sequences were used in PCR reactions with L. donovani gDNA as template. The resulting ~850 bp PCR product served as a probe to identify an L. donovani cosmid clone (16.3) for our subsequent isolation of the gene encoding the LdINV. Nucleotide sequencing of this cosmid clone revealed that the LdINV ORF consisted of 1932 bp. Analyses showed that this sequence was identical to that present on chromosome 4 of the L. donovani TriTryp genome database which was recently annotated as a putative/potential invertase enzyme (LdBPK_040300.1). Translation of the LdINV ORF showed that it encodes a polypeptide of 644 amino acids with a calculated molecular mass of 72,290 Da. Analysis of the LdINV-deduced protein using various structural algorithms indicated that it possessed features typical of a soluble/secreted glycoprotein (e.g., the presence of a putative N-terminal signal peptide for targeting to the ER; six potential N-linked glycosylation sites, overall hydrophilicity; absence of both, membrane anchor domains and ER retention motifs). These predicted structural properties are in good agreement with our experimental data demonstrating that the native wild-type ~70 kDa LdINV was glycosylated (i.e., mannosylated) and that it was constitutively released/secreted by L. donovani promastigotes during their growth in vitro. Further, these observations are in agreement with the earlier studies of Gontijo et al. who isolated an ~70 kDa released/secretory invertase-like (sic sucrase) protein from culture supernatants of L. amazonensis promastigotes. Results of Pfam analysis of the LdINV-deduced aa sequence showed that it had high homology with, and characteristics of, the glyco/hydrolase 32 protein family. Moreover, within this family, the LdINV possessed characteristic signature sequences of the invertase subfamily (i.e., proteins/enzymes which can hydrolyze sucrose into glucose and fructose).
Results of our molecular analysis indicated that we identified a gene which putatively encoded the L. donovani released/secreted invertase enzyme. The LdINV sequence was subsequently used as our reference to identify its homologs in several other Leishmania species [i.e., L. infantum (LinJ.04.0300), L. major (LmjF.04.0310), L. tarentolae (LtaP04.0290), L. mexicana (LmxM.04.0310), and L. braziliensis (LbrM.04.0350)] annotated within the TriTryp genome database (www.tritrypdb.org). We experimentally validated the presence of all of these LdINV gene homologs using PCR analysis and nucleotide sequencing. Further, genomic analyses showed that all of these LdINV homologs possessed similar high levels of structural synteny within their respective genomes. Thus, each of the LdINV homologs has been annotated respectively to chromosome number 4 (www.tritrypdb.org). Moreover, our analyses showed a very high level of conserved synteny across this region of chromosome 4 in which each upstream and downstream ORF was also conserved among the Leishmania species examined. The cumulative data of our alignments and TriTryp database annotations demonstrated that the LdINV gene homologs appear to be structurally conserved in the genomes of both Old and New World Leishmania species. The prevalence of the LdINV homologs among all Leishmanias presumably reflects parasite adaptation for survival, growth, and transmission within their sand fly vector hosts (e.g., Phlebotomus sp. and Lutzomyia sp.). In parallel to expression in all Leishmania species that have been tested, a variety of monoxenic insect and plant kinetoplastids have also been shown to constitutively release/secrete functional invertase activities (i.e., C. fasciculata, H. samuelpessoai, L. seymouri, and Phytomonas sp.). As anticipated, we found several LdINV-like homologs in the published genomes of two of these organisms [i.e., C. fasciculata and Phytomonas sp. (HART-1 strain)]. It contrast to the foregoing, it is significant to point out that no LdINV homologs have been identified/annotated within the genomes of either the pathogenic Old World African trypanosomes (i.e., T. brucei brucei, T. brucei rhodesiense, T. brucei gambiense, T. congolense, or T. vivax) nor the New World human pathogen T. cruzi. The absence of LdINV homologs from all of the Trypanosoma species suggests that this gene product is not necessary for their survival within their tsetse fly or triatoma insect vector hosts.
Results of Pfam analyses indicated that the LdINV-deduced protein contained a 28 aa signal peptide and cleavage at this site would result in the ~70 kDa secreted/released protein having an N-terminal end sequence of KDGVPYE. These predictions are in full agreement with our experimental results below. MAFFT alignment analyses showed that an analogous-predicted signal peptide was also present in each of the other leishmanial LdINV homologs. Those predictions are in good agreement with our experimental results which demonstrated that promastigotes of each of the Leishmania species tested produced released/secreted invertase activity during their growth in vitro.
Moreover, MAFFT analyses further showed that each of the LdINV homologs possessed a conserved 14 amino acid signature sequence characteristic of the glyco/hydrolase 32 protein subfamily of invertases, including the critical aspartic acid residue within the active site. In addition, each of the LdINV-deduced protein homologs possessed both a characteristic N-terminal domain (i.e., a 324 amino acid block) and a C-terminal domain (i.e., a 71 amino acid block) typically found in members of the glyco/hydrolase 32 protein (invertase) family. Interestingly, all of the LdINV-deduced protein homologs also possessed a common 142 amino acid sequence which is inserted between the two invertase family signature domains above. Further, all of the LdINV-deduced protein homologs also possessed a common 67 amino acid C-terminal extension sequence, terminating with a valine residue. While these two “inserted leishmanial sequences” were present in all of the LdINV-deduced protein homologs, neither sequence was present in any other known member of the glyco/hydrolase 32 protein family. Results of NetNGlyc analyses indicated that all of the LdINV-deduced proteins possessed six potential N-glycosylation sites. However, of these, only two were present in all of the LdINV-deduced protein homologs and those were restricted to the two amino acid domains unique to the Leishmania invertases (i.e., Asn396 and Asn628). The cumulative results of these structural analyses demonstrated a very high level of conservation in protein structure among all of the LdINV homologs. Presumably such structural conservation must be required for the function of this secretory enzyme. Further, this would suggest that this enzyme’s function must be critical to the survival and development of all of these Leishmania species within their insect vector hosts.
In light of the foregoing observations, it was necessary to actually demonstrate that a mature LdINV mRNA was in fact synthesized by these parasites. In that regard, the cumulative results of our Northern blot and RT-PCR analyses with L. donovani demonstrated that a mature message for the LdINV gene was in fact readily transcribed by promastigotes and to a much lesser degree by axenic amastigote developmental forms of this parasite. Interestingly, virtually identical Northern and RT-PCR results were obtained concerning the presence of mature message for LmexINV in L. mexicana promastigotes and axenic amastigotes. Thus, these results demonstrated that mature, translatable mRNA was transcribed from both the LdINV and LmexINV genes. Further, these data suggested that each of these genes was differentially/developmentally up-regulated in promastigote developmental forms in each of these species. These findings are in good agreement with results of our enzymatic assays which showed that promastigotes but not axenic amastigotes produced significant quantities of soluble/secretory invertase activity. Taken together, these observations underscore the significance of the idea that this enzyme must play an important role(s) in the survival, development, and transmission of Leishmania promastigotes within their insect vector host(s).
In this report, we generated a recombinant LdINV protein (rLdINV) in E. coli to determine whether this gene encoded a functional invertase activity. Results of enzymatic assays demonstrated that this rLdINV protein did in fact possess potent invertase activity. Subsequently, we generated a rabbit antibody (α-rLdINV) against the functional rLdINV protein. Results of Western blot analyses demonstrated that this α-rLdINV antibody specifically recognized both the ~70 kDa rLdINV protein as well as the ~72 kDa native released/secreted invertase produced by L. donovani promastigotes. Of equal importance were our results which showed that this α-rLdINV antibody also recognized the ~72 kDa native released/secretory invertase produced by L. mexicana promastigotes. Thus, these results showed that the LdINV and LmexINV proteins possessed common antigenic epitopes. Moreover, these cumulative results demonstrated that the LdINV and LmexINV genes actually encode the native secretory invertases produced by promastigotes of both of these parasite species. Further, it is of significance to point out that this antibody (α-rLdINV) also specifically recognized and reacted with the native ~72 kDa secretory invertases produced by a second geographically distinct L. donovani isolate (LRC-751) and in addition to multiple individual isolates of L. amazonensis, L. tropica, L. major, L. infantum, L. braziliensis, and L. tarentolae promastigotes used in this study. These experimental results are fully congruent with our molecular analyses which demonstrated the very high level of structural conservation noted among the various LdINV homologs analyzed in this report (i.e., LinJ.04.0300, LmjF.04.0310, LtaP04.0290, LmxM.04.0310, and LbrM.04.0350).
While the foregoing conclusions were compelling, we felt it was necessary to demonstrate that the L. donovani and L. mexicana INV genes in deed produced functional parasite secretory invertases. To do this, we generated two separate chimeric constructs: one contained the LdINV ORF and the second contained the LmexINV ORF. The 3′ end of each of these constructs contained the 9-aa HA epitope of the influenza virus. Promastigotes of both L. donovani and L. mexicana were episomally transfected with their homologous constructs. Subsequently, aliquots of culture supernatants from these transfectants were subjected to coupled IP and Western blotting analyses with rabbit α-HA and our rabbit α-E. colirLdINV antibodies. Taken together, the results of these experiments showed that: (1) the LdINV-HA and LmexINV-HA chimeric gene constructs were readily transcribed and translated into single ~72 kDa LdINV-HA and LmexINV-HA chimeric proteins in each of the transfected parasite species, respectively, (2) these chimeric proteins were actually released/secreted by the transfected parasites during their growth in vitro, and (3) the rabbit α-rLdINV antibody (i.e., made against the recombinantly expressed L. donovani invertase in E. coli) in fact recognized and immunoprecipitated both the LdINV-HA and the LmexINV-HA-expressed recombinant leishmanial proteins. Results of our qualitative agarose-gel-TTC reaction product capture assays demonstrated that the Ld- and LmexINV-HA-transfected promastigotes synthesized and released/secreted significantly enhanced levels of invertase activity versus their pKSNEO controls. These observations were verified and extended by results of our quantitative GO spectrophotometric assays. The latter showed that the LdINV-HA transfectants produced about ~100-fold higher levels of invertase activity than their pKSNEO control transfectants (i.e., ~10,000 vs. ~100 units of invertase activity). The levels of invertase activity observed with pKSNEO controls were found to be equivalent of those of non-transfected “wild-type” L. donovani promastigotes. Similar conclusions apply to the invertase activities observed with our LmexINV-HA and pKSNEO control transfected parasites. Cumulatively, these results demonstrated that the LdINV and LmexINV genes did in fact encode functional secretory invertases produced by these two parasite species. As indicated above, results of our preliminary experiments showed that all of the “wild-type” Old World Leishmania species characteristically produced ~10-fold higher levels of invertase activity compared to any of the New World species tested. Interestingly, this ~10-fold difference in invertase production between Old and New World Leishmania was also maintained and manifestly evident from the results of our over-expression studies using Ld- and LmexINV-HA-transfected parasites. These observations strongly suggest that even though driven by episomal expression, synthesis, and release/secretion of invertase by New World L. mexicana parasites must involve some level of endogenous/innate regulation.
As indicated above, the studies of Blum and Opperdoes were the first to report that L. donovani promastigotes produced and released/secreted an invertase-/sucrase-like activity [8]. Subsequently, Gontijo et al. reported that such invertases/sucrases (sic) were produced by promastigotes of a variety of New and Old World Leishmania species [9]. The results of the current study have confirmed those previous observations and extended them to include the molecular and structural characterization of the genes encoding these parasite secretory invertases. It is of significance to note that both the reports of Blum and Opperdoes and Gontijo et al. suggested that such secreted parasite invertases/sucrases might play important roles in the survival and development of promastigotes within the gut of their insect vector host(s). It is of relevance to point out that both Old and New World Leishmania parasites are transmitted to their respective phlebotomine sand fly vectors (i.e., Phlebotomus sp. in the Old World and Lutzomyia sps. in the New World) when these insects take a blood meal from an infected mammalian host. Within these infected insect vectors, Leishmania parasites multiply and move anteriorly in the sand fly alimentary tract. In order to sustain this anterior migration, these parasites must obtain nutrients/energy sources derived from the insect host gut. Interestingly, between their infectious blood meals, female sand flies characteristically feed on plant juice sugars including sucrose and other complex polysaccharides [7]. Following ingestion, such sugar meals are shunted and stored within the crop compartment of the insect’s gut. Hence, between infectious blood meal feeds, such sugars are regurgitated back into the sand fly anterior midgut where they would impact/interface with the anteriorly migrating Leishmania parasites. Within this milieu, the parasite extracellular secretory invertase could readily hydrolyze such plant sugars rendering them available for uptake by the parasites as nutrients/energy sources. Such uptake is imminently likely since Leishmania promastigotes possess multiple cell surface membrane sugar transporters (e.g., the highly active glucose/H+ symport system) [18, 19]. Thus, within this context, the secretory invertases would fulfill a vital role(s) in the survival, growth, development, and transmission of Leishmania parasites within their insect vector hosts.
It is tempting to speculate that these secretory invertases might also be part of a parasite chemi/osmotic sensory system which could stimulate/facilitate the anterior migratory behavior typically observed in Leishmania parasites within the insect vector gut. In that regard, Leishmania promastigotes have been shown experimentally to directionally orient and migrate within sugar and osmotic/chemical gradients similar to those potentially present in the insect vector midgut [20]. It is important to point out that the Leishmania apical flagellum is responsible for such directional orientation and movement of these parasites. Interestingly, two Leishmania cell surface transport proteins have recently been characterized [i.e., the L. mexicana glucose transporter 1 (LmxGT1) and a L. major aquaglyceroporin channel (LmjAQP1)] and shown to be localized to the flagellar membranes of their respective Leishmania sps. [21–23]. In these reports it was speculated that their respective specific proteins were involved with chemical/environmental sensing and transport regulation. Thus, in such a model, the hydrolysis products of the parasite secretory invertases, described herein, would change the osmotic microenvironment and could readily be sensed and transported by the latter flagellar transport systems to facilitate parasite motility and mobility within the insect vector gut.
As mentioned above, it has been reported that both Old and New World species of sand flies produce their own gut invertase-like activities [7]. It has been suggested that such endogenous gut invertases would aid the sand fly to digest complex polysaccharides they imbibed from their plant juice meals. Thus, in an infected (female) sand fly the leishmanial secretory invertase could be nutritionally beneficial to both the parasite and its insect vector. Presumably, the production of the parasite secretory invertases represents a symbiotic evolutionary adaptation to the plant juice feeding behavioral habits of their insect vector hosts. In that context, the ~10-fold difference observed in invertase production in the current study between Old and New World Leishmania species must reflect a symbiotic adaptation to the plant feeding preferences and habits of their respective geographically distinct sand fly vector hosts (i.e., Phlebotomus sps. vs. Lutzomyia sps.). The foregoing hypothesis seems logical within the context of the symbiotic relationships between the Leishmania parasites and their sand fly vector hosts. In addition to the secretory/released invertases described herein, a novel sucrose/H+ (proton) symport system has been recently reported to be present in the surface membranes of L. donovani promastigotes [24]. Presumably, the secretory invertases and this sucrose symport system would work in concert to fulfill the energy needs of Leishmania promastigotes within their insect vector gut. In contrast to Leishmania, no parallel host/parasite relationships can be espoused for either of their African or American Trypanosoma sp. relatives. In that regard, to date, no biochemical or molecular evidence exists concerning the presence of invertase or invertase-like enzymes or genes in any African or American Trypanosoma species.
In the current report, we have demonstrated that promastigotes of both Old and New World Leishmania species constitutively synthesized and released functional invertase enzyme activity into their cell culture supernatants. We assume that such invertase activity is released from these parasites via their endogenous secretory pathways similar to that described previously for their leishmanial secretory acid phosphatases [25, 26]. Recently; however, exosomes have been reported to exist as an alternative mechanism for the release of endogenous constituents by Leishmania parasites [27, 28]. While we cannot rule out that at least some invertase activity might leave these parasites via exosomes, the majority of the invertase activity that we measured in this study showed no functional latency (i.e., as mentioned in the “Results” section above).
In summary, results of the current study have shown that promastigotes of both Old and New World Leishmania species universally synthesize and release invertase enzymes during the course of their developmental life cycles. Further, we have identified the genes encoding these enzymes, shown that they are differentially/developmentally expressed and demonstrated the functional activity of their expressed proteins. The ability to now homologously over-express these genes in various Leishmania parasites should facilitate future studies concerning the role(s) of this enzyme in the developmental life cycles of these important human pathogens. Since invertases appear to be universally expressed by the promastigote (i.e., insect vector) forms of all Leishmania species tested, but not by amastigote forms (i.e., the parasite stage producing disease in humans), this enzyme might represent a logical target for disrupting their developmental life cycle and hence their transmission by their insect vector hosts.
Experimental procedures
Reagents
All chemicals used, unless specified, were of analytical grade and purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Enzymes used for molecular studies were obtained from Roche Molecular Biochemicals (Indianapolis, IN, USA); DNA and RNA molecular mass standards were from Life Technologies (Grand Island, NY, USA), and protein molecular mass standards were purchased from GE Healthcare Biosciences (Pittsburg, PA, USA).
Nomenclature
The designations used in this report for genes, proteins, and plasmids follow the nomenclature for Trypanosoma and Leishmania as outlined by [29].
Parasite cell lines
Multiple Leishmania cell lines which cause either visceral or cutaneous disease were used in this report. These included two visceral strains of L. donovani 1S-CL2D (WHO designation MHOM/SD/62/1S/CL2D originally isolated from a patient in Sudan) and LRC-751 (WHO designation MHOM/IN/93/BI2302/LRC-751 isolated from a visceral patient in India); two strains of L. infantum Spain (WHO designation MCAN/SP/00/FVM10001JL) and L. infantum LRC-639 (WHO designation MCAN/IL/94/Robi/LRC-639 isolated from a dog in Israel); two isolates causing cutaneous disease in the Old World: a single Old World isolate of L. major Friedlen (WHO designation MHOM/IL/81/Friedlin isolated from a patient with cutaneous disease); L. tropica WR683 (WHO designation MHOM/SU/58/OD/WR683); and various strains causing cutaneous disease in the New World: i.e., two cutaneous isolates of L. mexicana M379 (WHO designation MNYC/BZ/62/M379 an isolate from a rodent in Belize) and BEL21 (WHO designation MHOM/BZ/82/BEL21 isolated from Belize); one isolate of L. braziliensis WR668 (WHO designation MHOM/BR/76/LTB/300); two isolates of L. amazonensis PH8 (WHO designation VEC/BR/67/PH8) and GML584 (WHO designation MHOM/P/GML/584); one isolate of L. pifanoi WHOLL1 (WHO designation MHOM/VE/57/LL1); two isolates of L. panamensis WR470 (WHO designation MHOM/BR/470) and WHO594 (WHO designation MHOM/BR/L/594); and one isolate of L. guyanensis M4147 (WHO designation MHOM/BR/75/M4147), and a single isolate of L. tarentolae (Jena 101, Jena Bioscience, Jena, Germany an isolate of a gecko).
In vitro culture of parasites
Promastigote developmental forms of all of the above Leishmania species were grown at 26 °C in Medium M199 (Life Technologies), supplemented with 10 % (v/v) heat-inactivated fetal bovine serum (FBS, Gemini Bio-Products, Woodland, CA). In contrast, axenic amastigote forms for L. mexicana and L. donovani were grown at 32 and 37 °C, respectively, in medium RPMI 1640 supplemented with 20 % (v/v) FBS, as described previously [30]. For isolation of nucleic acids and proteins, both promastigote and axenic amastigote parasite cultures were harvested at ~mid-log phase (~1–2 × 107 cells ml−1) by centrifugation at 2100×g for 15 min at 4 °C. The resulting cell pellets were washed twice in ice-cold phosphate buffered saline (PBS, 10 mM sodium phosphate, 145 mM NaCl, pH 7.4) by centrifugation and finally re-suspended in the appropriate buffers for isolation of nucleic acids and proteins.
Short-term release assay for secretory proteins
In order to obtain useable quantities of proteins released/secreted by promastigotes during their growth in vitro, promastigotes of the various strains were cultured at 26 °C in a macromolecule-free, chemically defined medium (M199+) [31]. When such cultures reached a density of ~1.5 × 107 cells ml−1, the cells were examined by phase contrast microscopy to ensure >99.9 % cell viability and subsequently harvested by centrifugation at ~2100×g for 20 min at 4 °C. The cell pellets were washed (3×) by centrifugation as above using ice-cold HEPES/NaCl pH 7.4 [25] and resuspended at a density of 4 × 108 cells ml−1 in HEPES/NaCl pH 7.4 containing 1 mM glucose (as an energy source). Such cell suspension was incubated for 3 h at 26 °C on a rocking platform. During this period, secretory proteins previously synthesized by the cells were released into the buffered medium. At the end of incubation the cells were re-examined by phase contrast microscopy to ensure cell viability and subsequently harvested by centrifugation as described above. Such supernatants were finally recentrifuged at high speed (6000×g [25]) to ensure complete removal of cells. The resulting supernatants from several release assays (from each of the organisms tested) were pooled, dialyzed against several changes of buffer (50 mM Tris pH 7.4), and frozen for further analysis. One of these pools was subsequently subjected to anion exchange chromatography.
FPLC fractionation of released/secreted proteins
Dialyzed cell-free release assay supernatants obtained from L. donovani (1SCL-2D) promastigotes were loaded on to a Mono-Q column fitted onto a FPLC system (GE Healthcare Life Sciences). The column was extensively washed with buffer (50 mM Tris pH 7.4) and, subsequently, bound proteins were eluted using a Tris–NaCl gradient. Four ml fractions were collected, dialyzed, and concentrated. Aliquots from each fraction were separated on 10–15 % NUPAGE gels (Life Technologies) and silver stained using SilverXpress® silver staining kit (Life Technologies) according to manufacturer’s recommendations. Results of silver staining demonstrated that one of these fractions contained a single protein band with an apparent molecular mass of approximately 70 kDa. An aliquot of this selected fraction was separated by SDS-PAGE, blotted onto a PVDF membrane (Life Technologies), and stained with Coomassie Blue as described previously [32]. Subsequently, a single Coomassie Blue-stained protein band was excised from such blots and subjected to N-terminal Edman Sequencing at the Research Technologies Branch facility of the NIAID, NIH, Rockville, MD.
Generation of cell-free culture supernatants
In order to analyze, the secreted/released proteins (enzymes) produced by promastigotes of the various Leishmania species listed above were grown in a macromolecule-free, chemically defined medium (M199+) [31]. Such cultures were initiated as follows: mid-log phase promastigotes were inoculated into culture medium to a final density of ~1 × 106 ml−1 and allowed to grow for ~72 h at 26 °C. When such cultures reached a density of ~1.5 × 107 cells ml−1 they were examined by phase contrast microscopy to ensure >99.9 % cell viability and subsequently harvested by centrifugation at ~2100×g for 20 min at 4 °C. Following a high-speed re-centrifugation step, to ensure the complete pelleting of cells [33], the resulting cell-free (1×) culture supernatants were concentrated by ~100-fold (100×) using pressure ultrafiltration as described previously [34]. To evaluate the kinetics of the invertase enzyme activity produced/released by parasites during their in vitro growth, cultures as above were harvested at different time points during their growth. These were concentrated as described above and such samples were evaluated for their total enzyme activities.
Agarose-based qualitative assay for invertase
We developed an agarose-based activity assay in order to qualitatively determine the presence of invertase activity produced/secreted by parasites grown in vitro. Briefly 20 ml of a pre-warmed solution of 1 % (w/v) agarose (Sigma Aldrich) containing 0.2 M sodium acetate buffer (pH 5.0) and 25 mM sucrose was pipetted into a 100 mm petri dish. After cooling to RT, small wells were cut and the agarose plugs were extracted by vacuum. Aliquots of concentrated parasite culture supernatants from above were added to the wells of such plates and the plates were incubated for 4 h at 37 °C. The premise for these assays was based on the hypothesis that parasite released/secreted invertase activity would hydrolyze the sucrose substrate present in the agarose gel producing glucose and fructose as end products. One of the products of this reaction (fructose) was visualized in these plates by using the TTC (Sigma Aldrich) capture reagent. This was accomplished by flooding the plates with 0.1 % (w/v) TTC in 1 N NaOH and incubating them for 10–20 min at 45 °C. The resulting invertase activity was visualized in these agarose plates by the presence of a dark red–purple-colored ring of TTC-captured product radiating from the sample wells. Controls in these assays consisted of aliquots of unused parasite growth medium or short-term release assay buffer. Digital images of such plates were capture using a Nikon Coolpix 3500 camera (Nikon, Melville, NY, USA). Images were processed using Adobe Photoshop CS2 (Adobe Systems, Inc.).
Spectrophotometric assay of invertase activity
Invertase activity present in parasite culture supernatants was measured using sucrose as the substrate. The premise for these assays was also based on the hypothesis that parasite released/secreted invertase activity would hydrolyze the sucrose substrate into glucose and fructose. In these assays, we could quantitate the resulting glucose product. Briefly, aliquots of parasite culture supernatants or short-term release assays were incubated with 25 mM sucrose (final concentration) in a total volume of 0.5 ml in 0.1 M sodium citrate buffer, pH 5.5 at 37 °C. Following incubation, aliquots of the reaction mix (50–100 μl) were assayed for their glucose content using the GO assay kit (Sigma Aldrich) as per manufacturer’s instructions. Enzyme activity in these assays was expressed as μg of glucose produced by x number of parasites/ml per unit time. It was necessary to express activity in this fashion because the total amount of invertase (protein) actually secreted by these parasites into their culture medium was below current detection methods. Controls in these assays consisted of aliquots of unused parasite growth medium or short-term release assay buffer.
Oligonucleotide primers, PCR, and probe preparation
Degenerated oligonucleotide primers: a forward primer (PCR-Fwd) was designed based on the N-terminal sequence (aa: KDGVPYE) of the FPLC-purified L. donovani (1SCL-2D) ~70 kDa protein fraction, and a reverse primer (PCR-Rev) was designed to a relatively conserved consensus sequence (aa: FYASKTFYD, GenBank) common to numerous invertases [35]. These primers (PCR-Fwd: 5′-AAG GAC/TGGC/T/G GTG/C CCG/C TAC GAG-3′ and PCR-Rev: 5′-GTC GTA GAA C/GGT CTT C/G/AGA C/GGC GTA GAA-3′) were synthesized by β-cyanoethyl phosphoramidite chemistry using an Expedite™ nucleic acid synthesis system (PE Applied Biosystems: Eurofins MWG Operon, Huntsville, AL, USA) and used in PCR amplifications with L. donovani (1SCL-2D) genomic (g) DNA as a template. It is important to note that is was possible to directly use gDNA as template since trypanosomatid protozoans generally do not possess introns within the coding region of their ORFs [36, 37]. After an initial “hot start” at 94 °C for 2 min, the conditions used for amplification were 94 °C for 30 s, 50 °C for 30 s, 72 °C for 1 min (35 cycles), and a final step at 72 °C for 5 min. The 858 bp amplified product was cloned into the pCR®2.1-TOPO vector (Life Technologies) and the resulting plasmid (LdINV-PCR858) was subjected to nucleotide sequencing. Analyses of the sequence data obtained from the LdINV-PCR858 clone showed that it had very high sequence identity (~96 %) with the L. major Friedlin chromosome #4 left end sequence, obtained in the genome sequencing project (gi|150270 87|emb|AL389894.4|LMFLCHR4A). It is of pertinence to note that this L. major sequence was annotated as invertase (β-D fructofuranosidase).
Subsequently, the LdINV-PCR858 cloned PCR fragment was labeled with digoxigenin-dUTP (DIG) using the PCR Dig-Labeling Kit according to manufacturer’s instructions (Roche). The resulting DIG-labeled probe (LdINV-DIG856) was used to screen an L. donovani (1SCL-2D) cosmid library [38]. A positive cosmid clone (cosmid 16.3) was isolated and sequenced in the region of the putative LdINV gene. Results of such sequencing demonstrated that the LdINV consisted of an ORF of 1932 bp including a methionine start codon of ATG and a stop codon of TAG at the end. It is important to note that the LdINV-PCR858 sequence was found to have 100 % identity to that contained within the LdINV ORF.
The nucleotide sequence of the LdINV ORF was used in multiple sequence alignments of invertases from various organisms. These alignments were conducted using the MacVector 7.0 program and MAFFT [v6.860b] (myhits. isb-sib.ch) to determine cluster relationships among those sequences and to construct dendrograms representing cluster relationships. More importantly, such analyses were extended to assess whether an LdINV-like gene might also be present in the genomes of other related trypanosomatid parasites. To that end the LdINV sequence was used as our reference to examine the TriTryp/Leishmania genomes at www.tritrypdb.org.
Isolation of genomic DNA and Southern blot analysis
Total genomic gDNA was isolated from log-phase promastigotes of each parasite cell line using the Wizard Genomic DNA purification kit (Promega Corporation, Madison, WI, USA) according to manufacturer’s recommendations. For Southern blot analysis, such gDNA was digested with several restriction endonucleases and separated on 1 % agarose gels, transferred to positively charged Nylon membranes (Roche), and cross-linked to the membranes by UV irradiation using a Stratalinker® 2400 (Stratagene, Stratagene La Jolla, CA, USA). Subsequently, blots were hybridized under high stringency using a DIG-labeled DNA probe (i.e., LdINV-DIG856), corresponding to a portion of the L. donovani invertase ORF (i.e., nt 85–942). After high stringency washing (0.1× SSC, 0.1 % SDS at 65 °C), the hybridized fragments were visualized using an anti-digoxigenin-alkaline phosphatase conjugated antibody in conjunction with a chemiluminescent reagent (CSPD) according to manufacturer’s instructions (Roche). Images were captured from such blots using BIOMAX™-MR X-ray film (Kodak, Rochester, NY, USA) and processed using Adobe Photoshop CS2 (Adobe). In addition, DNA was also isolated from the LdINV-16.3 cosmid clone and similarly digested with several different restriction endonucleases. Such preparations were subsequently subjected to Southern blot analysis as above.
Isolation of RNA and Northern blot analysis
Total RNA was isolated from log-phase promastigote and axenic amastigote cultures using RNA STAT-60 according to the manufacturer’s instructions (Tel-Test, Inc., Friendswood, TX). Total RNA (10 μg) was separated in 1.2 % agarose gels using the formaldehyde method [39], transferred onto nylon membranes, and cross-linked by UV irradiation as above. Blots were hybridized under high stringency conditions using a DIG-labeled probe (LdINV-DIG858). After high stringency washing, such blots were subjected to immunological detection using anti-digoxigenin antibody conjugated to alkaline phosphatase and developed using the CSPD chemiluminescent reagent as described above.
Mapping of the 5′ spliced leader acceptor site
As indicated above, trypanosomatids generally do not possess introns within their ORFs. However, the pre-mRNAs in these organisms are joined to a 39 nt conserved spliced leader at their 5′-end by trans-splicing to generate mature, translatable mRNAs. To identify the 5′-splice acceptor site in the LdINV gene, RT-PCR analysis was performed essentially as described previously [40]. For these reactions, cDNA was generated from total RNAs isolated from both L. donovani 1S-Cl2D promastigotes and axenic amastigotes. Such cDNAs were used as template in PCR with a forward primer (i.e., SpliceFwd) based on the Leishmania-spliced leader sequence [40] 5′-AACGCTATATAAGTATCAGTTTCTGTACTTTATTG-3′ and an internal reverse primer (INV Rev: 5′-CCGTTGGGGTTG TACTGCATGTACAGGTGAATTTT-3′). The resulting PCR-amplified products obtained from these reactions (i.e., using cDNAs from both promastigote and axenic amastigotes) were cloned into the pCR®2.1-TOPO plasmid vector (Life Technologies), and sequenced using the vector-encoded M13 forward and reverse primers. Sequence data obtained from these two parasite developmental forms were compared. The results obtained from such analyses were also used to further verify the authenticity of the start site of the L. donovani invertase ORF.
Escherichia coli-recombinant LdINV expressed proteins and antibody production
An E. coli expression plasmid containing the wild-type LdINV gene [minus the N-terminal sequence (nt 1–75) encoding signal peptide] was made. To that end, the LdINV gene lacking a sequence encoding the putative signal peptide was amplified by PCR using the LdINV-16.3 cosmid as template and two primers. These included Primer-3 (5′-AAATGGATCCAgccgctcgcaaaggacggcgtgc-3′) which contained a BamHI restriction site (bold), and an extra nt A (underlined) to facilitate in-frame cloning in the expression vector, followed by 23 nucleotides of the LdINV gene sequence (lowercase), and Primer-5 (5′-ACAACTCGAGgac ggcgatgagggtgtcgagtaga-3′) which contained an XhoI restriction site (bold) followed by the last 25 nts of the LdINV gene sequence (excluding the stop codon, lowercase). The sequence of the resulting PCR product was cloned into pCR®2.1-TOPO (Life Technologies). The sequence of the cloned fragment was confirmed by nucleotide sequencing using the M13 forward and reverse primers. Subsequently the L. donovani invertase encoding fragment was excised from this plasmid by BamHI and XhoI digestion, purified and ligated into the BamHI and XhoI site of the pETBlue-2 expression plasmid (Novagen, Merck KGaA, Darmstadt, Germany). The resulting expression plasmid encoded the recombinant LdINV protein plus a C-terminal 6-His tag (rLdINV-His). The BL21DE3 pLacI E. coli host cells (Novagen) were transformed with the rLdINV-His containing plasmid from above and clones were selected following induction with 1 mM isopropylthio-β-galactoside. One such clone was selected for further analysis. As anticipated, results of such analysis demonstrated that the bulk of the expressed rLdINV-His protein was found to aggregate into inclusion bodies. The purification of the rLdINV-His proteins was performed under denaturing conditions and the denatured expressed proteins were bound to, and eluted from, nickel–nitrilotriacetic acid Superflow beads (Qiagen) in an FPLC (Amersham) according to manufacturer’s recommendations. The purified rLdINV-His protein was used to immunize a New Zealand White rabbit according to a standard protocol by Spring Valley Laboratories (Woodbine, MD, USA; i.e., primary immunization followed by three boosts). The resulting antiserum (i.e., the rLdINV antibody) was shown to specifically react against both the E. coli recombinant and the native parasite LdINV by Western blotting. This antibody was also used in subsequent immuneprecipitations and Western blot experiments as described below. Pre-immune serum (NRS) from this rabbit served as a control in all experiments.
Assessment of invertase activity of the E. coli-recombinant LdINV protein
Aliquots of recombinant (r) LdINV protein produced by E. coli were separated in 10 % SDS-PAGE gels and transferred on to PVDF membranes. Such membranes were washed extensively in several changes of Tris–HCl buffer, pH 7.4, containing 1 % (v/v) Triton X-100 (protein grade, Pierce Chemical, Thermo Fisher Scientific, Rockford, IL, USA) to remove bound SDS and re-nature the expressed rLdINV protein. Subsequently, such membranes were incubated for 1 h in 0.2 M acetate buffer pH 5.0 containing 25 mM sucrose as enzyme substrate. Activity of the rLdINV was demonstrated by the presence of its enzymatic reaction product (fructose) which was visualized by placing the gels in 0.1 % TTC in 1 N NaOH for 10–20 min at 45 °C. Subsequently, the reaction product demonstrating invertase activity appeared as a single brilliant red band at approximately 72 kDa. Images were captured from such blots using a Nikon Coolpix 3500 camera (Nikon, Melville, NY, USA) and processed using Adobe Photoshop CS2 (Adobe) as above.
Generation of an epitope-tagged LdINV expression construct
In this report pKSNEO, a leishmanial vector [41], was used to express a construct encoding an LdINV-hemagglutinin-tagged (-HA) chimeric protein in L. donovani (1SCL-2D) as well as analogous constructs in L. mexicana (M379). This vector has been used previously to express a variety of homologous and heterologous genes in both promastigotes and axenic amastigotes in several different species of Leishmania parasites [41–44]. For initial studies, a chimeric construct was designed that contained the complete ORF of the L. donovani invertase gene (including its 5′-end encoding the signal peptide) joined, at its 3′-end, with a sequence encoding a nine amino acid epitope of the influenza virus HA (Roche). For those studies, the LdINV-Cos16.3 cosmid was used as template in PCR reaction with a forward primer 5′-GATACTAGTATGGTGCAGAGGAGCTCACTT-3′ (containing an SpeI restriction endonuclease site shown in bold); and the reverse primer: 5′-CAAACTAGTTCACGCGTAGTCCGGCACGTCGTACGGGTATAGATCGCGGTC-3′ [containing an SpeI restriction endonuclease site shown in bold; stop codon in bold italics; and a -HA epitope tag (underlined sequence)]. The resulting amplified product was gel purified and cloned into the pCR2.1-TOPO vector (Life Technologies) to generate a pCR2.1::LdINV-HA plasmid. The sequence of the pCR2.1:: LdINV-HA construct was verified by nt sequencing. The insert was excised from this plasmid using SpeI endonuclease treatment. Subsequently, the excised fragment was ligated into the pKSNEO plasmid (linearized with SpeI) to generate the pKSNEO::LdINV-HA plasmid construct. The orientation of LdINV-HA in pKSNEO was verified using restriction endonuclease analysis and nt sequencing of the construct.
Transfection of pKSNEO expression plasmids into L. donovani parasites
Leishmania donovani (1SCL-2D) amastigotes were isolated from infected hamsters as described previously [30]. These in vivo derived amastigotes were allowed to transform into promastigotes at 26 °C in vitro, as described above. The promastigotes (i.e., in their second in vitro passage after transformation) were subsequently used in transfection experiments. Log-phase L. donovani promastigotes were transfected with either the pKSNEO control plasmid or the pKSNEO::LdINV-HA construct by electroporation using methods derived from Shakarian et al. 2002 [26]. To that end, L. donovani promastigotes were harvested, washed two times with 1×PBS by centrifugation (2100×g, 10 min at 4 °C), and re-suspended in ice-cold electroporation buffer (21 mM HEPES, 137 mM NaCl, 5 mM KCl, 0.7 mM Na2HPO4, 6 mM D-glucose, pH 7.0) at 108 cells ml−1. 500 μl aliquots of such cell suspensions were added to 2-mm gap electroporation cuvettes (BTX®Harvard Apparatus, Holliston, MA) to which 20 μl of purified plasmid DNA (at 1 μg/μl) in sterile 10 mM Tris, 2 mM EDTA, pH 8.0 (Quality Biological, Inc., Gaithersburg, MD) was added. The cells were electroporated using a single pulse (conditions: 475 V, 800 microfarads and 13 ohms) in a BTX ECM-600 electroporation system (BTX® Harvard Apparatus). Electroporated cells were incubated on ice for 10 min, transferred into 5 ml of complete culture medium (M199 containing 10 % [v/v] FBS) as described above and incubated at 26 °C for 24 h. Following recovery, the transfected cells were harvested by centrifugation as above and re-suspended in the same culture medium containing Geneticin® (G418, Life Technologies; at 15 μg ml−1 final concentration). Following initial drug selection, drug-resistant parasites were grown in increasing concentrations of G418 up to 50 μg/ml. Such cells were routinely maintained in culture using this antibiotic concentration or frozen and stored in liquid nitrogen. For some experiments, such transfected promastigotes were placed under conditions which permitted them to transform and grow as axenic amastigotes in the presence of G418 (i.e., in pH 5.5 medium at 37 °C), as described by [30]. Growth kinetics of both pKSNEO control and pKSNEO::LdINV-HA-transfected parasites were monitored at regular intervals during the time course of their growth in vitro. To that end, aliquots of such cultures were diluted appropriately with an isotonic buffer (ISOTON-II, Beckman–Coulter Particle Characterization, Miami, FL) and counted using a Model Z1 Coulter Counter (Beckman–Coulter) essentially as described by [30]. Both control and LdINV-HA transfectants were basically tested for their ability to produce/express LdINV-HA chimeric invertase proteins. In addition, cell lysates and culture supernatants from both pKSNEO and pKSNEO::LdINV-HA were tested for their functional invertase activity using both our agarose-based and spectrophotometric invertase assays as described above.
Identification of LdINV homologs in other Leishmania species
The TriTryp genomic database (www.tritrypdb.org) was examined to assess whether any other trypanosomatid parasites possessed any homologs of the LdINV gene. These databases were examined using the LdINV gene sequence as a prototype in various multiple sequence alignments conducted in MacVector 7.0 program and MAFFT [v6.860b] (myhits.isb-sib.ch). These analyses indicated that gene homologs were present in several Leishmania species (i.e., L. infantum, L. major, L. tarentolae, L. mexicana, and L. braziliensis). To validate whether such LdINV homologs were actually present in these various Leishmania species they were subjected to PCR analyses. For these assays, a forward oligonucleotide primer was synthesized reflecting the 5′ end of the LdINV ORF (i.e., from nt 85) and a reverse oligonucleotide primer was synthesized reflecting an internal sequence (i.e., from nt 850 within the LdINV ORF). PCR reactions were carried out with these oligonucleotides using gDNAs as templates isolated from L. donovani (1S-CL2D and LRC-751), L. amazonensis, L. mexicana, L. tropica, L. major, L. infantum, L. braziliensis, and L. tarentolae all as designated above. Products of these PCR reactions were separated on agarose gels and stained with ethidium bromide. An ~850 bp product was obtained for each of these reactions. Subsequently, each of these PCR products was cloned and subjected to nucleotide sequencing.
It is of importance to point out that L. infantum and L. major are the causative agents of human visceral and cutaneous disease, respectively, in the Old World; whereas, L. tarentolae is an Old World saurian (lizard) Leishmania. In contrast, L. mexicana and L. braziliensis are agents of human cutaneous and mucocutaneous diseases in the New World. It is of significance to note that results of preliminary experiments showed that the Old World LdINV gene encoded a functional invertase activity. In light of that, it was of interest, to ascertain whether one of these New World Leishmania gene homologs would also encode a similar functional enzyme activity. In that regard, we decided to investigate those possibilities using the L. mexicana LdINV homolog (LmexINV) as an example of an invertase enzyme present in an important New World Leishmania species.
Identification of an L. mexicana INV gene
As indicated above, the sequence of the LmexINV homolog was obtained from the TriTryp genome database (www.tritrypdb.org; GeneID: LmxM.04.0310). Using this sequence, we generated oligonucleotide primers reflecting the 5′ and 3′ end of this sequence: Forward Primer FLmex INVORF 5′-ATGCGCCGCGGGGTCATTCTGCTCCTCGTGGC-3′ and Reverse Primer RLmexINVORF 5′-CTAGACGTCGATGAGGGTGTCGAGTAGCGTGT-3′. These primers were used in PCR reactions with L. mexicana M379 promastigote gDNA as target using conditions similar to those as described above for the LdINV. Results of such reactions generated a 2 kb-amplified product which was cloned into the pCR®2.1TOPO vector and transfected into chemically competent TOP-10 cells (Invitrogen). Ten individual clones from those transfectants were randomly selected for nt sequencing. The resulting plasmid preparations from these were nt sequenced (Macrogen Corp., Rockville, MD) using vector encoded M13-F and M13-R primers. Results of these analyses showed that these plasmids contained an insert consistent with that of the LmexINV gene present in the Tri-Tryp genomic database.
Identification of mature mRNA encoding the LmexINV
Total RNA was obtained from ~107 L. mexicana M379 promastigotes or axenic amastigotes using STAT-60 (Tel-test, Inc., Friendswood, TX, USA) and processed according to the manufacturer’s recommendations [38]. The presence of mature LmexINV mRNA was assessed using RT-PCR analyses. As indicated above, trypanosomatid parasites generally do not possess introns within their ORFs. Further, pre-mRNAs in these organisms possess a 39 nt conserved SL at their 5′-end to generate mature translatable mRNAs [16, 45]. For these experiments, 1.5 μg of total RNA (pre-treated with DNase) from L. mexicana M379 Pro- and AxAm were reverse transcribed into cDNA using the Superscript® First Strand Synthesis System for RT-PCR (Invitrogen) and oligo d(t) primers as per manufacturer’s instructions. Subsequently, such cDNA was used as template in PCR with the forward L. mexicana SL sequence (i.e., nt 5–29 of the 39 nt SL) [46] primer 5′-AACGCTATATAAGTATCAGTTTCTG-3′) and an internal reverse primer based on a portion of the LmexINV ORF (i.e., RLmexINVRT 5′-CGCCAACTGCAGCGCCGAGTGC-3′). Controls for the RT-PRC reactions included no reverse transcriptase, no RNA, no SL primer and no internal invertase primer. The resulting PCR-amplified products obtained from these reactions using both Pro- and AxAm cDNAs were cloned into the pCR®2.1 TOPO plasmid vector (Invitrogen), nt sequenced, and analyzed. The sequence data for LmexINV obtained from each of the parasite developmental forms (i.e., promastigotes and axenic amastigotes) were compared.
Generation of an epitope-tagged construct for LmexINV
To examine the functional activity of the LmexINV, we generated an epitope-tagged construct of this gene and expressed it using the pKSNEO homologous expression system described above. To that end, an episomal construct was designed that contained the complete ORF of the LmexINV gene including the 5′-end encoding the putative signal peptide. A nine aa sequence encoding the influenza virus HA epitope was joined at the 3′-end of the above construct. This construct was generated by PCR using one of the pCR®2.1LmexINV clones above as template. The forward primer for this reaction was FLmexINVHA 5′-GATCACTAGTATGCGCCGCGGGGTCATTCTGC-3′ and the reverse primer was RLmexINVHA 5′-GATCACTAGTCTAGGCGTAGTCGGGGACGTCGTCGTAGGGGTAGACGTCGATGAGGGTGTCGAGT-3′. The PCR conditions used in these reactions were identical to those outlined above for the LdINV-HA reaction. The resulting 2 kb product obtained from these reactions was gel purified and cloned into pCR®2.1TOPO (Invitrogen) to generate pCR®2.1::LmexINV-HA. The insert was excised from the latter using SpeI digestion. Subsequently, this excised fragment was ligated into the SpeI site of the pKSNEO expression vector [41] to generate the pKSNEO::LmexINV-HA plasmid construct. The orientation of this plasmid was verified by restriction enzyme analysis and the nucleotide sequence was confirmed by DNA sequence analysis.
Transfection of pKSNEO expression plasmid into L. mexicana parasites
Leishmania mexicana wild-type M379 promastigotes were grown to mid-log phase and transfected with pKSNEO::L-mexINV-HA or a pKSNEO control plasmid construct by electroporation using the same conditions as described above. Subsequently, viable transfectants were selected in vitro using increasing drug concentrations of G418 up to 50 μg/ml as above. These transfectants were routinely maintained in culture using this antibiotic concentration or frozen and stored in liquid nitrogen. Further the growth of both the pKSNEO control and the pKSNEO::LmexINV-HA transfectants were monitored for their growth in vitro as above. For some experiments, such transfected promastigotes were placed under conditions which permitted them to transform and grow as axenic amastigotes in the presence of G418 (i.e., in pH 5.5 medium at 32 °C), as described by Joshi et al. 2007 [38]. Similar to the L. donovani transfectants above, the growth kinetics of both pKSNEO control and pKSNEO::LmexINV-HA-transfected parasites were monitored at regular intervals during the time course of their growth in vitro. In addition, the LmexINV-HA transfectants were tested for their ability to synthesize and release/secrete LmexINV-HA chimeric invertase protein.
Immunoprecipitations (IPs)
Lysates of pKSNEO control and transfected (pKSNEO::L-dINV-HA or pKSNEO::LmexINV-HA) promastigotes and their cell-free culture supernatants were subjected to IP assays with pre-washed rabbit anti-HA (Sigma Aldrich Co., St. Louis, MO, USA) antibody-bound Dynabeads® Protein A (Invitrogen, Carlsbad, CA, USA) as per manufacturer’s instructions. Briefly, 25 μl pellets of Dynabeads® Protein A were washed three times in 1× PBS/0.05 % Tween-20 (v/v) at room temperature (RT). Aliquots (25 μl pellet) of the washed Dynabeads were re-suspended in 100 μl of a 1:10 dilution of rabbit anti-HA antibody (Sigma) in 1× PBS/0.05 % Tween-20. Following agitation on an orbital rocker for 40 min at RT, the beads were washed five times with 1× PBS/0.05 % Tween-20. Subsequently, the anti-HA-bound Dynabeads were reacted with 100 μl of either parasite cell lysate or with an aliquot of 100× concentrated cell-free parasite culture supernatant. Such samples were incubated at 4 °C overnight on an orbital rocker. Subsequently, the Dynabeads–IP complexes were washed five times in 1× PBS/0.05 % Tween-20 at RT, re-suspended in 1× SDS-PAGE buffer, and heated to 90 °C for 10 min prior to separation in 10 % SDS-PAGE gels (Novex). Similar IP assays were also carried out with these samples using the rLdINV rabbit antibody and its NRS pre-immune serum in conjunction with Protein A Dynabeads under conditions similar to those used above. Immunoprecipitates obtained from these assays were further analyzed by SDS-PAGE and Western blotting as described below.
SDS-PAGE and western blotting
Log-phase promastigotes and axenic amastigotes of wild-type L. donovani (1SCL-2D) and L. mexicana (M379) were harvested and washed in PBS by centrifugation as above, and the resulting cell pellets were lysed in SDS-PAGE sample buffer and prepared for SDS-PAGE as previously described [40]. Prior to use, the protein concentrations of these samples were determined as previously described [40]. Aliquots of proteins (15 μg) were analyzed by SDS-PAGE, transferred onto nitrocellulose, and processed for Western blot analysis with the anti-LdINV antibody or the NRS control as described previously [30, 40]. In addition, aliquots of parasite culture supernatants, parasite release assays, and E. coli rLdINV proteins were prepared and subjected to SDS-PAGE and Western blot analysis. In addition to the above, cell lysates, parasite culture supernatants and immunoprecipitates obtained from both the pKSNEO::LdINV-HA and pKSNEO::LmexINV-HA transfectants and their respective controls were also subjected to SDS-PAGE analysis using 10 %, pre-cast, Tris–glycine polyacrylamide gels (Novex®, Invitrogen). Subsequently, proteins were transferred to polyvinylidene difluoride (PVDF) membranes using the iBlot® system (Invitrogen) as per manufacturer’s instructions. Membranes were blocked in a 5 % non-fat dry milk-TBST solution [0.05 % (v/v) Tween-20, 20 mM Tris–HCl, 150 mM NaCl, pH 8.0] and washed as previously described [43]. Such blots were reacted with either our rabbit anti-LdINV antibody, its NRS control or a mouse anti-HA.11 (Covance Research Products, Berkeley, CA, USA) or an appropriately matched purified mouse IgG1 κ chain control immunoglobulin (Sigma). Subsequently, blots were washed in TBST and incubated either with biotinylated goat anti-rabbit or goat anti-mouse antibodies (Invitrogen). After second set of washes with TBST, membranes were reacted with Q-Dots® (625 streptavidin conjugate nanoparticles, Invitrogen) for 30 min on an orbital shaker, washed three times at RT with TBST, and imaged using an Epichemi3 Darkroom (UVP Bioimaging Systems, Upland, CA, USA). The images were processed with Adobe Photoshop CS2 (Adobe Systems, Inc.).
Acknowledgments
This research was supported by the Intramural Research Program of the Division of Intramural Research, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH). Dr. Todd Lyda was supported by an Intramural Research Training Award Post-doctoral Fellowship from the NIAID, NIH. Dr. Manju Joshi was supported by an appointment through the Oak Ridge Institute for Science and Education (ORISE), Research Specialist Program at NIH. The program is administered by ORISE through an inter-agency agreement between the US Department of Energy and the NIH. Mr. Andrew Y. Kelada and Mr. Joshua P. Owings were supported by Postbaccalaureate Intramural Research Training Awards from the NIAID, NIH. We thank Dr. Greg Matlashewski, Department of Microbiology and Immunology McGill University, Montreal, CA for providing the pKSNEO leishmanial expression vector; Dr. Buddy Ullman, Department of Biochemistry and Molecular Biology, Oregon Health Sciences University, Portland, OR for providing the L. donovani cosmid library used in this study.
Footnotes
Contributor Information
Manju B. Joshi, Cell Biology Section, Laboratory of Parasitic Diseases, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892-0425, USA
John F. Andersen, Laboratory of Malaria and Vector Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Rockville, MD, USA
Andrew Y. Kelada, Cell Biology Section, Laboratory of Parasitic Diseases, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892-0425, USA
Joshua P. Owings, Cell Biology Section, Laboratory of Parasitic Diseases, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892-0425, USA
Paul A. Bates, Division of Biomedical and Life Sciences, Faculty of Health and Medicine, Lancaster University, Lancaster LA1 4YQ, UK
Dennis M. Dwyer, Cell Biology Section, Laboratory of Parasitic Diseases, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892-0425, USA
References
- 1.UNICEF/UNDP/World Bank/World Health Organization. Control of the leishmaniases: report of a meeting of the WHO Expert Committee on the Control of Leishmaniases. UNICEF/UNDP/World Bank/World Health Organization; Geneva: 2010. [Google Scholar]
- 2.Handman E. Leishmaniasis: current status of vaccine development. Clin Microbiol Rev. 2001;14:229–243. doi: 10.1128/CMR.14.2.229-243.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sacks DL, Perkins PV. Identification of an infective stage of Leishmania promastigotes. Science. 1984;223:1417–1419. doi: 10.1126/science.6701528. [DOI] [PubMed] [Google Scholar]
- 4.Sacks D, Kamhawi S. Molecular aspects of parasite–vector and vector–host interactions in leishmaniasis. Annu Rev Microbiol. 2001;55:453–483. doi: 10.1146/annurev.micro.55.1.453. [DOI] [PubMed] [Google Scholar]
- 5.Bates PA, Rogers ME. New insights into the developmental biology and transmission mechanisms of Leishmania. Curr Mol Med. 2004;4:601–609. doi: 10.2174/1566524043360285. [DOI] [PubMed] [Google Scholar]
- 6.Kamhawi S. Phlebotomine sand flies and Leishmania parasites: friends or foes? Trends Parasitol. 2006;22:439–445. doi: 10.1016/j.pt.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 7.Jacobson RL, Studentsky L, Schlein Y. Glycolytic and chitinolytic activities of Phlebotomus papatasi (diptera: Psychodidae) from diverse ecological habitats. Folia Parasitol (Praha) 2007;54:301–309. doi: 10.14411/fp.2007.039. [DOI] [PubMed] [Google Scholar]
- 8.Blum JJ, Opperdoes FR. Secretion of sucrase by Leishmania donovani. J Eukaryot Microbiol. 1994;41:228–231. doi: 10.1111/j.1550-7408.1994.tb01502.x. [DOI] [PubMed] [Google Scholar]
- 9.Gontijo NF, Melo MN, Riani EB, Almeida-Silva S, Mares-Guia ML. Glycosidases in Leishmania and their importance for Leishmania in phlebotomine sandflies with special reference to purification and characterization of a sucrase. Exp Parasitol. 1996;83:117–124. doi: 10.1006/expr.1996.0055. [DOI] [PubMed] [Google Scholar]
- 10.Stiles JK, Hicock PI, Shah PH, Meade JC. Genomic organization, transcription, splicing and gene regulation in Leishmania. Ann Trop Med Parasitol. 1999;93:781–807. doi: 10.1080/00034989957781. [DOI] [PubMed] [Google Scholar]
- 11.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alberto F, Bignon C, Sulzenbacher G, Henrissat B, Czjzek M. The three-dimensional structure of invertase (beta-fructosidase) from Thermotoga maritima reveals a bimodular arrangement and an evolutionary relationship between retaining and inverting glycosidases. J Biol Chem. 2004;279:18903–18910. doi: 10.1074/jbc.M313911200. [DOI] [PubMed] [Google Scholar]
- 13.Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 14.Gerber LD, Kodukula K, Udenfriend S. Phosphatidylinositol glycan (PI-G) anchored membrane proteins. Amino acid requirements adjacent to the site of cleavage and PI-G attachment in the COOH-terminal signal peptide. J Biol Chem. 1992;267:12168–12173. [PubMed] [Google Scholar]
- 15.Teasdale RD, Jackson MR. Signal-mediated sorting of membrane proteins between the endoplasmic reticulum and the golgi apparatus. Annu Rev Cell Dev Biol. 1996;12:27–54. doi: 10.1146/annurev.cellbio.12.1.27. [DOI] [PubMed] [Google Scholar]
- 16.Borst P. Discontinuous transcription and antigenic variation in trypanosomes. Annu Rev Biochem. 1986;55:701–732. doi: 10.1146/annurev.bi.55.070186.003413. [DOI] [PubMed] [Google Scholar]
- 17.Grimaldi G, Jr, Tesh RB. Leishmaniases of the new world: current concepts and implications for future research. Clin Microbiol Rev. 1993;6:230–250. doi: 10.1128/cmr.6.3.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zilberstein D, Dwyer DM. Glucose transport in Leishmania donovani promastigotes. Mol Biochem Parasitol. 1984;12:327–336. doi: 10.1016/0166-6851(84)90089-6. [DOI] [PubMed] [Google Scholar]
- 19.Zilberstein D, Dwyer DM, Matthaei S, Horuk R. Identification and biochemical characterization of the plasma membrane glucose transporter of Leishmania donovani. J Biol Chem. 1986;261:15053–15057. [PubMed] [Google Scholar]
- 20.Barros VC, Oliveira JS, Melo MN, Gontijo NF. Leishmania amazonensis: chemotaxic and osmotaxic responses in promastigotes and their probable role in development in the phlebotomine gut. Exp Parasitol. 2006;112:152–157. doi: 10.1016/j.exppara.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 21.Burchmore RJ, Rodriguez-Contreras D, McBride K, Merkel P, Barrett MP, Modi G, et al. Genetic characterization of glucose transporter function in Leishmania mexicana. Proc Natl Acad Sci USA. 2003;100:3901–3906. doi: 10.1073/pnas.0630165100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tran KD, Rodriguez-Contreras D, Shinde U, Landfear SM. Both sequence and context are important for flagellar targeting of a glucose transporter. J Cell Sci. 2012;125:3293–3298. doi: 10.1242/jcs.103028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Figarella K, Uzcategui NL, Zhou Y, LeFurgey A, Ouellette M, Bhattacharjee H, Mukhopadhyay R. Biochemical characterization of Leishmania major aquaglyceroporin LmAQP1: possible role in volume regulation and osmotaxis. Mol Microbiol. 2007;65:1006–1017. doi: 10.1111/j.1365-2958.2007.05845.x. [DOI] [PubMed] [Google Scholar]
- 24.Singh A, Mandal D. A novel sucrose/H+ symport system and an intracellular sucrase in Leishmania donovani. Int J Parasitol. 2011;41:817–826. doi: 10.1016/j.ijpara.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 25.Bates PA, Hermes I, Dwyer DM. Golgi-mediated post-translational processing of secretory acid phosphatase by Leishmania donovani promastigotes. Mol Biochem Parasitol. 1990;39:247–255. doi: 10.1016/0166-6851(90)90063-r. [DOI] [PubMed] [Google Scholar]
- 26.Shakarian AM, Joshi MB, Ghedin E, Dwyer DM. Molecular dissection of the functional domains of a unique, tartrate-resistant, surface membrane acid phosphatase in the primitive human pathogen Leishmania donovani. J Biol Chem. 2002;277:17994–18001. doi: 10.1074/jbc.M200114200. [DOI] [PubMed] [Google Scholar]
- 27.Silverman JM, Reiner NE. Exosomes and other microvesicles in infection biology: organelles with unanticipated phenotypes. Cell Microbiol. 2011;13:1–9. doi: 10.1111/j.1462-5822.2010.01537.x. [DOI] [PubMed] [Google Scholar]
- 28.Silverman JM, Reiner NE. Leishmania exosomes deliver preemptive strikes to create an environment permissive for early infection. Front Cell Infect Microbiol. 2012;1:26. doi: 10.3389/fcimb.2011.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clayton C, Adams M, Almeida R, Baltz T, Barrett M, Bastien P, et al. Genetic nomenclature for Trypanosoma and Leishmania. Mol Biochem Parasitol. 1998;97:221–224. doi: 10.1016/s0166-6851(98)00115-7. [DOI] [PubMed] [Google Scholar]
- 30.Debrabant A, Joshi MB, Pimenta PF, Dwyer DM. Generation of Leishmania donovani axenic amastigotes: their growth and biological characteristics. Int J Parasitol. 2004;34:205–217. doi: 10.1016/j.ijpara.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 31.McCarthy-Burke C, Bates PA, Dwyer DM. Leishmania donovani: use of two different, commercially available, chemically defined media for the continuous in vitro cultivation of promastigotes. Exp Parasitol. 1991;73:385–387. doi: 10.1016/0014-4894(91)90112-a. [DOI] [PubMed] [Google Scholar]
- 32.Valenzuela JG, Pham VM, Garfield MK, Francischetti IM, Ribeiro JM. Toward a description of the sialome of the adult female mosquito Aedes aegypti. Insect Biochem Mol Biol. 2002;32:1101–1122. doi: 10.1016/s0965-1748(02)00047-4. [DOI] [PubMed] [Google Scholar]
- 33.Bates PA, Hermes I, Dwyer DM. Leishmania donovani: immunochemical localization and secretory mechanism of soluble acid phosphatase. Exp Parasitol. 1989;68:335–346. doi: 10.1016/0014-4894(89)90115-x. [DOI] [PubMed] [Google Scholar]
- 34.Bates PA, Dwyer DM. Biosynthesis and secretion of acid phosphatase by Leishmania donovani promastigotes. Mol Biochem Parasitol. 1987;26:289–296. doi: 10.1016/0166-6851(87)90081-8. [DOI] [PubMed] [Google Scholar]
- 35.Benson DA, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. Genbank. Nucleic Acids Res. 2014;42:D32–D37. doi: 10.1093/nar/gkt1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schneider A, McNally KP, Agabian N. Splicing and 3′-processing of the tyrosine tRNA of Trypanosoma brucei. J Biol Chem. 1993;268:21868–21874. [PubMed] [Google Scholar]
- 37.Fong D, Lee B. Beta tubulin gene of the parasitic protozoan Leishmania mexicana. Mol Biochem Parasitol. 1988;31:97–106. doi: 10.1016/0166-6851(88)90149-1. [DOI] [PubMed] [Google Scholar]
- 38.Joshi MB, Dwyer DM. Molecular and functional analyses of a novel class I secretory nuclease from the human pathogen, Leishmania donovani. J Biol Chem. 2007;282:10079–10095. doi: 10.1074/jbc.M610770200. [DOI] [PubMed] [Google Scholar]
- 39.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1989. pp. 7.43–7.45. [Google Scholar]
- 40.Joshi MB, Hernandez Y, Owings JP, Dwyer DM. Diverse viscerotropic isolates of Leishmania all express a highly conserved secretory nuclease during human infections. Mol Cell Biochem. 2012;361:169–179. doi: 10.1007/s11010-011-1101-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Charest H, Zhang WW, Matlashewski G. The developmental expression of Leishmania donovani A2 amastigote-specific genes is post-transcriptionally mediated and involves elements located in the 3′-untranslated region. J Biol Chem. 1996;271:17081–17090. doi: 10.1074/jbc.271.29.17081. [DOI] [PubMed] [Google Scholar]
- 42.Ghedin E, Charest H, Zhang WW, Debrabant A, Dwyer D, Matlashewski G. Inducible expression of suicide genes in Leishmania donovani amastigotes. J Biol Chem. 1998;273:22997–23003. doi: 10.1074/jbc.273.36.22997. [DOI] [PubMed] [Google Scholar]
- 43.Debrabant A, Ghedin E, Dwyer DM. Dissection of the functional domains of the Leishmania surface membrane 3′-nucleotidase/nuclease, a unique member of the class I nuclease family. J Biol Chem. 2000;275:16366–16372. doi: 10.1074/jbc.M908725199. [DOI] [PubMed] [Google Scholar]
- 44.Zhang WW, Mendez S, Ghosh A, Myler P, Ivens A, Clos J, et al. Comparison of the A2 gene locus in Leishmania donovani and Leishmania major and its control over cutaneous infection. J Biol Chem. 2003;278:35508–35515. doi: 10.1074/jbc.M305030200. [DOI] [PubMed] [Google Scholar]
- 45.Agabian N. Trans splicing of nuclear pre-mRNAs. Cell. 1990;61:1157–1160. doi: 10.1016/0092-8674(90)90674-4. [DOI] [PubMed] [Google Scholar]
- 46.Fernandes O, Murthy VK, Kurath U, Degrave WM, Campbell DA. Mini-exon gene variation in human pathogenic Leishmania species. Mol Biochem Parasitol. 1994;66:261–271. doi: 10.1016/0166-6851(94)90153-8. [DOI] [PubMed] [Google Scholar]