


Abstract
Transformation-associated recombination (TAR) protocol allowing the selective isolation of full-length genes complete with their distal enhancer regions and entire genomic loci with sizes up to 250 kb from complex genomes in yeast S. cerevisiae has been developed more than a decade ago. However, its wide spread usage has been impeded by a low efficiency (0.5–2%) of chromosomal region capture during yeast transformants which in turn requires a time-consuming screen of hundreds of colonies. Here, we demonstrate that pre-treatment of genomic DNA with CRISPR-Cas9 nucleases to generate double-strand breaks near the targeted genomic region results in a dramatic increase in the fraction of gene-positive colonies (up to 32%). As only a dozen or less yeast transformants need to be screened to obtain a clone with the desired chromosomal region, extensive experience with yeast is no longer required. A TAR-CRISPR protocol may help to create a bank of human genes, each represented by a genomic copy containing its native regulatory elements, that would lead to a significant advance in functional, structural and comparative genomics, in diagnostics, gene replacement, generation of animal models for human diseases and has a potential for gene therapy.
INTRODUCTION
Transformation-associated recombination (TAR)-cloning is a unique method for isolating any large chromosomal region from mammalian genomes using yeast Saccharomyces cerevisiae without constructing a genomic library of random clones (1,2). TAR cloning is based on in vivo homologous recombination between a specific genome target and a linearized TAR cloning vector that contains terminal sequences (hooks) homologous to the targeted region. In TAR cloning, total genomic DNA is co-transformed into yeast cells along with a vector carrying the targeting sequences specific to the gene of interest. Upon co-transformation into yeast, homologous recombination occurs between the vector’ hooks and targeted genomic sequences flanking the gene of interest to form a circular YAC (Yeast Artificial Chromosome). This YAC readily propagates, segregates and can be selected for in yeast. TAR cloning produces YAC clones containing the desired insert at a frequency of 0.5–2% of all clones screened. Several dozen of human genes and specific chromosomal regions with sizes ranging to 250 kb have been isolated by TAR for functional and structural analyses (1–6). TAR cloning was also applied to characterize genome variations, including polymorphic structural rearrangements, mutations, evolution of genes and gene families and long-range haplotyping (1).
In this work, we describe a new protocol that greatly increases the efficiency of TAR cloning. We propose that in the standard method of genomic DNA preparation, the gene-specific sequence is represented within the transformation mixture as a population of overlapping DNA fragments, formed by random shearing of the genomic DNA during its isolation and manipulation. Consequently the distance between the targeted sequences and the DNA ends varies between DNA fragments. Probabilistically both 5′ and 3′ DNA ends of a desired fragment are unlikely to be near the targeted sequences.
In our previous works (1), we observed that homologous recombination is much more efficient between TAR vector hooks and targeted genomic sequences located closer to DNA ends compared to internally imbedded target sequences. Hence, we expect an increase in gene capture efficiency if double-stand breaks (DSBs) could be specifically introduced close (>100 bp) to the ends of the desired genomic fragment. In principle, these specific DSBs could be introduced in the regions flanking the gene of interest by rare cutting restriction enzyme(s). However, such an approach is very limited. In our attempts to clone genes larger than 40 kb from the human and other mammalian genomes, we found it practically impossible to choose suitable restriction enzymes that cleave near the 5′ and 3′ ends of a targeted gene without making additional cuts within the gene itself.
What we needed was a programmable endonuclease that could cleave at a user-defined sequence. A review of currently available commercial technologies yielded three possible candidates: ZFNs (engineered zinc-finger nucleases), TALENs (transcription activator-like effector nucleases) and CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats that are recognized by Cas9 nuclease, an enzyme specialized for cutting DNA, with two active cutting sites, one for each strand of the double helix) nucleases (7–10). Of these three, CRISPR-Cas9 technology is by far the cheapest and easiest to use. Cas9 is a family of bacterial, RNA-guided, double-stranded DNA endonucleases employed by type II CRISPR systems (7,8). In current art, the DNA target specificity of Cas9 is encoded by a 20 bp guide sequence located on the 5′ terminal of the gRNA, a small synthetic chimera of mature crRNA and tracrRNA, which is bound by Cas9. Target sequence recognition is mediated by RNA–DNA base pairing between the gRNA to the DNA target and an adjacent downstream DNA motif (NGG), the protospacer adjacent motif (PAM). As the gRNA's 5′ terminus can be cheaply and quickly modified using polymerase chain reaction (PCR) with synthetic oligomers, the Cas9 nuclease can be easily directed to any ∼20 bp sequence downstream of a PAM motif within complex genomes. It is thus not surprising that Cas9 has rapidly gained prominence in the field of genome editing. Recently programmable DNA cleavage by Cas9 has been demonstrated with purified genomic DNA (11).
To demonstrate the utility of Cas9 endonucleases in TAR cloning, we chose to clone the human NBS1 gene, mutations in which leads to Nijmegen breakage syndrome (NBS; MIM 251260). Human genomic DNA prepared in aqueous solution was used for TAR cloning as a source of the NBS1 sequence. The size of molecules in this DNA preparation varies between 30 and 200 kb. Previously we successfully used high-molecular DNA prepared from different human cell lines purified in aqueous solution or commercial human genomic DNA (Promega) for isolation of several genes with sizes up to 150 kb (1,12,13).
MATERIALS AND METHODS
Design of the CRISPR guide sequence
The DNA sequences, 1 kb upstream of the 5′ TAR-hook and 1 kb downstream of the 3′ TAR-hook sequences, were downloaded from USCS (http://genome.ucsc.edu/cgi-bin/hgGateway). The CRSIPR design tool (http://crispr.mit.edu/) was used to identify CRISPR guide sequences within the two 1 kb regions beginning with the 250 bp nearest to the hooks. This tool yields CRISPR guide sequences of three different qualities. Only high quality CRISPR guide sequences were considered for use. As an in-vitro transcription kit (New England BioLabs, cat. number # E2040S) based on the T7 RNA polymerase was later used, the CRISPR guide sequence had to conform to the rules governing efficient and precise initiation of the T7 promoter. Hence, the CRISPR guide must start with a ‘G’ (guanine), followed by a purine (G/A), with ‘G’ being significantly more preferred, and finally followed by an optional third ‘G’ (14). Thus, the CRISPR guide should start with ‘5-GGN’. A CRISPR guide sequence beginning with two ‘G's would normally be difficult to obtain. However, it has been reported that a ‘G’ may be added to the 5′ end of a 20 bp CRISPR guide sequence, forming a 21 bp sequence without ill effect on targeting (15). Additionally, it has also been reported that improved CRISPR-Cas9 specificity may be obtained by truncating the 20 bp guide to a sequence as short as 17 bp (16). Together, these two papers suggest that the 5′ terminal end of the CRISPR guide has sufficient flexibility to be modified for efficient transcription by the T7 promoter. Fortunately, in this study only one CRISPR guide sequence, gRNA-C, required a single base pair substitution (+1A→G) to make it conform to the ‘5-GG’ rule.
We then discarded all CRISPR guide sequences that had off target sites within the gene of interest. The off target sites are given by the CRSIPR design tool. We also used a Blat search (http://genome.ucsc.edu/cgi-bin/hgBlat) to further validate that the CRISPR guide candidates did not have other homologies within the gene of interest. Preference was given to the CRISPR guides located closest to the TAR-hooks and at least two CRISPR guides were selected for each TAR hook position. This gives redundancy should one guide be inefficient at DNA cleavage. The CRISPR guides for the 5′ TAR hook were named gRNA-5A and gRNA-5B, while the guides at 3′ TAR hook were named gRNA-3C and gRNA-3D. The cleavage sites of the CRISPR guides relative to the NBS1 gene are shown in Figure 1.
Figure 1.
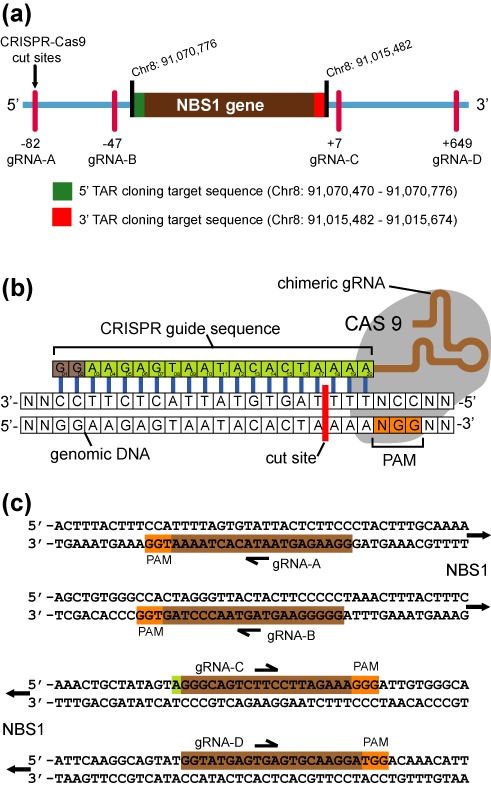
Locations of CRISPR guide induced cut sites relative to the NBS1 gene. (a) The NBS1-specific fragment is located at Chr8: 91,015,482–91,070,776 [Mar 2006 (NSB136/hg18)]. The sequences targeted by TAR cloning vector hooks are as indicated, 5′ target hook is in green while 3′ target hook is in red. The cut sites of Cas9-gRNA complexes A, B, C and D upstream and downstream of the hooks with their positions toward the hooks are as indicated. (b) Schematic representation of Cas9-gRNA binding to DNA. The CRISPR guide sequence is cloned into the 5′ end of the gRNA molecule. The guide sequence is any 20 bp DNA sequences upstream of a NGG, the Protospacer Adjacent Motif (PAM). The Cas9 cut site is 3 bp upstream from PAM. (c) Guide sequences used in this study are illustrated.
Synthesis of gRNA minigene
Next, the forward and reverse CRISPR guide primers were designed with the aid of an oligonucleotide calculator (http://www.basic.northwestern.edu/biotools/oligocalc.html). Melting temperatures of the primers were approximately 59°C. The forward guide primer consists of a 12 bp long 5′ guard sequence, followed by the T7 promoter sequence, then the CRISPR guide sequence and finally the template sequence to the gRNA from the pX330-U6-Chimeric_BB_CBh-hSpCas9 plasmid (Addgene, cat. number # 42230) (Supplementary Figure S1). The forward and reverse primers used to make the CRISPR minigenes are listed in Supplementary Table S1. In addition, primers were also designed to amplify a 1–2 kb region around the CRISPR guide target sites (Supplementary Table S1). This DNA fragment was later used to determine the cleavage efficiency of the Cas9-gRNA complexes (Figure 2a). This target DNA fragment should be designed such that upon Cas9-gRNA cleavage, it will yield two DNA fragments of distinct sizes. Furthermore, to avoid gRNA from hindering DNA visualization on the gel, the smallest DNA fragment must be no smaller than 400 bp.
Figure 2.
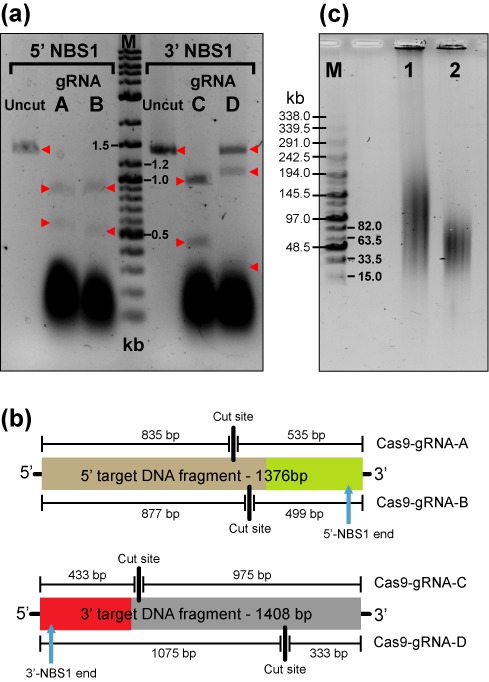
Efficiency of nuclease activity of different Cas9-gRNA complexes. (a) Cas9 was complexed with four gRNA created in this study, gRNA-A, gRNA-B, gRNA-C or gRNA-D and allowed 3 h to digest 200 ng of PCR amplified target DNA. Lane 1 is uncut 1376 bp 5′ target DNA fragment. Lane 2 is Cas9-gRNA-A cut 5′ target DNA, expected fragments are 835 bp and 535 bp. Lane 3 is Cas9-gRNA-B cut 5′ target DNA, expected fragments are 877 bp and 499 bp. Lane 4 is Marker which is 8 μl of DNA ladder (New England, BioLabs, N3200L). Lane 5 is uncut 1408 bp 3′ target DNA fragment. Lane 6 is Cas9-gRNA-C cut 3′ target DNA, expected fragments are 975 bp and 433 bp. Lane 7 is Cas9-gRNA-D cut 3′ target DNA, expected fragments are 1075 bp and 333 bp. (b) Cleavage positions of Cas9-gRNA complexes within their respective target DNA fragments and the expected fragment sizes generated after digest. Only Cas9 matched with gRNA-D failed to completely digest its target in this time frame, even though it was gRNA-C that has a (+1A→G) substitution. Cas9-gRNA-B and Cas9-gRNA-C pair was chosen for further experiments. (c) A contour-clamped homogeneous electric field (CHEF) electrophoresis was run to resolve the fragment size of the human genomic DNA Cas9-treared and untreated used in TAR cloning. Lane 1 is 1μg of uncut human genomic DNA (Promega, cat. #G304A). Lane 2 is 1μg of human genomic DNA digested overnight with 1 μl of Cas9 enzyme, 1 μl of gRNA-B and 1μl of gRNA-C in a total volume of 40 μl. M corresponds to the MidRange PFG Marker I (New England BioLabs).
Next, the CRISPR minigenes were made by PCR amplification using proof reading Phusion polymerase (New England BioLabs, cat. number # M0530S), the relevant pair of CRISPR guide primers (Supplementary Table S1) and 100 ng of XbaI-linearized pX330-U6-Chimeric_BB_CBh-hSpCas9 plasmid as a template. The PCR conditions used for all CRISPR guides are as follows: initial denaturation at 98°C for 30 s, denaturation at 98°C for 7 s, annealing at 62°C for 5 s, extension at 72°C for 10 s, 25 cycles. Final extension was at 72°C for 1 min. PCR mixture composition is as follows: 1.6 μl dNTPs (2.5 mM), 4.0 μl normal Phusion buffer, 1.0 μl template DNA (100 ng), 1.0 μl 10 μM primer F, 1.0 μl 10 μM primer R, 0.2 μl Phusion polymerase, 11.2 μl water. Total volume of the reaction mix was 20 μl. Four PCR replicates of each CRISPR minigene were made to give a sufficient material to allow any loses during subsequent steps.
Synthesis and test of gRNA-CRISPR complex
Two target DNA fragments, 5′ (1376 bp) and 3′ (1408 bp), were PCR amplified from human genomic DNA (Promega, cat. number # G3041) using Phusion polymerase and standard conditions recommended by the manufacturer. The primers used for amplification of the fragments are listed in Supplementary Table S1. The target DNA fragments contain the Cas9 cut sites and were used to determine the cutting efficiency of Cas9-gRNA complex. The target DNA fragments were gel purified from 1% agarose gel and isolated using gel extraction kit (Qiagen, cat. number # 28704) as per manufacturer protocols. The minigenes were purified from 3% agarose gel at 145 V (maximum voltage setting). To limit UV damage to the minigene DNA, guanosine (Sigma-Aldrich, cat. number # G6752) was added to the molten agarose gel at a final concentration of 1 mM, the UV transluminator was set to a long wave length setting and the desired DNA bands excised quickly from the gel. To avoid cross contamination, a new sterile scalpel blade was used for each DNA band. The minigenes were isolated from the agarose using gel extraction kit (Qiagen, cat. number # 28704). Each agarose plug was dissolved in 5 x volume of QG buffer at 50°C. For every 300 μl of gel-QG solution, 100 μl of isopropanol was added. The mixture was vortexed and passed through the column twice. Then the column was washed with 750 μl PE solution, and spun dried for 2 min at 14 000 rpm. 40 μl of warm EB buffer was added and the column was left to incubate at 50°C for 2 min. The column was then spun at 14 000 rpm to elute the minigene DNA solution.
Next, between 60 ng and 100 ng of minigene DNA was used as a template to transcribe CRISPR gRNA, using an in-vitro transcription kit (New England BioLabs, cat. number # E2040S). The buffer composition was: 2 μl ATP (100 mM), 2 μl GTP (100 mM), 2 μl CTP (100 mM), 2 μl UTP (100 mM), 2 μl reaction buffer, 2 μl T7 RNA polymerase mix, 5 μl minigene template, 10 μl RNAse/DNAse free water. The total volume of the reaction mixture was 27 μl. The reaction was incubated overnight at 37°C. The mixture was frozen at −20°C until use. No further purification was made.
The cleavage efficiency of each gRNA-Cas9 pair was evaluated by digesting ∼200 ng of the appropriate target DNA fragment. The digest composition was as follows: 1 μl of Cas9 (New England BioLabs, cat. number # M0386S), 3 μl CAS9 buffer, ∼4500 ng (1 μl) gRNA, 200 ng target DNA fragment and a sufficient water to attain a total volume of 30 μl. After 3 h, the digest was run directly on a 1.5% agarose gel and later stained with ethidium bromide. Based on our results, two pairs of nucleases could be equally chosen, i.e. either Cas9-gRNA-B and Cas9-gRNA-C or Cas9-gRNA-A and Cas9-gRNA-C (Figure 2a). Cas9-gRNA-D that did not completely digest the target DNA fragment was discarded. For our experiment we have chosen Cas9-gRNA-B and Cas9-gRNA-C pair. The predicted size of the target fragments after cutting by Cas9-gRNA-B and Cas9-gRNA-C are 499 bp/877 bp and 433 bp/975 bp, correspondingly.
Preparation of CRISPR-treated genomic DNA
Genomic DNA for TAR cloning was then cleaved using Cas9 with a gRNA-B for the 5′ and gRNA-C for 3′ TAR-hooks. The digest composition was as follows: 1 μg (5 μl) human genomic DNA, 1 μl of Cas9 (New England BioLabs, cat. number # M0386S), ∼4500 ng (1 μl) gRNA-5B for the 5′TAR hook, ∼4500 ng (1μl) gRNA-3C for the 3′TAR hook, 4 μl CAS9 buffer and 28 μl RNAse/DNAse free water. Total volume of the reaction mixture was 40 μl. The digest was incubated overnight at 37°C. The DNA was then ethanol precipitated with 5 μl of 3 M sodium acetate and 150 μl of 100% ethanol. The mixture was centrifuged at 14 000 rpm, 4°C for 12 min. The supernatant was then discarded. The precipitate was washed with 150 μl 70% ethanol and centrifuged at 14 000 rpm, 4°C for 12 min. The supernatant was discarded and the DNA pellet was left to air dry. Once the DNA pellet was dry, 20 μl of water was added and the pellet allowed to dissolve for several hours at 4°C. If the DNA was not used within the next 2 days, the DNA solution was frozen at −20°C.
Notably that genomic DNA may be isolated from human or other mammalian cell lines with either DNA Maxi kit (Qiagen, cat. number #51192) or Puregene DNA isolation kit (Gentra Systems, cat number # 158422). However, DNA isolated by this method does not allow TAR cloning of fragments larger than 150 kb. To isolate larger size fragments (up to 250 kb), the genomic DNA should be prepared in agarose blocks as described previously (17). In our experiments, we used commercial human gnomic DNA (Promega, cat. number # G3041).
Yeast strain and transformation
For spheroplasts transformations, the highly transformable S. cerevisiae strain VL6–48 (MATα, his3-Δ200, trp1-Δ1, ura3–52, lys2, ade2–101, met14) that has the HIS3 gene deleted was used. The TAR vector pVC-NBS1 was constructed as follows. The vector contains 5′ and 3′ sequences of the human NBS1 gene (hooks). The 306 bp ApaI- XmaI and 193 bp XmaI-XbaI fragments corresponding to 5′ and 3′ regions of NBS1 were inserted into the polylinker of the basic TAR vector pVC604 (2,17). ATG and stop codons are located approximately 5 kb upstream of the 5′ hook and approximately 1.5 kb downstream of the 3′ hook of NBS1, respectively. The 5′ and 3′ targeting sequences of NBS1 were designed based on the available information (March 2006, NCBI36/hg18) and corresponds to positions 91 070 470 to 91 070 776 and 91 015 482 to 91 015 674 on the chromosome 8 sequence. The size of the targeted genomic fragment containing the NBS1 gene is 55 294 bp. The targeting sequences were cloned into vector pVC604 in the same orientation as they occur in within the genome. Before transformation, the TAR vector was ‘activated’ by linearization with XmaI to release the recombinogenic sequences (hooks). The XmaI site is located between the targeting sequences.
To TAR-CRISPR clone the genomic copies of the NBS1gene, 10 aliquots of genomic DNA (20 μl each containing approximately 1 μg of high molecular weight human DNA treated by CRISPR/Cas9) were prepared. To each aliquot approximately 1 μg (∼5 μl) of the linearized TAR vector was added. 200 μl suspension of freshly prepared yeast spheroplasts (∼108) was then added to each mixture. In parallel experiments, 10 aliquots of untreated genomic DNA (5 μl each containing approximately 1 μg of high molecular weight human DNA) were mixed with 10 aliquots of the linearized TAR vector (∼5 μl each containing approximately 1 μg of the linearized TAR vector DNA) and 200 μl of freshly prepared yeast spheroplasts added as described previously in detail (2,17). Cell density of the culture of yeast used to prepare spheroplasts is critical, and ∼2 × 107 cells per ml is optimal. The OD660 of the culture was about 3.0. Yeast transformants were selected on synthetic complete medium plates lacking histidine (His−). After transformation experiments were carried out, the His+ colonies appeared after 3 days and allowed to grow for another 4–5 days. Approximately 30–50 His+ colonies per plate were obtained (Supplementary Table S2). To identify NBS1 gene-containing clones, 570 transformants obtained with untreated human genomic DNA and 100 transformants obtained with human DNA treated by CRISPR/Cas9 were streaked on the His− plates. An example of a plate with streaked His+ transformants is shown on Figure 3c. 570 transformants from untreated genomic DNA were combined into 19 pools, each containing 30 transformants. Yeast genomic DNA from each pool was isolated following the previously described protocol (2) and then screened by PCR with diagnostic primers E13s that were specific to the exon 13 sequence of NBS1 but absent in the TAR vector hooks (Supplementary Table S1). 12 gene-positive pools were identified, and individual clones from each positive pool were PCR-screened again. A total of 12 colonies containing the NBS1 gene was identified, one for each gene-positive pool. Amongst His+ transformants obtained with CRISPR/Cas9 treated genomic DNA, 32 out of 100 screened colonies were gene-positive (Supplementary Table S2).
Figure 3.
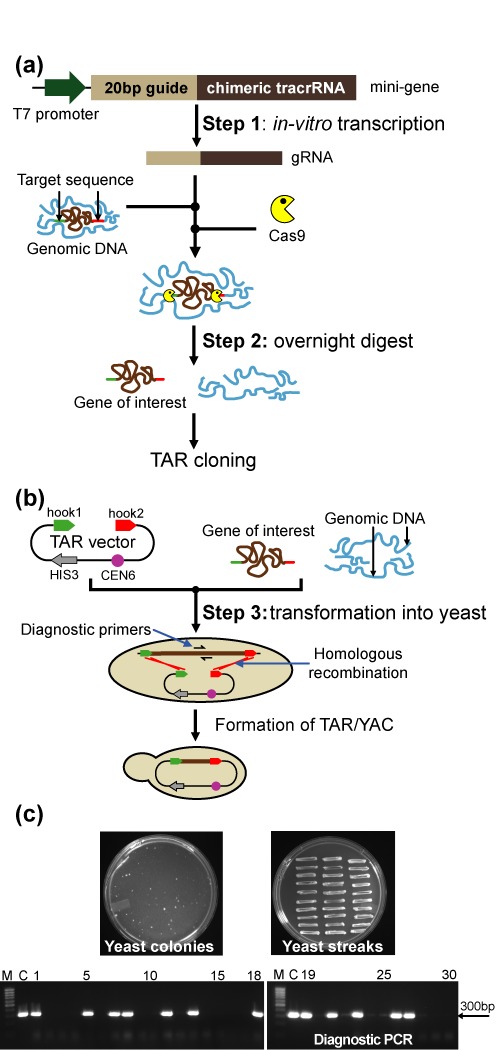
A scheme of the TAR-CRISPR technology for selective isolation of a gene of interest, the NBS1 gene, from the human genome. (a) Human genomic DNA was cleaved at positions close to the 5′ and 3′ ends of NBS1 by the Cas9-gRNA complexes. The 126 bp CRISPR minigene may be generated by PCR as was done in this study or artificially synthesized. Step 1: The minigene was in vitro transcribed to make gRNA. gRNA gives sequence specificity to the Cas9 nuclease (New England BioLabs). Step 2: A digestion mix containing gRNAs, Cas9 and genomic DNA was prepared and left to digest overnight. (b) Step 3: Yeast spheroplasts were transformed with CRISPR/Cas9 treated genomic human DNA along with the linearized TAR-NBS1 cloning vector. Homologous recombination between the targeting sequences (hook 1 and hook 2) in the vector and the targeted NBS1 genomic DNA fragment ends leads to the establishment of a circular YAC. The diagnostic primers used to find the NBS1 gene-positive transformants are E13F/E13R for the exon 13 sequence of the NBS1. The close proximity of the DNA ends to the targeted sequence dramatically enhances the efficiency of recombination. CEN corresponds to the yeast chromosome VI centromere, and HIS3 is a yeast selectable marker. (c) A representative His− plate with yeast His+ transformants obtained with 1 μg of genomic DNA and 1 μg of the TAR vector after 5 days of growth and the plate with 30 streaked randomly chosen His+ transformants after one day of growth and analysis of 30 His+ transformants by PCR for the presence of the human NBS1 sequences using a pair of diagnostic primers for exon 13 (Supplementary Table S1) are shown. M-GeneRuler 1kb DNA Ladder; C-control PCR with human genomic DNA.
Conversion of TAR-CRISPR-isolated NBS1-containing YACs into BACs
A diagram of the pJBRV1 vector and retrofitting of a circular YAC into a BAC molecule is shown in Supplementary Figure S2. Vector pJBRV1 contains the 3′ end of the HPRT gene, a loxP sequence, and two short targeting hooks (∼300 bp each), separated by the unique BamHI site, that flank the ColE1 origin of replication in the pVC604-based TAR cloning vector (2,17). The hooks are homologous to the vector sequences of pVC604. Recombination of the BamHI-linearized pJBRV1 vector with a YAC in yeast leads to replacement of the ColE1 origin of replication in the TAR cloning vector by a cassette containing the F factor origin replication, the chloramphenicol acetyltransferase (CmR) gene and the URA3 yeast selectable marker. A standard lithium acetate transformation procedure was used to retrofit YACs into BACs. The transformants were selected on synthetic complete medium plates lacking uracil (Ura−). YAC retrofitting was highly efficient, more than 95% of Ura+ His+ transformants obtained with pJBRV1 contained retrofitted YACs. The YAC/BACs were moved to Escherichia coli by electroporation. In brief, yeast chromosome-size DNA was prepared in agarose plugs. Then the agarose plugs were melted and β-agarase treated. Finally, the DNA fragments were electroporated into DH10B bacterial competent cells using a Bio-Rad Gene Pulser as described previously (17).
Physical characterization of NBS1-containing YAC and BAC clones
Several approaches were taken to establish the integrity and stability of the cloned material in the TAR isolates obtained from CRISPR/Cas9 treated total human DNA. The NBS1 coding regions of TAR/YAC clones were examined by PCR with specific pairs of primers for gene exonic regions (Supplementary Table S1). All PCR products were sequenced and found to match the expected NBS1 sequences. To identify fragments containing Alu sequences (Alu profiles), 1 μg of total yeast DNA containing a YAC was digested to completion with TaqI. Samples were resolved by gel electrophoresis, transferred to a nylon membrane and hybridized with an Alu probe as described previously (17,18). To check the size of the BAC inserts, chromosomal-size DNAs from three E. coli isolates were digested with BsiW1, separated by CHEF and stained with EtBr. Also, BAC DNA samples were digested by EcoRI and HindIII and run in 1% agarose gel. BAC clones correspond to the YAC clone, containing the NBS1 gene, retrofitted by pJBRV1.
Reverse-transcriptase PCR (RT-PCR)
Transcription of the NBS1 gene was detected after transient transfection of the BAC/NBS1-containing DNA into hypoxanthine phosphoribosyltransferase (HPRT)-deficient Chinese hamster ovary (CHO) cells by RT-PCR. CHO cells in a 6-well plate at 70% confluent were transfected with 2 μg of BAC/NBS1 DNA (∼70 kb) with 6 μl of XtremeGENE 9 (Roche, Cat# 06–365–787–001) in 200 μl of Optimem (Life Technologies, Cat# 11058021). Cells were scraped off the plate in PBS, 48 h after transformation. Whole RNA was extracted using a RNasy Mini Kit (Qiagen, Cat# 74104) with RNAse-Free DNAse Set (Qiagen, Cat# 79254). The protocol used was as recommended by the manufacturer. Cells were lysed using a syringe and a 27 gauge needle. cDNA was made using M-MLV Reverse Transcriptase (Life Technologies, Cat# 28025–013). RT-PCR used human specific NSB1 primers listed in Supplementary Table S1. The PCR products were sequenced by ACGT Inc.
Statistics
Statistical analysis was made using Prism (GraphPad Software Inc., La Jolla, CA, USA).
RESULTS
Design of Cas9-gRNA complexes
Four programmable Cas9 complexes, Cas9-gRNA-A, Cas9-gRNA-B, Cas9-gRNA-C and Cas9-gRNA-D, corresponding respectively to the 5′ and 3′ NBS1 targeting hooks in the TAR vector were assembled in vitro (Figure 1; Supplementary Figure S1). Then efficiency of nucleases activity was examined as described in Materials and Methods and Supplementary Figure S1. Two complexes, Cas9-gRNA-B and Cas9-gRNA-C, were chosen for further experiments (Figure 2a and b). Cas9-gRNA-B recognizes and cuts the genome 47 bp upstream of the 5′ NBS1 targeting hook while Cas9-gRNA-C recognizes and cuts the genomic sequence 7 bp downstream of the 3′ hook (Figure 1a). Before TAR cloning, total human genomic DNA was treated simultaneously in vitro by both Cas9-gRNA-B and Cas9-gRNA-C complexes to induce DSBs near the targeted genomic sequences (Figure 2c).
TAR-CRISPR cloning of the human NBS1 gene
The TAR cloning procedure itself was described in detail previously (2). All steps of TAR-CRISPR cloning are shown in Figure 3. In our study, a linearized TAR-NBS1 vector was combined with either Cas9-treated or untreated human genomic DNA (as a control) and individually presented to yeast spheroplasts (Figure 3a and b). The TAR vector contains short targeting hooks specific to 5′ and 3′ sequence ends of the NBS1 gene (see Materials and Methods). In addition to the targeting hooks, this vector also contains a YAC cassette that includes the HIS3 marker for selection in yeast and the CEN6 centromere for proper segregation in yeast cells. In vivo recombination between the hooks and genomic DNA fragment containing the NBS1 gene would result in isolation of the gene as a circular YAC with an insert size of 55 294 bp (Figure 3b). The cloned NBS1 locus contains approximately 5 kb sequence upstream from the ATG start codon and 1.5 kb sequence downstream of the stop codon that includes all known 5′ and 3′ regulatory elements. After 5–6 days His+ transformants were selected. The number of transformants per μg DNA was comparable between Cas9-treated and non-treated genomic DNA. Between 20 and 50 His+ colonies were typically obtained when a mixture containing 1 μg of genomic DNA and 1 μg of the TAR vector was used. To identify transformants containing the NBS1 gene, 570 transformants from experiments with untreated genomic DNA and 100 transformants with Cas9-treated genomic DNA were examined by PCR using a pair of diagnostic primers E13F/E13R (Supplementary Table S1) that amplified the exon 13 sequence, approximately 12 kb upstream of the 3′ NBS1 hook.
Twelve NBS1 gene positive colonies were identified among 570 His+ transformants when untreated genomic DNA was used for yeast transformation. Such a frequency (∼2%) was expected for isolation of a single copy gene (1–6). A screen of transformants from two pools, one gene-positive and one gene-negative, is shown in Supplementary Figure S3. In contrast, the analysis of 100 His+ transformants obtained with the Cas9-treated human genomic DNA, yielded 32 colonies positive for the NBS1 sequence (32%). An example of the PCR screen for 30 randomly chosen transformants is presented in Figure 3c (12 among 30 transformants were gene-positive that is equal to ∼40%). The cloned material may be analyzed either in a YAC form (19) or BAC form after YAC conversion into BAC molecules to simplify DNA isolation (17).
Physical analysis of TAR-CRISPR isolated clones containing the NBS1 gene
Several approaches were taken to establish the integrity of the NBS1-TAR-CRISPR-cloned material derived from the Cas9-treated human genomic DNA. The presence of NBS1 exon sequences in three TAR/YAC-containing transformants was confirmed by PCR using primers for each exon on total yeast DNA (Supplementary Table S1). An example of PCRs for gene exonic regions 1, 3, 6, 7, 8, 9, 11, 14 and 16 is presented in Figure 4a and b. Next, Alu profiles were determined for the two TAR/YACs and they were found to be identical (Figure 4c). Next, one NBS1-containing YAC was retrofitted into a YAC/BAC using the retrofitting vector pJBRV1 (13) and transferred to E. coli by electroporation as previously described (17) (Supplementary Figure S2). The presence of NBS1 coding regions in eight randomly chosen E. coli colonies were examined by PCR using pairs of exon primers (Supplementary Figure S4; Supplementary Table S1). BAC DNAs were purified from three E. coli colonies by a standard method. All BACs have the same size insert (∼55 kb) after digestion with BsiWI that eliminates the vector sequence. The BACs also had identical restriction profiles after digestion by either EcoRI or HindIII, indicating no structural rearrangements (Figure 4d and e). To demonstrate that the CRISPR/TAR-isolated NBS1 is functional, we confirmed expression of the full-size cDNA in hamster CHO cells by RT-PCR (Figure 4f and g) using specific primers listed in Supplementary Table S1. The PCR product was sequenced and the sequence matched human cDNA of NBS1 (Supplementary Figure S5). To summarize, these analyses demonstrate a high level of integrity for the NBS1 sequences in the TAR-CRISPR-isolated clones.
Figure 4.
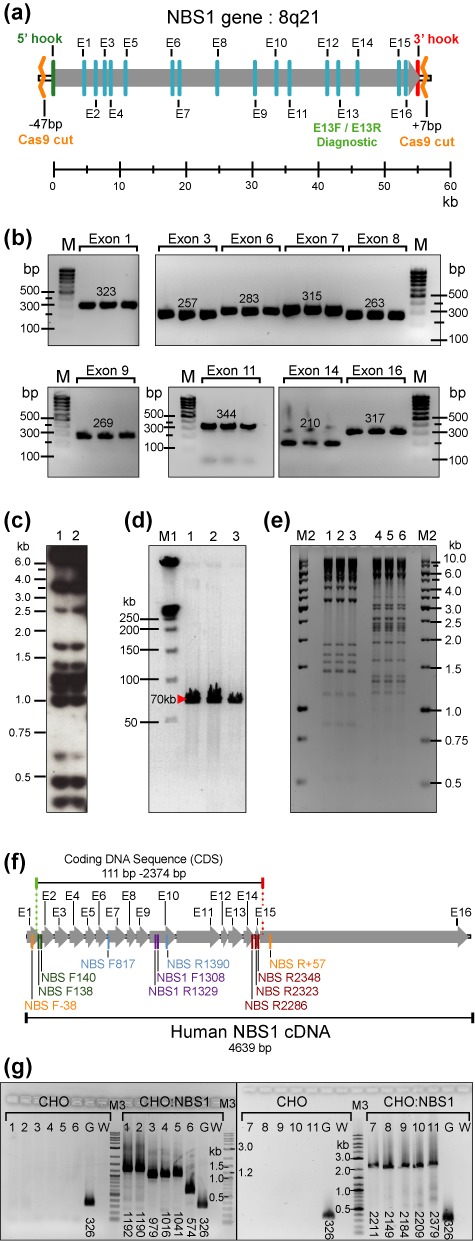
Physical characterization of NBS1-containing YAC and BAC clones obtained by TAR-CRISPR cloning from total human DNA. (a) The structure of the human NBS1 gene. The NBS1 gene is mapped at 8q21 and contains 16 exons. Arrow indicates the direction of NBS1 transcription. E13F/E13R are diagnostic primers for the exon 13 sequence of the NBS1 gene used to screen yeast transformants to find gene-positive clones. Location of all primers for the NBS1 gene (Supplementary Table S1) corresponds to positions of exon sequences of NBS1. Cas9 sites relative to 5′ and 3′ hooks are indicated. (b) PCR analysis of three independent His+ TAR-CRISPR/YAC clones containing NBS1. The presence of the NBS1 coding regions in the YAC clones was examined by PCR using pairs of exon specific primers (Supplementary Table S1). Presented in this figure are PCR products obtained for nine exons of three independent gene-positive yeast transformants (exons 1, 3, 6, 7, 8, 9, 11, 14 and 16). (We note that comparable results were obtained for the other exon specific primers too, in that the PCR products with the YAC clones matched the sequences of the human NBS1 DNA). M corresponds to GenRuler DNA Ladder Low Range. (c) Alu-profile characterization of two independently obtained YACs containing NBS1 obtained by TAR-CRISPR cloning from human genomic DNA. Two gene-positive isolates were characterized and compared in terms of Alu profiles of restriction fragments. (d) Characterization of circular NBS1-BACs. TAR-CRISPR/YACs were retrofitted into BACs by pJBRV1 vector and moved to E. coli cells. BAC DNAs were isolated, digested by BsiW1 for linearization, separated by CHEF and visualized by staining with ethidium bromide. Predicted and identical size of the NBS1-containing fragments was observed (∼70 kb that corresponds to ∼55 kb NBS1 plus 15 kb vector sequences). These three BAC clones (lanes 1–3) corresponding to one independent TAR/YAC His+ isolate were analyzed. M1 corresponds to CHEF DNA Size Standard (BIO-RAD) (e) Restriction digestion profiles of BAC DNAs. BAC DNA samples were digested by HindIII (lanes 1–3) and EcoRI (lanes 4–6). Identical profiles were observed for each restriction endonuclease. M2 corresponds to GenRuler 1 kb DNA Ladder. (f) The 4639 bp human NBS1 cDNA and positions of exons and primers used for RT-PCR. Primer sequences are listed in Supplementary Table S1. Human NBS1 coding DNA sequence (CDS) starts at position 111 and ends at position 2374. (g) RT-PCR analysis using human specific NBS1 primers on reverse transcribed RNA purified from either naive CHO cells or CHO cells transiently transfected with human NBS1-containing BAC DNA. Primers in lanes 1–6 amplify parts of the human NBS1 CDS. Primers in lanes 7–10 amplify nearly the complete CDS. Primers in lane 11 amplify the complete CDS. Primers in lane 1 are NBS F138 / NBS1 R1329, expected product size is 1192 bp; lane 2 - NBS F140 / NBS1 R1329 primers, expected size is 1190 bp; lane 3 - NBS R2286 / NBS1 F1308 primers, expected size is 979 bp; lane 4 - NBS R2323 / NBS1 F1308 primers, expected size is 1016 bp; lane 5 - NBS R2348/ NBS1 F1308 primers, expected size is 1041 bp; lane 6 - NBS F817 / NBS R1390 primers, expected size is 584 bp; lane 7 - NBS F138 / NBS R2348 primers, expected size is 2211 bp; lane 8 - NBS F138 / NBS R2286 primers, expected size is 2149 bp; lane 9 - NBS F140 / NBS R2323 primers, expected size is 2184 bp; lane 10 - NBS F140 / NBS R2348 primers, expected size is 2209 bp; lane 11 NBS F-38 / NBS R+57 primers, expected size is 2379 bp. Lanes G are positive controls, with primers rt-mh GAPDH-F / rt-mh GAPDH-R which amplify both human and hamster GAPDH cDNAs. Expected product size is 326 bp. Lanes W are negative controls which do not contain primers. M3 is DNA ladder (New England BioLabs, N3200L).
To conclude, we demonstrated that the yield of gene-positive clones significantly increased [Fisher's exact test, two-tailed (P < 0.0001)] by 16-fold from 2% up to 32% after treatment of genomic DNA by a pair of Cas9-gRNA nucleases which made double strand breaks near the targeted sequences.
DISCUSSION
The most palpable effect of TAR-CRISPR approach is the decreased workload of the experimenter in isolating gene-positive clones. To obtain a 95% probability of isolation at least 1 gene-positive clone, only 8 colonies have to be screened when the success rate is 32%, in contrast to 148 colonies at 2% success rate (Supplementary Figure S6). This is very important because the main stumbling block to successful TAR cloning using the standard protocol (2) is the need for highly efficient yeast spheroplasts transformation (i.e. ∼108 colonies per one microgram of ARS-containing vector), so that hundreds of colonies have to be obtained and screened to find a few gene-positive clones. This in turn means that the previous TAR protocol required significant experience working with yeast. With our new procedure, only a dozen or less colonies need to be screened to obtain gene-positive clones. Hence, even people inexperienced in yeast work can successfully TAR-CRISPR-clone their gene of interest.
It is also worth noting that the pJBRV1retrofitting vector used to convert YACs into BACs contains a 3′ HPRT-loxP cassette allowing an isolated TAR-CRISPR-gene to be loaded into a unique loxP site within the alphoidtetO-HAC gene delivery vector by Cre-loxP mediated recombination (13,20). HAC vectors (Human Artificial Chromosomes) are a unique system for delivery and expression of full-length genes in recipient human cells, with the potential to overcome many problems caused by using viral-based vectors (21–23). The alphoidtetO-HAC represents one of the most advanced HACs for delivery and expression of full-length human genes as it has a regulated centromere, which allows the unique opportunity of a three way comparison between the phenotypes of human cell lines, without a functional copy of a gene, with a functional HAC-encoded gene copy and one ‘cured’ of the HAC (Supplementary Figure S7). This provides a real control for phenotypic changes attributed to expression of a HAC-encoded gene by returning the mutant cell line to its original state following ‘curing’ of the HAC (13,24)
This novel TAR-CRISPR technology could in principle be used to create a bank of all annotated human genes, each represented by a genomic copy containing coding regions (exons), 5′ upstream, 3′ downstream regulatory sequences and introns. At present only cDNA collections are available. A bank of full-length genes with their native regulatory elements offers previously unheard-of possibilities for functional and structural genomics, in diagnostic and gene therapy and would launch a new generation of genomic research products. The ability to clone genes with their distal 5′ and 3′ enhancers could provide a revolutionary approach to analyze genomic changes detected in GWAS (Genome-Wide Associated Study). We could examine the expression of the entire locus in the HAC vector and compare the wild type with the GWAS-identified mutations, which would provide an exact functional test.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, USA (V.L. and N.K). Funding for open access charge: Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, USA.
Conflict of interest statement. None declared.
REFERENCES
- 1.Kouprina N., Larionov V. TAR cloning: insights into gene function, long-range haplotypes and genome structure and evolution. Nat. Rev. Genet. 2006;7:805–812. doi: 10.1038/nrg1943. [DOI] [PubMed] [Google Scholar]
- 2.Kouprina N., Larionov V. Selective isolation of genomic loci from complex genomes by transformation-associated recombination cloning in the yeast Saccharomyces cerevisiae. Nat. Protoc. 2008;3:371–377. doi: 10.1038/nprot.2008.5. [DOI] [PubMed] [Google Scholar]
- 3.Annab L.A., Kouprina N., Solomon G., Cable P.L., Hill D.E., Barrett J.C., Larionov V., Afshari C.A. Isolation of a functional copy of the human BRCA1 gene by transformation-associated recombination in yeast. Gene. 2000;250:201–208. doi: 10.1016/s0378-1119(00)00180-3. [DOI] [PubMed] [Google Scholar]
- 4.Kim J.H., Leem S.H., Sunwoo Y., Kouprina N. Separation of long-range human TERT gene haplotypes by transformation-associated recombination cloning in yeast. Oncogene. 2003;22:2452–2456. doi: 10.1038/sj.onc.1206316. [DOI] [PubMed] [Google Scholar]
- 5.Kouprina N., Pavlicek A., Collins N.K., Nakano M., Noskov V.N., Ohzeki J., Mochida G.H., Risinger J.I., Goldsmith P., Gunsior M., et al. The microcephaly ASPM gene is expressed in proliferating tissues and encodes for a mitotic spindle protein. Hum. Mol. Genet. 2005;14:2155–2165. doi: 10.1093/hmg/ddi220. [DOI] [PubMed] [Google Scholar]
- 6.Kouprina N., Pavlicek A., Noskov V.N., Solomon G., Otstot J., Isaacs W., Carpten J.D., Trent J.M., Schleutker J., Barrett J.C., et al. Dynamic structure of the SPANX gene cluster mapped to the prostate cancer susceptibility locus HPCX at Xq27. Genome Res. 2005;15:1477–1486. doi: 10.1101/gr.4212705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mali P., Esvelt K.M., Church G.M. Cas9 as a versatile tool for engineering biology. Nat. Methods. 2013;10:957–963. doi: 10.1038/nmeth.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu P.D., Lander E.S., Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wijshake T., Baker D.J., van de Sluis B. Endonucleases: new tools to edit the mouse genome. Biochim. Biophys. Acta. 2014;1842:1942–1950. doi: 10.1016/j.bbadis.2014.04.020. [DOI] [PubMed] [Google Scholar]
- 10.Kim H., Kim J.S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014;15:321–334. doi: 10.1038/nrg3686. [DOI] [PubMed] [Google Scholar]
- 11.Karvelis T., Gasiunas G., Siksnys V. Programmable DNA cleavage in vitro by Cas9. Biochem. Soc. Trans. 2013;41:1401–1406. doi: 10.1042/BST20130164. [DOI] [PubMed] [Google Scholar]
- 12.Leem S.H., Londono-Vallejo J.A., Kim J.H., Bui H., Tubacher E., Solomon G., Park J.E., Horikawa I., Kouprina N., Barrett J.C., et al. The human telomerase gene: complete genomic sequence and analysis of tandem repeat polymorphisms in intronic regions. Oncogene. 2002;21:769–777. doi: 10.1038/sj.onc.1205122. [DOI] [PubMed] [Google Scholar]
- 13.Kim J.H., Kononenko A., Erliandri I., Kim T.A., Nakano M., Iida Y., Barrett J.C., Oshimura M., Masumoto H., Earnshaw W.C., et al. Human artificial chromosome (HAC) vector with a conditional centromere for correction of genetic deficiencies in human cells. Proc. Natl. Acad. Sci. U.S.A. 2011;108:20048–20053. doi: 10.1073/pnas.1114483108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Imburgio D., Rong M., Ma K., McAllister W.T. Studies of promoter recognition and start site selection by T7 RNA polymerase using a comprehensive collection of promoter variants. Biochemistry. 2000;39:10419–10430. doi: 10.1021/bi000365w. [DOI] [PubMed] [Google Scholar]
- 15.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu Y., Sander J.D., Reyon D., Cascio V.M., Joung J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kouprina N., Noskov V.N., Koriabine M., Leem S.H., Larionov V. Exploring transformation-associated recombination cloning for selective isolation of genomic regions. Methods Mol. Biol. 2004;255:69–89. doi: 10.1385/1-59259-752-1:069. [DOI] [PubMed] [Google Scholar]
- 18.Larionov V., Kouprina N., Solomon G., Barrett J.C., Resnick M.A. Direct isolation of human BRCA2 gene by transformation-associated recombination in yeast. Proc. Natl. Acad. Sci. U.S.A. 1997;94:7384–7387. doi: 10.1073/pnas.94.14.7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noskov V.N., Chuang R.Y., Gibson D.G., Leem S.H., Larionov V., Kouprina N. Isolation of circular yeast artificial chromosomes for synthetic biology and functional genomics studies. Nat. Protoc. 2011;6:89–96. doi: 10.1038/nprot.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kononenko A.V., Bansal R., Lee N.C., Grimes B.R., Masumoto H., Earnshaw W.C., Larionov V., Kouprina N. A portable BRCA1-HAC (human artificial chromosome) module for analysis of BRCA1 tumor suppressor function. Nucleic Acids Res. 2014;42:e164. doi: 10.1093/nar/gku870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kouprina N., Earnshaw W.C., Masumoto H., Larionov V. A new generation of human artificial chromosomes for functional genomics and gene therapy. Cell. Mol. Life Sci. 2013;70:1135–1148. doi: 10.1007/s00018-012-1113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kouprina N., Tomilin A.N., Masumoto H., Earnshaw W.C., Larionov V. Human artificial chromosome-based gene delivery vectors for biomedicine and biotechnology. Expert Opin. Drug Deliv. 2014;11:517–535. doi: 10.1517/17425247.2014.882314. [DOI] [PubMed] [Google Scholar]
- 23.Kazuki Y., Oshimura M. Human artificial chromosomes for gene delivery and the development of animal models. Mol. Ther. 2011;19:1591–1601. doi: 10.1038/mt.2011.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iida Y., Kim J.H., Kazuki Y., Hoshiya H., Takiguchi M., Hayashi M., Erliandri I., Lee H.S., Samoshkin A., Masumoto H., et al. Human artificial chromosome with a conditional centromere for gene delivery and gene expression. DNA Res. 2010;17:293–301. doi: 10.1093/dnares/dsq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.