

Abstract
Blocking the immunoinhibitory PD-1:PD-L1 pathway using monoclonal antibodies has led to dramatic clinical responses by reversing tumor immune evasion and provoking robust and durable antitumor responses. Anti-PD-1 antibodies have now been approved for the treatment of melanoma, and are being clinically tested in a number of other tumor types as both a monotherapy and as part of combination regimens. Here, we report the development of DNA aptamers as synthetic, nonimmunogenic antibody mimics, which bind specifically to the murine extracellular domain of PD-1 and block the PD-1:PD-L1 interaction. One such aptamer, MP7, functionally inhibits the PD-L1-mediated suppression of IL-2 secretion in primary T-cells. A PEGylated form of MP7 retains the ability to block the PD-1:PD-L1 interaction, and significantly suppresses the growth of PD-L1+ colon carcinoma cells in vivo with a potency equivalent to an antagonistic anti-PD-1 antibody. Importantly, the anti-PD-1 DNA aptamer treatment was not associated with off-target TLR-9-related immune responses. Due to the inherent advantages of aptamers including their lack of immunogenicity, low cost, long shelf life, and ease of synthesis, PD-1 antagonistic aptamers may represent an attractive alternative over antibody-based anti PD-1 therapeutics.
Keywords: cancer, DNA aptamer, immunotherapy, PD-1
Introduction
Cancer cells evade immune surveillance through multiple mechanisms including increased secretion of immunosuppressive cytokines (i.e., interleukin (IL)-10 and tumor growth factor (TGF)-β), reduced expression of major histocompatibility antigens on their cell surface, the enhanced differentiation of immune effector cells to a regulatory phenotype, as well as an influx of myeloid-derived suppressor cells and tumor associated macrophages.1 Importantly, tumors also limit immune responses by activating immune inhibitory checkpoint pathways by the cell surface expression of coinhibitory ligands (i.e., PD-L1, B7-H4, VISTA) within the tumor microenviroment which bind to paired coinhibitory receptors on infiltrating effector T-cells and limit antitumor immune responses.2 The combined action of all these mechanisms results in the creation of an immunosuppressive tumor microenvironment that explains in part the lack of clinical effectiveness of present-day cancer vaccines aimed at generating tumor antigen-specific T-cell responses.3
One key immune inhibitory pathway involves PD-1 interacting with either PD-L1 or PD-L2 (refs. 4,5). While PD-L2 expression is limited to cells of the hematopoietic lineage,5 PD-L1 is inducibly expressed on most tissues and is a key player in maintaining peripheral tolerance.4,6 In cancer, PD-L1 is often adaptively expressed by cells derived from many tumor types including melanoma, lung, ovarian, and colon in response to IFNγ.7 The engagement of PD-1 on T-cells by its ligands present on either tumor cells, cells of the tumor microenvironment, or antigen presenting cells, signals for the recruitment of SHP-2 to the phosphorylated tyrosine of ITIM and ITSM motifs within the PD-1 cytoplasmic tail.8 The recruited SHP2 functions to suppress T-cell receptor signaling thereby inducing anergy, exhaustion, or apoptosis of tumor-specific T-cells.8,9 Supportingly, PD-1 signaling induces IL-10 secretion by purified T-cells undergoing activation in vitro, while decreasing IL-2 and IFNγ secretion.4,10 Moreover, this signaling pathway has been shown to be bidirectional in nature, as PD-L1 signaling within tumor cells delivers an antiapoptotic signal to resist cytotoxic lysis by tumor-infiltrating CTLs.11
PD-1:PD-L1 blockade by monoclonal antibodies has proven to be an effective modality to overcome immune evasion and mobilize specific antitumor responses in mouse models and clinically in patients.12,13 For instance, 38% of 135 advanced melanoma patients treated with the anti-PD-1 antibody lambrolizumab (Keytruda, MK-3475) displayed objective response outcomes.14 Furthermore, 15–25% objective response has been reported in an anti-PD-1 monotherapy phase trial in non–small-cell lung cancer patients, a tumor type previously thought to be unresponsive to immune therapy.15 Currently, over 50 clinical trials are evaluating anti-PD-1 antibodies as a single agent, or in a combination setting, across greater than 25 tumor types.
However, monoclonal antibody therapy inherently carries a number of disadvantages such as their immunogenicity (following repeated administration) and high production costs which may limit their use and broad availability to patients.16,17,18 In contrast, short single-stranded synthetic oligonucleotides termed aptamers, can be rapidly identified to bind molecular targets such as PD-1 with affinity and specificity features rivaling that of antibodies.19,20,21 Importantly, DNA aptamers have been shown to lack immunogenicity, and are synthesized using scalable, low-cost solid-phase chemistry, avoiding the requirement for biological cultivation, protein expression, and the subsequent purification which is associated with antibody production.19,22,23,24 Thus, anti-PD-1 DNA aptamers may represent novel therapeutic entities with improved characteristics relative to anti-PD-1 monoclonal antibodies. In this study, we report as a proof of principle the development of DNA aptamers which bind specifically to the extracellular domain of murine PD-1 with nanomolar affinity. One of them, MP7, functionally blocks PD-1 from interacting with PD-L1, and its PEGylated form suppresses the tumor growth of a PD-L1 positive murine colon carcinoma in vivo.
Results
Identification of murine PD-1-binding DNA aptamers
In vitro-based SELEX was performed to identify DNA aptamers which specifically recognize the extracellular domain of murine PD-1 (mPD-1) by screening a random DNA library against a recombinant chimera consisting of the mPD-1 extracellular domain fused to a histidine-tagged human Fc domain (hereafter referred to as mPD-1.FcHIS). Five rounds of enrichment were performed consisting of a selection step towards mPD-1.FcHIS and a counter selection step toward an irrelevant isotype matched hIgG. Next-generation sequencing (NGS) was then performed on the enriched sublibrary of DNA oligonucleotides, revealing a significant enrichment of aptamer families harboring sequences with high sequence identity to a parental highly enriched sequence. Specifically, the two highest enriched sequence families called MP5 and MP7 (Table 1) were observed to be present at 11.7 and 13% respectively of the total sequences before a substantial decrease in enrichment (<1%) was observed for another family MP23. A pull down experiment was performed where biotinylated versions of these aptamers immobilized onto magnetic streptavidin beads was used to immunoprecipitate mPD-1.FcHIS, confirming that these sequences were enriched from the bulk library during selection due to their ability to bind mPD-1.FcHIS (Supplementary Figure S1).
Table 1. Sequence enrichment of anti-PD-1 DNA aptamers from SELEX.
Anti-PD-1 DNA aptamers MP5 and MP7 bind to mPD-1.FcHIS with nanomolar affinity
The highest enriched parental sequences MP5 and MP7 were chosen for further evaluation. Structural predictions performed on these aptamer sequences illustrate a complex hairpin-bulge folding state (Figure 1a,b). Neither of these sequences contain a high prevalence of GG repeats and are not predicted to fold into a G-quadruplex structure. Nitrocellulose filter binding assay demonstrates that MP5 and MP7 bind to mPD-1.FcHIS in solution with dissociation constants (KD) of 112 and 167 nmol/l respectively (Figure 1c,d). In contrast, the reverse complement of these aptamers (Rev-MP5 and Rev-MP7) and a negative control sequence (cSeq: CCTGTGTGAGCCTCCTAACCAGAACAGTTAAAGTGTCCCACTCCCATGCTT ATTCTTGTCTCTC) did not bind mPD-1.FcHIS to a comparable extent.
Figure 1.

Secondary structure and binding affinities of highly enriched anti-PD-1 aptamer sequences MP5 and MP7. Mfold40 secondary structure predictions of the top two enriched aptamer sequences MP5 (a) and MP7 (b) identified by next-generation sequencing after five rounds of SELEX toward mPD-1.FcHIS. (c,d) Concentration-dependent binding of 32P-labeled aptamers MP5 (c) and MP7 (d) to mPD-1.FcHIS captured in a nitrocellulose filter binding assay. Filled symbols represent the % DNA bound of the full-length aptamer at each mPD-1.FcHIS concentration while empty symbols represent the % DNA Bound of the respective reverse complement of each aptamer used as a negative binding control.
Anti-PD-1 DNA aptamers bind specifically to the extracellular region of murine PD-1
Mouse and human isoforms of PD-1 are highly homologous with 67% amino acid identity within their extracellular binding domain. Similarly, the IgV domains of these molecules share significant structural homology with each other and other members of the IgV superfamily (Figure 2a,b).25 Furthermore, it has been demonstrated that cross-species binding between mPD-1 and hPD-L1, or hPD-1 and mPD-L1 occurs with comparable affinity as the same species interaction.26 Yet, surface plasmon resonance (SPR)-based binding studies performed with aptamers MP5 and MP7 immobilized onto a solid sensor chip confirmed that both aptamers bound with specificity towards mPD-1.FcHIS with no detectable binding to hPD-1.Fc injected at the same concentration (50 nmol/l) (Figure 2c). Importantly, these aptamers did not bind to an isotype-matched hIgG (500 nmol/l), the IgV superfamily member mPD-L1.Fc (50 nmol/l) nor to the IgV-related domain of CEA-N.HIS (500 nmol/l) confirming that both selected aptamers bind specifically to the extracellular region of mPD-1 as opposed to binding the histidine tag, the Fc region, or to structurally-related IgV domains (Figure 2c). Additionally, fluorescently labeled versions of MP5 and MP7 as well as two anti-PD-1 monoclonal antibodies, but not cSeq or isotype control antibodies, bound to PD-1 expressed on the surface of P815 mouse mastocytoma cells (Supplementary Figure S2). Apparent avidity constants for the aptamer-mPD-1 interaction were determined by SPR to be <1 nmol/l for both MP5 and MP7 as they reflect the nonphysiological, bivalent binding of the mPD-1.FcHIS chimera to the aptamers immobilized on the solid surface (data not shown).
Figure 2.
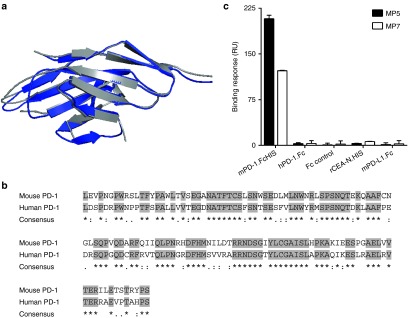
Specificity of anti-PD-1 aptamers MP5 and MP7 towards the mPD-1 extracellular domain. (a) Ribbon structure overlays highlighting similarities between the mouse (blue) and human (gray) PD-1 extracellular IgV-like domain. (b) Aligned primary sequences emphasizing homology between the extracellular regions of mouse (PDB ID: 1NPU) and human (PDB ID: 3RRQ) PD-1 (refs. 41,42). Gray highlights represent conserved primary sequences. (c) Surface plasmon resonance binding responses of mPD-1.FcHIS, hPD-1.Fc, an irrelevant Fc control, histidine-tagged rCEA-N domain, and mPD-L1.Fc toward immobilized MP5 (filled bars) or MP7 (empty bars). Histograms represent mean binding response of duplicate injections ± SD.
Aptamer MP7 functionally antagonizes PD-1/PD-L1-mediated immunosuppression
An IL-2-based enzyme-linked immunospot (ELISPOT) assay was performed to determine if aptamers MP5 or MP7 functionally antagonize PD-1/PD-L1 signaling in primary cells. In this assay, PD-L1.Fc, but not an isotype matched hIgG Fc control significantly suppresses IL-2 secretion by primary murine T-cells stimulated with a polyclonal TCR-signal from plate bound anti-CD3 antibody (Figure 3a). Addition of aptamer MP7 (250 nmol/l) or a known blocking anti-PD-1 antibody (RMPI-14 mAb) to the culture, but not a control aptamer sequence (cSeq) nor MP5, significantly restored IL-2 secretion in the presence of PD-L1.Fc (Figure 3b). Importantly, addition of MP7 to splenocytes stimulated with anti-CD3 in the absence of PD-L1 did not increase the amount of IL-2 secretion above that of anti-CD3 stimulus alone (Supplementary Figure S3) suggesting that the increase in IL-2 in the presence of PD-L1 is due to the specific blocking of the PD-1:PD-L1 inhibitory signal. Finally, the ability of aptamer MP7, but not MP5, to restore PD-L1 suppressed proliferation of anti-CD3 stimulated lymphocytes was confirmed using a CFSE dilution assay (Supplementary Figure S4).
Figure 3.

Anti-PD-1 DNA aptamer MP7 antagonizes PD-1/PD-L1 mediated suppression of IL-2 secretion in vitro. (a) IL-2 ELISPOT assay to compare the effects of PD-L1.Fc or an isotype matched control Fc on the IL-2 secretion by splenocytes stimulated with anti-CD3 antibody. PD-L1.Fc (15 µg/ml) reduced IL-2 SFU by 51% as compared to the Fc control. Each bar represents the mean SFU/2 × 105 cells from at least three replicate wells ± SEM. **P < 0.05 relative to anti-CD3 + Fc-control group. (b) Phosphate-buffered saline, cSeq, anti-PD-1 aptamers (250 nmol/l) or an antagonistic anti-PD-1 antibody (RMPI-14 mAb, 125 nmol/l) were added to splenocytes in wells coated with anti-CD3 + PD-L1.Fc to monitor the blockage of PD-L1-mediated suppression of IL-2 secretion. Bars represent % Blockage of PD-L1 suppressed IL-2 Secretion where the IL-2 spot forming units (SFU) in wells without aptamer/antibody are set to 0% and IL-2 SFU in wells supplemented with anti-PD-1 blocking antibody are set to 100% as anti-PD-1 antibody restored IL-2 secretion to levels 20% above that of anti-CD3 stimulation alone. Each histogram bar represents the mean value ± SEM (n = 5). NS, not significant.
PEGylated MP7 directly blocks PD-1 binding to PD-L1
Unmodified DNA aptamers exhibit short in vivo half-lives (<1 hour) owing to the rapid renal filtration of such relatively small molecules (~8–25 kDa).19 It has been demonstrated that conjugation of aptamers to high molecular weight polyethylene glycol (PEG) can limit rate of filtration and extend half-life up to 24–48 hours.27,28 Thus, control and antagonistic anti-PD-1 DNA aptamers were PEGylated prior to evaluating their in vivo antitumor protective properties. Specifically, aptamers MP5, MP7, and cSeq were modified at their 5' termini with a 40 kDa PEG and the conjugated aptamers recovered by RP-HPLC (Figure 4a and Supplementary Figure S5).
Figure 4.

PEGylated MP7 directly blocks PD-1/PD-L1 binding. (a) Reaction scheme of aptamer conjugation to a 40 kDa polyethylene glycol (PEG) at the 5' termini. (b) The ability of PEGylated anti-PD-1 aptamers (PEG-MP5, PEG-MP7), PEG-cSeq, RPMI-14 mAb, or an isotype matched IgG control to inhibit the binding of soluble mPD-1.FcHIS to plate bound mPD-L1.Fc was determined using a competitive enzyme-linked immunosorbent assay utilizing an anti-HIS-HRP conjugated antibody. Each histogram bar represents the mean % of PD-1 blocked from binding PD-L1 ± SD (n = 3).
The ability of the PEGylated and non-PEGylated aptamers to directly block the binding of PD-1 with PD-L1 was assessed using a competitive enzyme-linked immunosorbent assay (ELISA)-based assay where the binding of soluble mPD-1.FcHIS to immobilized mPD-L1.Fc is inhibited by the addition of aptamer. Consistent with the IL-2 ELISPOT experiments, both PEG-MP7 and RMPI-14 mAb were able to significantly block >75% of PD-1/PD-L1 binding in this assay confirming that aptamer MP7 functions as a PD-1 antagonist (Figure 4b). In contrast, neither an isotype matched antibody nor PEG-MP5 inhibited PD-1 binding to PD-L1 while the cSeq weakly blocks ~20% of the interaction, a value that is not statistically significant in comparison to wells where no aptamer was added. Notably, PEG-MP7 blocked the PD-1/PD-L1 interaction in this assay to a comparable extent as unmodified MP7 (80 versus 94%, Supplementary Figure S6) indicating that PEGylation does not overtly alter the structure or inhibit its antagonistic function in vitro.
PEGylated anti-PD-1 aptamer suppresses growth of disseminated MC38.CEA colon carcinoma cells
Release of PD-1:PD-L1 immunosuppression by anti-PD-1 or PD-L1 antibodies has been shown to provoke remarkable antitumor responses to slow growth of tumors in both animal models and in human clinical trials. To evaluate the ability of our anti-PD-1 aptamers to similarly promote in vivo antitumor responses, we used a tumor mouse model where murine colon carcinoma MC38 cells stably expressing human CEA (MC38.CEA) as a heterologous antigen were injected intraperitoneally to wild-type C57Bl/6 mice. Consistent with previous studies using MC38 cells,29 we found that MC38.CEA cells express low basal levels of PD-L1, which is upregulated 10-fold by stimulation with IFNγ (Figure 5a). After implantation, the mice were treated with the PEGylated aptamers MP7 (n = 5), cSeq (n = 4), and as a positive control the RMPI-14 mAb (n = 5) or an isotype matched irrelevant IgG (n = 5) (Figure 5b). The monotherapy PD-1 blockade using either the mAb or aptamer MP7 significantly suppressed tumor burden as measured by the number of peritoneal nodules formed (Figure 5c,d) or the total cumulative volume of all tumors within each animal (Figure 5c,e). Impressively, animals treated with PEG-MP7 (on average 0.6 nodules/animal with 46 mm2 cumulative volume/animal) displayed an equivalent or higher antitumor efficacy as the mAb (3.2 nodules/animal, 210 mm2 cumulative volume). As expected, the injection of an irrelevant PEGylated oligonucleotide sequence (PEG-cSeq) did not alter tumor progression. Notably, we did not observe any overt toxicity upon aptamer treatment such as splenomegaly or organ hyperplasia in the liver or lymphoid organs. Furthermore, in a similar but more aggressive tumor model where animals were fed a higher fat diet reaching an endpoint within 14 days, PEG-MP7 significantly suppressed tumor growth when compared to animals receiving buffer alone (phosphate-buffered saline (PBS)) or an anti-adhesive PEGylated DNA aptamer specific to carcinoembryonic antigen (CEA)30 (PEG-N54; a DNA aptamer shown to block CEA-mediated, MC38.CEA implantation in the peritoneal cavity) (Supplementary Figure S7).
Figure 5.

PEGylated MP7 suppresses growth of disseminated PD-L1+ colon carcinoma MC38.CEA cells in vivo. (a) Flow cytometry analysis of MC38.CEA cells costained with FITC-conjugated anti-CEA antibody and APC-conjugated anti-PD-L1 antibody confirming low basal PD-L1 expression which is upregulated by IFNγ stimulation (500 units/ml, 24 hours). (b) Experimental outline of in vivo tumor model. MC38.CEA cells were injected i.p. into groups of C57Bl/6 mice (n = 5; day 0) and animals were subsequently treated with either RMPI-14 mAb, an isotype matched IgG (Isotype), PEG-MP7 or PEG-cSeq on days 1,3,5, and 7. (c) Photographs highlighting tumor nodules found in the intraperitoneal cavity of treated or control mice upon necropsy at day-21 post-tumor implantation. The total number of tumor nodules in each animal (d) and their cumulative tumor volume (e) was quantified upon necropsy to evaluate the antitumor effect of each PD-1:PD-L1 blocking reagent.
PEGylated MP7 is not cytotoxic and does not trigger TLR9-innate immune signaling
Experiments were carried out to confirm that the reduction in tumor growth in animals treated with PEG-MP7 is due to its PD-1 antagonistic activity and not as a result of off-target PD-1 independent effects. First, we confirmed that aptamer MP7 was not cytotoxic toward MC38.CEA cells grown in-vitro, excluding the possibility that reduction in tumor growth is a result of off-target aptamer-induced cytotoxicity of tumor cells (Figure 6a). Second, oligodeoxynucleotides (ODN) containing CpG motifs have been shown to promote Th1 type immune responses by triggering innate immune signaling through TLR9 expressed on human plasmacytoid DC and B cells.31,32 To test whether MP7, which contains several CG repeats, can trigger TLR9 innate immune signaling, PEG-MP7 or a known TLR-9 agonistic CpG ODN were injected in the peritoneal cavity of naive mice at a dosage which mirrored the amount used in our tumor model and the serum levels TNFα and IL-6 were quantified by ELISA 3 hours later. The CpG ODN significantly increased serum levels of TNFα and IL-6, meanwhile PEG-MP7 did not induce cytokine production to levels that are detectable by these assays, excluding the possibility of this aptamer behaving as a strong TLR9 agonist (Figure 6b,c). Furthermore, real-time polymerase chain reaction performed on cultured RAW264.7 mouse macrophages treated with PEG-MP7 or a CpG ODN confirmed that PEG-MP7 does not induce IFNα or IL-12 transcription in vitro relative to a specific CpG ODN (Supplementary Figure S7). Taken together, our data suggest that that the anti-PD-1 DNA aptamer PEG-MP7 functions in vivo as a specific PD-1 antagonist to promote strong antitumor immune responses.
Figure 6.

Anti-PD-1 aptamer PEG-MP7 is not cytotoxic and enhances tumor-specific T-cell responses in vivo without induction of a TLR9-mediated innate immune response. (a) Cell viability assay was quantified using (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) dye after a 24-hour incubation of MC38.CEA cells with 5 µmol/l of PEG-MP7, PEG-cSeq, MP7, or 8M urea. Bars represent the mean OD490 nm ± SD from replicate wells. ***P < 0.001. Aptamer-induced TLR-9 signaling was assayed by injection of PEG-cSeq, PEG-MP7, PBS, or a control CpG ODN into naive C57Bl/6 mice (n = 3) and 3 hours later the sera levels of TNFα (b) and IL-6 (c) were quantified by ELISA. Only injection of the CpG ODN yielded signal above the lower limit of detection (dotted line). Each symbol represents the average cytokine levels from each individual animal measured in duplicate.
Discussion
Immune responses are heavily regulated at the cellular level by a number of ligand: receptor interactions which transduce either proinflammatory costimulatory signals or negative coinhibitory signals.2 These orchestrated signals finely balance the immune system by provoking appropriate responses to danger signals, while the coinhibitory ligands, otherwise known as immunological checkpoints, are key in promoting self-tolerance and preventing autoimmunity2,6. However, malignant cancer cells often usurp these checkpoint pathways to limit antitumor immune responses and evade immunological deletion.1,2 Thus, checkpoint blockade by monoclonal antibodies has become a viable therapeutic intervention functioning to prevent evasion and provoke strong and durable antitumor immune responses. The first in-class therapeutic used in this manner was an anti-CTLA-4 monoclonal antibody which functions by antagonizing the T-cell inhibitory signals delivered through the CTLA-4:B7-1/B7-2 pathway resulting in increased activation of CD4+ and CD8+ effector cells.13 More recently, antagonistic anti-PD-1 monoclonal antibodies have shown potent antitumor effect with objective response rates of 38% in 135 patients tested with advanced melanoma.14 Supportingly, another trial using a distinct anti-PD-1 antibody showed objective responses in 28% (26 of 94) of advanced melanoma patients.15 Collectively, the preclinical and clinical data validate that therapeutic intervention to block to PD-1 inhibitory signal delivered by either PD-L1+ tumor cells, antigen presenting cells, or cells within the tumor microenvironment results in mobilization of powerful antitumor immune responses capable of eradicating malignant disease.
To date, all biotherapeutics directed at blocking the PD-1:PD-L1 pathway are protein-based agents including monoclonal antibodies as well as a PD-L2 Fc-fusion chimera. These reagents are being clinically validated in over 50 trials, across several cancers, as monotherapies or in combination with other therapeutic reagents or interventions. However, as these protein-based reagents become widely used, there are key limitations which may impact their effectiveness and use. First, the repeated use of monoclonal antibodies (including humanized and fully human antibodies) can still engender humoral immune responses which may become problematic and limit therapeutic efficacy.17,18 Another important limitation which will be realized as these reagents are broadly used—in some cases as a front line therapy—is the cost associated with manufacturing protein biologics for human use.16,17,19 Consequently, there remains a need for novel, preferably synthetic agents which target immune inhibitory pathways while being cost-effective and nonimmunogenic. In the present study, we report for the first time the development of short synthetic anti-PD-1 DNA aptamers. In contrast to antibodies, aptamers have been shown to be nonimmunogenic, are readily synthesized in a scalable low-cost process with minimal batch to batch variation, and are reversibly denatured with long shelf-life, all properties which inherently make DNA aptamers attractive alternatives as biotherapeutics.19
As a proof-of principle, we have identified DNA aptamers using traditional in vitro SELEX that are specific to the murine extracellular domain of PD-1. By coupling our SELEX searches with NGS technology, we have identified a number of highly enriched sequence families (Table 1), which led to the identification of high affinity PD-1 binding ligands after five rounds of selection. The two most highly enriched aptamers, termed MP5 and MP7, were synthesized and found to bind to mPD-1.FcHIS with high affinity, and high specificity, with no detectable binding observed to homologous proteins including human PD-1 (Figures 1 and 2). In contrast to aptamer MP5 which did not block PD-L1 induced signal despite its ability to bind PD-1, aptamer MP7 was found to inhibit PD-L1 mediated immunosuppression (i.e., suppression of IL2 secretion in response to polyclonal anti-CD3 stimulus) on primary T-cells (Figure 3), and was also able to antagonize direct PD-1/PD-L1 binding in a competitive ELISA (Figure 4 and Supplementary Figure S5). A major disadvantage of aptamers as therapeutic entities is their poor pharmacokinetic profiles, as these short DNA strands are rapidly removed from circulation due to renal filtration.19 However, the conjugation of aptamers to high molecular weight polymers such as polyethylene glycol (PEG) has been reported to dramatically increase their half-life up to 24–48 hours in some cases. Thus, a 40 kDa PEG moiety was attached to the 5' amino end of MP7, the most effective PD-1 antagonistic aptamer in our in vitro studies, with a view to evaluate its therapeutic potential in vivo (Figure 4 and Supplementary Figure S4). Finally, the ability of PEG-MP7 to restore antitumor immune responses in vivo and slow growth of disseminated tumors was illustrated in a syngeneic mouse model using immunocompetent animals (Figure 5). In this model, we monitored the growth of murine MC38 colon carcinoma cells in syngeneic mice, a model in which tumor growth has been previously reported to be susceptible to PD-1 blockade.29 However, to improve antigenic immune responses provoked by PD-1 blockade as a monotherapy, we used MC38 cells transfected to stably express human carcinoembryonic antigen (MC38.CEA cells) as a heterologous tumor antigen. Similar to MC38 cells, the MC38.CEA cells retained low basal levels of PD-L1 expression which is upregulated by inflammatory stimuli (i.e., IFNγ). Notably, treatment of animals with PEG-MP7 reduced tumor burden to a similar extent as a known antagonistic antibody (Figure 5d,e). Lastly, aptamer MP7 did not reduce tumor burden due to a direct cytotoxic effect toward tumor cells, nor through an off-target TLR9 innate immune triggering (Figure 6).
In summary, we have shown that DNA aptamers directed at PD-1 are capable of antagonizing PD-L1 signaling, and similar to antibodies, can provoke strong antitumor immune responses to eradicate aggressive disseminated tumors. In addition to PD-1, we and others have developed functional aptamers (acting as agonists or as antagonists) to other immune regulatory molecules such as CTLA-4 (ref. 33), 4-1BB34, CD28 (ref. 35), TNFα36, and CD200R1 (ref. 37). Importantly, we have demonstrated for CD200R1 agonistic aptamers that PEGylation can readily improve the therapeutic potential by improving the pharmacokinetic properties of aptamers, readily addressing a past limitation associated with their in vivo use. Future studies are ongoing to identify human-specific anti-PD-1 aptamers and to determine and optimize the in vivo biodistribution and pharmacokinetic properties of these reagents.
Materials and Methods
Mice and tumor cell line. C57BL/6 mice (6–12 weeks old) were kept under pathogen-free conditions at the Sunnybrook Health Sciences Center Comparative Research Animal facility. All experiments were performed under the approval of the local animal welfare committee in accordance with the rules and regulations of the Canadian Council for Animal Care. The murine colon carcinoma cell line MC38 expressing the human carcinoembryonic antigen (MC38.CEA) was a kind gift from Dr. Jeffrey Schlom (National Cancer Institute, Bethesda, MD).
Fusion proteins and monoclonal antibodies. A chimeric protein consisting of the murine PD-1 extracellular domain fused to a human IgG constant region with a C-terminal histidine tag (mPD-1.FcHIS) was purchased from Sino Biological (Beijing, China) and used for aptamer selection and in vitro experiments. Recombinant purified hPD-1.Fc and mPD-L1.Fc were purchased from R&D Systems (Minneapolis, MN). A blocking anti-mPD-1 antibody (clone RMPI-14) and isotype control 2A3 were purchased from BioXcell (West Lebanon, NH). Flow cytometry was performed with APC labeled anti-mPD-L1 antibody (clone 10F.9G2, BioLegend, San Diego, CA), FITC-labeled anti-CEACAM5 antibody (clone C365D3 (NCRC23), Cedarlane, Canada), Fluorescein isothiocyanate (FITC) -labeled anti-PD-1 antibody (clone 29F.1A12, BioLegend), or the RMPI-14 anti-PD-1 monoclonal antibody conjugated to FITC (4 °C overnight in 100 mmol/l NaCO3, pH 8.3) yielding 10 fluorophores per antibody. Histidine-tagged CEA-N domain (CEA-N.HIS) was expressed and purified as previously described.38
DNA aptamer selection. Single-stranded DNA aptamers recognizing murine PD-1 were identified using the in vitro SELEX method.20,21 A randomized ssDNA library constructed to have a 25 nucleotide (nt) internal variable sequence flanked by 25nt 5′ and 3′ constant primer regions regions (5′-GACGATAGCGGTGACGGCACAGACGNNNNNNNNNNNNNNNNNNNNNNNNCGTATGCCGCTTCCGTCCGTCGCTC-3′) was synthesized by Integrated DNA Technologies. A 4nmol aliquot of this library, representing ~2.5 × 1015 sequences, was counter-selected three times against MagneHis Ni-Particles (Promega, Madison, WI) at 37 °C for 1 hour in selection buffer (10 mmol/l (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES), 150 mmol/l NaCl, 0.05% Tween-20, 1 mmol/l MgCl2, 1 mmol/l CaCl2) followed by counter selection to an irrelevant human IgG immobilized onto Protein A beads (Life Technologies, Burlington, ON). Subsequently, the counter-selected library was incubated with recombinant mPD-1.FcHIS immobilized on the Ni-Particles for 1 hour at 37 °C in binding buffer. After three washes, bound aptamers were eluted by denaturation at 95 °C for 5 minutes. The enriched library was amplified for the next round of SELEX by asymmetric PCR using a 20:1 forward: reverse primer ratio. Five rounds of SELEX were carried forward as described above, each round consisting of counter selection to both the MagneHIS particles and human IgG followed by selection to mPD-1.FcHIS. Selection stringency was increased by halving the concentration of mPD-1.FcHIS from 10 µg/cycle during rounds 1–3 to 5 µg/cycle in the remaining rounds, and by increasing wash time from 10 to 15 minutes after cycle 2.
NGS. To identify sequences after five rounds of selection, adaptor sequences were ligated to the enriched library using the Ion Plus Fragment Library Kit (Life Technologies) and NGS performed using Ion Torrent PGM (SRI Genomics Core Facility) with the Ion 314 chip (Life Technologies) and data processed using Galaxy and FASTAaptamer software.39
Nitrocellulose filter binding assay. The binding affinity of selected aptamers toward mPD-1.FcHIS was determined using a double membrane filter binding assay as previously described.37 Briefly, 32P-labeled aptamers were incubated with a serial dilutions of mPD-1.FcHIS prepared in selection buffer supplemented with 0.1% bovine serum albumin at 37 °C for 1 hour before being filtered through a double membrane system. Protein-bound aptamers adsorbed to the nitrocellulose membrane and unbound aptamers captured on the lower nylon membrane were detected by exposure to x-ray film.
Aptamer flow cytometry. Fluorescently labeled aptamers MP7, MP5, and cSeq were synthesized with a 3' terminal FAM (IDT). 1 × 106 P815 mastocytoma cells were preincubated in 100 µl PBS supplemented with 1% fetal bovine serum, 0.09% NaN3, 1 mmol/l MgCl2, 1 mmol/l CaCl2, 100 µg/ml salmon sperm DNA, 100 µg/ml yeast tRNA, and 1 µg Fc blocker (BioLegend) for 15 minutes before addition of 2.5 µmol/l aptamer or FITC-labeled antibody. After a 30-minute incubation at 37 °C each sample was washed with PBS and flow cytometry performed using a FACScalibur Cell Analyser (BD Biosciences, San Jose, CA.)
SPR. Aptamer specificity to the extracellular domain of mPD-1 was evaluated by SPR binding experiments using the Biacore T200 (GE Healthcare Mississauga, ON, Canada). Aptamers were synthesized with a 5' biotin and immobilized onto separate flow cells of a streptavidin-coated sensor chip reaching 50-100RU. Binding analyses were performed by exposing each immobilized aptamer as well as a blank reference flow cell to injections (120 seconds, 30 µl/minute) of mPD-1.FcHIS (50 nmol/l), hPD-1.Fc (50 nmol/l), mPD-L1.Fc (50 nmol/l), hIgG (500 nmol/l), or CEA-N.HIS (500 nmol/l) dissolved in HBS-P running buffer (10 mmol/l HEPES pH 7.4, 150 mmol/l NaCl, 0.05% v/v Tween 20). Flow cells were regenerated before each cycle by a 60-second pulse with 10 mmol/l NaOH/1M NaCl followed by a 5-minute stabilization period.
IL-2 ELISPOT assay. The antagonistic activity of mPD-1 aptamers was assessed by their ability to restore IL-2 secretion by splenocytes which was suppressed by PD-L1 signaling. C57BL/6 splenocytes (2 × 105 cells/well) were cultured in microtiter wells precoated with anti-IL2 capture antibody, anti-CD3 antibody (1 µg/ml) and either mPD-L1.Fc (15 µg/ml) or an irrelevant hIgG-Fc isotype control (15 µg/ml). To monitor the effect of PD-1 blockage by anti-PD-1 aptamers (250 nmol/l, 1.15 µg, 200 µl) or antibody (125 nmol/l, 3.75 µg, 200 µl) were added to the appropriate wells. Levels of secreted IL-2 were detected after 48 hours of culture using a biotinylated anti-IL-2 detection antibody and the ELISPOT Blue Color Module (R&D Systems, Minneapolis, MN). After color development, spots were enumerated using an automated CTL Immunospot Analyser (Cellular Technology, Shaker Heights, OH). Proliferation experiments were performed by stimulating CFSE-labeled splenocytes in microtiter plates coated as described above. Splenocytes were labeled with 5 µmol/l CFSE as recommended by the manufacturer (Life Technologies) and dilution profiles analyzed 3 days after culture using a FACScalibur Cell Analyser. When used, aptamers were present in culture at a final concentration of 1.25 µmol/l.
Competitive ELISA The ability of anti-PD-1 aptamers or antibody to block the PD-1/PD-L1 interaction was evaluated using a competitive ELISA. Recombinant mPD-L1.Fc (3 µg/ml) precoated onto wells of a 96-well microtiter plate was incubated with recombinant mPD-1.FcHIS (3 µg/ml, 130 nmol/l) in competition with aptamer (2.5 µmol/l) or antibody (1 µmol/l) and bound mPD-1.FcHis detected using an anti-HIS HRP antibody followed by color development with TMB as substrate. The amount of mPD-1.FcHIS bound in each well was quantified using a standard curve generated using a dilution series of mPD-1.FcHIS and used to determine the percent of PD-1 blocked by each aptamer or antibody.
PEGylation of DNA aptamers. DNA aptamers with a 5' hexylamine modifier (IDT) were incubated with a 10-fold excess of a 40kDa mPEG-succinimidyl glutarate ester (NOF America, White Plains, NY) added drop wise over 3 hours to a 0.5 mmol/l solution of aptamer dissolved in 0.1M NaHCO3/CH3CN (1:1, pH 9.0). PEGylated aptamers were prepared in 0.1M triethylammonium acetate (TEAA, pH 7.0) and purified by reverse phase high-performance liquid chromatography using a C18 OST semipreparative column (Waters, Milford, MA). The bound PEGylated aptamers were eluted using a linear gradient going from 5–90% CH3CN over a 60-minute period.
MC38.CEA tumor challenge MC38.CEA colon carcinoma cells (5 × 105 cells) were implanted intraperitoneally (i.p.) into C57Bl/6 mice. Mice were subsequently treated i.p. with either PEGylated aptamers (40 µg oligonucleotide weight, 1.3 nmol) or antibody (200 µg, 1.3 nmol) on days 1, 3, 5, and 7 following tumor implantation. After 21 days, animals were sacrificed for necropsy and their tumor burden examined by blinded investigators. Longitudinal diameters of tumor masses were measured using callipers and a modified ellipsoid formula was used to determine tumor volume where
Tumor volume = ½ ((greatest longitudinal diameter) × (smaller longitudinal diameter)2)
Aptamer-induced TLR9 immune responses. PEGylated aptamers (40 µg, 1.3 nmol) or a TLR9 ligand CpG ODN (ODN 1826, Invivogen, CA) were administered i.p. into naive C57BL/6 mice (n = 3). Mice were sacrificed 3 hours later and serum levels of TNFα and IL-6 were quantified using commercial ELISA duosets (R&D Systems). For in vitro studies, RAW 264.7 mouse macrophages (American Type Culture Collection, Manassas, VA) were treated with 3 µmol/l CpG ODN 1585 or PEG-MP7 for 3 hours. RNA was harvested using an RNeasy Mini Kit (Qiagen, Valencia, CA) and one-step real-time polymerase chain reaction performed following manufacturers recommendations with optimized primers and conditions.
Statistics. P values for ELISPOT and ELISA assays were calculated using Student's t-test or analysis of variance where appropriate. GraphPad PRISM 6 software was used to plot all data, perform statistical analysis, and fit binding curves using a one site binding model.
SUPPLEMENTARY MATERIAL Figure S1. Enriched aptamers pull down mPD-1.FcHIS. Figure S2. Aptamers MP5 and MP7 bind PD-1 expressing cells. Figure S3. Anti-PD-1 DNA Aptamers do not stimulate IL-2 secretion in the absence of PD-L1 signalling. Figure S4. MP7 restores anti-CD3 induced PD-L1 suppressed lymphocyte proliferation. Figure S5. Purification of PEGylated aptamers. Figure S6. PEGylation does not disrupt the ability of MP7 to block the PD-1:PD-L1 interaction. Figure S7. PEGylated MP7 does not induce transcription of genes associated with TLR-9 triggering.
Acknowledgments
This work was financially supported by operating grants from the Canadian Institutes of Health Research and the Canadian Breast Cancer Foundation to J.G. A.P. is the recipient of an Ontario Graduate Studentship. The authors would like to thank the Sunnybrook Research Institute – Genomic Core Facility for performing next-generation sequencing. The authors would also like to thank Khalid K. Alam and Professor Donald H. Burke (University of Missouri) for providing FASTAptamer software and assisting with aptamer next-generation sequencing data analysis. The authors declare no conflict of interest.
Supplementary Material
References
- Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol. 2006;90:51–81. doi: 10.1016/S0065-2776(06)90002-9. [DOI] [PubMed] [Google Scholar]
- Ceeraz S, Nowak EC, Noelle RJ. B7 family checkpoint regulators in immune regulation and disease. Trends Immunol. 2013;34:556–563. doi: 10.1016/j.it.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaddanapudi K, Mitchell RA, Eaton JW. Cancer vaccines: Looking to the future. Oncoimmunology. 2013;2:e23403. doi: 10.4161/onci.23403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 2012;209:1201–1217. doi: 10.1084/jem.20112741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanmamed MF, Chen L. Inducible expression of B7-H1 (PD-L1) and its selective role in tumor site immune modulation. Cancer J. 2014;20:256–261. doi: 10.1097/PPO.0000000000000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- Azuma T, Yao S, Zhu G, Flies AS, Flies SJ, Chen L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood. 2008;111:3635–3643. doi: 10.1182/blood-2007-11-123141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65:1089–1096. [PubMed] [Google Scholar]
- Callahan MK, Wolchok JD. At the bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy. J Leukoc Biol. 2013;94:41–53. doi: 10.1189/jlb.1212631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs. 2010;2:256–265. doi: 10.4161/mabs.2.3.11641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9:767–774. doi: 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157:220–233. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nat Rev Drug Discov. 2010;9:537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- White RR, Sullenger BA, Rusconi CP. Developing aptamers into therapeutics. J Clin Invest. 2000;106:929–934. doi: 10.1172/JCI11325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyetech Study Group Anti-vascular endothelial growth factor therapy for subfoveal choroidal neovascularization secondary to age-related macular degeneration: Phase II study results. Ophthalmol. 2003;110:979–986. doi: 10.1016/S0161-6420(03)00085-X. [DOI] [PubMed] [Google Scholar]
- Eyetech Study Group Preclinical and phase IA clinical evaluation of an anti-VEGF pegylated aptamer (EYE001) for the treatment of exudative age-related macular degeneration. Retina. 2002;22:143–152. doi: 10.1097/00006982-200204000-00002. [DOI] [PubMed] [Google Scholar]
- Lázár-Molnár E, Yan Q, Cao E, Ramagopal U, Nathenson SG, Almo SC. Crystal structure of the complex between programmed death-1 (PD-1) and its ligand PD-L2. Proc Natl Acad Sci USA. 2008;105:10483–10488. doi: 10.1073/pnas.0804453105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Veverka V, Radhakrishnan A, Waters LC, Muskett FW, Morgan SH, et al. Structure and interactions of the human programmed cell death 1 receptor. J Biol Chem. 2013;288:11771–11785. doi: 10.1074/jbc.M112.448126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Pieve C, Blackshaw E, Missailidis S, Perkins AC. PEGylation and biodistribution of an anti-MUC1 aptamer in MCF-7 tumor-bearing mice. Bioconjug Chem. 2012;23:1377–1381. doi: 10.1021/bc300128r. [DOI] [PubMed] [Google Scholar]
- Healy JM, Lewis SD, Kurz M, Boomer RM, Thompson KM, Wilson C, et al. Pharmacokinetics and biodistribution of novel aptamer compositions. Pharm Res. 2004;21:2234–2246. doi: 10.1007/s11095-004-7676-4. [DOI] [PubMed] [Google Scholar]
- Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T, et al. IFN-á directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J Immunol. 2011;186:2772–2779. doi: 10.4049/jimmunol.1003208. [DOI] [PubMed] [Google Scholar]
- Orava EW, Abdul-Wahid A, Huang EH, Mallick AI, Gariépy J. Blocking the attachment of cancer cells in vivo with DNA aptamers displaying anti-adhesive properties against the carcinoembryonic antigen. Mol Oncol. 2013;7:799–811. doi: 10.1016/j.molonc.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–760. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- Krug A, Rothenfusser S, Hornung V, Jahrsdorfer B, Blackwell S, Ballas ZK, et al. Identification of CpG oligonucleotide sequences with high induction of IFN-alpha/beta in plasmacytoid dendritic cells. Eur J Immunol. 2001;31:2154–2163. doi: 10.1002/1521-4141(200107)31:7<2154::aid-immu2154>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Santulli-Marotto S, Nair SK, Rusconi C, Sullenger B, Gilboa E. Multivalent RNA aptamers that inhibit CTLA-4 and enhance tumor immunity. Cancer Res. 2003;63:7483–7489. [PubMed] [Google Scholar]
- McNamara JO, Kolonias D, Pastor F, Mittler RS, Chen L, Giangrande PH, et al. Multivalent 4-1BB binding aptamers costimulate CD8+ T cells and inhibit tumor growth in mice. J Clin Invest. 2008;118:376–386. doi: 10.1172/JCI33365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor F, Soldevilla MM, Villanueva H, Kolonias D, Inoges S, de Cerio AL, et al. CD28 aptamers as powerful immune response modulators. Mol Ther Nucleic Acids. 2013;2:e98. doi: 10.1038/mtna.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orava EW, Jarvik N, Shek YL, Sidhu SS, Gariépy J. A short DNA aptamer that recognizes TNFá and blocks its activity in vitro. ACS Chem Biol. 2013;8:170–178. doi: 10.1021/cb3003557. [DOI] [PubMed] [Google Scholar]
- Prodeus A, Cydzik M, Abdul-Wahid A, Huang E, Khatri I, Gorczynski R, et al. Agonistic CD200R1 DNA Aptamers Are Potent Immunosuppressants That Prolong Allogeneic Skin Graft Survival. Mol Ther Nucleic Acids. 2014;3:e190. doi: 10.1038/mtna.2014.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdul-Wahid A, Huang EH, Lu H, Flanagan J, Mallick AI, Gariépy J. A focused immune response targeting the homotypic binding domain of the carcinoembryonic antigen blocks the establishment of tumor foci in vivo. Int J Cancer. 2012;131:2839–2851. doi: 10.1002/ijc.27582. [DOI] [PubMed] [Google Scholar]
- Alam KK, Chang JL, Burke DH. FASTAptamer: A Bioinformatic Toolkit for High-throughput Sequence Analysis of Combinatorial Selections. Mol Ther Nucleic Acids. 2015;4:e230. doi: 10.1038/mtna.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar-Molnar E, Ramagopal UA, Nathenson SG, Almo SC. Crystal structure of the extracellular domain of human PD-1. 2011.
- Zhang X, Schwartz JC, Guo X, Bhatia S, Cao E, Lorenz M, et al. Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity. 2004;20:337–347. doi: 10.1016/s1074-7613(04)00051-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.