

Abstract
Candida albicans, the major invasive fungal pathogen of humans, can cause both debilitating mucosal infections and fatal invasive infections. Understanding the complex nature of the host–pathogen interaction in each of these contexts is essential to developing desperately needed therapies to treat fungal infections. RNA-seq enables a systems-level understanding of infection by facilitating comprehensive analysis of transcriptomes from multiple species (e.g., host and pathogen) simultaneously. We used RNA-seq to characterize the transcriptomes of both C. albicans and human endothelial cells or oral epithelial cells during in vitro infection. Network analysis of the differentially expressed genes identified the activation of several signaling pathways that have not previously been associated with the host response to fungal pathogens. Using an siRNA knockdown approach, we demonstrate that two of these pathways—platelet-derived growth factor BB (PDGF BB) and neural precursor-cell-expressed developmentally down-regulated protein 9 (NEDD9)—govern the host–pathogen interaction by regulating the uptake of C. albicans by host cells. Using RNA-seq analysis of a mouse model of hematogenously disseminated candidiasis (HDC) and episodes of vulvovaginal candidiasis (VVC) in humans, we found evidence that many of the same signaling pathways are activated during mucosal (VVC) and/or disseminated (HDC) infections in vivo. Our analyses have uncovered several signaling pathways at the interface between C. albicans and host cells in various contexts of infection, and suggest that PDGF BB and NEDD9 play important roles in this interaction. In addition, these data provide a valuable community resource for better understanding host-fungal pathogen interactions.
The pathogenic fungus, Candida albicans, frequently grows asymptomatically on mucosal surfaces as part of the normal microbiota. However, when the host becomes immunocompromised or when the bacterial microbiota are disrupted by the use of broad-spectrum antibiotics, C. albicans can cause mucosal diseases, such as oropharyngeal candidiasis (OPC) (Sangeorzan et al. 1994; Rhodus et al. 1997; Revankar et al. 1998; Redding et al. 1999; Willis et al. 1999) and vulvovaginal candidiasis (VVC) (Sobel 1985). C. albicans also has the ability to disseminate through the bloodstream and infect most organs in the body (Klepser 2006). Hematogenously disseminated candidiasis (HDC) (Rangel-Frausto et al. 1999; Bader et al. 2004; Hajjeh et al. 2004) has a 47% mortality rate despite administration of antifungals (Gudlaugsson et al. 2003).
In both the mucosal and disseminated forms of disease, C. albicans must adhere to and invade nonphagocytic host cells. For example, during the initiation of OPC, C. albicans invades oral epithelial cells. During HDC, this organism invades endothelial cells in order to escape from the vasculature and infect the deep tissues. The observation that invasion-defective C. albicans mutants display attenuated virulence in murine models of OPC and HDC is evidence of the critical importance of invasion to virulence (Phan et al. 2000; Sanchez et al. 2004; Park et al. 2005; Chiang et al. 2007). Invasion into both cell types occurs in part by endocytosis, which is induced when the C. albicans invasins Als3p and Ssa1p interact with receptors expressed on the surface of endothelial and epithelial cells (Phan et al. 2007; Sun et al. 2010). These receptors include N- and E-cadherin, HSP90B1, the epidermal growth factor receptor (EGFR), and ERBB2 (also known as HER2) (Phan et al. 2005, 2007; Liu et al. 2011; Zhu et al. 2012).
As endothelial and epithelial cells are among the first host cells that interact with C. albicans during the course of infection, a comprehensive analysis of how these cells respond to the pathogen, and vice versa, is essential to understanding the pathogenesis of OPC and HDC. Previous studies have used microarrays to individually examine the transcriptional response of C. albicans to endothelial or epithelial cells (Müller et al. 2007; Barker et al. 2008; Lim et al. 2011; Ikuta et al. 2012; Moyes et al. 2014), as well as the transcriptional response of these host cells to infection by C. albicans (Sandovsky-Losica et al. 2006; Zakikhany et al. 2007; Park et al. 2009; Martin et al. 2011; Wachtler et al. 2011). Although these studies have contributed to our understanding of the interaction between pathogen and host, the full catalog of transcriptional changes is not complete owing to the limitations of microarrays, which include limited dynamic range and poor sensitivity to analyze low abundance transcripts.
RNA-seq (deep-sequencing of cDNA) is not subject to the limitations associated with microarray-based transcriptional assays. Gene expression profiling with RNA-seq is consistent with results obtained by microarrays (Liu et al. 2007; Marioni et al. 2008; Bradford et al. 2010; Malone and Oliver 2011) but is significantly more sensitive, with a much greater dynamic range (Wang et al. 2009). These traits and the probe-independent nature of RNA-seq make it possible to analyze the transcriptomes of multiple species (e.g., pathogen and host) simultaneously (Tierney et al. 2012; Westermann et al. 2012; Humphrys et al. 2013).
In this study, we analyzed the transcriptional profile both of C. albicans and host cells during in vitro infection of oral epithelial and vascular endothelial cells. Network analysis of the data set uncovered several signaling proteins that were not previously associated with the host response to any pathogenic fungus. We functionally validated the role of two of these pathways, platelet-derived growth factor BB (PDGF BB) and neural precursor cell expressed developmentally down-regulated protein 9 (NEDD9) using a siRNA approach. RNA-seq analysis of a murine model of HDC and vaginal samples collected longitudinally from women as they progressed from health to disease suggested that many of the same signaling pathways are activated during the course of mucosal and/or disseminated infections in vivo.
Results
RNA-seq of in vitro C. albicans infections
To understand the complex interaction that takes place between a fungal pathogen and host cells, we performed high-throughput sequencing of cDNA (RNA-seq) prepared from poly(A)-enriched RNA isolated from monolayers of primary human umbilical vein endothelial cells (HUVECs) and immortalized oral epithelial cells (the OKF6/TERT-2 cell line) (Dickson et al. 2000) that had been infected with C. albicans. The RNA preparations contained a mixture of mRNAs expressed by C. albicans as well as mRNAs expressed by the host cells. In order to reduce strain-specific responses, we infected each type of host cell individually with yeast-phase organisms of two different clinical isolates of C. albicans for which genome sequences are available, SC5314 and WO-1. After being added to the host cells, both isolates germinated and formed hyphae that progressively elongated (Fig. 1A). Exposure of C. albicans to the tissue culture medium also triggers hyphal morphogenesis as well as a corresponding transcriptional change (Bensen et al. 2004; Bruno et al. 2010). To ensure that the observed fungal gene expression changes were due to interaction with the host cell and not simply a response to the medium, we performed RNA-seq on time-matched controls in which each C. albicans isolate was grown in each type of tissue culture medium in the absence of host cells. Similarly, we performed RNA-seq on control, uninfected endothelial and epithelial cells. All sequencing reads were aligned to the human genome (Ensembl GRCh38) (Cunningham et al. 2015) as well as the reference genome sequences for each of the C. albicans strains (SC5314, CGD; WO-1, Broad Institute), and the samples were processed as depicted in Supplemental Figure S1 and described in Methods. From all of the experiments combined, we obtained a total of 4 × 109 reads that mapped to the human genome (average 160.2 × 106 reads per sample) and 4.5 × 109 reads that mapped to the C. albicans reference genomes (averaging 105.3 × 106 reads per sample) (Supplemental Table S1). Our ability to obtain equivalent amounts of reads from the different organisms despite the considerable difference in genome size is a function of (1) the fact that we added five yeast cells to the infection for every human cell; and (2) the fact that C. albicans forms multinucleated hyphae over the course of the in vitro infection (Fig. 1A). Our ability to obtain approximately 100 million reads that mapped to each of the genomes in each of the mixed samples indicates that RNA-seq is a suitable approach to comprehensively analyze the transcriptome of both fungal pathogen and host in the context of an in vitro infection.
Figure 1.

Interaction of C. albicans strains with each of the host cell types. (A) Microscopic images of endothelial cells and epithelial cells infected with each C. albicans strain for 1.5, 5, or 8 h in vitro. (B) A quantification of endocytosis for each C. albicans strain in each of the host cell lines at 1.5, 5, and 8 h post-infection. Results are the mean ± SD of three experiments, each performed in triplicate.
The transcriptional response of the fungi and host cells was determined after 1.5, 5, and 8 h. The 1.5-h time point corresponded to the initiation of host cell invasion (Fig. 1B), the 5-h time point corresponded to the induction of host cell gene expression, and the 8-h time point corresponded to fungal induction of host cell damage.
We defined differentially expressed genes as those with a minimum of twofold change in gene expression (P-value <0.05) between C. albicans infected host cells compared to either uninfected host cells or C. albicans cells grown in the corresponding tissue culture medium in the absence of host cells. Despite the significant sequencing coverage that we obtained (between 91% and 95% of open reading frames in each sample had an RPKM >0.1), we observed a minimal transcriptional response in C. albicans. Surprisingly, we found very few C. albicans genes (127 from SC5314; 181 from WO-1) that were differentially expressed in response to either of the host cell types at any of the time points. The number of genes differentially expressed during individual experiments ranged from 15 to 67 for strain SC5314 (Supplemental Table S2) and from four to 86 for strain WO-1 (Supplemental Table S3). Notably, only 11 genes were differentially expressed in both SC5314 and WO-1 in the same direction under the same conditions across both host cell types and all three time points (Supplemental Table S4). Taken together, these results suggest that C. albicans does not mount a robust core transcriptional response to the presence of these host cells and that many of the genes required for the interaction with host cells are expressed during growth in tissue culture media alone.
Analysis of host cell gene expression revealed 792 genes that were differentially expressed during at least one time point of the experiments. Three hundred eighty-eight genes were differentially expressed during the course of infection by endothelial cells, and 478 genes were differentially expressed during the course of infection by epithelial cells (Supplemental Table S5). The transcriptional responses of each of the human cell lines was relatively distinct, as evidenced by the small number of genes (up-regulated: nine, 27, and 15; down-regulated: one, one, and two, at 1.5, 5, and 8 h, respectively) that were differentially expressed in both of the cell lines at any given time point (Supplemental Fig. S2A,B). We were unable to identify human genes that were differentially spliced in response to the fungal infection in vitro model.
Upstream regulator analysis of host gene expression
We reasoned that gene expression changes in the host could be used to identify novel signal transduction pathways that are modulated in response to C. albicans infection, and that these pathways might be relevant to pathogenesis. We used the Upstream Regulator Analytic from the Ingenuity Pathway Analysis software (Ingenuity Systems; http://www.ingenuity.com) to identify signaling proteins whose downstream signaling pathways were potentially activated or repressed during the course of infection. This software assesses the overlap between experimentally derived gene lists and an extensively curated database of target genes for each of several hundred known regulatory proteins. It then uses the statistical significance of the overlap and the direction of the differential gene expression to make predictions about activation or repression of these regulatory proteins.
Analysis of differentially expressed gene lists from each time point of each host cell line predicted the modulation of a total of 230 different signaling proteins with most of the activation taking place at the 5- and 8-h time points during endothelial cell infection and during the 1.5- and 5-h time points during epithelial cell infection (Fig. 2; Supplemental Table S6). Figure 2 depicts the signaling proteins with the highest predicted regulations. The utility of this approach was verified by our identification of multiple pathways that are already known to be activated by C. albicans infection. Known pathways predicted to be activated in one or both cell types included TNF (tumor necrosis factor), MAPK1/3 (mitogen-activated protein kinase 1/3), IL1A (interleukin 1 alpha), IL1B (interleukin 1 beta), ERBB2 (erb-b2 tyrosine kinase 2), and NFKB (nuclear factor kappa B), TLR7 (toll-like receptor 7), CD40 ligand, IL6 (interleukin 6), MAP2K1 (mitogen-activated protein kinase kinase 1), EGF (epidermal growth factor), MAPK8 (mitogen-activated protein kinase 8), IFNG (interferon gamma) and PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha) (Orozco et al. 2000; Cannom et al. 2002; Villar et al. 2005; Moyes et al. 2010, 2014; Zhu et al. 2012;). Collectively, these results suggest that infection with C. albicans activates multiple signaling pathways, some cell-type-specific, in both endothelial cells and oral epithelial cells.
Figure 2.

Upstream regulator analysis of host gene expression. Each regulator was predicted by Ingenuity Pathway Analysis (IPA) to be activated (green, Z-score > 2) or repressed (red, Z-score < −2) during infection with C. albicans in either endothelial cells or epithelial cells during at least one time point. Depicted are the regulators with the highest predicted regulations (Z-score). Values represent −log10(P-value) for the overlap between the target gene set of each regulator and the list of differentially expressed genes. Black indicates no predicted activation or repression. Red circles indicate regulators that have not been previously associated with the host response to C. albicans infection.
The analysis also predicted the regulation of several signaling pathways that have not previously been reported to play a role in the host response to infection by any fungal pathogen (Fig. 2; Supplemental Table S6). Specifically, upstream regulators that are predicted to be activated by infection included FOXL2 (forkhead box L2), NEDD9 (neural precursor cell expressed, developmentally down-regulated 9), PDGF BB (platelet-derived growth factor BB homodimer), PAF1 (RNA polymerase II associated factor), SELPLG (selectin P ligand), SYVN1 (synovial apoptosis inhibitor 1), IGF1 (insulin-like growth factor 1), NUPR1, (nuclear protein, transcriptional regulator 1), and PPRC1 (peroxisome proliferator-activated receptor gamma coactivator-related 1). Upstream regulators that are predicted to be repressed by infection included COL18A1 (collagen type XVIII alpha 1), NKX2-3 (NK2 homeobox 3), and S100A6 (S100 calcium binding protein A6). We chose two significantly up-regulated pathways, NEDD9 and PDGF BB, for functional follow-up. In endothelial cells, both the NEDD9 and PDGF BB pathways were predicted to be activated at both the 5- and 8-h time points. In epithelial cells, both pathways were predicted to be activated at all three time points (Figs. 2, 3A,B; Supplemental Table S6).
Figure 3.
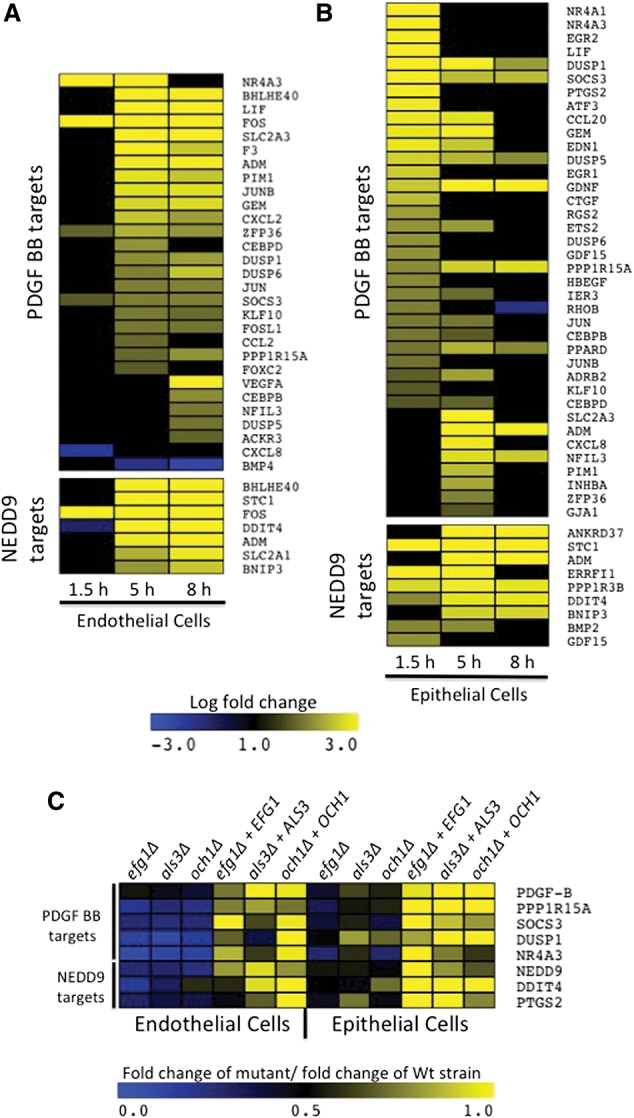
Regulation of PDGF- and NEDD9-pathway targets during in vitro infection. (A,B) Differential expression of PDGF- and NEDD9-target genes during infection of endothelial cells (A) and epithelial cells (B). Values represent log fold change expression relative to uninfected controls (RNA-seq expression values during infection divided by expression during uninfected mock infection). Yellow indicates increased expression during infection; blue indicates reduced expression during infection. (C) Differential expression of PDGF- and NEDD9-target genes during infection with invasion-defective C. albicans mutants. Values represent fold change differences in induction by mutant strain divided by induction by appropriate wild-type strain as determined by qRT-PCR.
PDGFR and NEDD9 govern uptake of C. albicans into endothelial and epithelial cells
We next performed a series of experiments to test if the PDGF and NEDD9 pathways govern the interaction between C. albicans and host cells. Using qRT-PCR, we examined the expression of a subset of genes that are downstream from PDGF BB (PPP1R15A, SOCS3, DUSP1, and NR4A3) or NEDD9 (DDIT4 and PTGS2) following infection with C. albicans mutants that had homozygous deletions in EFG1, ALS3, and OCH1. Deletion of each of these genes confers decreased uptake into endothelial and/or epithelial cells (Park et al. 2005; Phan et al. 2007; S Filler and Q Phan, unpubl.). Consistent with our RNA-seq results, infection with wild-type C. albicans cells for 5 h induced the expression of each of these genes. Furthermore, infection with each of the mutants resulted in reduced activation of the PDGF BB- and NEDD-target genes compared to both the matched wild-type and complemented strains (Fig. 3C). Our observation that invasion-defective mutants have decreased ability to induce PDGF- and NEDD9-target genes is consistent with a model in which activation of the PDGF BB and NEDD9 pathways is required for maximum invasion of C. albicans.
One mechanism by which C. albicans invades endothelial and epithelial cells is induced endocytosis (Filler et al. 1995; Phan et al. 2000; Park et al. 2005). We used a siRNA knockdown approach to determine if the PDGF BB and NEDD9 pathways are required for the endocytosis of C. albicans by host cells. The PDGF BB signaling pathway is activated when PDGF BB binds to the PDGF receptor beta (PDGFRB) (Battegay et al. 1994). Knockdown of either PDGFRB or NEDD9 resulted in a significant reduction in endocytosis of C. albicans by both endothelial cells and epithelial cells (Fig. 4A,B). We also attempted to knockdown both PDGFRB and NEDD9 in the same cells. However, although knockdown of either PDGFRB or NEDD9 alone caused no detectable cytotoxicity, combined knockdown of both proteins was synthetically lethal, which precluded determination of the effects of this knockdown on fungal endocytosis.
Figure 4.
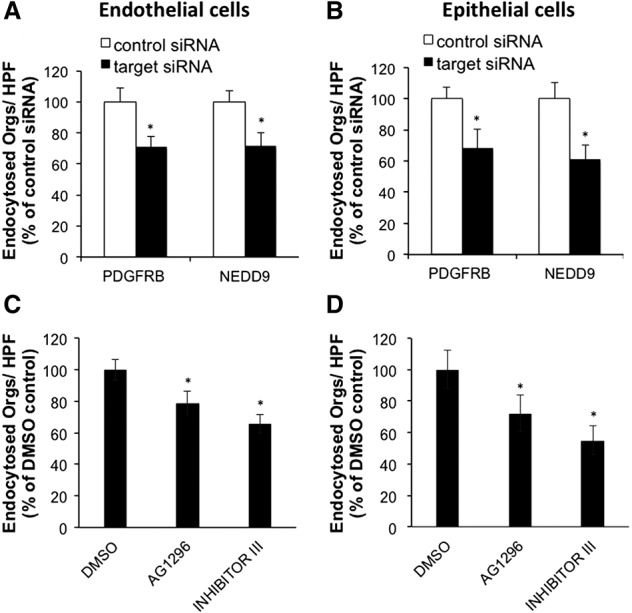
PDFGRB and NEDD9 mediate endocytosis of C. albicans by host cells. Effects of siRNA knockdown of PDGFRB or NEDD9 on endocytosis of C. albicans by endothelial cells (A) and oral epithelial cells (B). The effect of pharmacological inhibition of the PDGF receptor on endocytosis by endothelial cells (C) and oral epithelial cells (D) was also tested. Results are the mean ± SD of three experiments, each performed in triplicate. (*) P < 0.00003 versus controls; n = 12 from four independent experiments.
To confirm our finding that signaling through PDGFRB was required for maximal host cell endocytosis of C. albicans, we analyzed the effects of two different small molecule PDGFR inhibitors on the endocytosis of C. albicans. Both AG1296 (Kovalenko et al. 1997) and PDGFR Tyrosine Kinase Inhibitor III (CAS 205254-94-0) (Matsuno et al. 2002) significantly reduced the endocytosis of C. albicans by endothelial cells and epithelial cells (Fig. 4C,D). Collectively, these results demonstrate the key role of PDGFRB signaling in the endocytosis of C. albicans.
E-cadherin and N-cadherin function as host cell receptors for C. albicans and are required for efficient endocytosis by host cells into epithelial and endothelial cells, respectively (Phan et al. 2005, 2007). We tested whether the decrease in endocytosis that we observe by knockdown of PDGFRB or NEDD9 could be simply the result of reduced expression of the cadherins. Neither E- nor N-cadherin expression was affected by knockdown of PDGFRB or NEDD9 (Supplemental Fig. S3). Collectively, our results suggest that C. albicans activates the NEDD9 and PDGF BB signaling pathways in both endothelial and epithelial cells, which subsequently facilitates invasion into these cells in a manner that is independent of cadherin expression.
Several of the pathways, including PDGF BB and NEDD9, are also regulated in mice with hematogenously disseminated candidiasis
Since C. albicans must invade endothelial cells in order to escape from the vasculature and infect the deep tissues during the development of disseminated disease, in vitro infection of endothelial cells serves as a well-established model to understand the pathogenesis of HDC. To test whether PDGF BB and NEDD9 are activated during in vivo HDC infections, we performed RNA-seq analysis on kidneys of mice 24 h after they were inoculated intravenously with C. albicans. We obtained an average of 587.4 × 106 reads from each of four kidneys (two infected and two uninfected controls). Expression analysis revealed that 1024 genes were up-regulated in the infected kidney relative to uninfected control and 661 genes that were down-regulated (Supplemental Table S7). Among the newly identified C. albicans response pathways that we found to be regulated by C. albicans infection in vitro, our upstream regulator analysis predicted the regulation of several of these during the in vivo kidney infection (Supplemental Table S8), including FOXL2, PAF1, SELPLG, SYVN1, NUPR1, COL18A1, and S100A6. Notably, our analysis also predicted the activation of both the PDGF BB (P-value of overlap = 1.62 × 10−26) and NEDD9 (P-value of overlap = 1.09 × 10−7) pathways (Supplemental Table S8). Figure 5 depicts the increase in gene expression of PDGF BB- and NEDD9-target genes in infected kidneys relative to uninfected kidneys. These results indicate that C. albicans infection activates both the PDGF BB and NEDD9 pathways in mice with disseminated candidiasis.
Figure 5.
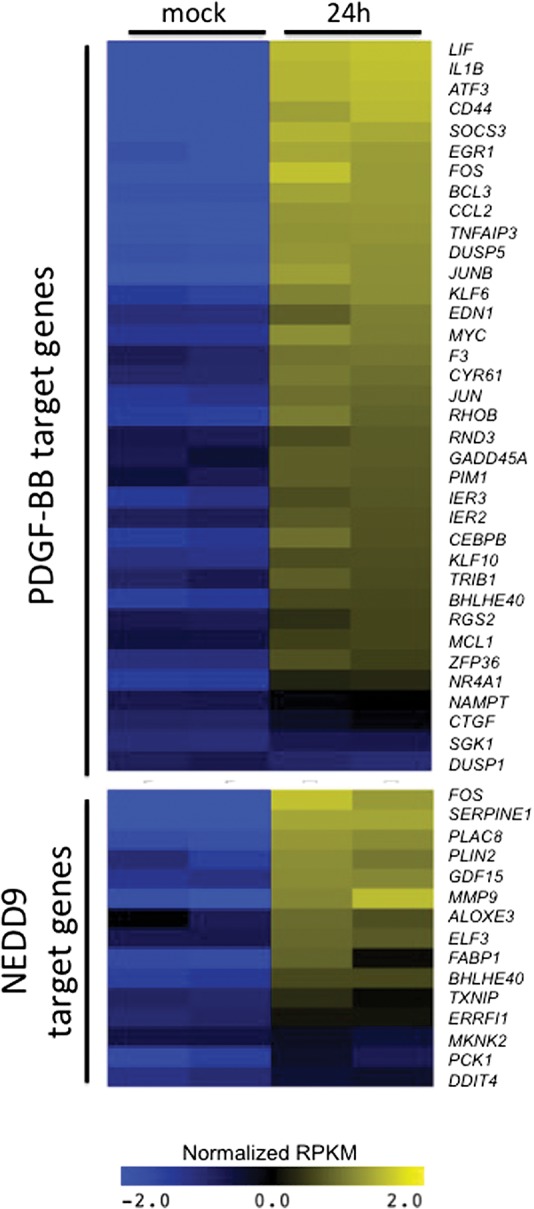
PDGF BB and NEDD9 are activated during disseminated candidiasis. Differential expression of PDGF- and NEDD9- target genes, as indicated by IPA analysis, in two uninfected mouse kidneys (mock) and two C. albicans-infected mouse kidneys 24 h after injection (24 h). Plotted are log transformed RPKM values that have been normalized across all four samples. Yellow indicates high gene expression; blue indicates low expression.
Transcriptomic evidence for activation of PDGF BB and ERBB2 pathways during vaginal candidiasis in humans
We next investigated whether the PDGF BB and NEDD9 pathways would be activated during C. albicans infection in humans. The best way to perform such an experiment would be to assay the same tissue from the same individual both prior to and during an infection. To this end, we took advantage of a preexisting clinical study focused on understanding the dynamics of the vaginal microbiome. In this study, vaginal samples were collected daily from 135 nonpregnant women over the course of 10 wk (Ravel et al. 2013). To test if the PDGF BB and NEDD9 pathways are activated during clinical episodes of vaginal candidiasis, we performed RNA-seq using RNA isolated from vaginal swabs that were collected as part of this study. Our analysis included samples from two women who were clinically diagnosed with an episode of vaginal candidiasis at least once during the course of the study (Ravel et al. 2013). In each case, the first sample (healthy) was collected when the subject was experiencing no vaginal symptoms and the second sample was collected when the subject was experiencing a clinically diagnosed episode of vaginal candidiasis (Fig. 6). An average of 18 × 106 reads per sample were obtained (Supplemental Table S9). In a focused analysis of these data, we found that 26 known PDGF BB-target genes were up-regulated during infection in Subject 12, and 17 of PDGF BB-target genes were up-regulated in Subject 40 (Fig. 6). We also found up-regulation of many target genes from two of the novel upstream regulators (SYVN1 and SELPLG) (Supplemental Fig. S4) that were predicted to be activated in both our in vitro infections (Fig. 2) and our in vivo mouse infections (Supplemental Table S8). A similar analysis did not indicate that the NEDD9 pathway was activated during vaginal candidiasis. These results suggest that the PDGF BB, SYVN1, and SELPLG pathways are activated during vaginal candidiasis in humans, whereas the NEDD9 pathway may only be activated during disseminated infections.
Figure 6.
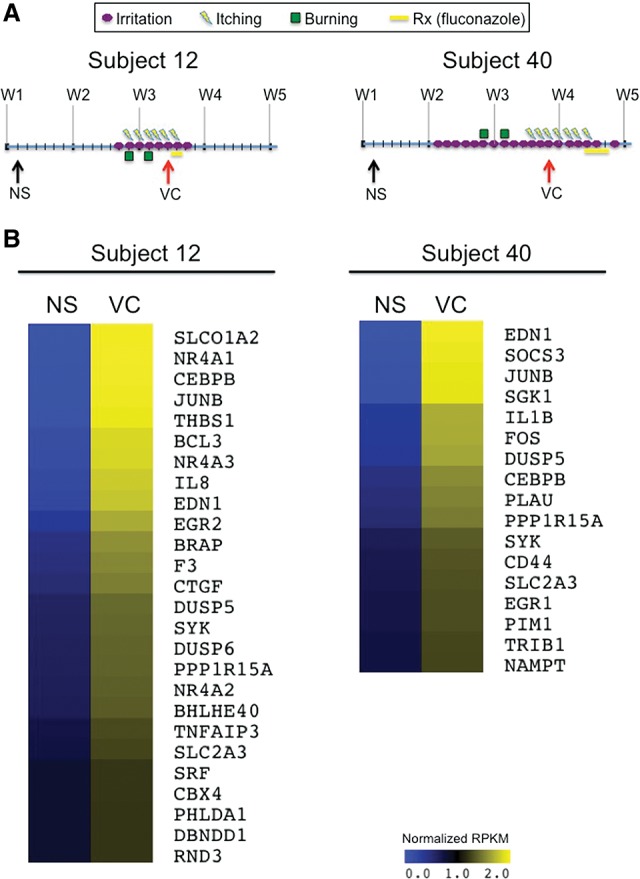
PDGF-target genes are induced during clinical episodes of vaginal candidiasis. (A) Schematic representing the symptomology and treatment of both subjects over the course of 5 wk. The arrows indicate the samples that were subject to RNA-seq. (NS) No symptoms were observed during sample collection; (VC) the sample was collected during an episode of vaginal candidiasis. (B) Heatmaps depicting the expression (normalized RPKMs) of PDGF BB-target genes with a minimum 1.5-fold change between the healthy samples (NS) and diseased (VC).
Previous work has demonstrated that the host receptor kinase ERBB2 is autophosphorylated in oral epithelial cells during interaction with C. albicans, and this receptor plays a key role in the interactions of C. albicans with oral epithelial cells, both in vitro and in the mouse model of oropharyngeal candidiasis (Zhu et al. 2012). This pathway was also predicted to be activated in our in vitro infections (Fig. 2) and in vivo mouse infections (Supplemental Table S8). To search for evidence that the ERBB2 pathway is activated in vaginal candidiasis, we searched for known ERBB2-target genes that might be differentially expressed in our longitudinal samples. We observed that 36 and 19 known ERBB2-target genes in Subjects 12 and 40, respectively, had increased expression during episodes of vaginal candidiasis. Therefore, these data indicate that the ERBB2 pathway is also activated during vaginal candidiasis in humans.
Discussion
In this study, we performed the most comprehensive transcriptomic analysis of host–pathogen interaction, for any bacterial or eukaryotic pathogen, to date. By performing RNA-seq on mixtures of RNA isolated from human endothelial and epithelial cells infected with C. albicans, we generated a data set of 8.5 billion mapped reads (4 billion for human and 4.5 billion for C. albicans) that will serve as an excellent genomic resource for the study of host–pathogen interaction in general. We used network analysis of our data set to identify host signaling pathways that are activated by C. albicans infection and identified ones that have not been previously tied to fungal infection. Our in-depth analysis of two of these novel pathways provides evidence that PDGF BB is activated during both disseminated and mucosal infection in vivo, whereas the NEDD9 pathway is activated during HDC.
To our knowledge, only two other studies, both microarray-based, examined the response of epithelial cells or endothelial cells to C. albicans using a similar time-matched negative control sample that consisted of fungal cells incubated in tissue culture media without host cells (Park et al. 2009; Wachtler et al. 2011). The list of genes that were found to be differentially expressed does not overlap significantly with either of these previous studies. For example, of the 127 genes that were differentially expressed during at least one time point in one cell type in the current study, five were reported by Park et al. (2009) and 11 were reported by Wachtler et al. (2011) to have changed in the same direction. There are several possible explanations for the poor concordance. For the Wachtler study, the analysis was done with a different cell line (polarized TR146 oral epithelial cells) at 20, 60, and 180 min post-infection (Wachtler et al. 2011). The authors observed the most robust transcriptional response at 20 min post-infection with the majority of the gene expression changes disappearing by 60 min post-infection. Therefore, the most likely explanation for the discordance with this study is the difference in host cells and the timing of the experiment. For the Park study, the analysis was done on FaDu oral epithelial cells at 45, 90, and 180 min post-infection with clinical isolates that differ from the C. albicans strain, SC5314 (Park et al. 2009). So, the most likely explanation for the discordance with this study is the combination of different host cells as well as the different C. albicans strains examined.
Because of the large number of host genes whose transcript levels changed in response to C. albicans infection, we focused our analysis on upstream regulators, which we define as any protein or protein complex that is known to govern the mRNA abundance of another gene. These molecules play important roles in governing many cellular pathways including those involved in the host response to microbial infection. Their potential to regulate the expression of functionally related or redundant genes makes them a valuable starting point for functional follow-up studies of large transcriptomic data sets. To identify upstream regulators in the host that might be activated or repressed in response to infection by C. albicans, we performed network analysis of the gene expression data using the Upstream Regulator Analytic from the Ingenuity Pathway Analysis software. This approach was validated by the prediction of many signaling proteins that have already been demonstrated to respond to infection by C. albicans.
We observed a significant overlap in predicted pathway activation between the two cell lines at the later time points (Fig. 2) despite a very small overlap in coregulated genes at the same time points (Supplemental Fig. S2). In order to get a positive hit in our analysis, we need a statistically significant overlap between our data set and the list of target genes for a given pathway curated in the IPA database. The high level of overlap in the predicted pathways is the result of the endothelial cells and epithelial cells each expressing a different subset of genes in each of the pathways, and the list of differentially expressed target genes in each cell line is large enough to allow for each of the two minimally or nonoverlapping sets to still be statistically significant.
We focused our functional follow-up on the two novel pathways that were significantly activated during our in vitro infection experiments: PDGF BB and NEDD9 PDGFs are serum proteins that stimulate cellular migration and have well-established roles in embryonic development and human diseases such as cancer and atherosclerosis (Kohler and Lipton 1974; Ross et al. 1974; Westermark and Wasteson 1976; Demoulin and Essaghir 2014). The PDGFs are encoded by four different genes—PDGFA, PDGFB, PDGFC, and PDGFD—and function as secreted homodimeric proteins with distinct biological roles (Demoulin and Essaghir 2014). We found significant overlap between the set of genes that are induced in response to C. albicans infection and the set of genes that are regulated by the interaction of the PDGF BB homodimer with its receptor. This significant overlap, along with the directionality of the gene expression changes, is consistent with activation of cellular signaling via the PDGFRB subunit. We determined that siRNA knockdown of the PDGFRB subunit in both endothelial and epithelial cells significantly reduced the capacity of each cell type to endocytose C. albicans cells. In addition, invasion-defective C. albicans mutants were less effective at inducing the expression of PDGF BB dependent genes, as assayed by qRT-PCR. These results suggest that C. albicans uses PDGF signaling as a mechanism to invade barrier cells by induced endocytosis. This mechanism of invasion has not been described for a fungal pathogen; however, one study suggests that PDGFRB promotes cellular uptake of the intracellular pathogen Chlamydia trachomatis (Elwell et al. 2008).
Although we observed significant reduction of the endocytosis upon knockdown of PDGFRB, host cell invasion was not completely abrogated. This can be explained by the fact that C. albicans is known to interact with other cellular receptors to induce its own endocytosis including ERBB2, EGFR (Zhu et al. 2012), and N-cadherin and E-cadherin (Phan et al. 2007). Furthermore, although PDGFRB is the principle receptor for PDGF BB, this molecule can also bind to the PDGFRA subunit homodimer or PDGFR A/B heterodimer. Therefore, the possibility exists that C. albicans can also signal through the alpha subunit (Demoulin and Essaghir 2014). The PDGFR inhibitors that we used in our experiments inhibit both PDGFRA and PDGFRB, which may explain why they caused a greater reduction in host cell endocytosis of C. albicans than siRNA knockdown of PDGFRB.
NEDD9 is a member of the Cas family of scaffolding proteins upon which many signaling pathways converge, and it is implicated in many aspects of cancer biology (Cabodi et al. 2010). Although NEDD9 is known previously to be expressed by epithelial cells and T cells, we found that it is also expressed by endothelial cells. Several lines of evidence point toward a scenario in which NEDD9 transduces C. albicans-stimulated signals to promote induced endocytosis in both endothelial and epithelial cells. First, there was significant overlap between the C. albicans-induced genes and known NEDD9-target genes; and the directionality of these expression changes are consistent with activation of NEDD9 by C. albicans infection. Second, siRNA knockdown of NEDD9 resulted in reduced uptake of C. albicans by both endothelial and epithelial cells. Third, invasion-defective C. albicans mutants simulated the expression of NEDD9-target genes only weakly. NEDD9 has been shown to be essential for transduction of signals that result from stimulation by PDGF (Natarajan et al. 2006) as well as overexpression of ERBB2 (Little et al. 2014). Our data suggest that NEDD9 may govern the host cell response to C. albicans by transducing signals from both the PDGF BB and ERBB2 pathways. On the other hand, our finding that dual knockdown of NEDD9 and PDGFRB was synthetically lethal indicates that these two proteins also function in parallel pathways that together are required for cell survival.
The results of our in vitro experiments suggest that PDGF BB and NEDD9 play a role in the host response to C. albicans in vascular endothelial cells, which are relevant to HDC, and oral epithelial cells, which are relevant to mucosal infections. To look for evidence that these pathways are activated during in vivo infection, we characterized the host response to C. albicans infection using a murine model of HDC and in women with vaginal candidiasis using RNA-seq. We found significant transcriptomic evidence of PDGF BB activation in both the murine HDC model and during the clinical episodes of vaginal candidiasis. These results indicate that C. albicans activates PDGFRB, either directly or indirectly during both disseminated and mucosal candidiasis. On the other hand, our in vivo transcriptomic analyses suggested that C. albicans activates NEDD9 during HDC but not during vaginal candidiasis. However, the lack of detection could be due to the relatively low coverage of the human genome that we were able to achieve in the vaginal samples. The low coverage is most likely the result of a significant number of sequencing reads originating from the bacterial microbiota. Whether disseminated infection with other microbial pathogens also activates the NEDD9 pathway is currently unknown.
A previous study has raised the question as to whether the response of mice to a pathogen has any relationship to the response of humans (Seok et al. 2013). However, the work presented here shows that the human response to VVC has significant similarity to both the mouse response to HDC and the in vitro response of epithelial and endothelial cells to C. albicans infection. Therefore, our results demonstrate the power of RNA-seq and network analysis, across multiple infection models, including murine models, to identify novel pathways that govern the interaction between fungal pathogens and the human host during invasive and mucosal infections. To our knowledge, this is the first study to analyze the host response to microbial infection in longitudinally collected clinical vaginal samples. Specifically, these results suggest that at least PDGF BB and NEDD9 have important roles in Candida–host interactions. Additionally, our significant data set will serve as a valuable community resource to better understand host–fungal pathogen interactions as well as to improve the human genome annotation.
Methods
Fungal strains and host cells
Both of the wild-type C. albicans strains used in this study (SC5314 and WO-1) were derived from clinical infections and are therefore both human derived. Strain SC5314 was originally recovered from a patient with generalized candidiasis at the Columbia College of Physicians and Surgeons, New York City (Odds et al. 2004). Strain WO-1 was isolated from the blood and lungs of a patient suffering from systemic candidiasis at University Hospitals, Iowa City, Iowa, in 1984 (Slutsky et al. 1987). The genetically engineered C. albicans strains used in this study are listed in Supplemental Table S10. Human umbilical vein endothelial cells (HUVECs) were harvested from umbilical cord veins and cultured as previously described (Filler et al. 1995). The human oral epithelial cell line, OKF6/TERT-2, was kindly provided by J. Rheinwald (Harvard University, Cambridge, Massachusetts) (Dickson et al. 2000) and was cultured as previously described (Phan et al. 2007).
Isolation of RNA from epithelial and endothelial cells
HUVEC and OKF6/TERT-2 cells were grown in a 10 × 15-mm tissue culture dish. For HUVECs, the medium was M199 (Life Technologies) containing 10% fetal calf serum and 10% fetal bovine serum. For OKF6/TERT-2 cells, the medium was DMEM (Life Technologies) without serum. The host cells incubated 5% CO2 at 37°C until confluent, at which time 3 × 107 C. albicans yeast were added to each dish for a multiplicity of infection of five yeast cells for every one human cell. After 90 min, 5 h, and 8 h of infection, the cells were scraped from the plate and the human and fungal RNA was isolated using the Ribopure Yeast Kit (Ambion), according to the manufacturer's instructions. As a negative control, 3 × 107 C. albicans yeast were added to the tissue culture plates containing medium alone without host cells and processed in parallel.
Murine model of disseminated candidiasis
Male BALB/c mice were infected via the lateral tail vein with 106 yeast cells of C. albicans SC5314. After 24 h of infection, two infected mice and two uninfected mice were killed. Their kidneys were harvested, snap frozen in liquid nitrogen, and stored at −80°C. To isolate the RNA, the kidneys were homogenized and then subjected to bead beating with zirconium beads. Next, total RNA was extracted using the RNeasy mini kit (Qiagen; Cat #74104) using the washing and elution steps in the Qiagen Quick-Start Protocol. The animal experiments were approved by the Animal Care and Use Committee at the Los Angeles Biomedical Research Institute.
Processing of vaginal swabs
After obtaining informed consent, vaginal swabs were prospectively collected daily for 10 wk from 135 women (Ravel et al. 2013). Two samples were from each of two women who developed vaginal candidiasis prior to the development of symptoms and at the time of symptoms but prior to treatment. The swabs were immediately stored in RNAlater (Qiagen) after collection and stored at −20°C for a week and then at −80°C until RNA extraction. To isolate total RNA, the cells were rinsed from the swabs with ice-cold PBS, incubated with acid phenol, and lysed by bead beating. The nucleic acids were purified chloroform-isoamyl alcohol and ethanol precipitated. The samples were treated with the TURBO DNA-free DNase Treatment kit (Ambion), after which RNA was purified using the RNeasy Mini Kit. Samples are enriched for mRNA using a combination of the Ribo-Zero rRNA Removal Kits for gram-negative, gram-positive, and human/mouse/rat RNA (Epicentre Biotechnologies). Absence of the DNA contamination in these samples was verified by PCR using 16S rRNA primers. The clinical study protocol was approved by the Institutional Review Board of the University of Alabama at Birmingham and the University of Maryland School of Medicine. Written informed consent was appropriately obtained from all participants.
RNA-seq and gene expression analysis
All RNA-seq libraries (non-strand-specific, paired end) were prepared with the TruSeq RNA Sample Prep kit (Illumina). The total RNA samples isolated from the in vitro infections and the murine kidneys were subject to poly(A) enrichment as part of the TruSeq protocol. One hundred nucleotides of sequence was determined from each end of each cDNA fragment using the HiSeq platform (Illumina) per the manufacturer's protocol. Sequencing reads were annotated and aligned to the Ensembl GRCh38 (Cunningham et al. 2015) of the human reference genome, the Ensembl GRCm38 of the mouse reference genome, or the C. albicans reference genomes (SC5314 or WO-1) using TopHat2 (Kim et al. 2013). The alignment files from TopHat2 were input to Cufflinks to calculate the gene expression for each gene in each sample and differential expression across samples was determined using Cuffdiff (Trapnell et al. 2012). For the differential expression analysis of host gene expression, we took a more conservative approach and combined the RNA-seq data from the infections with SC5314 and WO-1, for each host cell type, to increase the statistical power. Both C. albicans strains were endocytosed at similar rates in each of the host cell types (Supplemental Fig. S1). For differential expression of the C. albicans genes, the data for the different strains and host cell types were kept separate. A gene was considered differentially expressed if the false discovery rate (FDR) for differential expression was <0.05.
qRT-PCR analysis
Prior to performing real-time PCR, cDNA was prepared using the RETROscript reverse transcriptase kit (Life Technologies). Real-time PCR was performed in triplicate in optical 96-well plates using the Power SYBRGreen PCR Master (Life Technologies). The primers used are listed in Supplemental Table S11. The mRNA levels of NEDD9- and PDGF BB-target genes were normalized against the internal GAPDH mRNA control using the ΔΔCt method (Kubista et al. 2006).
Identification of signal transduction pathways
We used the Upstream Regulator Analytic of IPA (Ingenuity Systems; http://www.ingenuity.com) to identify signaling proteins that are potentially activated or repressed during the course of infection. This software assesses the overlap between experimentally derived gene lists and an extensively curated database of target genes for each of several hundred known regulatory proteins. It then uses the statistical significance of the overlap and the direction of the differential gene expression to make predictions about activation or repression of these regulatory proteins. The database has target lists for individual proteins as well as for protein complexes (Fig. 2; Supplemental Tables S6, S8).
PDGFRB and NEDD9 siRNA
HUVECs and OKF6/TERT-2 cells were grown to 80% confluence in six-well plates and then transfected with siRNA for PDGFRB (Thermo Fisher Scientific) and NEDD9 (Qiagen) using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. Control cells were transfected with random control siRNA (Qiagen) in parallel. After 72 h post-transfection, the extent of PDGFRB and NEDD9 knockdown was monitored by immunoblotting of whole-cell lysates with anti-PDGFRB antibodies (Clonetech: Cat. # 22B1 for HUVECs; Cell Signaling: Cat. # C82A3 for OKF6/TERT-2 cells) and anti-NEDD9 antibodies (Cell Signaling: Cat. # 2G9). At least 80% knockdown was achieved in all experiments.
Measurement of host cell endocytosis
The endocytosis of C. albicans cells by HUVECs and OKF6/TERT-2 cells was determined by our standard differential fluorescence assay as described previously (Park et al. 2005; Phan et al. 2007). Briefly, the host cells were grown on fibronectin-coated glass coverslips and infected with 105 cells of C. albicans in RPMI 1640 medium. After a 90-min (HUVECs) or 180-min (OKF6/TERT-2 cells) incubation, the cells were rinsed twice with PBS and then fixed with 3% paraformaldehyde. The nonendocytosed organisms were stained with an anti-C. albicans rabbit serum (Biodesign International) conjugated with Alexa 568 (Molecular Probes). Afterward, the cells were rinsed extensively with PBS and then permeabilized with 0.05% (vol/vol) Triton X-100 in PBS. Next, the cell-associated organisms (the endocytosed plus nonendocytosed organisms) were stained with the anti-C. albicans rabbit serum conjugated with Alexa 488 (Molecular Probes). The coverslips were observed with an epifluorescence microscope. The number of organisms endocytosed by the host cells was determined by subtracting the number of cell-associated organisms (labeled with Alexa 568, which fluoresces red) from the total number of organisms (labeled with Alexa 488, which fluoresces green). At least 100 organisms were counted on each coverslip, and all experiments were performed in triplicate.
In some experiments, the host cells were incubated with the PDGFR inhibitors, AG1296 (Selleck Chemicals) (Kovalenko et al. 1997), and PDGFR Tyrosine Kinase Inhibitor III (CAS 205254-94-0, Santa Cruz Biotechnology) (Matsuno et al. 2002) at a final concentration of 10 µM. The inhibitors were added to the host cells 1 h before the C. albicans and were present for the duration of the experiment. Control cells were exposed to an equal concentration of the diluent (0.2% DMSO).
Data access
All of the raw sequencing reads from this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession numbers SRP011085 and SRP028748. All of the raw and processed gene expression data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE56093.
Competing interest statement
S.G.F. is a shareholder in and consultant for NovaDigm Therapeutics, Inc.
Supplementary Material
Acknowledgments
This project was funded in part with federal funds from the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Department of Health and Human Services under contract number HHSN272200900009C. V.M.B., C.M.F., and S.G.F. were supported by U19AI110820. S.G.F. was supported by grants R01AI054928 and R01DE017088. A.P.M. was supported by grant R21 DE023311A1. The human vaginal candidiasis RNA-seq transcriptome was supported by the NIAID under award number UH2AI083264 (J.R.). The endothelial cells used in these studies were isolated from human umbilical cords, which were collected by the pediatric, perinatal, and mobile unit of the UCLA Clinical and Translational Science Institute at LA BioMed/Harbor-UCLA Medical Center (UL1TR000124). We thank Julie Dunning-Hotopp and David Rasko for critical reading of this manuscript.
Author contributions: Y.L., S.G.F., and V.M.B. conceived and designed the experiments. Y.L., J.A.S., L.L.B., and W.X. performed the experiments. Y.L., A.C.S., J.A.S., P.K., S.G.F., and V.M.B. analyzed the data. A.M., A.P.M., J.R., C.M.F., and S.G.F. contributed reagents, material, or analysis tools. S.G.F. and V.M.B. wrote the paper. Y.L., J.A.S., A.P.M., J.R., C.M.F., S.G.F., and V.M.B. reviewed and edited the paper. All authors read and approved the final manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.187427.114.
References
- Bader MS, Lai SM, Kumar V, Hinthorn D. 2004. Candidemia in patients with diabetes mellitus: epidemiology and predictors of mortality. Scand J Infect Dis 36: 860–864. [DOI] [PubMed] [Google Scholar]
- Barker KS, Park H, Phan QT, Xu L, Homayouni R, Rogers PD, Filler SG. 2008. Transcriptome profile of the vascular endothelial cell response to Candida albicans. J Infect Dis 198: 193–202. [DOI] [PubMed] [Google Scholar]
- Battegay EJ, Rupp J, Iruela-Arispe L, Sage EH, Pech M. 1994. PDGF-BB modulates endothelial proliferation and angiogenesis in vitro via PDGF β-receptors. J Cell Biol 125: 917–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensen ES, Martin SJ, Li M, Berman J, Davis DA. 2004. Transcriptional profiling in Candida albicans reveals new adaptive responses to extracellular pH and functions for Rim101p. Mol Microbiol 54: 1335–1351. [DOI] [PubMed] [Google Scholar]
- Bradford JR, Hey Y, Yates T, Li Y, Pepper SD, Miller CJ. 2010. A comparison of massively parallel nucleotide sequencing with oligonucleotide microarrays for global transcription profiling. BMC Genomics 11: 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno VM, Wang Z, Marjani SL, Euskirchen GM, Martin J, Sherlock G, Snyder M. 2010. Comprehensive annotation of the transcriptome of the human fungal pathogen Candida albicans using RNA-seq. Genome Res 20: 1451–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabodi S, del Pilar Camacho-Leal M, Di Stefano P, Defilippi P. 2010. Integrin signalling adaptors: not only figurants in the cancer story. Nat Rev Cancer 10: 858–870. [DOI] [PubMed] [Google Scholar]
- Cannom RR, French SW, Johnston D, Edwards JE Jr, Filler SG. 2002. Candida albicans stimulates local expression of leukocyte adhesion molecules and cytokines in vivo. J Infect Dis 186: 389–396. [DOI] [PubMed] [Google Scholar]
- Chiang LY, Sheppard DC, Bruno VM, Mitchell AP, Edwards JE Jr, Filler SG. 2007. Candida albicans protein kinase CK2 governs virulence during oropharyngeal candidiasis. Cell Microbiol 9: 233–245. [DOI] [PubMed] [Google Scholar]
- Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al. 2015. Ensembl 2015. Nucleic Acids Res 43: D662–D669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demoulin JB, Essaghir A. 2014. PDGF receptor signaling networks in normal and cancer cells. Cytokine Growth Factor Rev 25: 273–283. [DOI] [PubMed] [Google Scholar]
- Dickson MA, Hahn WC, Ino Y, Ronfard V, Wu JY, Weinberg RA, Louis DN, Li FP, Rheinwald JG. 2000. Human keratinocytes that express hTERT and also bypass a p16INK4a-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol Cell Biol 20: 1436–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwell CA, Ceesay A, Kim JH, Kalman D, Engel JN. 2008. RNA interference screen identifies Abl kinase and PDGFR signaling in Chlamydia trachomatis entry. PLoS Pathog 4: e1000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filler SG, Swerdloff JN, Hobbs C, Luckett PM. 1995. Penetration and damage of endothelial cells by Candida albicans. Infect Immun 63: 976–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudlaugsson O, Gillespie S, Lee K, Vande Berg J, Hu J, Messer S, Herwaldt L, Pfaller M, Diekema D. 2003. Attributable mortality of nosocomial candidemia, revisited. Clin Infect Dis 37: 1172–1177. [DOI] [PubMed] [Google Scholar]
- Hajjeh RA, Sofair AN, Harrison LH, Lyon GM, Arthington-Skaggs BA, Mirza SA, Phelan M, Morgan J, Lee-Yang W, Ciblak MA, et al. 2004. Incidence of bloodstream infections due to Candida species and in vitro susceptibilities of isolates collected from 1998 to 2000 in a population-based active surveillance program. J Clin Microbiol 42: 1519–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrys MS, Creasy T, Sun Y, Shetty AC, Chibucos MC, Drabek EF, Fraser CM, Farooq U, Sengamalay N, Ott S, et al. 2013. Simultaneous transcriptional profiling of bacteria and their host cells. PLoS One 8: e80597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikuta T, Bhawal UK, Tsushima K, Aoki A, Kuboyama N, Abiko Y. 2012. Identification by DNA microarray of genes involved in Candida albicans-treated gingival epithelial cells. J Oral Pathol Med 41: 769–778. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klepser ME. 2006. Candida resistance and its clinical relevance. Pharmacotherapy 26: 68S–75S. [DOI] [PubMed] [Google Scholar]
- Kohler N, Lipton A. 1974. Platelets as a source of fibroblast growth-promoting activity. Exp Cell Res 87: 297–301. [DOI] [PubMed] [Google Scholar]
- Kovalenko M, Rönnstrand L, Heldin CH, Loubtchenkov M, Gazit A, Levitzki A, Böhmer FD. 1997. Phosphorylation site-specific inhibition of platelet-derived growth factor β-receptor autophosphorylation by the receptor blocking tyrphostin AG1296. Biochemistry 36: 6260–6269. [DOI] [PubMed] [Google Scholar]
- Kubista M, Andrade JM, Bengtsson M, Forootan A, Jonak J, Lind K, Sindelka R, Sjoback R, Sjogreen B, Strombom L, et al. 2006. The real-time polymerase chain reaction. Mol Aspects Med 27: 95–125. [DOI] [PubMed] [Google Scholar]
- Lim CS, Rosli R, Seow HF, Chong PP. 2011. Transcriptome profiling of endothelial cells during infections with high and low densities of C. albicans cells. Int J Med Microbiol 301: 536–546. [DOI] [PubMed] [Google Scholar]
- Little JL, Serzhanova V, Izumchenko E, Egleston BL, Parise E, Klein-Szanto AJ, Loudon G, Shubina M, Seo S, Kurokawa M, et al. 2014. A requirement for Nedd9 in luminal progenitor cells prior to mammary tumorigenesis in MMTV-HER2/ErbB2 mice. Oncogene 33: 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Jenssen TK, Trimarchi J, Punzo C, Cepko CL, Ohno-Machado L, Hovig E, Kuo WP. 2007. Comparison of hybridization-based and sequencing-based gene expression technologies on biological replicates. BMC Genomics 8: 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Mittal R, Solis NV, Prasadarao NV, Filler SG. 2011. Mechanisms of Candida albicans trafficking to the brain. PLoS Pathog 7: e1002305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone JH, Oliver B. 2011. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol 9: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. 2008. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 18: 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin R, Moran GP, Jacobsen ID, Heyken A, Domey J, Sullivan DJ, Kurzai O, Hube B. 2011. The Candida albicans-specific gene EED1 encodes a key regulator of hyphal extension. PLoS One 6: e18394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuno K, Ichimura M, Nakajima T, Tahara K, Fujiwara S, Kase H, Ushiki J, Giese NA, Pandey A, Scarborough RM, et al. 2002. Potent and selective inhibitors of platelet-derived growth factor receptor phosphorylation. 1. Synthesis, structure-activity relationship, and biological effects of a new class of quinazoline derivatives. J Med Chem 45: 3057–3066. [DOI] [PubMed] [Google Scholar]
- Moyes DL, Runglall M, Murciano C, Shen C, Nayar D, Thavaraj S, Kohli A, Islam A, Mora-Montes H, Challacombe SJ, et al. 2010. A biphasic innate immune MAPK response discriminates between the yeast and hyphal forms of Candida albicans in epithelial cells. Cell Host Microbe 8: 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyes DL, Shen C, Murciano C, Runglall M, Richardson JP, Arno M, Aldecoa-Otalora E, Naglik JR. 2014. Protection against epithelial damage during Candida albicans infection is mediated by PI3K/Akt and mammalian target of rapamycin signaling. J Infect Dis 209: 1816–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller V, Viemann D, Schmidt M, Endres N, Ludwig S, Leverkus M, Roth J, Goebeler M. 2007. Candida albicans triggers activation of distinct signaling pathways to establish a proinflammatory gene expression program in primary human endothelial cells. J Immunol 179: 8435–8445. [DOI] [PubMed] [Google Scholar]
- Natarajan M, Stewart JE, Golemis EA, Pugacheva EN, Alexandropoulos K, Cox BD, Wang W, Grammer JR, Gladson CL. 2006. HEF1 is a necessary and specific downstream effector of FAK that promotes the migration of glioblastoma cells. Oncogene 25: 1721–1732. [DOI] [PubMed] [Google Scholar]
- Odds FC, Brown AJ, Gow NA. 2004. Candida albicans genome sequence: a platform for genomics in the absence of genetics. Genome Biol 5: 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco AS, Zhou X, Filler SG. 2000. Mechanisms of the proinflammatory response of endothelial cells to Candida albicans infection. Infect Immun 68: 1134–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Myers CL, Sheppard DC, Phan QT, Sanchez AA, Edwards JE, Filler SG. 2005. Role of the fungal Ras-protein kinase A pathway in governing epithelial cell interactions during oropharyngeal candidiasis. Cell Microbiol 7: 499–510. [DOI] [PubMed] [Google Scholar]
- Park H, Liu Y, Solis N, Spotkov J, Hamaker J, Blankenship JR, Yeaman MR, Mitchell AP, Liu H, Filler SG. 2009. Transcriptional responses of Candida albicans to epithelial and endothelial cells. Eukaryot Cell 8: 1498–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan QT, Belanger PH, Filler SG. 2000. Role of hyphal formation in interactions of Candida albicans with endothelial cells. Infect Immun 68: 3485–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan QT, Fratti RA, Prasadarao NV, Edwards JE Jr, Filler SG. 2005. N-cadherin mediates endocytosis of Candida albicans by endothelial cells. J Biol Chem 280: 10455–10461. [DOI] [PubMed] [Google Scholar]
- Phan QT, Myers CL, Fu Y, Sheppard DC, Yeaman MR, Welch WH, Ibrahim AS, Edwards JE Jr, Filler SG. 2007. Als3 is a Candida albicans invasin that binds to cadherins and induces endocytosis by host cells. PLoS Biol 5: e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel-Frausto MS, Wiblin T, Blumberg HM, Saiman L, Patterson J, Rinaldi M, Pfaller M, Edwards JE Jr, Jarvis W, Dawson J, et al. 1999. National epidemiology of mycoses survey (NEMIS): variations in rates of bloodstream infections due to Candida species in seven surgical intensive care units and six neonatal intensive care units. Clin Infect Dis 29: 253–258. [DOI] [PubMed] [Google Scholar]
- Ravel J, Brotman RM, Gajer P, Ma B, Nandy M, Fadrosh DW, Sakamoto J, Koenig SS, Fu L, Zhou X, et al. 2013. Daily temporal dynamics of vaginal microbiota before, during and after episodes of bacterial vaginosis. Microbiome 1: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redding SW, Zellars RC, Kirkpatrick WR, McAtee RK, Caceres MA, Fothergill AW, Lopez-Ribot JL, Bailey CW, Rinaldi MG, Patterson TF. 1999. Epidemiology of oropharyngeal Candida colonization and infection in patients receiving radiation for head and neck cancer. J Clin Microbiol 37: 3896–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revankar SG, Dib OP, Kirkpatrick WR, McAtee RK, Fothergill AW, Rinaldi MG, Redding SW, Patterson TF. 1998. Clinical evaluation and microbiology of oropharyngeal infection due to fluconazole-resistant Candida in human immunodeficiency virus–infected patients. Clin Infect Dis 26: 960–963. [DOI] [PubMed] [Google Scholar]
- Rhodus NL, Bloomquist C, Liljemark W, Bereuter J. 1997. Prevalence, density, and manifestations of oral Candida albicans in patients with Sjögren's syndrome. J Otolaryngol 26: 300–305. [PubMed] [Google Scholar]
- Ross R, Glomset J, Kariya B, Harker L. 1974. A platelet-dependent serum factor that stimulates the proliferation of arterial smooth muscle cells in vitro. Proc Natl Acad Sci 71: 1207–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez AA, Johnston DA, Myers C, Edwards JE Jr, Mitchell AP, Filler SG. 2004. Relationship between Candida albicans virulence during experimental hematogenously disseminated infection and endothelial cell damage in vitro. Infect Immun 72: 598–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandovsky-Losica H, Chauhan N, Calderone R, Segal E. 2006. Gene transcription studies of Candida albicans following infection of HEp2 epithelial cells. Med Mycol 44: 329–334. [DOI] [PubMed] [Google Scholar]
- Sangeorzan JA, Bradley SF, He X, Zarins LT, Ridenour GL, Tiballi RN, Kauffman CA. 1994. Epidemiology of oral candidiasis in HIV-infected patients: colonization, infection, treatment, and emergence of fluconazole resistance. Am J Med 97: 339–346. [DOI] [PubMed] [Google Scholar]
- Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, et al. 2013. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci 110: 3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slutsky B, Staebell M, Anderson J, Risen L, Pfaller M, Soll DR. 1987. “White-opaque transition”: a second high-frequency switching system in Candida albicans. J Bacteriol 169: 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel JD. 1985. Epidemiology and pathogenesis of recurrent vulvovaginal candidiasis. Am J Obstet Gynecol 152: 924–935. [DOI] [PubMed] [Google Scholar]
- Sun JN, Solis NV, Phan QT, Bajwa JS, Kashleva H, Thompson A, Liu Y, Dongari-Bagtzoglou A, Edgerton M, Filler SG. 2010. Host cell invasion and virulence mediated by Candida albicans Ssa1. PLoS Pathog 6: e1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tierney L, Linde J, Müller S, Brunke S, Molina JC, Hube B, Schöck U, Guthke R, Kuchler K. 2012. An interspecies regulatory network inferred from simultaneous RNA-seq of Candida albicans invading innate immune cells. Front Microbiol 3: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar CC, Kashleva H, Mitchell AP, Dongari-Bagtzoglou A. 2005. Invasive phenotype of Candida albicans affects the host proinflammatory response to infection. Infect Immun 73: 4588–4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachtler B, Wilson D, Haedicke K, Dalle F, Hube B. 2011. From attachment to damage: defined genes of Candida albicans mediate adhesion, invasion and damage during interaction with oral epithelial cells. PLoS One 6: e17046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. 2009. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10: 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermark B, Wasteson A. 1976. A platelet factor stimulating human normal glial cells. Exp Cell Res 98: 170–174. [DOI] [PubMed] [Google Scholar]
- Westermann AJ, Gorski SA, Vogel J. 2012. Dual RNA-seq of pathogen and host. Nat Rev Microbiol 10: 618–630. [DOI] [PubMed] [Google Scholar]
- Willis AM, Coulter WA, Fulton CR, Hayes JR, Bell PM, Lamey PJ. 1999. Oral candidal carriage and infection in insulin-treated diabetic patients. Diabet Med 16: 675–679. [DOI] [PubMed] [Google Scholar]
- Zakikhany K, Naglik JR, Schmidt-Westhausen A, Holland G, Schaller M, Hube B. 2007. In vivo transcript profiling of Candida albicans identifies a gene essential for interepithelial dissemination. Cell Microbiol 9: 2938–2954. [DOI] [PubMed] [Google Scholar]
- Zhu W, Phan QT, Boontheung P, Solis NV, Loo JA, Filler SG. 2012. EGFR and HER2 receptor kinase signaling mediate epithelial cell invasion by Candida albicans during oropharyngeal infection. Proc Natl Acad Sci 109: 14194–14199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}