

Abstract
Recombinant adenoviruses containing a double-stranded DNA genome of 26–45 kb were broadly explored in basic virology, for vaccination purposes, for treatment of tumors based on oncolytic virotherapy, or simply as a tool for efficient gene transfer. However, the majority of recombinant adenoviral vectors (AdVs) is based on a small fraction of adenovirus types and their genetic modification. Recombineering techniques provide powerful tools for arbitrary engineering of recombinant DNA. Here, we adopted a seamless recombineering technology for high-throughput and arbitrary genetic engineering of recombinant adenoviral DNA molecules. Our cloning platform which also includes a novel recombination pipeline is based on bacterial artificial chromosomes (BACs). It enables generation of novel recombinant adenoviruses from different sources and switching between commonly used early generation AdVs and the last generation high-capacity AdVs lacking all viral coding sequences making them attractive candidates for clinical use. In combination with a novel recombination pipeline allowing cloning of AdVs containing large and complex transgenes and the possibility to generate arbitrary chimeric capsid-modified adenoviruses, these techniques allow generation of tailored AdVs with distinct features. Our technologies will pave the way toward broader applications of AdVs in molecular medicine including gene therapy and vaccination studies.
INTRODUCTION
To broadly study biological and clinical features of large DNA viruses, efficient generation of genetically modified viral DNA molecules is mandatory. Adenoviruses are non-enveloped double-stranded DNA viruses and more than 60 human adenoviruses types with different type-dependent clinical pictures were identified. Recombinant adenoviruses were broadly explored in various applications including vaccination studies, oncolytic virotherapy and therapeutic gene transfer. However, limitations of currently used recombinant adenoviruses, especially those which were employed in preclinical and clinical trials, originate in the limited number of distinct adenovirus types which are available as vectors and the limited possibilities to modify the available vector backbones.
Most adenoviral vectors (AdVs) are based on the human adenovirus type 5 with a distinct tropism and a high sero-prevalence strongly limiting the usage of these vectors (1,2). For construction of early generation vectors associated with cellular toxicity due to induction of innate and adoptive immune responses, the early gene region E1 or in addition the early regions E2, E3 or E4 were replaced by the genetic cargo sequence (3). In contrast, newest generation high-capacity adenoviral vectors (HCAdV) are devoid of all viral coding sequences (4–6). Their production is dependent on a helper virus (HV), which provides viral capsid proteins in trans for efficient capsid assembly and release of viral particles. However, packaging of helper virus genomes is inhibited due to recombinase-mediated excision of the packaging signal. This enables packaging of HCAdV genomes containing cargo sequences flanked by minimal 5′ and 3′ sequences of the adenoviral genome with a maximized capacity for cargo sequences (up to 36 kb) and reduced vector-related toxicity and immune responses (7–9). Thus, HCAdVs represent attractive tools for gene and cell therapy as well as for vaccination approaches and basic research applications. Due to their large genome they offer new perspectives regarding applications for transfer of complex expression systems such as inducible systems or transgenes under control of endogenous regulatory sequences. In addition, a limited amount of capsid-modulations was exploited utilizing early generation AdVs allowing significant improvements in transduction efficiencies for specific cell types, in vivo targeting and escape from neutralizing anti-adenoviral antibodies (10,11). However, up to now the full potential of HCAdV was not accessible due to the major challenge of cloning and modulating HCAdV and HV genomes.
For improving biological, preclinical and clinical features of recombinant adenoviruses, the establishment of molecular methods allowing high-throughput genetic manipulation of virus genomes is mandatory. Currently used methods for genetic manipulation of adenoviruses are based on either rare endonucleases or homologous recombination in mammalian cells or bacteria of Escherichia coli strain BJ5183, which constitutively express λ recombinases (12–14). However, these methods are work-intensive and time-consuming. Moreover, generation is dependent on complex constructions of intermediate clones containing either respective endonuclease binding sites or long homologous sequences (>200 bp) (15). Previously, an elegant recombination-mediated genetic engineering (recombineering) method was established allowing traceless modification of bacterial artificial chromosomes (BACs). Herein, modifications were incorporated by two successive homologous recombinations at high efficiencies (16) based on the selection marker galK which can be used for positive and negative selection. In this system, recombinases are encoded endogenously by bacteria of E. coli strain SW102, which express high levels upon heat-induction. In contrast to previous methods, small homologous regions (<50 bp) are sufficient for efficient recombination, which can be generated by PCR utilizing adequate primers. Therefore, time-consuming and challenging construction of intermediate clones containing the modified sequence flanked by long homologies or endonuclease binding sites can be avoided. In a previous study it was shown that this technique works efficiently for inserting transgenes into early generation vectors (17). Here, we adopted this recombineering technique for high-throughput generation of AdVs including HCAdV and HV facilitating development of novel vectors with improved features.
MATERIALS AND METHODS
Cloning of complete adenoviral vector genomes from different sources
For cloning of complete adenoviral genomes we adopted a DNA recombination method. This technology only requires short homology arms (50 bp) which can for instance be linked to a PCR primer. A first description of this methodology for modulation of BACs is provided in a previous publication (16). In brief, this method is based on SW102 strain of E. coli in which λ recombinases can be induced by heat shock at 42°C for 15 min. After this heat shock, SW102 bacteria containing the target BAC can be used for preparation of electro-competent cells and 1.5 μg of the linear DNA for homologous recombination can be transformed into these bacteria. For isolation of single clones, bacteria were selected on agar plates containing the respective selection marker.
As a first option for cloning of complete adenoviral genomes, purified adenovirus particles can be used. These particles were isolated from crude cell lysate containing amplified adenovirus by caesium chloride (CsCl) gradients. According to our previous publication (6), a step gradient and continuous CsCl gradient were performed. For removal of CsCl a dialysis was performed as described previously (6). Final vector aliquots were stored at −80°C until usage. After titration (6), viral DNA was isolated from 2 to 4 × 1010 purified adenoviral particles. After proteinase K digest a phenol–chloroform extraction was performed and adenoviral DNA precipitated by ice-cold 100% ethanol. After a washing step with 70% ethanol, adenoviral DNA was dissolved in H2O. It is of note that for all steps required for DNA isolation from virions shearing of the DNA should be avoided by treating the DNA gently and by avoiding repeated pipetting or vortexing. For recombineering, we used the target BAC pB-GK which contains 5′ and 3′ inverted temrinal repeats (ITRs) of human adenovirus type 5 separated by a galK-Kan cassette encoding kanamycin and galactokinase as selection markers (18). Note that this BAC vector can be used for cloning of any human adenovirus type 5 based genome. This target BAC was transformed into SW102 cells and electro-competent bacteria were prepared. Next 1.5 μg of the adenovirus genome purified from virions were transformed into these bacteria and λ recombinases were induced by heat shock at 42°C for 15 min. For isolation of single clones, bacteria were selected on agar plates containing the respective selection markers.
If plasmids were used as a source for adenoviral genomes as a second option, respective BACs were generated by backbone-exchange, replacing the plasmid backbone with the BAC-specific backbone. Especially, commercially available kits for adenovirus production deliver large plasmids containing the major part of the adenoviral genome representing perfect sources for adenoviral DNA contained in plasmids allowing access to the BAC-platform. Here, the BAC-specific backbone containing a chloramphenicol resistance marker gene was amplified by PCR utilizing primer for generation of regions homologous to the 5′- and 3′-end of adenovirus type 5 genome. The purified PCR-product was transformed into SW102 bacteria with the plasmid pHCAdV5-CMV/eGFP containing an adenoviral genome of a HCAdV encoding GFP under the control of the CMV promoter. Homologous recombination was induced and bacteria with BACs containing the adenoviral genome were isolated by positive selection for chloramphenicol resistance. Note, that this method can be performed for any adenoviral genome which was cloned as a plasmid. For construct-specific PCR, primer pairs and their sequences please refer to Supplementary Table S1.
Traceless genetic modification of hexon and fiber contained in adenoviral genomes which were cloned as BACs
For construction of BACs encoding for hexon- and fiber-modified adenoviral genomes we utilized cloning techniques based on BACs for which the basic principle was described elsewhere (16). The BACs (BAC-AdV5 and BAC-AdV5-HV) were transformed into SW102 E. coli. In parallel, a galK/Amp cassette expressing galactokinase and amplicillin selection markers was PCR-amplified using pT-GA as template with additional sequences at 5′ and 3′ ends homologous to the fiber and hexon gene sequences upstream and downstream (for primer sequences please refer to Supplementary Table S2). 1.5 μg of respective PCR products was transformed into SW102 containing pB-HV and after heat-shock activated homologous recombination and selection for the ampicillin intermediate clone BAC-AdV5-HV-HVR-galK/Amp was isolated. Subsequently, chimeric hexon sequences generated by synthesis or regions of respective adenoviral genomes coding for the surface domains (SDs) of the hexon were PCR-amplified (for primer sequences please refer to Supplementary Table S2). Purified PCR products were used for BAC cloning replacing the galk/Amp sequence with chimeric hexon sequences by homologous recombination and single clones were isolated by negative selection against galK. Next, all hexon-modified helper-virus BACs were modulated replacing the stuffer DNA in the E3 region by a reporter gene expression cassette encoding a eGFP-firefly luciferase fusion protein under the control of the CMV-enhancer/EF1α promoter. Positive clones were purified and used for reconstitution of respective hexon-modified adenoviral helper-viruses (HV).
Reconstitution, amplification and purification of hexon- and fiber-modified helper-viruses
HV were generated and amplified according to protocol published previously (6). In brief, respective helper virus BAC DNA was digested with PacI and transfected into HEK293 cells with Fugene 6 (Roche). After cytopathic effect occurred 10–20 days post-transfection, cells were harvested and virus particles were released by three consecutive freeze–thaw steps. HV was amplified by infection of increasing numbers of HEK293 cells. For the final amplification step, 4 × 108 HEK293 cells were infected and generated HV particles were purified by two successive CsCl gradients. For application in vitro and in vivo toxic CsCl solutions with virus were exchanged with a physiological buffer by dialysis and vector aliquots were stored at −80°C until usage.
Recombination pipeline and cloning of the high-capacity adenovirus HCAdV5-2indsys
The recombination pipeline is a valuable tool if complex DNA sequences are combined or if complex expression systems need to be designed. Therefore, we developed a novel recombination pipeline which allows combining various DNA sequences using a cloning method which is solely based on liquid bacteria culture. For a detailed protocol describing the recombination pipeline, refer to the supplementary material part. Here, we provide a step-by-step protocol for a single cloning step within the recombination pipeline.
We used this method to generate a complex HCAdV for transient cell marking to track differentiation or dedifferentiation processes. Toward that end two independent inducible and tissue-specific expression systems were incorporated into the HCA genome resulting in the vector HCA-2indsys. For details regarding all sequences contained in HCA-2indsys, refer to the ‘Results’ section. The mifepristone inducible system was based on the GeneSwitch Mammalian Expression Kit provided by Life Technologies. All primers used for PCRs to amplify DNA sequences used for the recombination pipeline to generate the vector HCAdV5-2indsys are summarized in Supplementary Table S1.
PCR
For all PCRs high-fidelity KOD Hot Start DNA Polymerase (Novagen, Darmstadt) was used except for amplification of the BAC-backbone, which was performed with the PRECISOR polymerase (Biocat, Heidelberg). All used primers and templates for respective PCRs are listed in Supplementary Table S1. Specific primers for control PCRs to verify hexon chimeras with precisely exchanged hypervariable regions (HVRs) were designed to bind to sequences of HVR5 and HVR7 of respective hexon genes (for primer sequences, refer to Supplementary Table S2). Replacements of the complete SDs were determined by primers used for generation of respective plasmids for expression in mammalian cells (for primer sequences, refer to Supplementary Table S2). For verification of sequences, vector genomes were sequenced by Eurofins MWG Operon (Ebersberg, Germany) utilizing adequate primers listed in Supplementary Table S2.
Cell culture
Human embryonic kidney cells (HEK293 cells) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS, PAA Laboratories). One hundred sixteen cells used for production of HCA were grown in modified Eagle's medium (MEM) supplemented with 10% FBS (PAA Laboratories) and hygromycin B (100 μg/ml). All media were also supplemented with penicillin and streptomycin provided as a 100-fold concentrated stock solution (PAA Laboratories).
Luciferase assay
Quantification of firefly luciferase expression was performed utilizing the Dual-Luciferase® Reporter Assay System kit (Promega). In detail, HEK293 cells seeded in six-well plates were infected with unmodified or hexon-modified helper-virus (1 μl/well). Forty-eight hours post-infection, cells were washed with Dulbecco's Phosphate-Buffered Saline (DPBS) and harvested in 500 μl passive lysis buffer provided by the kit. Five microliters of the cell lysate were mixed with 35 μl luciferase assay reagent II, respectively, and subsequently firefly luciferase activity was measured for 5 s at 492 nm utilizing a luminometer (Microlumat Plus LB 96V, Berthold).
LacZ staining
HEK293 cells were seeded in six-well plates and transduced with the capsid-modified HCA containing the lacZ expression cassette. Forty-eight hrous post-transduction cells were washed with Dulbecco's phosphate buffered saline (PBS, 1×) and fixed with glutaraldehyd (0.8%). After 10 min, cells were washed with Dulbecco's PBS (1×) and stained with the staining solution (1 mg/ml X_Gal, 10 mM FeK4(CN)6, 10 mM FeK3(CN)6, 2 mM MgCl2) for 10 h.
Fluorescence microscopy
HEK293 cells were seeded in six-well plates and transduced with the HCAdV containing the eGFP expression cassette. Forty-eight hrous post-transduction eGFP expression was detected with an inverse fluorescence microscope (Leica DM IRB).
Construction of plasmids for chimeric hexon expression in mammalian cells
For generation of plasmids encoding hexon chimeras with completely exchanged surface regions, a master clone (pCI-M) was constructed (Supplementary Figure S1). To generate this master clone, conserved 5′ and 3′ sequences were amplified by PCR and subcloned into the vector pCR-BluntII-Topo (Invitrogen) generating vectors pT-hex5-5′ with AarI and BglII binding sites at the 3′ end and pT-hex5-3′ with BglII and BspQI binding sites at the 5′ end (for primer sequences, refer to Supplementary Table S2). Both vectors were cut with BglII and NotI and fragments were ligated resulting in pT-M. Subsequently conserved hexon regions were excised from pT-M with restriction enzymes SpeI and NotI and cloned into mammalian expression vector pCI (Promega) which was cut with XbaI and NotI. Constructed vector pCI-M contained conserved 5′ and 3′ regions of human adenovirus type 5 hexon separated by restriction enzyme binding sites AarI, BglII and BspQI. Recognition sites for endonucleases type II AarI and BspQI are directed toward the conserved hexon sequences. Digests with AarI and BspQI enabled cutting in the conserved regions generating sticky ends and excision of binding sites. For generation of hexon chimeras, sequences of hexon genes encoded by viral genomes of respective adenovirus serotypes were amplified by PCR (for primer sequences, refer to Supplementary Table S2). PCR products contain AarI binding sites at the 5′ end and BspQI binding sites at the 3′ end, with respective cutting sites in the amplified hexon-specific region. Amplified sequences were subcloned into the plasmid pCR-BluntII-Topo resulting in vectors pT-hex5-SD4, -SD7, -SD12, -SD13 and -SD41. After excising inserts with AarI and BspQI, fragments were subcloned into the master plasmid pCI-M digested with AarI and BspQI resulting in mammalian expression plasmids encoding for hexon chimera. To construct plasmid pCI-100K-pr, vector pCI was linearized with SmaI and dephosphorylated with alkaline phosphatase. In parallel, the gene encoding adenoviral 100K-protein was PCR-amplified utilizing viral genomic DNA isolated from purified hAd5 particles as template (for primer sequences, refer to Supplementary Table S2). Purified PCR product was phosphorylated with T4 polynucleotide kinase and blunt-end ligated with SmaI-cutted pCI vector (T4 DNA Ligase) resulting in vector pCI-100K-pr.
Synthesis of sequences encoding chimeric hexon surface domains
For synthesis of chimeric hexon SDs sequences of HVRs located within the human adenovirus type 5, hexon gene sequence at positions 403–495 (HVR1), 562–582 (HVR2), 634–657 (HVR3), 742–783 (HVR4), 802–843 (HVR5), 913–945 (HVR6) and 1255–1353 (HVR7) were precisely exchanged with respective sequences of human adenovirus types 12, 41, 44, 48 and 50. Sequence synthesis was performed by GeneArt (Regensburg, Germany) and for delivery sequences were incorporated into a standard vector.
Detection of Cre-mediated excision of the packaging signal
HEK293 cells and 116 HCAdV producer cells based on HEK293 cells were seeded in six-well plates. At 90% confluency, cells were infected with the helper virus containing a floxed packaging signal at MOI 3. Thirty-six hours post transduction DNA was extracted from harvested cells, purified with phenol–chloroform and precipitated with isopropanol. DNA samples were solved in water and used as templates for PCR analysis (for primer sequences, refer to Supplementary Table S1).
Western blot analysis
HEK 293 cells were seeded in 60 mm dishes and co-transfected at 60% confluency with mammalian expression plasmids pCI-100K-pr encoding adenoviral 100K-protein and pCI-hex5-SD4, -SD7, -SD12, -SD13 and -SD41 for expression of the chimeric hexon protein. Seventy-two hours post-transfection, cells were harvested in 200 μl lysis buffer (50 nM Tris–HCl pH 8.0, 150 nM NaCl, 1% NP-40) and incubated for 30 min on ice. For western blot analysis, 5 μl loading dye (5×) was added to 15 μl cell lysate, respectively, and subsequently boiled at 95°C for 5 min or cooled at 4°C. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) utilizing a 10% SDS-polyacrylamide gel and transferred by semi-dry blotting on a methanol-activated polyvinylidene difluoride (PVDF) membrane. The membrane was treated over night with blocking buffer (20 mM pH 7.5, 500 mM NaCl, 5% milk powder) and after three washing steps with TBST (20 mM, Tris pH 7.5, 500 mM NaCl, 0.05% Tween, 0.2% Triton-X100), the membrane was incubated for 1 h with the polyclonal anti-adenovirus antibody PA1-7201 (Dianova, final concentration 1:500) diluted in blocking buffer. Subsequently, the membrane was washed three times and incubated with the monoclonal peroxidase-conjugated rabbit anti-goat antibody (Jackson ImmunoResearch Laboratories Inc., final dilution 1:10 000) diluted with blocking buffer. For protein detection, blots were washed three times, ECL reagent (GE Healthcare) was added and peroxidase activity detected according to manufacturer's instructions.
Production of high-capacity adenoviral vectors (HCAdV)
Production of HCAdVs was performed in the producer cell line 116 as described earlier (5,6). In brief, the HCAdV genome was released from the BAC and DNA was transfected into a 6 cm tissue culture dish with 116 cells. After 24 h, cells are co-infected the helper virus and harvested 72 h after transfection. HCAdV capsids were released by repeated freeze/thaw steps and used for three further amplification steps. In contrast, for large-scale amplification 10 confluent 15 cm tissue culture dishes of 116 cells were transferred into 1 l of medium placed in a spinner flask. Cells were grown for 5 days increasing the volume of culture medium to up to 3 l. At day 5, cells were harvested by centrifugation, resuspended in 150 ml medium, and co-infected with 100% of the lysate derived from 8 × 107 116 producer cells and AdNG163R-2 helper virus (5) (one infectious unit per cell). Virus infection was performed at 37°C on a magnetic stir plate for 2 h. Subsequently, medium (MEM supplemented with 5% FBS) was added to a final volume of 2 l. Co-infected cells were harvested 48–72 h later for unmodified and fiber-modified HV and 96 h later for hexon-modified HV. Cells were lysed and released HCAs were purified by ion exchange columns or CsCl gradients.
Purification of adenoviral particles with ion-exchange columns and CsCl gradients
Purification of small-scale AdV preparations was performed with ion-exchange columns (Sartorius, Göttingen, Germany) according to manufacturer's instructions. To reduce the volume of cell lysate (80 ml) to the volume recommended by the manual, cells were harvested by centrifugation before lysis and resuspended in 20 ml of the supernatant. Large-scale amplified HCAs were purified by two successive CsCl gradients (6). For in vitro and in vivo applications toxic CsCl solutions with virus were exchanged with a physiological buffer by dialysis (6). Final vector aliquots were stored at −80°C until usage.
Determination of physical titers of hexon-modified helper-viruses and sequencing
Detailed procedures to determine physical titers of an AdV preparation were described elsewhere (6). In brief, for determination of number of packaged viral particles (physical titer) we diluted 5 μl of the vector preparation with 95 μl Tris-EDTA (TE) buffer supplemented with 0.1% sodium dodecylsulfate. After shaking for 30 min at 37°C, OD260 was measured five times and mean value was used to calculate the physical titer. Sequencing for verification of modified hexon regions was performed by Eurofins MWG Operon (Ebersberg, Germany) utilizing sequencing primers (Supplementary Table S2).
Flow cytometric analysis
HEK293 cells were seeded in six-well plates and infected with unmodified or hexon-modified helper-virus (1 μl/well). After 48 h incubation at 37°C, cells were washed with Dulbecco's Phosphate-Buffered Saline (DPBS) and treated with trypsin. Detached cells were harvested and resuspended in DPBS supplemented with 0.1% FBS. Subsequently, each sample was analyzed for eGFP expression by flow cytometer FACS Canto II and data derived from 20 000 detected events were used for evaluation, respectively.
RESULTS
A BAC platform for cloning and traceless genetic modification of adenoviral DNA
Currently used methods for genetic manipulation of adenoviruses are work-intensive and time-consuming and are dependent on complex construction of intermediate clones containing either respective endonuclease binding sites or long homologous sequences. Here, we adopted a different recombination method which was first described for modulation of BACs (16). For efficient cloning of AdVs, this recombineering technology only relies on short homology arms (50 bp) which can be added to a PCR product using respective PCR primers. We explored recombineering based on a positive/negative selection system as an elegant method for genetic engineering allowing seamless genetic modification of adenoviral genomes contained in BACs (16,19–20). This unique BAC-platform enables access to different sources of adenovirus DNA such as purified virions or plasmids containing full-length adenovirus DNA molecules (Figure 1a). The cloning strategy is based on an intermediate BAC which contains galK flanked by respective homologous regions and in the presence of the linear adenoviral DNA homologous recombination occurs in SW102 bacteria. The same recomineering strategy can be utilized for traceless genetic modification of adenoviral genomes as schematically shown in Figure 1b. In a first step the selection marker including galK and an antibiotic resistance gene (e.g. ampicillin, kanamycin, zeocin) is PCR amplified using PCR primers which will flank the PCR product by homologous regions which are also represent in the target BAC. During homologous recombination, these selection markers will be inserted into the intermediate BAC. In the next step, a second PCR product containing the desired DNA to be inserted into the BAC flanked by respective homologous regions can then replace the selection marker using negative selection based on galK (Figure 1b).
Figure 1.
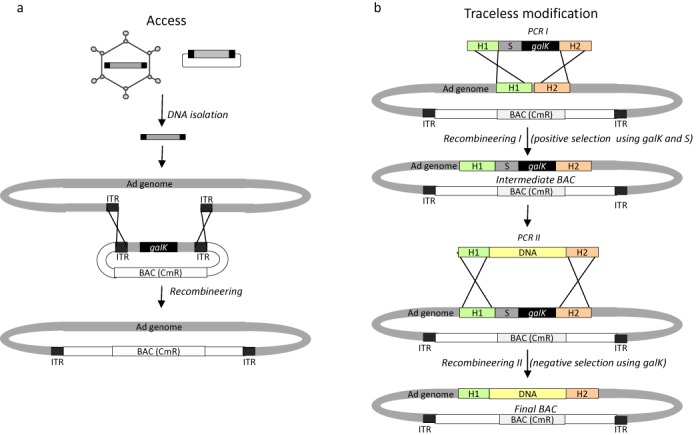
The galK technology for cloning of recombinant adenoviral genomes contained in BACs. (a) Access to adenoviral genomes from different sources. DNA extracted from purified adenoviral particles or plasmids can be used as a source of adenoviral genomes. Isolated viral DNA was transformed into SW102 bacteria containing an intermediate BAC containing the 5′- and 3′- inverted terminal repeats (ITRs) of an adenoviral genome, a chloramphenicol resistance (CmR) and the selection marker gene galaktokinase (galK) allowing positive and negative selection. In addition, this minimal BAC contains two homologous regions (H1 and H2) and after induction of homologous recombination of 5′- and 3′-ends, bacteria with the BAC containing the adenoviral genome were isolated upon negative selection against galK. (b) Traceless modification of adenoviral genomes. A BAC containing a chloramphenicol resistance gene and the adenoviral genome and a PCR product (PCR I) with galK allowing positive and negative selection and another positive selection marker (S) for instance kanamycin or ampicillin are con-transformed into SW102 bacteria. The PCR product is flanked by homologous regions (H1 and H2) also contained in the adenoviral genome and after recombineering (recombineering I) the resulting intermediate BAC is used for the next step. A second PCR product (PCR II) flanked by the homologous regions H1 and H2 is used for the second recombineering (recombineering II) step for which negative galK selection is used leading to the final BAC.
To show integrity of the recombineering system, we exemplify access to adenoviral DNA by direct cloning of an E1/E3-deleted adenoviral DNA molecule which was isolated from purified virions (Figure 2a). For that purpose, the target BAC pB-GK, which contains 5′ and 3′ ITRs of human adenovirus type 5 separated by a galK-Kan cassette encoding kanamycin and galactokinase as selection markers (18), was transformed into SW102 E. coli. Next, purified adenoviral DNA molecules were transformed into these bacteria and λ recombinases were induced by heat shock at 42°C for 15 min. For isolation of single clones, bacteria were selected on agar plates containing the respective selection markers. The resulting BAC (BAC-AdV5) was used for further traceless genetic modification. We introduced capsid modifications resulting into the two cloned capsid–chimeric recombinant AdVs BAC-AdV5-Fib35 and BAC-AdV5-HVR48 (Figure 2b). BAC-AdV5-Fib35 contains fiber knob domains from human adenovirus type 35 (species B) allowing transduction of cells, which are not accessible for vectors with unmodified fibers such as hematopoietic stem cells (11,21). BAC-AdV5-HVR48 contains precisely exchanged HVRs from human adenovirus type 48 (species D) in the major capsid protein hexon and it was shown that this capsid modification escapes neutralizing anti-adenoviral antibodies directed against human adenovirus type 5 (10). For incorporation of fiber and hexon chimeric sequences, we transformed BAC-AdV5 into SW102 E. coli and for homologous recombination a PCR fragment containing the galK/Amp cassette expressing galactokinase and amplicillin selection markers was PCR-amplified with additional sequences at 5′ and 3′ ends homologous to respective fiber and hexon gene sequences. DNA sequences contained in the BACs were verified by PCR and diagnostic restriction enzyme pattern (Figure 2b) and respective viruses (AdV5-Fib35 and AdV5-HVR48) could be reconstituted in HEK293 cells.
Figure 2.
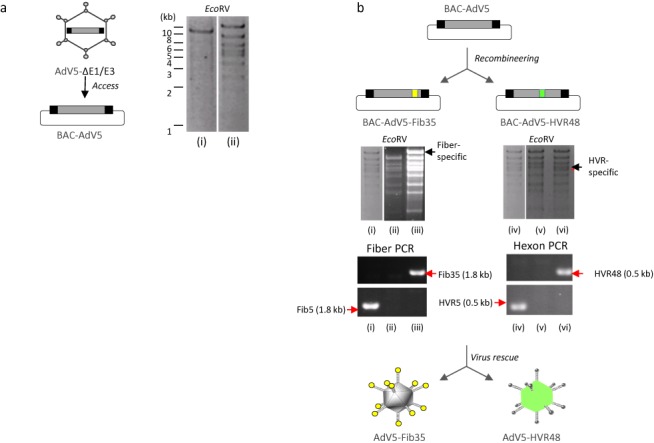
Cloning of E1/E3-deleted adenoviral genomes from virions (access) and traceless hexon and fiber genetic modification. (a) Cloning of a first generation E1/E3-deleted adenoviral genome from purified virions. Verification of the cloned adenovirus genome from purified virus particles as BAC showing the expected DNA fragmentation pattern after EcoRV restriction enzyme digest of isolated BAC-DNA. (i) Intermediate BAC containing galK, (ii) final BAC (BAC-AdV5) with cloned adenovirus genome. (b) Homologous recombination for generation of fiber- and hexon-modified E1/E3-deleted adenoviral genomes. For generation of the intermediate fiber-modified adenovirus BAC (BAC-AdV5-Fib-galK/Amp) and the intermediate hexon-modified adenovirus BAC (BAC-AdV5-HVR-galK/Amp), the sequence encoding the fiber knob and the hexon-derived HVRs were replaced by inserting the ampicillin resistance gene (Amp) as a positive-selection marker and galactokinase (galK) as a positive/negative-selection marker. For traceless modification, PCR-amplified DNA-fragments containing the coding sequence for the fiber knob from adenovirus type 35 and a synthesized DNA-fragment with HVRs precisely replaced with respective sequences of adenovirus type 48 were incorporated replacing the positive/negative selection marker combination. Integrity of final BACs (BAC-AdV5-Fib35 and BAC-AdV5-HVR48) was verified by EcoRV restriction enzyme digests and specific PCRs amplifying the respective modified capsid regions. (i) BAC-AdV5; (ii) BAC-AdV5-Fib-galK/Amp; (iii) BAC-AdV-fib5/35; (iv) original BAC-AdV5; (v) BAC-AdV5-HVR-galK/Amp; (vi) BAC-AdV-hex5/48. PCRs specific for unmodified and modified fiber (Fib5) and hexon (HVR5) genes amplify 1.8 and 0.5 kb fiber- and hexon-specific DNA fragments, respectively. After transfection of the linearized constructs into HEK293 cells, viruses (AdV5-Fib35 and AdV5-HVR48) were rescued.
A novel recombination pipeline for generation of complex adenoviral vectors
To further exploit our BAC-platform, we used capsid chimeric rAdVs as HV which carry a floxed packaging signal required for production of capsid modified HCAdVs encoding ß-galactosidase (Figure 3a). Respective HVs (AdV5-HV, AdV5-HV-Fib35, AdV5-HV-HVR48) were produced at normal titers and excision of the packaging signal was shown by PCR (Figure 3a). Subsequently, a HCAdV encoding ß-galactosidase was produced in the HCAdV producer cell line (116 cells) and functionality of the produced HCAdV (HCAdV5-lacZ, HCAdV5-Fib35-lacZ, HCAdV5-HVR48-lacZ) was demonstrated by transgene expression after infection (Figure 3a).
Figure 3.
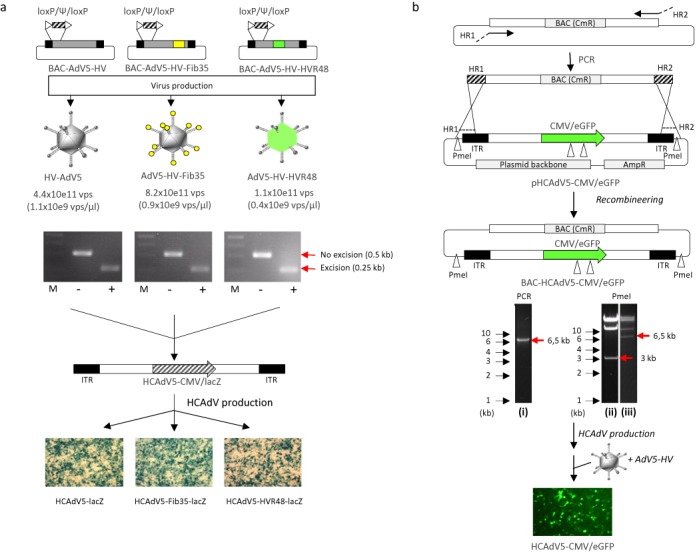
Exploring the BAC platform for cloning and production of HCAdV. (a) Generation of capsid-modified HV with a floxed packaging signal using the BAC platform. A floxed packaging signal (loxP/Ψ/loxP) was inserted into unmodified and capsid-modified BACs (BAC-AdV5, BAC-AdV5-Fib35, BAC-AdV5-HVR48) (see also Figure 2) by recombineering resulting into BAC-AdV5-HV, BAC-AdV5-HV-Fib35, and BAC-AdV5-HV-HVR48. Respective helper viruses were produced and excision of the packaging signal was shown by PCR. Agarose gels show PCR products after infection with HVs of cells either lacking Cre recombinase (−) or stably encoding Cre recombinase (+). HVs were used to produce a capsid modified HCAdV (HCAdV5-CMV-lacZ) lacking all viral coding sequences which contains a transgene expressing ß-galactosidase (lacZ) under the control of the cytomegalovirus (CMV) promoter. The lower panel shows ß-galactosidase of cells infected with unmodified (HCAdV-lacZ) and capsid-modified HCAdVs (HCAdV5-Fib35-lacZ and HCAdV5-HVR48-lacZ). ITR: adenoviral inverted terminal repeats, M: DNA ladder. (b) For available plasmids containing adenoviral genomes (e.g. the HCAdV genome), respective BACs can be generated by backbone-exchange, replacing the plasmid backbone with the BAC-specific backbone containing a chloramphenicol resistance marker gene (CmR). Here, the BAC-specific backbone was amplified by PCR utilizing primers to generate regions homologous to the 5′- and 3′-end of adenoviral genome (HR1 and HR2). For recombineering the purified PCR-product was transformed into SW102 bacteria with the plasmid containing the adenoviral genome. Bacteria with BACs containing the adenoviral genome were isolated by positive selection for chloramphenicol resistance. The left agarose gel shows amplification of the PCR-product used for transformation (i). Successful backbone-exchange was verified by PmeI digest (PmeI restriction enzyme recognition sites are indicated by white arrows) of the plasmid (ii) and the generated BAC (iii) containing the genome of a high-capacity AdV genome with an eGFP expression cassette (BAC-HCAdV5-CMV/eGFP). Note that the 700 bp band after PmeI digest is not visible on the agarose gel. Fragments specific for respective backbones are marked with red horizontal arrows. The corresponding HCAdV (HCAdV5-CMV/eGFP) was produced using the helper virus HV-AdV5 and infection of HEK293 cells with the purified HCAdV resulted in eGFP expression.
To enable genetic modification of HCAdV genomes which allow packaging of transgenes of up to 36 kb, we cloned a HCAdV genome as BAC by backbone exchange (Figure 3b). We used a plasmid carrying a HCAdV genome and simply exchanged the plasmid backbone with a BAC backbone which was amplified by PCR (Figure 3b). After recombineering integrity of the cloned BAC (BAC-HCAdV5-CMV/eGFP) was confirmed by diagnostic restriction enzyme pattern and the resulting HCAdV (HCAdV5-CMV/eGFP) was produced using a commonly used HV (AdV5-HV) based on adenovirus type 5 (Figure 3b).
To design complex expression systems to be delivered by AdV we established a novel DNA recombination pipeline only based on liquid bacteria culture enabling smooth combination of DNA sequences. This pipeline is based on iterative BAC cloning steps successively incorporating sequences coupled with alternating positive selection markers into the same BAC (Figure 4a). For that purpose, a PCR product is generated containing a positive selection marker and the first sequence of interest and transformed into bacteria with the target BAC. The second PCR product contains another positive selection marker and the second sequence of interest flanked by sequences, which are homologous to 5′- and 3′-sequences of the first selection marker. Homologous recombination and selection for the second selection marker enables efficient site-directed incorporation of the second sequence of interest into the BAC (Figure 4a and Supplementary Material section). This process can be repeated for fast combination of sequences within a single BAC. In addition, DNA can be isolated from liquid culture and utilized as template for PCR amplification. Therefore, sequences combined by iterative homologous recombination steps could be amplified by PCR and incorporated into another BAC. Combinations of sequences with alternating positive selection markers, which are necessary for the recombination pipeline, are either present in adequate plasmids or generated by subcloning, overlapping PCR or ligation of PCR-amplified sequences. Furthermore, iterative PCRs can be used to attach small sequences such as minimal polyadenylation signals or minimal internal ribosomal entry sites (IRES), to the amplified template-derived sequences. Therefore, the recombination pipeline enables construction of complex genome fragments by smooth modulation of basic BACs utilizing repetitive cloning steps without the need of isolation of intermediates.
Figure 4.
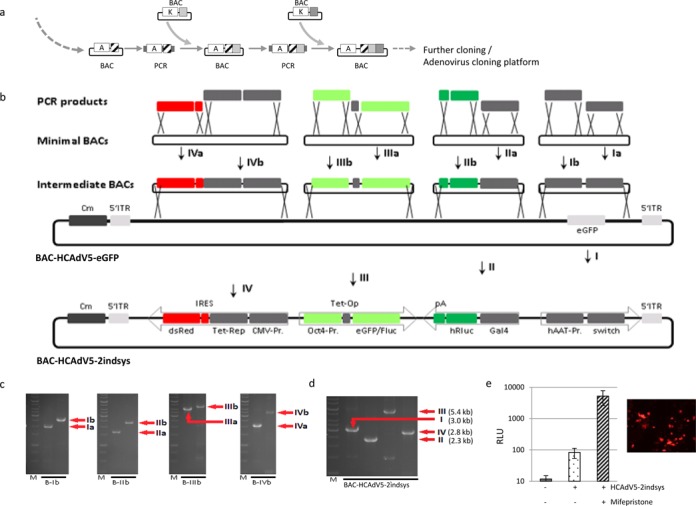
Generation of the complex high-capacity AdV HCAdV5–2indsys utilizing the novel recombination pipeline. (a) Recombination pipeline for cloning of complex transgenes solely based on liquid bacteria culture. Different DNA sequences can be sequentially combined using different selection markers (e.g. A: ampicillin, K: kanamycin). As a starting source of DNA a plasmid containing the DNA fragment of interest can be used and PCR performed containing an additional selection marker. In the next step, an additional DNA fragment can be added by insertion of a different selection marker. Using this method, complex DNA sequences can be assembled step by step. After recombineering, these assembled DNA sequences can get access to the BAC-platform or other applications. (b) Strategy for generation of the complex high-capacity adenovirus genome HCA-2indsys. The DNA recombination pipeline was used to generate four intermediate BACs containing different expression cassettes. Please note that positive selection markers are not shown. For each intermediate BAC, two sequences were combined, respectively (steps Ia-IVa and Ib-IVb). Furthermore, additional sequences were generated by extension of the respective PCR products (IIb: pA-signal, IIIa: Tet-Operator, IVa: IRES). Subsequently, expression cassettes encoded by the intermediate BACs were amplified by PCR incorporated into BAC-HCAdV5-CMV/eGFP resulting in the BAC BAC-HCAdV5–2indsys (steps I–IV). DNA sequences contained in the final BAC BAC-HCAdV5–2indsys are shown at the bottom. dsRed: red fluorescent protein, Tet-Rep: Tet-repressor, CMV-Pr.: cytomegalovirus promoter, Oct4: octamer transcription factor binding factor 4, eGFP: green fluorescent protein, Fluc: firefly luciferase, hRlus: renilla luciferase, Gal4: regulatory protein Gal4, hAAT-Pr.: alpha-1-antitrypsin promoter, switch: switch protein. (c) A specific PCR detecting the respective cloned DNA fragments was performed. DNA was isolated from liquid cultures of intermediate clones B-Ib–B-IVb and the final BAC BHCA-2indsys. Sizes of PCR products: Ia: 2.0 kb, Ib: 2.5 kb, IIa: 1.6 kb, IIb: 2.4 kb, IIIa: 4 kb, IIIb: 4.5 kb, IVa: 2.1 kb, IVb: 3.5 kb. (d) The final BAC BAC-HCAdV5-2indsys was analysed for inserted sequences by transgene specific PCRs (I–IV). (e) Functionality of complex vector genomes exemplified by HCAdV5-2indsys with multiple transgenes generated by the recombination pipeline. The HCAdV encodes dsRed (right panel) and mifepristone-inducible expression expressed as relative light units (RLU) in hepatocytes is shown in the left panel.
Here, we used this recombination pipeline to generate a complex HCAdV genome for transient cell marking and to track differentiation or dedifferentiation processes. Two independent inducible and tissue-specific expression systems were incorporated into the HCAdV genome resulting into the BAC and corresponding viral vector BAC-HCAdV5-2indsys and HCAdV5-2indsys, respectively (Figure 4b). The first tissue-specific inducible system consists of the switch protein under control of a liver-specific promoter construct containing the apolipoprotein E (ApoE) enhancer element in combination with the human α1-antitrypsin (hAAT) promoter and the Renilla luciferase gene under control of a hybrid promoter. This hybrid promoter contains the Saccharomyces cerevisiae GAL4 upstream activating sequences (UAS) linked to an adenoviral TATA box derived from the early adenoviral gene E1b (Figure 4b). The fusion protein pSwitch consists of the yeast GAL4 DNA binding domain (DBD), a truncated human progesterone receptor ligand binding domain (hPR-LBD), and the human p65 activation domain (AD) from NF-κB. Therefore, transduced hepatocytes express the switch protein and upon administration of mifepristone this protein is activated mediating specific transcription of the Gal4-promoter controlled Renilla luciferase. The second inducible system was realized by two expression cassettes encoding the Tet-suppressor protein under control of the CMV-promotor as well as an eGFP/Firefly luciferase fusion protein under control of the endogenous oct-4 promoter and four Tet-operator sequences (Figure 4b). Thus, after transduction of cells expressing the stem cell-specific transcription factor oct4, this factor can bind to the oct4-promoter. However, transcription is blocked by binding of highly expressed Tet-suppressor proteins to the Tet-operator sequences. Only in presence of doxycycline, which interacts with the Tet-suppressor protein and therefore inhibits binding to Tet-operator sequences, the fusion protein eGFP/FLuc is transcribed efficiently. In addition, fluorescence marker dsRed was coupled to constitutive expression of Tet-repressors via an IRES enabling detection of transduction rates. PCR products used for the recombination pipeline are shown in Figure 4c and the final BAC (BAC-HCAdV5–2indsys) was also verified by PCR BAC-specific PCRs (Figure 4d). After reconstitution of the respective virus HCAdV5-2indsys, we checked the liver-specific inducible system contained in the vector. As expected we found that transduced hepatocytes express the switch protein and upon administration of mifepristone this protein is activated mediating specific transcription from the Gal4-promoter (Figure 4e).
High-throughput generation of hexon-modified adenoviral vectors based on recombineering
Already a limited number of single hexon-modifications had a major impact on understanding adenovirus biology and uptake of AdV virions in vivo (10,22). However, over the recent years this research field was limited due to the lack of a reliable method to introduce hexon-modifications in a high-throughput manner. Thus to exploit the full potential of the adenoviral recombineering technology, chimeric hexon gene regions were generated in which either the HVRs were precisely exchanged or complete SDs were replaced by SDs derived from other human adenovirus types (Figure 5a). We synthesized coding sequences of hexons with HVRs precisely exchanged with respective sequences of human adenovirus types 12, 41, 44, 48 and 50. In parallel, a cloning system based on type II endonuclease was used to generate chimeric hexon genes replacing complete hexon SDs with respective sequences from human adenovirus types 4, 7, 12, 13 and 41 (Supplementary Figure S1). These chimeric hexon proteins form trimers in the presence of adenoviral chaperone 100K-protein (Supplementary Figure S1) which is a prerequisite for virion assembly. All modified chimeric hexon genes were incorporated into the genome of a E1/E3-deleted adenovirus type 5 genome by DNA recombineering (Figure 5b). To show that further genetic modification in subsequent steps is feasible, we exemplified this by inserting a marker gene expression cassette encoding firefly luciferase and eGFP into the E3 region of the hexon modified AdVs. The cloning strategy and characterization of the final BAC for virus reconstitution is exemplarily shown for the hexon modified AdV5-HV-HVR48 in Supplementary Figure S2a and Sb. Furthermore, the cloned hexon-modified viruses contained a floxed packaging signal enabling production of respective hexon-modified HCAdVs. Final constructs were analyzed by restriction enzyme digests resulting in a specific pattern of fragments of respective viral genomes (Supplementary Figure S2c). Furthermore, incorporated hexon modifications were verified by PCRs specific for respective modifications (Figure 5c) and in parallel contaminations with unmodified helper-virus BACs were excluded by PCR analysis with primers specific for the unmodified hexon gene (Figure 5d).
Figure 5.
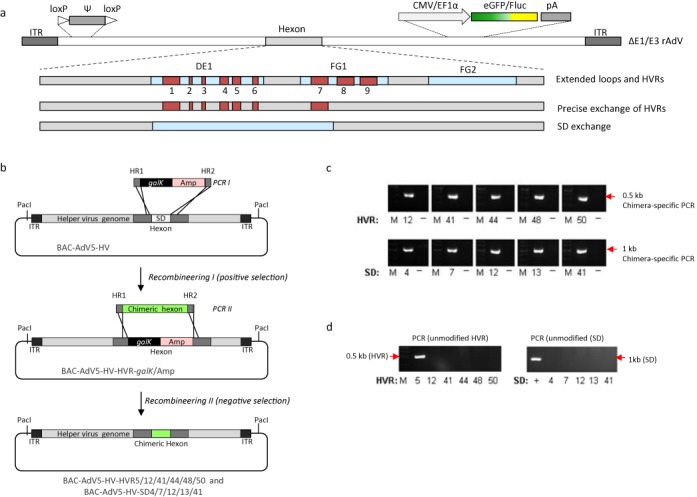
Incorporation of chimeric hexon genes into E1 and E3 deleted adenoviral HV genomes embedded in BACs. (a) A scheme of precise exchanges of HVRs 1–7 and SDs in the adenoviral capsid is shown. DE1, FG1 and FG2 display extended loops. The packaging signal is flanked by Cre recombinase recognition sites (loxP/Ψ/loxP) and the HV contains a bicistronic transgene encoding firefly luciferase (Fluc) and eGFP. (b) Hexon-modified helper-virus genomes were generated by traceless modulation of a BAC utilizing homologous recombination based on the PCR I based product which was flanked by homologous regions HR1 and HR2. The positive–negative selection marker cassette containing galactokinase and ampicillin (galK/Amp) was incorporated into BAC-AdV5-HV containing the helper-virus genome replacing the region encoding SDs of the hexon gene. Intermediate clone BAC-AdV5-HV-HVR-galK/Amp was isolated after selection with ampicillin. Subsequently, PCR-amplified chimeric hexon sequences (sequences for chimeric hexon genes containing HVRs and SDs from other adenovirus types) replaced the selection cassette and final BACs were obtained upon selection against galactokinase. (c) Incorporation of chimeric hexon sequences was verified by specific PCRs detecting the respective chimeric hexon. The original BAC-AdV5-HV was used as negative control. (d) Contaminations with unmodified helper-virus BACs were excluded by PCR specifically detecting the SDs of the original hexon from adenovirus type 5. Unmodified BAC-AdV5-HV as well as the revertant BAC-AdV5-HV-HVR5 were used as a positive control. M: 1 kb DNA ladder.
For reconstitution of hexon modified viruses, linearized BACs with chimeric hexon-modified adenoviral genomes were transfected into HEK293 cells. We successfully rescued hexon-modified viruses AdV5-HV-HVR12, AdV5-HV-SD12 and AdV5-HV-SD4 (Figure 6a). We checked stability of hexon-modified genomes after virus amplification and purification by PCR specifically detecting hexon chimeras (Figure 6b) and we verified hexon sequences by sequencing. Viral particle numbers (Figure 6c) and infectious titers of final vector preparations were comparable to unmodified virus as demonstrated by measuring eGFP expression and luciferase expression levels after infection (Figure 6d and e).
Figure 6.
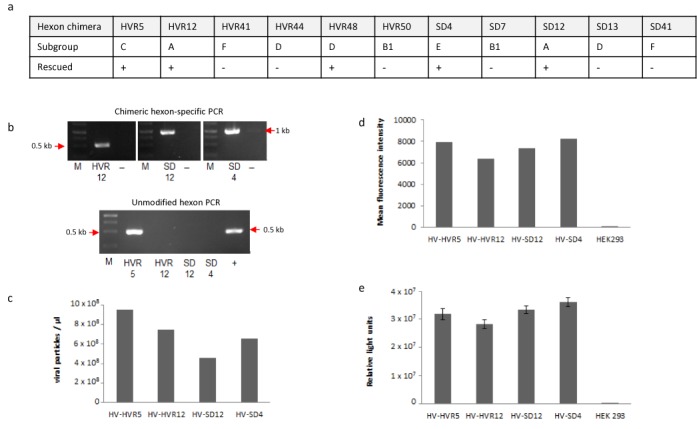
Rescue and characterization of hexon chimeric adenoviruses. (a) Summary of cloned hexon chimeric helper viruses and the adenovirus subgroup these hexon modifications belong to. (+): Rescue of adenovirus successful; (−) rescue of recombinant unsuccessful. (b) Characterization of final preparations of reconstituted hexon-modified helper-viruses. Vector genomes derived from purified viral particles of final vector preparations of AdV5-HV-HVR12, AdV-HV-SD12 and AdV-HV-SD4 were analyzed by PCRs specific for respective hexon-modification to verify chimeric hexon genes. The lower panel shows PCR reactions specific for unmodified hexon gene to exclude contamination. (c) Physical titers (viral particles per μl) for these vector preparations. (d) Functionality of reporter gene eGFP-FLuc was tested by infection of HEK293 cells (1 μl each). eGFP expression was determined by flow cytometry and (e) by luciferase assay for firefly luciferase activity.
DISCUSSION
Here we introduced a novel BAC platform utilizing traceless cloning strategies enabling smooth design of arbitrary DNA-modified adenoviruses. We demonstrate that this BAC cloning technology is a suitable method for rapid assembly of recombinant adenoviral genomes containing a variety of mutations at a single genomic location (Figure 7). A flow chart of the technology and a general time line are schematically shown in Supplementary Figure S3. This allows switching between different adenovirus types such as cloned wild type viruses, E1- and E1/E3-deleted AdVs, HCAdVs including their helper viruses for HCAdV production, and oncolytic rAd used in biotechnology, preclinical and clinical studies.
Figure 7.
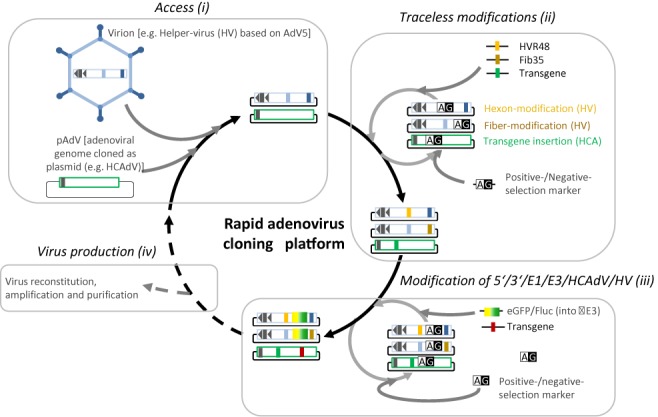
Rapid adenovirus cloning platform for generation of arbitrary genetically modified adenoviruses in a high-throughput manner. Shown is the BAC-platform allowing (i) access to different sources of adenovirus DNA, (ii) arbitrary traceless genetic modification of cloned viral genomes and (iii) further modifications at the 5′ and 3′ ends of the adenoviral genome, at the early adenoviral genes E1 and E3 regions, within a HCAdV genome and cloned HV used for production of high-capacity AdVs. After production (iv) and characterization of recombinant adenoviruses, these can be further modified using the BAC-pipeline from the beginning. The cloning procedure is based on different selection markers such as galK (G) which can be used for positive and negative selection and other resistance genes such as ampicillin (A). Cloned recombinant adenoviruses can either be used for virus production or if desired further modified using the BAC-platform. Access to different sources of adenovirus DNA is exemplified by cloning of HV from adenovirus virions and HCAdV from a plasmid. Traceless modification is exemplified by hexon-modification (HVR48: hexon from adenovirus types 5 with HVRs from adenovirus type 48), fiber-modification (Fib35: fiber from adenovirus type 5 with fiber knob from adenovirus type 35), and modification of HCAdV by adding a transgene. Further modification of the adenovirus genome is exemplified by replacing E3 with a transgene encoding eGFP and firefly luciferase (Fluc) and by adding an additional transgene to the HCAdV genome.
Compared to conventional cloning techniques our recombineering platform is less time-consuming and challenging construction of intermediate clones with DNA flanked by long homologies or endonuclease binding sites can be avoided. In combination with the recombination pipeline the adenoviral genome can be directly modified in a stepwise and traceless manner. Commonly used techniques for generation of recombinant adenoviral genomes are based on either rare endonucleases or homologous recombination relying on long homologous sequences (>200 bp) (15) in mammalian cells or bacteria of E. coli strain BJ5183 (12–14). In contrast our recombineering technology allows efficient homologous recombination based on small homologous regions (<50 bp), which can be generated by PCR utilizing appropriate primers. As another major advantage our technique allows getting direct access to complete adenoviral genomes which can be cloned as BACs in a single step. In the present study we focused on cloning and engineering of human adenovirus genomes. However, it needs to be emphasized that our technology can also be used to clone adenoviruses from other species.
Furthermore, a newly invented recombination pipeline was demonstrated to allow complex modulations of high-capacity adenoviral genomes. It is of note that utilization of the recombination pipeline could be limited by the size of the PCR product. However, it was shown that amplification of PCR products of up to 10 kb is feasible when utilizing specific proof-reading polymerases. Nevertheless, larger sequences such as the dystrophin gene used for treatment of patients with muscular dystrophy (23) could be split in two fragments and sequentially incorporated into the target BAC. Furthermore, incorporation of sequences with homologies to the target BAC smaller than 50 bp appeared to be challenging due to unspecific recombination events. Especially with respect to sequences often used in expression cassettes such as polyadenylation signals and some promoter- or enhancer-elements, careful planning of the construction procedure is essential to avoid problems caused by this phenomenon. In contrast, two homologous regions of more than 500 bp, which were part of the same BAC, were stable even after heat-induction of recombinases (data not shown).
To reconstitute hexon-modified HV, genomes were excised from respective HV BACs and HEK293 cells were transfected with linear adenoviral DNA. After cytopathic effect was observed, cells were harvested and used for further amplification steps. However, reconstitution remained challenging, because no stable protocol could be established and no clear bottleneck could be identified for the reconstitution procedure. Purity of linearized viral genome DNA utilized for transduction of adenovirus producer cells as well as high transfection efficiencies may represent critical factors. Nevertheless, several viral vectors with exchanged HVRs as well as with completely replaced SDs were reconstituted, indicating that not the size of the inserted sequence but rather the protein folding of the chimeric hexon could play a crucial for reconstitution (24). In concordance with this assumption, complete trimerization of protein chimeras in cell culture demonstrated, that steric hindrance did not inhibit early assembly steps and that hexon trimer stability was not significantly reduced. Furthermore, serotype-specific HVR sequences are similar in size in comparison with the original sequence derived from human adenovirus type 5. Therefore, steric hindrance causing inhibition of interactions between hexon trimers and neighboring hexon trimers or other proteins of the capsid shell is unlikely.
In summary, we introduced improved techniques based on recombineering allowing arbitrary genetic modification of AdVs. We developed a method which allows introducing any traceless genetic modification (insertion, deletion or replacement of viral DNA) at any location of the adenovirus genome. In this study we cloned numerous AdVs with different features underlining the potential for high-throughput applications. Our method enables efficient generation of AdV for applications in biotechnology and in therapeutic and vaccination approaches. It may pave the way towards finding novel insights into adenovirus biology and developing improved therapeutic AdVs including AdVs for genome editing based on HCAdVs (25). Therefore, new resources are opened for current approaches in gene therapy or vaccination as well as for new areas like stem cell technology, tissue engineering or basic research.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors would like to thank Philip Ng (Baylor College of Medicine, Houston, TX, USA) for providing the producer cell line 116 and the original helper virus for high-capacity adenovirus production.
Author contributions: M.M.-H., M.S., W.Z. and A.E. designed and performed experiments. M.M.-H. performed the majority of experiments. Z.R. introduced the initial BAC cloning technology to us and provided respective material. A.E. wrote the manuscript.
Footnotes
Present address: Martin Mück-Häusl, Institute of Virology, Helmholtz Zentrum München, Munich, Germany.
FUNDING
Deutsche Forschungsgemeinschaft (DFG) [EH 192/5-1]; EU Framework Programme 7 (Persist, Project Nr. 222878) (to A.E.). Funding for open access charge: University Witten/Herdecke.
Conflict of interest statement. None declared.
REFERENCES
- 1.Sullivan D.E., Dash S., Du H., Hiramatsu N., Aydin F., Kolls J., Blanchard J., Baskin G., Gerber M.A. Liver-directed gene transfer in non-human primates. Hum. Gene Ther. 1997;8:1195–1206. doi: 10.1089/hum.1997.8.10-1195. [DOI] [PubMed] [Google Scholar]
- 2.Benihoud K., Yeh P., Perricaudet M. Adenovirus vectors for gene delivery. Curr. Opin. Biotechnol. 1999;10:440–447. doi: 10.1016/s0958-1669(99)00007-5. [DOI] [PubMed] [Google Scholar]
- 3.Lusky M., Christ M., Rittner K., Dieterle A., Dreyer D., Mourot B., Schultz H., Stoeckel F., Pavirani A., Mehtali M. In vitro and in vivo biology of recombinant adenovirus vectors with E1, E1/E2A, or E1/E4 deleted. J. Virol. 1998;72:2022–2032. doi: 10.1128/jvi.72.3.2022-2032.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parks R.J., Chen L., Anton M., Sankar U., Rudnicki M.A., Graham F.L. A helper-dependent adenovirus vector system: removal of helper virus by Cre-mediated excision of the viral packaging signal. Proc. Natl. Acad. Sci. U.S.A. 1996;93:13565–13570. doi: 10.1073/pnas.93.24.13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palmer D., Ng P. Improved system for helper-dependent adenoviral vector production. Mol. Ther. 2003;8:846–852. doi: 10.1016/j.ymthe.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 6.Jager L., Hausl M.A., Rauschhuber C., Wolf N.M., Kay M.A., Ehrhardt A. A rapid protocol for construction and production of high-capacity adenoviral vectors. Nat. Protoc. 2009;4:547–564. doi: 10.1038/nprot.2009.4. [DOI] [PubMed] [Google Scholar]
- 7.Morral N., Parks R.J., Zhou H., Langston C., Schiedner G., Quinones J., Graham F.L., Kochanek S., Beaudet A.L. High doses of a helper-dependent adenoviral vector yield supraphysiological levels of alpha1-antitrypsin with negligible toxicity. Hum. Gene Ther. 1998;9:2709–2716. doi: 10.1089/hum.1998.9.18-2709. [DOI] [PubMed] [Google Scholar]
- 8.Muruve D.A., Cotter M.J., Zaiss A.K., White L.R., Liu Q., Chan T., Clark S.A., Ross P.J., Meulenbroek R.A., Maelandsmo G.M., et al. Helper-dependent adenovirus vectors elicit intact innate but attenuated adaptive host immune responses in vivo. J. Virol. 2004;78:5966–5972. doi: 10.1128/JVI.78.11.5966-5972.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ehrhardt A., Xu H., Dillow A.M., Bellinger D.A., Nichols T.C., Kay M.A. A gene-deleted adenoviral vector results in phenotypic correction of canine hemophilia B without liver toxicity or thrombocytopenia. Blood. 2003;102:2403–2411. doi: 10.1182/blood-2003-01-0314. [DOI] [PubMed] [Google Scholar]
- 10.Roberts D.M., Nanda A., Havenga M.J., Abbink P., Lynch D.M., Ewald B.A., Liu J., Thorner A.R., Swanson P.E., Gorgone D.A., et al. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature. 2006;441:239–243. doi: 10.1038/nature04721. [DOI] [PubMed] [Google Scholar]
- 11.Shayakhmetov D.M., Papayannopoulou T., Stamatoyannopoulos G., Lieber A. Efficient gene transfer into human CD34(+) cells by a retargeted adenovirus vector. J. Virol. 2000;74:2567–2583. doi: 10.1128/jvi.74.6.2567-2583.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chartier C., Degryse E., Gantzer M., Dieterle A., Pavirani A., Mehtali M. Efficient generation of recombinant adenovirus vectors by homologous recombination in Escherichia coli. J. Virol. 1996;70:4805–4810. doi: 10.1128/jvi.70.7.4805-4810.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McGrory W.J., Bautista D.S., Graham F.L. A simple technique for the rescue of early region I mutations into infectious human adenovirus type 5. Virology. 1988;163:614–617. doi: 10.1016/0042-6822(88)90302-9. [DOI] [PubMed] [Google Scholar]
- 14.Mizuguchi H., Kay M.A. Efficient construction of a recombinant adenovirus vector by an improved in vitro ligation method. Hum. Gene Ther. 1998;9:2577–2583. doi: 10.1089/hum.1998.9.17-2577. [DOI] [PubMed] [Google Scholar]
- 15.Degryse E. In vivo intermolecular recombination in Escherichia coli: application to plasmid constructions. Gene. 1996;170:45–50. doi: 10.1016/0378-1119(95)00858-6. [DOI] [PubMed] [Google Scholar]
- 16.Warming S., Costantino N., Court D.L., Jenkins N.A., Copeland N.G. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005;33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stanton R.J., McSharry B.P., Armstrong M., Tomasec P., Wilkinson G.W. Re-engineering adenovirus vector systems to enable high-throughput analyses of gene function. Biotechniques. 2008;45:664–668. doi: 10.2144/000112993. [DOI] [PubMed] [Google Scholar]
- 18.Ruzsics Z., Lemnitzer F., Thirion C. Engineering adenovirus genome by bacterial artificial chromosome (BAC) technology. Methods Mol. Biol. 2014;1089:143–158. doi: 10.1007/978-1-62703-679-5_11. [DOI] [PubMed] [Google Scholar]
- 19.Copeland N.G., Jenkins N.A., Court D.L. Recombineering: a powerful new tool for mouse functional genomics. Nat. Rev. Genet. 2001;2:769–779. doi: 10.1038/35093556. [DOI] [PubMed] [Google Scholar]
- 20.Sharan S.K., Thomason L.C., Kuznetsov S.G., Court D.L. Recombineering: a homologous recombination-based method of genetic engineering. Nat. Protoc. 2009;4:206–223. doi: 10.1038/nprot.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shayakhmetov D.M., Carlson C.A., Stecher H., Li Q., Stamatoyannopoulos G., Lieber A. A high-capacity, capsid-modified hybrid adenovirus/adeno-associated virus vector for stable transduction of human hematopoietic cells. J. Virol. 2002;76:1135–1143. doi: 10.1128/JVI.76.3.1135-1143.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waddington S.N., McVey J.H., Bhella D., Parker A.L., Barker K., Atoda H., Pink R., Buckley S.M., Greig J.A., Denby L., et al. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell. 2008;132:397–409. doi: 10.1016/j.cell.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 23.Kawano R., Ishizaki M., Maeda Y., Uchida Y., Kimura E., Uchino M. Transduction of full-length dystrophin to multiple skeletal muscles improves motor performance and life span in utrophin/dystrophin double knockout mice. Mol. Ther. 2008;16:825–831. doi: 10.1038/mt.2008.23. [DOI] [PubMed] [Google Scholar]
- 24.Bradley R.R., Maxfield L.F., Lynch D.M., Iampietro M.J., Borducchi E.N., Barouch D.H. Adenovirus serotype 5-specific neutralizing antibodies target multiple hexon hypervariable regions. J. Virol. 2012;86:1267–1272. doi: 10.1128/JVI.06165-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holkers M., Maggio I., Henriques S.F., Janssen J.M., Cathomen T., Goncalves M.A. Adenoviral vector DNA for accurate genome editing with engineered nucleases. Nat. Methods. 2014;11:1051–1057. doi: 10.1038/nmeth.3075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.