

Abstract
Assemblies of huntingtin (HTT) fragments with expanded polyglutamine (polyQ) tracts are a pathological hallmark of Huntington's disease (HD). The molecular mechanisms by which these structures are formed and cause neuronal dysfunction and toxicity are poorly understood. Here, we utilized available gene expression data sets of selected brain regions of HD patients and controls for systematic interaction network filtering in order to predict disease-relevant, brain region-specific HTT interaction partners. Starting from a large protein–protein interaction (PPI) data set, a step-by-step computational filtering strategy facilitated the generation of a focused PPI network that directly or indirectly connects 13 proteins potentially dysregulated in HD with the disease protein HTT. This network enabled the discovery of the neuron-specific protein CRMP1 that targets aggregation-prone, N-terminal HTT fragments and suppresses their spontaneous self-assembly into proteotoxic structures in various models of HD. Experimental validation indicates that our network filtering procedure provides a simple but powerful strategy to identify disease-relevant proteins that influence misfolding and aggregation of polyQ disease proteins.
Huntington's disease (HD) is an inherited neurodegenerative disorder characterized by motor impairment and progressive neuropsychiatric changes (Harper 1996). It is caused by an expanded polyglutamine (polyQ) tract in huntingtin (HTT), a large, ubiquitously expressed, multidomain protein with multiple cellular functions (Harjes and Wanker 2003). The elongated polyQ sequence is believed to confer a toxic gain of function to HTT, leading to a selective neurodegenerative disease phenotype primarily in patient brains (DiFiglia et al. 1995).
A hallmark of HD pathology is the accumulation of insoluble aggregates that are formed of N-terminal HTT fragments (DiFiglia et al. 1997). Aggregates of HTT in the brains of patients and transgenic animals are sequestered into nuclear and cytoplasmic inclusions. These structures also contain molecular chaperones or components of the ubiquitin proteasome system involved in cellular defense mechanisms (Ciechanover and Brundin 2003). This association, as well as the fact that the formation of inclusion bodies correlates with motor impairment in HD transgenic mice, suggests that inclusions with large insoluble HTT aggregates are harmful for neuronal cells (Yamamoto et al. 2000). Alternatively, evidence has been provided that the toxic species in HD may be diffusible oligomeric or protofibrillar HTT aggregates with pathogenic polyQ tracts (Hands and Wyttenbach 2010). In contrast to large inclusion bodies, these smaller structures are described as highly mobile and able to associate with numerous cellular proteins, leading to abnormal protein–protein interactions and dysfunction of a range of important cellular processes (Miller et al. 2011).
In any case, the molecular mechanisms by which toxic, polyQ-containing HTT protein complexes and aggregates form in neuronal cells and cause selective dysfunction and toxicity remain unclear (Hands and Wyttenbach 2010). Various studies have demonstrated that polyQ tracts with about 40 or more glutamine residues drive the spontaneous misfolding and aggregation of N-terminal HTT fragments in cell-free and cell-based assays (Scherzinger et al. 1997; Waelter et al. 2001). This is supported by investigations in Caenorhabditis elegans, which show that GFP-tagged fusion proteins with long polyQ tracts (more than 40 glutamines) form inclusion bodies much more efficiently than fusion proteins with short polyQ tracts (20–35 glutamines) (Morley et al. 2002). Initially, it was suggested that the spontaneous misfolding and aggregation of HTT fragments is initiated by hydrogen bonds that stabilize intermolecular interactions between polyQ tracts (Perutz et al. 1994). More recent studies, however, indicate that polyQ flanking sequences, rather than the polyQ tracts themselves, initiate spontaneous HTT protein aggregation (Saunders and Bottomley 2009). For instance, it was shown that the first 17 amino acids in HTT, which adopt an α-helical conformation, can initiate the spontaneous aggregation of pathogenic polyQ tracts in cell-free assays (Thakur et al. 2009). The importance of this polyQ flanking sequence for HTT misfolding is supported by biochemical studies with a chaperonin, which directly binds to the first 17 amino acids of HTT and blocks the de novo aggregation of N-terminal HTT fragments (Tam et al. 2009). Thus, proteins that directly associate with polyQ flanking regions appear to strongly influence the spontaneous polymerization of disease proteins.
Potential modifiers of mutant HTT aggregation expressed in neurons may be waiting to be discovered in the large number of proteins that associate with N-terminal polyQ-containing HTT fragments in yeast two-hybrid (Y2H) or coimmunoprecipitation assays (Goehler et al. 2004; Kaltenbach et al. 2007). The challenge has been to distinguish modifiers with relevance to disease from other interaction partners. Several observations may be helpful in meeting this challenge. First, neuronal cell death in HD occurs predominantly in particular brain regions. Second, disease-causing mutations can lead to perturbations in the PPI networks in affected tissues, thereby influencing pathogenesis. Changes in functional protein interactions are frequently linked to changed protein levels in cells; both phenomena have been shown to favor spontaneous protein misfolding and are indicators that a protein might influence HTT aggregation and HD pathogenesis (Pechmann et al. 2009; Vavouri et al. 2009). Therefore, we reasoned that HTT interaction partners displaying abnormal changes in abundance in regions of the brain that are particularly vulnerable to HD may be disease-relevant modifiers that influence mutant HTT misfolding and aggregation.
To identify them, we developed a simple but powerful computational procedure. We exploited published gene expression data obtained from specific brain regions and HD patients to carry out a step-by-step filtering of a large HTT PPI network that includes more than 500 known interaction partners directly or indirectly linked to HTT. By applying sequential interaction network filtering, we defined 13 HTT-associated human proteins whose expression in the caudate nucleus of HD patient brains is likely to be abnormal. Evaluation of available data and focused confirmation studies enabled the discovery of the protein collapsin response mediator protein 1 (CRMP1), a neuron-specific phosphoprotein that plays a central role in neuronal development and communication (Charrier et al. 2003). Levels of CRMP1 transcription were abnormally low in the brains of HD patients. Experimental studies in cell model systems revealed that overproduction of CRMP1 significantly reduces both misfolding and toxicity of mutant HTT, whereas down-regulating the protein using siRNA had the opposite effect. We confirmed these findings in a Drosophila melanogaster model of HD. Further investigations in cell-free assays revealed that CRMP1 directly targets aggregation-prone HTT molecules and reduces their propensity to spontaneously self-assemble into insoluble amyloid structures. A mutant, nonfunctional CRMP1 protein (D408V) did not produce this effect. Thus, our results indicate that CRMP1 acts as a chaperone-like protein in neuronal cells, where it associates with polyQ-containing HTT and reduces its conversion into proteotoxic protein aggregates. This has implications for HD pathogenesis and the development of therapeutic strategies. More generally, our experimental results confirm our computational network filtering procedure to be highly valuable to identify disease-relevant proteins. It can be applied for interaction network analysis and the identification of aggregation modulators in a wide variety of diseases.
Results
Application of a sequential interaction network filtering strategy to predict brain region-specific, dysregulated HTT-associated proteins
To identify disease-relevant proteins that influence mutant HTT misfolding and aggregation in neurons among the large number of already published HTT interaction partners (Chaurasia et al. 2009), we developed a computational procedure for prioritization of interactions. It is based on the systematic integration of protein interaction data with gene expression data obtained from different brain regions of healthy controls and HD patients (Supplemental Tables S1–S9). Starting from a large HTT interaction network with more than 500 partner proteins, we performed a three-step interaction network filtering procedure in order to arrive at a relatively small and focused HTT interaction network, in which potentially dysregulated HTT interaction partners should be enriched (Fig. 1A,B). Such a network should facilitate focused follow-up validation studies and the identification of modulator proteins that are supposed to directly influence HTT misfolding and aggregation.
Figure 1.

Predicting caudate nucleus-specific, dysregulated HTT-associated proteins by interaction network filtering. (A) Data integration strategy for interaction network filtering by differentially expressed genes. By systematic integration (three filtering steps) of protein interaction and gene expression data (PPI1-3 and ΔEG1-3), a caudate nucleus-specific HD network (PPI4) with potentially dysregulated, disease-relevant HTT-associated proteins was created. (B) Potential function of HTT-associated proteins predicted though systematic HTT interaction network filtering. In PPI4, 13 potentially dysregulated proteins are directly or indirectly linked to HTT. The orange ring indicates known HD therapy targets (HDTTs). (C) Effects of the computationally predicted proteins CRMP1, KLF6, HMGA1, and CFLAR on mutant HTT aggregation in cell-based assays. Potential modulator proteins were coproduced as mCherry-tagged fusions with YFP-tagged HTTex1Q79 fusion protein in SH-EP neuroblastoma cells. Formation of YFP-HTTex1Q79 aggregates was quantified after 48 h by high-content fluorescence imaging. Data are represented as mean ± SEM. (***) P ≤ 0.001, two-sided t-test with unequal variance.
In step I, we created a HTT-centric protein interaction network (PPI1), using information from the literature (Goehler et al. 2004; Chaurasia et al. 2009). This network comprised 1319 interactions in which 509 proteins were directly or indirectly linked to HTT (Supplemental Table S2A,B). In order to generate a network of human proteins that are potentially altered in their abundance in comparison to nonbrain tissues, we filtered PPI1 for HTT interaction partners that are differentially expressed in healthy human brain compared to nonbrain tissues (ΔEG1, P1 < 10−4) (Supplemental Table S3). This resulted in the brain-specific network PPI2 (Fig. 1A), which consists of 113 interactions. In PPI2, 88 human proteins are directly or indirectly linked to the disease protein HTT (Supplemental Table S4A,B).
HD affects the whole brain, but certain areas such as the caudate nucleus are more vulnerable than others (Vonsattel et al. 1985). We therefore generated a brain region-specific protein interaction network in step II of the filtering procedure. We defined genes as differentially expressed in the caudate nucleus (CN) by comparison to motor cortex (MC), prefrontal cortex (PFC), and cerebellum (CE), again using publicly available gene expression profiles for healthy brain regions (ΔEG2, P2 < 10−3) (Fig. 1A; Supplemental Table S5). This filtering step yielded PPI3, a network with caudate nucleus proteins that are potentially altered in their abundance in comparison to other brain regions. This network connects 66 human proteins via 84 interactions directly or indirectly to HTT (Fig. 1A; Supplemental Table S6A,B).
Previous studies suggested that dysregulated HTT-interacting proteins might influence the propensity of HTT to misfold and self-assemble into insoluble protein aggregates (Goehler et al. 2004; Behrends et al. 2006). In step III of the filtering procedure we therefore searched for HTT-associated proteins dysregulated in HD patient brains. In a comparison of gene expression profiles from 38 HD patients and 32 healthy individuals (Supplemental Table S1), we identified human genes that are differentially expressed in the caudate nucleus of HD patients and healthy individuals (ΔEG3, P3 < 10−6) (Fig. 1A; Supplemental Table S7). This set was then utilized to filter the PPI3 network to yield the final, HD dysregulated caudate-nucleus-specific HTT interaction network PPI4 (Fig. 1A; Supplemental Table S8A,B). The result is a network of 13 proteins that are directly or indirectly linked to the disease-causing protein HTT and that might be dysregulated in their abundance based on the available gene expression data in HD patients compared to controls (Fig. 1B). The transcripts of seven proteins were found to be abnormally up-regulated in brains of HD patients, whereas six were down-regulated (Fig. 1B; Supplemental Table S9).
The significance thresholds (P1, P2, P3) for defining differential expression (steps I–III) (Fig. 1A) were derived by optimization with respect to an independently generated reference set of validated HD therapy targets (HDTTs) from the HD Research Crossroads database (Kalathur et al. 2012). This reference set was curated by experts in the field and constitutes the most comprehensive collection of HDTTs to date. For optimization of the significance thresholds, we hypothesized that the relative number of HDTTs in the final PPI4 data set is a measure of the potential relevance of the network to disease. Additionally, we required that the final interaction network PPI4 should consist of >50% HDTTs. Through an exhaustive search procedure (as described in Methods), we found that the combination of thresholds with P1 < 10−4 (step I), P2 < 10 × 10−3 (step II), and P3 < 10−6 (step III) produced the largest network that fulfilled these requirements (Fig. 1A,B). In PPI4, seven of 13 (>50%) HTT-associated proteins are known HDTTs. This enrichment of a large number of known HDTTs in PPI4 suggests that the other proteins in the network are also relevant to disease and might influence mutant HTT misfolding and aggregation in model systems.
A comparison of the performance of single- and multistep interaction network filtering approaches
We utilized the manually curated set of HDTTs to examine the effect of the number of consecutive data integration steps on the outcome of our network filtering procedure. First, the enrichment of HDTTs in the interaction data sets (PPI1–PPI4) and the selected gene expression data sets (ΔEG1, ΔEG2, and ΔEG3) was determined (Fig. 1A; Supplemental Fig. S1A). HDTTs were more enriched in the interaction network PPI1 compared to data sets of differentially expressed genes. This provides evidence that focused disease protein interaction networks contain relevant information for disease research, in line with previous studies (Goehler et al. 2004). We also observed that procedures of one- or two-step interaction filtering only modestly increase the relative number of HDTTs in the PPI2 and PPI3 networks. However, a large increase was observed after three filtering steps (PPI4) (Supplemental Fig. S1A), supporting our expectations that a three-step procedure is most productive in identifying disease-relevant HTT-associated proteins.
To further investigate whether all three filtering steps are required to obtain maximum enrichment of HDTTs, we also filtered the initial HTT network (PPI1) directly with a set of genes that are differentially expressed in HD patients and healthy individuals (ΔEG3). This has been the computational approach most commonly used to create disease-relevant interaction networks (Chuang et al. 2007; Zhao et al. 2011). This procedure yielded the interaction network PPID (Supplemental Fig. S1A; Supplemental Table S10A,B). Remarkably, the relative enrichment of HDTTs in PPID (23% HDTTs) was considerably lower than in PPI4 (54% HDTTs), supporting our view that the step-by-step integration of tissue expression data with PPI data helps in the identification of disease-relevant proteins (Fig. 1A; Supplemental Fig. S1A). This difference was also observed when the significance threshold was optimized independently for PPID (Supplemental Fig. S1B). Thus, our studies indicate that consecutive filtering of PPI data with multiple sets of differentially expressed genes is a valuable approach to create focused, disease protein-centered interaction networks.
Interaction network filtering reveals known dysregulated HTT-associated proteins
Utilizing information available in the literature via PubMed, we searched for descriptions of dysregulated HTT interacting proteins. We found that the abundance of five HTT-associated proteins (CASP6, PACSIN1, DNM1, SP1, and PRPF40A) in PPI4 is altered in brains of HD patients or transgenic mice (Fig. 1B; Supplemental Table S9). In addition, evidence was reported that the levels of the proteins HMGB1 and HMGB2, which are close homologs of HMGA1 (Fig. 1B), are reduced in HD cell lines (Qi et al. 2007). These investigations support the assumption that systematic network filtering is a useful approach to select dysregulated HTT-associated proteins.
Previous studies indicate that alterations in gene transcription, synaptic vesicle endocytosis, cytoskeletal reorganization, apoptotic pathways, and RNA metabolism are critical for the development of HD (Truant et al. 2007; Caviston and Holzbaur 2009; Fossale et al. 2011). Remarkably, all these processes can be associated with proteins in the derived PPI4 network. Thus, our systematic network filtering studies support these previous observations (Fig. 1B), suggesting that abnormal interactions in these processes contribute to pathogenesis.
Interaction network filtering enables the identification of novel HTT aggregation modulators
We hypothesized that the network PPI4 should contain proteins that influence polyQ-mediated HTT misfolding and aggregation in model systems. To test this hypothesis, we focused on the proteins KLF6, HMGA1, CFLAR, and CRMP1, which previously have not been associated with HD pathogenesis (Fig. 1B). We coexpressed the mCherry-tagged proteins KLF6, HMGA1, CFLAR, and CRMP1 together with a YFP-tagged HTT exon-1 fragment (YFP-HTTex1Q79) in SH-EP neuroblastoma cells and quantified the formation of YFP-HTTex1Q79 aggregates through high-content fluorescence imaging (Abraham et al. 2004). We observed that three of the four selected proteins (HMGA1, CFLAR, and CRMP1) efficiently suppressed YFP-HTTex1Q79 aggregation in SH-EP cells (Fig. 1C), supporting our assumption that aggregation modulators are enriched by network filtering. A similar effect was observed with a well-established suppressor of mutant HTT aggregation DNAJB1 (Muchowski et al. 2000), which was used as a positive control protein. The expression of YFP- and mCherry-tagged proteins was confirmed by SDS-PAGE and immunoblotting (Supplemental Fig. S1C,D). Thus, our cell-based studies suggest that interaction network filtering with gene expression data and the generation of disease protein-centered networks is a suitable strategy to predict potential modulators of HTT misfolding and aggregation.
For more detailed validation studies (see below), we focused on the HTT interacting protein CRMP1, whose transcript in comparison to the other genes was found to be most significantly up-regulated in brain compared to nonbrain tissues (P = 5.3 × 10−11). This supports previous observations that CRMP1 is predominantly expressed in neurons (Bretin et al. 2005), suggesting that abnormal dysregulation of the protein in HD brains might contribute to the neurodegenerative disease phenotype. A potential importance of CRMP1 for neuronal function under physiological conditions is also supported by gene knockdown studies in mice, indicating that CRMP1 function is critical for learning and memory (Su et al. 2007). Thus, for further more detailed experimental investigations, we focused our efforts on the protein CRMP1, which likely is a neuron-specific modulator of mutant HTT misfolding and aggregation in brains of HD patients.
CRMP1 expression levels are altered in brains of HD transgenic mice
To investigate potential changes of CRMP1 protein levels in HD animal models, we studied the expression of the protein in brains of HD transgenic mice by SDS-PAGE and immunoblotting. For detection, an antibody was applied that is specific to CRMP1 and does not recognize the related proteins DPYSL2, DPYSL3, DPYSL4, and DPYSL5 (Supplemental Fig. S2A,B). First, striatal tissues prepared from 12-mo-old YAC128 HD transgenic mice that express a full-length human HTT protein with an expanded polyQ tract of 128 glutamines were examined (Slow et al. 2003). We found that levels of CRMP1 protein were significantly reduced in brains of HD transgenic mice compared to age-matched controls (Fig. 2A,B). Next, the expression of CRMP1 was assessed in R6/2 HD transgenic mice, which produce a truncated HTT exon-1 fragment with a pathogenic polyQ tract of ∼150–200 glutamines (Mangiarini et al. 1996). Protein lysates were prepared from 4-, 8-, and 14-wk-old R6/2 transgenic mouse brains and wild-type controls and analyzed by SDS-PAGE and immunoblotting. In 14-wk-old transgenic R6/2 mice, which show a severe disease phenotype (Labbadia et al. 2011), levels of CRMP1 were significantly reduced compared to age-matched controls (Fig. 2C,D), consistent with the results from the YAC128 HD model (Fig. 2A,B). The reduced CRMP1 protein levels in R6/2 mice also correspond to decreased Crmp1 transcript levels in brains of 12-wk-old R6/2 mice compared to age-matched controls (Supplemental Fig. S2C).
Figure 2.
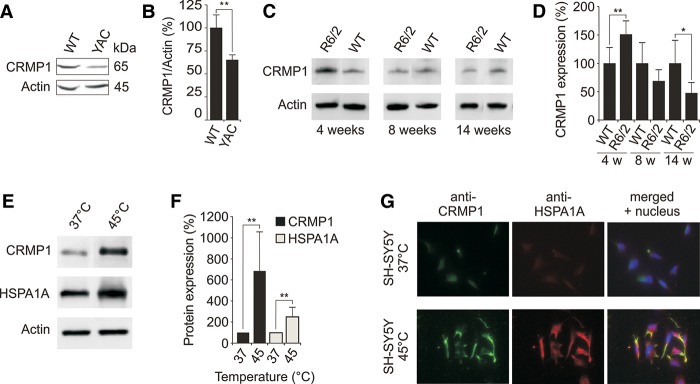
Expression of CRMP1 in brains of HD transgenic mice and cell models. (A) Endogenous CRMP1 protein levels are reduced in brain tissues of 12-mo-old YAC128 HD transgenic mice compared to controls. Protein extracts prepared from striatal tissues of wild-type and transgenic HD mice were analyzed by SDS-PAGE and immunoblotting using the anti-CRMP1 antibody 504-518. (B) Quantification of ∼65 kDa CRMP1 bands in A. Values represent the means of three independent experiments. (**) P ≤ 0.01, two-sided t-test with unequal variance. (C) Endogenous CRMP1 levels are altered in brains of R6/2 HD transgenic mice compared to controls. Protein extracts were analyzed by SDS-PAGE and immunoblotting using the anti-CRMP1 antibody 504-518. (D) Quantification of CRMP1 protein bands shown in C. Values represent the means of three independent experiments. (*) P < 0.1; (**) P ≤ 0.01, two-sided t-test with unequal variance. (E) Heat stress induces endogenous CRMP1 protein levels in SH-SY5Y neuroblastoma cells. Cell lysates were analyzed by SDS-PAGE and immunoblotting. (F) Quantification of CRMP1 and HSPA1A protein bands shown in E. Values represent the means of three independent experiments. (**) P ≤ 0.01, two-sided t-test with unequal variance. (G) Analysis of CRMP1 and HSPA1A protein expression in SH-SY5Y cells under heat stress conditions by immunofluorescence microscopy. Data in B, D, and F are represented as mean ± SEM.
Interestingly, we found that CRMP1 levels were enhanced in 4-wk-old presymptomatic R6/2 transgenic mice, which suggests that protein levels are up-regulated during early stages of pathogenesis (Fig. 2C,D). Increased CRMP1 levels at early disease stages suggest that stress conditions may induce the expression of the protein. To address this question, we analyzed the promoter region of the CRMP1 gene (600 bp) for potential transcription factor binding sites (Supplemental Fig. S2D; Supplemental Table S11). We observed multiple conserved heat shock factor (HSF1/HSF2) binding sites in the cis-regulatory region upstream of the transcriptional start site, suggesting that CRMP1 gene transcription is heat shock inducible. We next tested this hypothesis using a SH-SY5Y neuroblastoma cell line (Fig. 2E–G). Cells grown at 37°C were incubated for 30 min at 45°C, and then analyzed by SDS-PAGE and immunoblotting. We found that CRMP1 protein levels are increased at 45°C (Fig. 2E,F). Similar results were obtained for the well-known heat shock-inducible protein HSPA1A (positive control). The findings were confirmed by immunofluorescence microscopy studies (Fig. 2G), indicating that CRMP1, similar to HSPA1A, is a heat-inducible stress protein.
Altered CRMP1 levels affect HTT aggregation and toxicity in a PC12 cell model
To investigate whether CRMP1 influences the aggregation and toxicity of mutant HTT in mammalian cells, we increased or reduced the protein's abundance in a PC12 HD cell model overproducing a pathogenic HTTQ103-EGFP fusion protein (Apostol et al. 2006). Cells were treated with CRMP1 siRNA, and HTTQ103-EGFP aggregation was analyzed after 40 h using the filter retardation assay (Wanker et al. 1999). To quantify HTTQ103-EGFP-induced cellular toxicity, we used a caspase 3/7 assay. Finally, CRMP1 and HTTQ103-EGFP protein levels were examined by SDS-PAGE and immunoblotting. We observed that siRNA-mediated reduction of CRMP1 protein levels increased both the aggregation and toxicity of HTTQ103-EGFP in PC12 cells (Fig. 3A–D). In contrast, a significant reduction of both HTTQ103-EGFP aggregation and toxicity was detected in cells overproducing a hemagglutinin (HA)-tagged CRMP1 fusion protein (Fig. 3E–H), confirming our observations from neuroblastoma cells (Fig. 1C).
Figure 3.

CRMP1 protein production influences polyQ-mediated HTT aggregation and toxicity in PC12 cells. (A,E) SDS-PAGE and immunoblot analysis of PC12 cells overproducing HTTQ103-EGFP fusion protein. Cells treated with si-CRMP1 RNA (A) or overproducing a HA-tagged CRMP1 protein (E) were analyzed. (B,F) Detection of insoluble HTTQ103-EGFP aggregates in si-CRMP1-treated (B) and HA-CRMP1 overproducing (F) PC12 cells. Insoluble aggregates (black dots) were detected by filter retardation assay. (C,G) Quantification of insoluble HTTQ103-EGFP aggregates shown in B and F by image analysis: n = 3; (***) P ≤ 0.001, two-sided t-test. (D,H) Quantification of caspase-3/7 activity in si-CRMP1-treated (D) and HA-CRMP1 overproducing (H) PC12-HTTQ103-EGFP cells: n = 3; (***) P ≤ 0.001, two-sided t-test. Data in C, D, G, and H are represented as mean ± SEM.
Overproduction of CRMP1 reduces toxicity and aggregation of mutant HTT in HD transgenic flies
Next, we examined whether CRMP1 influences HTT-mediated neurotoxicity in a Drosophila model of HD (Kaltenbach et al. 2007). In this model, an N-terminal HTT fragment with a pathogenic polyQ tract of 128 glutamines (HTT336Q128) is overproduced in photoreceptor neurons. We found that coproduction of human CRMP1 and its Drosophila homolog CRMP significantly reduced HTT336Q128-induced photoreceptor degeneration in HD transgenic flies (Fig. 4A–G), supporting results from the cell-based assays (Figs. 1C, 3H). In contrast, the control protein GFP had no effect on photoreceptor damage (Fig. 4D,G). The production of HTT336Q128 and CRMP1 in Drosophila eye imaginal discs was confirmed by immunoblotting and immunofluorescence microscopy (Supplemental Fig. S3A–F).
Figure 4.
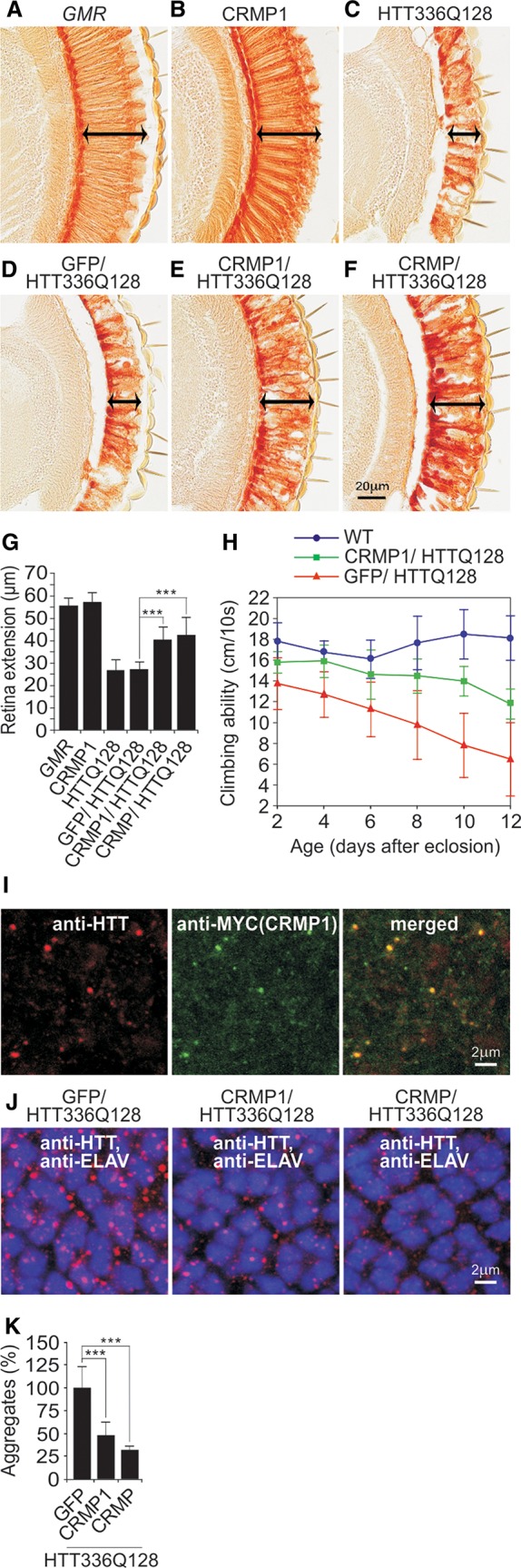
CRMP1 overproduction mitigates mutant HTT-induced photoreceptor degeneration and motor impairment in HD transgenic flies. (A–F) Investigation of eye sections. Analysis of eye sections in control flies (A) and transgenic flies overproducing CRMP1 (B) or HTT336Q128 (C) alone. Coproduction of the control protein GFP does not influence HTT336Q128-induced retina degeneration (D), whereas coproduction of human CRMP1 (E) or Drosophila CRMP (F) mitigates HTT336Q128-induced retinal degeneration. Arrows indicate retinal degeneration. (G) Quantification of retinal degeneration in control and HD transgenic flies (A–F): n ≥ 9 flies per genotype; (***) P ≤ 0.001, two-sided t-test with unequal variance. (H) CRMP1 significantly improves climbing abilities of HTT336Q128 overproducing HD transgenic flies. Data were analyzed using Graphpad Prism 5: n ≥ 16 flies per genotype; P = 0.0017; linear regression followed by F-test. (I–J) Immunofluorescence analysis of eye imaginal discs of HD transgenic flies. (I) MYC-CRMP1 colocalizes with HTT336Q128 in multiple inclusion bodies with a diameter of 0.2–0.4 μm. (J) In contrast to the control protein GFP, coproduction of the proteins CRMP1 and CRMP reduced both the number as well as the average size of HTT336Q128 inclusion bodies. (K) Quantification of the number of HTT336Q128 inclusion bodies in HD flies coproducing modulator proteins. An area of 35.71 μm2 in the middle of the posterior third of eye imaginal discs was systematically analyzed: n ≥ 5; (***) P ≤ 0.001, two-sided t-test with unequal variance. Data in G, H, and K are represented as mean ± SEM.
In order to investigate whether CRMP1 influences motor function, we examined the climbing ability of HD transgenic flies. We discovered that HD transgenic flies display an impairment of motor activity compared to wild-type controls. Climbing was significantly improved in CRMP1-overproducing HD transgenic flies, whereas the control protein GFP showed no effect (Fig. 4H). Hence, our climbing studies with HD transgenic flies further support a protective function of CRMP1 in neuronal cells.
Next, we used immunofluorescence microscopy (IFM) to assess whether CRMP1 colocalizes with insoluble HTT336Q128 aggregates in HD transgenic flies. Analysis of eye imaginal discs of late third instar larvae revealed colocalization of CRMP1 with HTT336Q128 in inclusion bodies (Fig. 4I), indicating that CRMP1 is recruited to insoluble HTT aggregates in HD transgenic flies. A similar result was obtained from brains of HD transgenic mice expressing an N-terminal, aggregation-prone HTT fragment (Supplemental Fig. S3G), supporting the results from the Drosophila experiments.
Finally, we examined whether overproduction of CRMP1 influences the formation of insoluble HTT336Q128 aggregates in photoreceptor neurons. An IFM analysis of eye imaginal discs of late third instar larvae demonstrated that HD flies that overproduce human CRMP1 or its Drosophila homolog CRMP exhibit significantly lower numbers of HTT336Q128 inclusion bodies compared to control flies (Fig. 4J,K), confirming the results obtained in neuroblastoma and PC12 cells (Figs. 1C, 3F,G).
The missense mutation D408V suppresses the effect of CRMP1 on mutant HTT aggregation and toxicity in model systems
Previous studies in Drosophila indicate that an exchange of an aspartic acid at position 430 for a valine residue alters the function of CRMP (Rawls 2006), suggesting that exchanging the corresponding amino acid in human CRMP1 might also influence its function. We found that the aspartic acid at position 408 is highly conserved in human CRMP1 (Fig. 5A). Subsequently, we engineered the human protein CRMP1-D408V for functional studies in cell model systems. We coproduced mCherry-tagged fusion proteins (for example mCherry-CRMP1-D408V) together with YFP-HTTex1Q79 in SH-EP neuroblastoma cells and systematically examined the formation of insoluble YFP-HTTex1Q79 aggregates. Protein aggregates were quantified after 48 h by measuring YFP fluorescence in intracellular inclusion bodies using a high-content fluorescence imaging technology. We found that coproduction of wild-type CRMP1 efficiently reduced YFP-HTTex1Q79 aggregation in SH-EP cells (Fig. 5B). In contrast, the mutant protein CRMP1-D408V behaved like the control protein luciferase and did not reduce YFP-HTTex1Q79 aggregation. A reduction of YFP-HTTex1Q79 aggregation was also observed in conjunction with the molecular chaperone DNAJB6, whereas the control protein GIT1 showed the opposite effect, confirming previously published results (Goehler et al. 2004; Hageman et al. 2010).
Figure 5.

A mutant variant of CRMP1 (D408V) shows impaired activity in cell-based assays and HD transgenic flies. (A) Multiple sequence alignment of CRMP1 protein sequences. The aspartic acid residue 408 in human CRMP1 is conserved in a large number of related proteins (yellow box). (B) Effects of mCherry-tagged CRMP1 and CRMP1-D408V fusion proteins on aggregation of YFP-tagged HTTex1Q79 in cell-based assays. Formation of insoluble YFP-HTTex1Q79 aggregates in SH-EP cells was quantified by high-content fluorescence imaging. Aggregates were detected by YFP fluorescence of HTT aggregates or by immunostaining using the anti-HTT antibody MW8. MW8 preferentially detects insoluble HTT aggregates. Data are represented as mean ± SEM. (***) P ≤ 0.001, two-sided t-test with unequal variance. (C) Retinal degeneration in control flies carrying the GMR-GAL4 driver (GMR) alone and in flies coexpressing HTT336Q128 with CRMP1-D408V or the GFP control protein under control of the GMR driver. Analysis of eye sections reveals that in contrast to wild-type CRMP1, neither GFP nor CRMP1-D408V affects HTT336Q128-induced retinal degeneration compared to the GMR flies. (D–E) Effects of mCherry-tagged CRMP1 or CRMP1-D408V on aggregation of YFP-tagged ATXN1Q82 (D) or TARDBP (E) in cell-based assays. (D) YFP-ATXN1Q82 or (E) YFP-TARDBP aggregates were detected in SH-EP cells by YFP fluorescence and quantified by high-content fluorescence imaging. Data are represented as mean ± SEM. (***) P ≤ 0.001, two-sided t-test with unequal variance.
To examine whether protein expression levels influence the effects on YFP-HTTex1Q79 aggregation, we quantified mCherry- and YFP-tagged fusion proteins in transiently transfected SH-EP cells. We obtained similar ratios for all mCherry- and YFP-tagged fusion proteins in cotransfected cells (Supplemental Fig. S4A), indicating that the CRMP1-mediated reduction of HTT aggregation cannot be explained by a simple protein dosage effect.
Next, we studied whether imaging of YFP fluorescence indeed is suitable to detect insoluble YFP-HTTex1Q79 aggregates in SH-EP cells. We performed additional imaging studies utilizing the monoclonal antibody MW8 to detect YFP-HTTex1Q79 inclusion bodies. This antibody preferentially recognizes insoluble HTT protein aggregates in cells but not monomers or small oligomers (Ko et al. 2001). The quantification of YFP fluorescence and MW8 immunoreactivity yielded very similar results (Fig. 5B), confirming that insoluble YFP-HTTex1Q79 aggregates are detected by YFP imaging.
Finally, we investigated whether CRMP1-D408V influences mutant HTT-induced photoreceptor degeneration in the Drosophila HD model. We found that the mutant protein, like the control GFP, did not rescue photoreceptor degeneration in Drosophila eyes (Fig. 5C; Supplemental Fig. S4B), whereas the wild-type CRMP1 protein did (Fig. 4E,G). Thus, our studies with cell models and HD transgenic flies indicate that the missense mutation D408V in CRMP1 is sufficient to suppress its effect on mutant HTT aggregation and toxicity.
CRMP1 reduces the aggregation of ATXN1 and TARDBP in cell-based assays
We hypothesized that CRMP1 might display properties similar to molecular chaperones such as HSPA1A or crystallins (Morimoto 2008; Robertson et al. 2010), which target several neurodegenerative disease proteins and suppress their aggregation propensity. To address this question, we examined the effect of CRMP1 on the disease proteins ATXN1Q82 (a toxic polyQ-expanded ATXN1) and TARDBP, which form insoluble protein aggregates in patients with spinocerebellar ataxia type 1 (SCA1) and frontotemporal dementia, respectively (Neumann et al. 2006). YFP-tagged ATXN1Q82 or TARDBP fusion proteins were coproduced with mCherry-tagged CRMP1 in SH-EP cells. After 48 h, the formation of insoluble protein aggregates was quantified using high-content fluorescence imaging (Fig. 5D,E). We observed that wild-type CRMP1 potently reduces the aggregation of both YFP-ATXN1Q82 and YFP-TARDBP in cell-based assays, whereas the mutant protein CRMP1-D408V did not exert this effect (Fig. 5D,E). Reduction in aggregation of YFP-ATXN1Q82 and YFP-TARDBP was also detected with the molecular chaperone DNAJB1, which was used as a positive control. Protein expression was verified by SDS-PAGE and immunoblotting (Supplemental Fig. S4C,D). Thus, our cell-based studies suggest that CRMP1 functions as a stress-inducible molecular chaperone in neuronal cells.
CRMP1 directly targets mutant HTT fragments and reduces their spontaneous self-assembly in cell-free assays
We also investigated whether CRMP1 directly influences polyQ-mediated HTT aggregation in cell-free assays. GST- and His-tagged fusion proteins (GST-HTTex1Q49, His-CRMP1, and His-CRMP1-D408V) were produced in E. coli and purified to >90% homogeneity by affinity chromatography (Fig. 6A). Soluble GST-HTTex1Q49 fusion protein was incubated with PreScission protease (PP) and either His-CRMP1 or His-CRMP1-D408V. We quantified the spontaneous formation of insoluble HTTex1Q49 aggregates over time using the filter retardation assay (Wanker et al. 1999). PP was added to reactions to remove the GST-tag from the GST-HTTex1Q49 fusion protein and to initiate spontaneous HTTex1Q49 aggregation (Supplemental Fig. S5A). We found that spontaneous HTTex1Q49 aggregation was slowed down through addition of wild-type His-CRMP1, whereas the mutant protein His-CRMP1-D408V had no effect (Fig. 6B). A reduction of HTTex1Q49 aggregation was also observed with the molecular chaperone HSPA1A, confirming previously published results (Muchowski et al. 2000).
Figure 6.
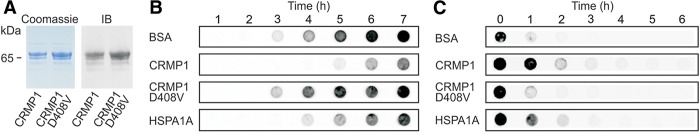
A mutant variant of CRMP1 (D408V) shows impaired activity in cell-free HTT aggregation assays. (A) The proteins His-CRMP1 and His-CRMP1-D408V were produced in E. coli and purified to >90% homogeneity by affinity chromatography. Protein expression and purity was confirmed by Coomassie staining of SDS gels or immunoblotting (IB). (B) Analysis of spontaneous HTTQ49 aggregation in the presence of modulator proteins in cell-free assays by filter retardation assay. SDS-insoluble HTTQ49 protein aggregates were detected on filter membranes using the anti-HTT antibody CAG53b. Representative results from three independent experiments are shown. (C) Analysis of spontaneous HTTQ49 aggregation in the presence of modulator proteins by dot-blot assays. The conversion of soluble HTTQ49 into insoluble protein aggregates was monitored using the antibody 3B5H10. Representative results from three independent experiments are shown.
Next, we studied whether the soluble HTTex1Q49 protein is a direct target of CRMP1 in cell-free aggregation assays. To detect soluble HTTex1Q49 molecules we used the monoclonal antibody 3B5H10, which exclusively recognizes expanded polyQ tracts in soluble HTT molecules (Miller et al. 2011). An analysis of time-resolved HTTex1Q49 aggregation assays with dot-blot assays revealed that soluble 3B5H10-reactive HTT molecules disappeared at a slower rate in the presence of wild-type CRMP1 than in the presence of mutant CRMP1-D408V (Fig. 6C). A similar effect was observed with the molecular chaperone HSPA1A, which directly targets soluble, aggregation-prone HTT molecules and reduces their aggregation propensity (Muchowski et al. 2000). Thus, it can be concluded that CRMP1 directly targets soluble HTTex1Q49 molecules with exposed polyQ sequences and reduces their propensity to spontaneously convert into insoluble protein aggregates.
Previous studies indicate that functional high molecular weight CRMP1 oligomers are formed under nondenaturating conditions (Deo et al. 2004). This suggests that a change in the structure or stability of CRMP1 oligomers through the missense mutation D408V could be responsible for the reduced activity of CRMP1 on HTT aggregation (Fig. 6B). This view is supported by our computational structure analysis of mouse CRMP1, which suggests that aspartic acid D408 is critical for oligomer assembly and stability (Supplemental Fig. S5B,C). In order to address whether the mutation D408V influences the stability of CRMP1 oligomers, the recombinant proteins His-CRMP1 and His-CRMP1-D408V were incubated with different concentrations of urea and analyzed by blue-native (BN) PAGE and immunoblotting. We observed that both wild-type and mutant CRMP1 form tetramers under nondenaturating conditions, migrating at ∼480 kDa (Supplemental Fig. S5D). This confirms previously published results, indicating that CRMP1 proteins form stable oligomers (Deo et al. 2004). Interestingly, analysis by BN-PAGE revealed that wild-type CRMP1 oligomers are less stable and dissociated with lower urea concentrations than mutant CRMP1-D408V oligomers (Supplemental Fig. S5D). This suggests that increased oligomer stability might be responsible for the reduced activity of the CRMP1-D408V protein in HTT aggregation assays. This result is supported by previously published studies indicating that flexibility and reorientation of domains in chaperones are critical for their function (Haslbeck et al. 2004; White et al. 2006; Quan et al. 2014). Analysis by SDS-PAGE and Coomassie staining confirmed that similar amounts of both proteins were analyzed (Supplemental Fig. S5E). Finally, we also observed a difference between the proteins His-CRMP1 and His-CRMP1-D408V when they were examined for their intrinsic tryptophan fluorescence (Supplemental Fig. S5F). Recent studies indicate that oligomerization and aggregation of proteins is associated with an increase of intrinsic protein fluorescence (Chan et al. 2014), supporting our results by BN-PAGE that CRMP1-D408V but not the wild-type protein forms higher molecular weight oligomers (HMWOs) (Supplemental Fig. S5D). Thus, our biochemical and biophysical studies suggest that the mutation D408V alters the stability CRMP1 oligomers and promotes their abnormal oligomerization.
Discussion
This study reports the development and application of a computational approach utilizing protein–protein interaction and gene expression data to identify disease-relevant proteins that are associated with HTT and influence its aggregation propensity. The method was designed to select proteins that are dysregulated in the caudate nucleus in HD brains from a large, HTT-centered PPI data set. The caudate nucleus displays severe neuropathological changes at an early stage of disease, suggesting that it plays a critical role in the development and progression of HD (Aylward 2007). Our protein interaction network filtering approach greatly reduced the complexity of the initially very large HTT-centered PPI network in a stepwise manner. For filtering of PPI data, we used information on genes that are differentially expressed in (1) brain compared to nonbrain tissues; (2) caudate nucleus compared to other brain tissues; and (3) brains of HD patients compared to healthy individuals. This finally resulted in the generation of a focused network with potentially dysregulated HTT-associated proteins (PPI4) that might influence HTT misfolding and aggregation in neurons.
We designed our protein interaction network filtering procedure to create a focused HTT interaction network that contains a high percentage of already known HD therapy targets (HDTTs). These targets were shown in previous studies to influence the pathogenesis in various HD model systems (Kalathur et al. 2012), suggesting that the other proteins in the network PPI4 (Fig. 1B) might also affect the disease process in HD. Through our network filtering approach, four proteins (HMGA1, KLF6, CFLAR, and CRMP1) were selected that have not previously been linked to HD pathogenesis. They subsequently were systematically examined in cell-based assays to determine whether they influence mutant HTT aggregation. Strikingly, we found that three (HMGA1, CFLAR, and CRMP1) of four computationally predicted proteins are potent modulators of mutant HTT aggregation (Fig. 1C), supporting our initial hypothesis that among dysregulated HTT interaction partners, aggregation modulators can be identified. Intriguingly, two of them are located in chromosomal regions (CFLAR: 2q33; CRMP1: 4p16), which harbor genes that potentially influence HD age of onset (Kalathur et al. 2012), suggesting that they are candidates for genetic modifiers.
For more detailed experimental validation studies, we focused our efforts on the protein CRMP1, whose transcript levels are significantly diminished in HD brains (Fig. 1B). CRMP1 is a cytoplasmic phosphoprotein that is predominantly expressed in neurons (Goshima et al. 1995). Previous studies revealed that mice lacking CRMP1 show impairment of learning and memory (Su et al. 2007), suggesting that a reduction of CRMP1 levels in HD brains might contribute to the disease phenotype (Paulsen et al. 2013). This view is also supported by studies in cell model systems, indicating that reduced levels of CRMP1 promote death of spinal cord neurons (Kurnellas et al. 2010). Therefore, previously published studies support the selection of CRMP1 for more detailed experimental validation studies in HD model systems.
Our studies with cell-based assays and HD transgenic flies revealed that CRMP1 influences both aggregation and toxicity of mutant HTT in mammalian cells (Figs. 3, 4). While overproduction of CRMP1 significantly decreased misfolding and toxicity of mutant HTT, down-regulating the protein levels through siRNA treatment showed the opposite effect. Together, these results suggest that in neurons with high levels of CRMP1, abnormal mutant HTT aggregation is suppressed; whereas in neurons with low CRMP1 protein levels, the protein forms insoluble aggregates. Intriguingly, we noted that the expression of CRMP1 is significantly lower in the caudate nucleus compared to other brain regions (P = 4.7 * 10−28)—an observation which could help to explain the region specific vulnerability in HD brains.
Currently, the molecular mechanisms that lead to a decrease of CRMP1 protein levels in brains of HD patients and transgenic mice (Fig. 2A–D) are unclear. Experimental evidence indicates that pathogenesis in HD is associated with altered gene expression in patient brains (Cha 2000; Ross and Thompson 2006), suggesting that reduced gene transcription could be responsible for the decrease of CRMP1 protein. This is also supported by the reduction of Crmp1 transcript levels in transgenic HD mice (Supplemental Fig. S2C). However, a reduction of CRMP1 protein levels could also be caused by the recruitment of the protein into insoluble HTT inclusion bodies (Fig. 4I; Supplemental Fig. S3G), enhanced aggregation in response to cellular stress (Meriin et al. 2001; Hipp et al. 2014), or enhanced protein degradation during HD progression. Additional studies will be necessary to address this question in more detail.
Our experiments with cell-free assays indicate that CRMP1 directly targets soluble, aggregation-prone HTT molecules and reduces their propensity to spontaneously self-assemble into insoluble, amyloidogenic protein structures (Fig. 6B,C). This suggests that CRMP1 acts as a molecular chaperone, which directly binds to unfolded or partially folded aggregation-pone proteins. Molecular chaperones are classified either as “foldases” or “holdases.” Although foldases (e.g., HSPA1A and DNAJB1) promote correct folding in an ATP-dependent process, holdases prevent the aggregation of misfolded proteins in an ATP-independent manner (Haslbeck et al. 2005). Our studies indicate that CRMP1 functions as a holdase that does not require ATP hydrolysis to suppress mutant HTT aggregation (Figs. 2E–G, 5B,C, 6B,C). This is also supported by preliminary studies indicating that CRMP1, in contrast to HSPA1A, is unable to refold denatured luciferase in cell-based assays. We propose a model in which a CRMP1 protein with high flexibility directly binds soluble, unfolded, aggregation-prone polyQ-containing HTT molecules and stabilizes their aggregation-incompetent conformation (Kim et al. 2009). This effect, however, is diminished when CRMP1 harbors the mutation D408V, which enhances oligomer stability in vitro (Supplemental Fig. S5D). Previous studies indicate that flexibility of oligomers and reorganization of domains are critical for chaperone activity. Hsp26, a heat-sensitive small heat shock protein, e.g., forms nonfunctional oligomers under normal physiological conditions, but can undergo structural rearrangements at higher temperatures to facilitate the formation of functional oligomers with chaperone activity. This includes the dissociation of larger oligomers into dimers, which is required for the efficient interaction with nonnative proteins (Haslbeck et al. 2004; White et al. 2006). Furthermore, it was demonstrated that the protein Spy for high chaperone activity requires a high oligomer flexibility (Quan et al. 2014). A chaperone function of CRMP1 is also supported by our observation that it reduces the spontaneous aggregation of the disease proteins ATXN1 and TARDBP in cell-based assays (Fig. 5D,E). Thus, we suggest that CRMP1 can function under physiological conditions as a neuron-specific molecular chaperone that directly binds to unfolded, aggregation-prone proteins and prevents their abnormal deposition into insoluble inclusion bodies.
In conclusion, we propose that step-by-step filtering of protein interaction networks with differentially expressed genes is a powerful strategy to identify novel proteins that control misfolding and proteotoxicity of mutant HTT. The approach is scalable and can be expanded to include additional data types. More importantly, it can be applied to other aggregation-prone disease proteins, given that high-quality PPI and gene expression data from affected tissues of patients and healthy individuals are available. The efficiency of our filtering strategy is underscored by the discovery of the neuron-specific protein CRMP1, which functions as a potent suppressor of HTT aggregation and toxicity in various in vitro and in vivo disease model systems.
Methods
Data sets
Protein interaction data were extracted from the UniHI database (http://www.unihi.org/). For the HD network, data from a previous Y2H-screen for HTT interactors were additionally included (Goehler et al. 2004). Gene expression data sets utilized for filtering of protein interaction networks can be found in Supplemental Table S1. Manually annotated HD therapy targets (HDTTs) were obtained from the curators of the HD Research Crossroads database. This database provides the most comprehensive collection of potential therapeutic molecular targets for HD to date (Kalathur et al. 2012).
Analysis of gene expression data
For detection of differentially expressed genes in brain versus nonbrain tissues (ΔEG1), gene expression data of normal tissues were obtained from the SymAtlas database (http://biogps.org) (see also Supplemental Table S1). Statistical significance of differential expression was determined using a local pooled error approach (Jain et al. 2003). Derived P-values were corrected for multiple comparisons using the Benjamini-Hochberg procedure (Benjamini and Hochberg 1995).
For the generation of networks that are specific to the caudate nucleus expression profiles obtained from normal tissue, samples of different brain regions were compared (Hodges et al. 2006). Similarly, samples from HD patients compared to healthy individuals were used to derive disease specificity. The significance of differential gene expression was assessed using a linear model, as it allowed the correction for collection sites, gender, and age in the case of the HD study (Smyth 2004; Hodges et al. 2006). P-values were subsequently adjusted by applying the Benjamini-Hochberg procedure (Benjamini and Hochberg 1995).
Optimization of selection thresholds for the generation of the dysregulated HTT network PPI4
For optimization of thresholds P1, P2, and P3, a curated list of HDTTs was used. In this list, each HDTT is assigned with a target validation score ranging from 0 to 5 describing the status of the experimental validation of its disease-modifying effects. The highest score of 5.0 is assigned if a drug or a gene therapy targeting the gene has shown efficacy in a Phase 3 clinical trial, whereas a score of 4 states that an improvement of the HD phenotype has been observed in rodent models upon targeting a gene by drugs or genetic intervention. A score of 3 is assigned to genes and proteins where a causal relationship with HD was observed in a validated cell culture or lower organism model of HD upon genetic or pharmacologic modification. Lower scores are assigned if genes show altered functional activity in HD, changes in expression, binding to HTT, or are linked to HD-relevant biological processes. To ensure that the set of HDTTs constitutes a truly independent set, i.e., is not biased toward HTT interactors or differentially expressed genes in HD, we required that HDTTs have score values equal to or larger than 3. Thus, information about huntingtin interaction partners or gene expression changes in tissues of HD patients was not utilized for the definition of HDTTs. Altogether, a set of 476 unique HDTTs passed this requirement.
To determine an optimal set of significance thresholds, the network filtering strategy was repeatedly applied to the PPI networks. Thresholds for detection of differentially expressed genes (P1, P2, and P3) were independently varied using a range of values between 5 × 10−8 and 1 × 10−2. All possible combinations of these thresholds were evaluated, and the relative number of HDTTs as well as the total number of proteins in the final PPI4 data set was recorded after iterative network filtering. In total, 2197 ( = 133) combinations of threshold values were tested to optimize the relative percentage of included HDTTs in the final network (PPI4). We demanded that the precision or positive predictive value [PPV = (TP − 1)/(P − 1), in which TP is the number of HDTTs, and P is the total number of proteins included in the network] is larger than 0.5. The factor (−1) is to exclude HTT from the calculation. The thresholds with P1 = 10−4 for the first (ΔEG1), P2 = 10−3 for the second (ΔEG2), and P3 = 10−6 for the third filtering step (ΔEG3) were selected as optimal, since they resulted in the highest relative number of HDTTs in PPI4 with a PPV > 0.5.
Other statistical methods
Experimental data in most experiments were analyzed with two-sided t-tests; details can be found in the figure legends. Additionally, we applied linear regression followed by an F-test to analyze whether CRMP1 significantly improves motor dysfunction in HTT336Q128 transgenic flies.
Analysis of cis-regulatory sequences of CRMP1
We used the sequence NM_001313 and exon 1 to define the transcriptional start site of the gene encoding CRMP1. Subsequently the ConTra v2 software (Broos et al. 2011) was applied to identify potential heat shock factor (HSF) binding sites. Default software parameters were used for the analyses. Position weight matrices were obtained from the TRANSFAC database (version 10.4). Individual HSF binding sites are described in Supplemental Table S11.
Plasmids
The destination plasmids pGEX-6p-D21, pQLinkHD, pTLHA1-D-48, pmCherryGW, pTHW, and pTMW were utilized for the expression of recombinant proteins in E. coli, mammalian cells, and flies. cDNAs were shuttled into destination plasmids using the Gateway technology (Invitrogen). Entry plasmids harboring the genes encoding the proteins CRMP1, CRMP1-D408V, DPYSL2, DPYSL3, DPYSL4, DPYSL5, DNAJB1, HSPA1A, or luciferase were utilized. For high-content fluorescence imaging experiments, a cDNA fragment encoding HTTex1Q79 was cloned into the expression vector pdEYFP-C1Amp (Imagenes). P{EPgy2} (Kalathur et al. 2012) was used in Drosophila for overexpression of endogenous CRMP. The plasmid for expression of HTT336Q128 in Drosophila was described previously (Kaltenbach et al. 2007).
Antibodies
Polyclonal rabbit anti-CRMP1 antibodies were generated using the KLH-MBS-conjugated linear peptides 504-YEVPATPKYATPAPS-518 and 513-ATPAPSAKSSPSKHQ-527 (Eurogentec). The antibodies were affinity purified on an AF-Amino-TOYOPEARL 650M (1 mL) column, containing ACH sepharose (0.375 g) and CNBR-activated sepharose (0.375 g) by Eurogentec. HTT aggregates were detected by monitoring YFP fluorescence as well as MW8 antibody immunoreactivity. A complete list of the antibodies used can be found in Supplemental Table S12.
Silencing and overexpression of CRMP1 in PC12 cells
For CRMP1 silencing, PC12 cells (Apostol et al. 2006) were transfected (Lipofectamine 2000) with 50 pmol CRMP1-specific siRNA or 50 pmol nontargeting siRNA (On-target Plus smart pool L-090088, On-target plus siCONTROL D-001810; Dharmacon, Inc.). For CRMP1 overproduction, PC12 cells were transfected with the plasmid pTLHA1-D-CRMP1 encoding a HA-tagged CRMP1 protein and with the empty control plasmid pTLHA1-D-48. In CRMP1 knockdown and overexpression experiments, HTTQ103-EGFP expression was induced 12 h post-transfection with 5 µM ponasterone A (PonA). HTTQ103-EGFP aggregates were quantified using a filter retardation assay (Wanker et al. 1999) and a LAS-3000 imaging system (Fujifilm). Cellular toxicity was analyzed using the Apo-ONE homogenous caspase 3/7 assay (Promega).
Effect of heat stress on CRMP1 and HSPA1A protein expression in SH-SY5Y cells
SH-SY5Y cells were seeded on coverslips in 24-well cell culture plates for immunofluorescence microscopy studies (50,000 cells/well). For SDS-PAGE and immunoblotting, SH-SY5Y cells were seeded in 5-cm cell culture petri dishes (1 billion cells/well). After 24 h, cell culture media were exchanged with prewarmed media at 45°C or 37°C. After 30 min incubation at 45°C or 37°C, media were exchanged, and cells were incubated for another 4 h at 37°C. Endogenous protein levels were examined by confocal microscopy and immunoblotting. For confocal microscopy, cells were fixed in 4% paraformaldehyde, washed with PBS, permeabilized for 2 min with 0.2% Triton X-100 in PBS, washed, and blocked for 45 min in blocking buffer (1% BSA in PBS). After 60 min incubation with primary CRMP1 and HSPA1A antibodies, cells were washed with PBS before addition of secondary antibodies (goat anti-mouse or anti-rabbit coupled with Alexa Fluor 488 or Alexa Fluor 594; 1:200 in blocking buffer) for 45 min, followed by DNA staining with Hoechst 33342 and several washing steps with PBS. Images were recorded using a Leica SP2 confocal microscope. Microscope settings were carefully chosen to be able to compare the fluorescence intensities in the different samples.
In vitro HTT aggregation assays
The proteins His-CRMP1, His-CRMP1-D408V, and His-HSPA1A were expressed in E. coli BL21-Codon PlusTM RP (Stratagene) and purified to ∼95% homogeneity using Ni-NTA agarose beads. For monitoring the effects of modulator proteins on spontaneous polyQ-mediated HTT aggregation, purified GST-HTTQ49 fusion protein was centrifuged for 40 min (200,000g) at 4°C to remove potential preformed insoluble protein aggregates. Then, the fusion protein (2 µM) was incubated at 20°C (300 rpm) in aggregation buffer (50 mM Tris/HCl, pH 7.4, 150 mM NaCl and 1 mM EDTA, 1 mM DTT) with PreScission Protease (2 units/µL; GE Healthcare) as well as with potential modifier proteins (BSA, CRMP1, CRMP1-D408V or HSPA1A; 16 µM). Formation of SDS-stable HTTQ49 protein aggregates was quantified by filter retardation assays (Wanker et al. 1999). For dot-blot analysis, 3 µL (250 ng) of each aggregation reaction was filtered onto a nitrocellulose membrane (Whatman, Protran BA85 0.2 µm), and insoluble HTTQ49 aggregates retained on filter membranes were detected using the CAG53b antibody (1:5000). Soluble, HTTQ49 molecules were detected with the monoclonal antibody 3B5H10 (1:5000; Sigma). Signal intensities were quantified using the AIDA Image Analyzer v3.21 software.
High-content fluorescence microscopy
SH-EP cells were grown at 37°C and 5% CO2 in Dulbecco's modified Eagle medium (DMEM) supplemented with 4.5 g/L D-glucose, 10% fetal calf serum, penicillin (100 units/mL) and streptomycin (100 μg/mL) in 96-well cell culture plates (10,000 cells/well) and cotransfected (GeneJet, SignaGen) with an equimolar mix of cDNAs encoding YFP-HTTex1Q79 and mCherry-tagged modifiers (100 ng each). Forty-eight hours post-transfection, arrayed cells were fixed with 4% paraformaldehyde in 1× PBS, containing Hoechst 33342 adjusted to a final concentration of 1:2500. After 15 min incubation at room temperature, cells were washed twice with 1× PBS. Afterward, cells were permeabilized with 0.2% Triton X-100 for 2 min. Then, cells were blocked with 1% BSA in 1× PBS for 45 min and incubated with the MW8 antibody (1:500, diluted in 1% BSA) overnight at 4°C. After overnight incubation, cells were washed three times with 1× PBS to remove unbound primary antibody. Thereafter, cells were incubated with the goat anti-mouse Cy5-conjugated secondary antibody (Invitrogen) (1:200, diluted in 1% BSA) for 1 h at room temperature. Finally, cells were washed three times with 1× PBS and analyzed on the Cellomics instrument, named ArrayScan VTI (Thermo Scientific) using the ArrayScan VTI software. Individual SH-EP cells were identified through the fluorescence of the nucleus dye Hoechst 33342. Afterward, double-transfected cells were identified via the YFP fluorescence. Besides measuring the total YFP fluorescence of the YFP-HTTex1Q79 protein, YFP-HTTex1Q79 aggregates were quantified via the Spot YFP fluorescence method in the imaging analysis protocol. In addition, the mCherry fluorescence was measured to quantify mCherry-tagged proteins in cells. The Cy5 fluorescence of a secondary Cy5 conjugated antibody was measured to quantify YFP-HTTex1Q79 aggregates via the MW8 antibody.
Drosophila experiments
For analysis of retina degeneration, heads of 2- to 3-d-old females (10–20 heads per genotype) were fixed for 48 h (Bouin's fixative; Sigma-Aldrich), and vertical 10-µm semithin paraffin sections were analyzed to measure the absolute retina extension using light microscopy.
For detection of HTT aggregates in flies, eye/antennal imaginal discs of third instar wandering larvae were prepared in Ringer's solution and fixed in 4% formaldehyde for 30 min. Subsequently, imaginal discs were washed 5 × 5 min and 1 × 30 min with PBT. After preincubation in PBT/NGS for 1 h, primary antibodies (1-82, 7E8A10, and rabbit anti-MYC) were added (1:200) for overnight incubation. HTT aggregates were detected using the mouse monoclonal anti-HTT antibody 1-82 (Millipore). Neuronal photoreceptor precursor cells were identified using the rat monoclonal anti-elav antibody 7E8A10 (Developmental Studies Hybridoma Bank), and MYC-CRMP1 protein was recognized using a rabbit anti-MYC antibody. Then, imaginal discs were washed again in PBT and incubated for 4 h in PBT/NGS, including secondary antibodies (anti-rabbit Alexa Fluor 488, anti-mouse Alexa Fluor 555 [Invitrogen], anti-rat Cy5 [Dianova], 1:200). After washing, DNA was stained with Hoechst 33258 (Sigma-Aldrich), and HTT aggregates were determined in a field of 35.71 µm2 in the middle of the posterior third of the eye/antennal imaginal disc using a Zeiss LSM 510 META confocal microscope. One field was analyzed per individual.
For analysis of motor activity, flies with clipped wings were tested individually in a glass cylinder. Starting with 2-day-old females, the distance flies can readily climb up in a glass tube within 10 sec was quantified in triplicates every 2 d.
A detailed description of the Drosophila stocks used and genotypes is provided in the Supplemental Material and Methods.
Data access
We have established a web server (http://artemis.mdc-berlin.de/crmp1/ppi_search.php) to highlight network proteins and their direct and indirect interaction partners within interaction networks (Supplemental Tables S1–S12).
Supplementary Material
Acknowledgments
We thank M. Zenkner, A. Redel, K. Rau, S. Plaßmann, U. Stelzl, M. Lalowski, and A. Arumughan for technical assistance, data analysis, and helpful comments; R. Luthi-Carter for providing the original R script for HD microarray data analysis; and R. Hodge for critical reading of the manuscript. This work was supported by grants from DFG (SFB740, 740/2-11 and SFB618, 618/3-09), BMBF (NGFN-Plus; NeuroNet, 01GS08169-73; MooDS, 01GS08150; Mutanom, 01GS08108; GoBio), HDSA Coalition for the Cure, EU (EuroSpin, Health-F2-2009-241498 and SynSys, HEALTH-F2-2009-242167), and the Helmholtz Association (MSBN, HelMA, HA-215) to E.E.W.; by DFG grant SFB/TRR43 A7 to J.P.; and FCT grant IF/00881/2013 to M.E.F.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.182444.114.
References
- Abraham VC, Taylor DL, Haskins JR. 2004. High content screening applied to large-scale cell biology. Trends Biotechnol 22: 15–22. [DOI] [PubMed] [Google Scholar]
- Apostol BL, Illes K, Pallos J, Bodai L, Wu J, Strand A, Schweitzer ES, Olson JM, Kazantsev A, Marsh JL, et al. 2006. Mutant huntingtin alters MAPK signaling pathways in PC12 and striatal cells: ERK1/2 protects against mutant huntingtin-associated toxicity. Hum Mol Genet 15: 273–285. [DOI] [PubMed] [Google Scholar]
- Aylward EH. 2007. Change in MRI striatal volumes as a biomarker in preclinical Huntington's disease. Brain Res Bull 72: 152–158. [DOI] [PubMed] [Google Scholar]
- Behrends C, Langer CA, Boteva R, Böttcher UM, Stemp MJ, Schaffar G, Rao BV, Giese A, Kretzschmar H, Siegers K, et al. 2006. Chaperonin TRiC promotes the assembly of polyQ expansion proteins into nontoxic oligomers. Mol Cell 23: 887–897. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Methodol 57: 289–300. [Google Scholar]
- Bretin S, Reibel S, Charrier E, Maus-Moatti M, Auvergnon N, Thevenoux A, Glowinski J, Rogemond V, Prémont J, Honnorat J, et al. 2005. Differential expression of CRMP1, CRMP2A, CRMP2B, and CRMP5 in axons or dendrites of distinct neurons in the mouse brain. J Comp Neurol 486: 1–17. [DOI] [PubMed] [Google Scholar]
- Broos S, Hulpiau P, Galle J, Hooghe B, Van Roy F, De Bleser P. 2011. ConTra v2: a tool to identify transcription factor binding sites across species, update 2011. Nucleic Acids Res 39 (Web Server issue): W74–W78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviston JP, Holzbaur EL. 2009. Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol 19: 147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JH. 2000. Transcriptional dysregulation in Huntington's disease. Trends Neurosci 23: 387–392. [DOI] [PubMed] [Google Scholar]
- Chan FTS, Pinotsi D, Kaminski Schierle GS, Kaminski CF. 2014. Structure-specific intrinsic fluorescence of protein amyloids used to study their kinetics of aggregation. In Bio-nanoimaging: protein misfolding and aggregation (ed. Uversky V, Lyubchenko Y), pp. 147–155 Elsevier, Amsterdam. [Google Scholar]
- Charrier E, Reibel S, Rogemond V, Aguera M, Thomasset N, Honnorat J. 2003. Collapsin response mediator proteins (CRMPs): involvement in nervous system development and adult neurodegenerative disorders. Mol Neurobiol 28: 51–64. [DOI] [PubMed] [Google Scholar]
- Chaurasia G, Malhotra S, Russ J, Schnoegl S, Hänig C, Wanker EE, Futschik ME. 2009. UniHI 4: new tools for query, analysis and visualization of the human protein–protein interactome. Nucleic Acids Res 37 (Database issue): D657–D660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang HY, Lee E, Liu YT, Lee D, Ideker T. 2007. Network-based classification of breast cancer metastasis. Mol Syst Biol 3: 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, Brundin P. 2003. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40: 427–446. [DOI] [PubMed] [Google Scholar]
- Deo RC, Schmidt EF, Elhabazi A, Togashi H, Burley SK, Strittmatter SM. 2004. Structural bases for CRMP function in plexin-dependent semaphorin3A signaling. EMBO J 23: 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, Martin E, Vonsattel JP, Carraway R, Reeves SA, et al. 1995. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron 14: 1075–1081. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. 1997. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277: 1990–1993. [DOI] [PubMed] [Google Scholar]
- Fossale E, Seong IS, Coser KR, Shioda T, Kohane IS, Wheeler VC, Gusella JF, MacDonald ME, Lee JM. 2011. Differential effects of the Huntington's disease CAG mutation in striatum and cerebellum are quantitative not qualitative. Hum Mol Genet 20: 4258–4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehler H, Lalowski M, Stelzl U, Waelter S, Stroedicke M, Worm U, Droege A, Lindenberg KS, Knoblich M, Haenig C, et al. 2004. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington's disease. Mol Cell 15: 853–865. [DOI] [PubMed] [Google Scholar]
- Goshima Y, Nakamura F, Strittmatter P, Strittmatter SM. 1995. Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature 376: 509–514. [DOI] [PubMed] [Google Scholar]
- Hageman J, Rujano MA, van Waarde MA, Kakkar V, Dirks RP, Govorukhina N, Oosterveld-Hut HM, Lubsen NH, Kampinga HH. 2010. A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol Cell 37: 355–369. [DOI] [PubMed] [Google Scholar]
- Hands SL, Wyttenbach A. 2010. Neurotoxic protein oligomerisation associated with polyglutamine diseases. Acta Neuropathol 120: 419–437. [DOI] [PubMed] [Google Scholar]
- Harjes P, Wanker EE. 2003. The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci 28: 425–433. [DOI] [PubMed] [Google Scholar]
- Harper PS. 1996. New genes for old diseases: the molecular basis of myotonic dystrophy and Huntington's disease. The Lumleian Lecture 1995. J R Coll Physicians Lond 30: 221–231. [PMC free article] [PubMed] [Google Scholar]
- Haslbeck M, Ignatiou A, Saibil H, Helmich S, Frenzl E, Stromer T, Buchner J. 2004. A domain in the N-terminal part of Hsp26 is essential for chaperone function and oligomerization. J Mol Biol 343: 445–455. [DOI] [PubMed] [Google Scholar]
- Haslbeck M, Franzmann T, Weinfurtner D, Buchner J. 2005. Some like it hot: the structure and function of small heat-shock proteins. Nat Struct Mol Biol 12: 842–846. [DOI] [PubMed] [Google Scholar]
- Hipp MS, Park SH, Hartl FU. 2014. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol 24: 506–514. [DOI] [PubMed] [Google Scholar]
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, et al. 2006. Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet 15: 965–977. [DOI] [PubMed] [Google Scholar]
- Jain N, Thatte J, Braciale T, Ley K, O'Connell M, Lee JK. 2003. Local-pooled-error test for identifying differentially expressed genes with a small number of replicated microarrays. Bioinformatics 19: 1945–1951. [DOI] [PubMed] [Google Scholar]
- Kalathur RK, Hernández-Prieto MA, Futschik ME. 2012. Huntington's disease and its therapeutic target genes: a global functional profile based on the HD Research Crossroads database. BMC Neurol 12: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltenbach LS, Romero E, Becklin RR, Chettier R, Bell R, Phansalkar A, Strand A, Torcassi C, Savage J, Hurlburt A, et al. 2007. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet 3: e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MW, Chelliah Y, Kim SW, Otwinowski Z, Bezprozvanny I. 2009. Secondary structure of Huntingtin amino-terminal region. Structure 17: 1205–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Ou S, Patterson PH. 2001. New anti-huntingtin monoclonal antibodies: implications for huntingtin conformation and its binding proteins. Brain Res Bull 56: 319–329. [DOI] [PubMed] [Google Scholar]
- Kurnellas MP, Li H, Jain MR, Giraud SN, Nicot AB, Ratnayake A, Heary RF, Elkabes S. 2010. Reduced expression of plasma membrane calcium ATPase 2 and collapsin response mediator protein 1 promotes death of spinal cord neurons. Cell Death Differ 17: 1501–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J, Cunliffe H, Weiss A, Katsyuba E, Sathasivam K, Seredenina T, Woodman B, Moussaoui S, Frentzel S, Luthi-Carter R, et al. 2011. Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J Clin Invest 121: 3306–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, et al. 1996. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87: 493–506. [DOI] [PubMed] [Google Scholar]
- Meriin AB, Mabuchi K, Gabai VL, Yaglom JA, Kazantsev A, Sherman MY. 2001. Intracellular aggregation of polypeptides with expanded polyglutamine domain is stimulated by stress-activated kinase MEKK1. J Cell Biol 153: 851–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J, Arrasate M, Brooks E, Libeu CP, Legleiter J, Hatters D, Curtis J, Cheung K, Krishnan P, Mitra S, et al. 2011. Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat Chem Biol 7: 925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto RI. 2008. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev 22: 1427–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley JF, Brignull HR, Weyers JJ, Morimoto RI. 2002. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci 99: 10417–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchowski PJ, Schaffar G, Sittler A, Wanker EE, Hayer-Hartl MK, Hartl FU. 2000. Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc Natl Acad Sci 97: 7841–7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. 2006. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314: 130–133. [DOI] [PubMed] [Google Scholar]
- Paulsen JS, Smith MM, Long JD, PREDICT HD investigators and coordinators of the Huntington Study Group. 2013. Cognitive decline in prodromal Huntington Disease: implications for clinical trials. J Neurol Neurosurg Psychiatry 84: 1233–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechmann S, Levy ED, Tartaglia GG, Vendruscolo M. 2009. Physicochemical principles that regulate the competition between functional and dysfunctional association of proteins. Proc Natl Acad Sci 106: 10159–10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perutz MF, Johnson T, Suzuki M, Finch JT. 1994. Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci 91: 5355–5358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi ML, Tagawa K, Enokido Y, Yoshimura N, Wada Y, Watase K, Ishiura S, Kanazawa I, Botas J, Saitoe M, et al. 2007. Proteome analysis of soluble nuclear proteins reveals that HMGB1/2 suppress genotoxic stress in polyglutamine diseases. Nat Cell Biol 9: 402–414. [DOI] [PubMed] [Google Scholar]
- Quan S, Wang L, Petrotchenko EV, Makepeace KA, Horowitz S, Yang J, Zhang Y, Borchers CH, Bardwell JC. 2014. Super Spy variants implicate flexibility in chaperone action. eLife 3: e01584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawls JM Jr. 2006. Analysis of pyrimidine catabolism in Drosophila melanogaster using epistatic interactions with mutations of pyrimidine biosynthesis and β-alanine metabolism. Genetics 172: 1665–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson AL, Headey SJ, Saunders HM, Ecroyd H, Scanlon MJ, Carver JA, Bottomley SP. 2010. Small heat-shock proteins interact with a flanking domain to suppress polyglutamine aggregation. Proc Natl Acad Sci 107: 10424–10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Thompson LM. 2006. Transcription meets metabolism in neurodegeneration. Nat Med 12: 1239–1241. [DOI] [PubMed] [Google Scholar]
- Saunders HM, Bottomley SP. 2009. Multi-domain misfolding: understanding the aggregation pathway of polyglutamine proteins. Protein Eng Des Sel 22: 447–451. [DOI] [PubMed] [Google Scholar]
- Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. 1997. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90: 549–558. [DOI] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, et al. 2003. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet 12: 1555–1567. [DOI] [PubMed] [Google Scholar]
- Smyth GK. 2004. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article3. [DOI] [PubMed] [Google Scholar]
- Su KY, Chien WL, Fu WM, Yu IS, Huang HP, Huang PH, Lin SR, Shih JY, Lin YL, Hsueh YP, et al. 2007. Mice deficient in collapsin response mediator protein-1 exhibit impaired long-term potentiation and impaired spatial learning and memory. J Neurosci 27: 2513–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam S, Spiess C, Auyeung W, Joachimiak L, Chen B, Poirier MA, Frydman J. 2009. The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat Struct Mol Biol 16: 1279–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur AK, Jayaraman M, Mishra R, Thakur M, Chellgren VM, Byeon IJ, Anjum DH, Kodali R, Creamer TP, Conway JF, et al. 2009. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat Struct Mol Biol 16: 380–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truant R, Atwal RS, Burtnik A. 2007. Nucleocytoplasmic trafficking and transcription effects of huntingtin in Huntington's disease. Prog Neurobiol 83: 211–227. [DOI] [PubMed] [Google Scholar]
- Vavouri T, Semple JI, Garcia-Verdugo R, Lehner B. 2009. Intrinsic protein disorder and interaction promiscuity are widely associated with dosage sensitivity. Cell 138: 198–208. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr. 1985. Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 44: 559–577. [DOI] [PubMed] [Google Scholar]
- Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. 2001. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell 12: 1393–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanker EE, Scherzinger E, Heiser V, Sittler A, Eickhoff H, Lehrach H. 1999. Membrane filter assay for detection of amyloid-like polyglutamine-containing protein aggregates. Methods Enzymol 309: 375–386. [DOI] [PubMed] [Google Scholar]
- White HE, Orlova EV, Chen S, Wang L, Ignatiou A, Gowen B, Stromer T, Franzmann TM, Haslbeck M, Buchner J, et al. 2006. Multiple distinct assemblies reveal conformational flexibility in the small heat shock protein Hsp26. Structure 14: 1197–1204. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R. 2000. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell 101: 57–66. [DOI] [PubMed] [Google Scholar]
- Zhao J, Yang TH, Huang Y, Holme P. 2011. Ranking candidate disease genes from gene expression and protein interaction: a Katz-centrality based approach. PLoS One 6: e24306. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.