

Abstract
The mitochondrial replicative helicase Twinkle is involved in strand separation at the replication fork of mitochondrial DNA (mtDNA). Twinkle malfunction is associated with rare diseases that include late onset mitochondrial myopathies, neuromuscular disorders and fatal infantile mtDNA depletion syndrome. We examined its 3D structure by electron microscopy (EM) and small angle X-ray scattering (SAXS) and built the corresponding atomic models, which gave insight into the first molecular architecture of a full-length SF4 helicase that includes an N-terminal zinc-binding domain (ZBD), an intermediate RNA polymerase domain (RPD) and a RecA-like hexamerization C-terminal domain (CTD). The EM model of Twinkle reveals a hexameric two-layered ring comprising the ZBDs and RPDs in one layer and the CTDs in another. In the hexamer, contacts in trans with adjacent subunits occur between ZBDs and RPDs, and between RPDs and CTDs. The ZBDs show important structural heterogeneity. In solution, the scattering data are compatible with a mixture of extended hexa- and heptameric models in variable conformations. Overall, our structural data show a complex network of dynamic interactions that reconciles with the structural flexibility required for helicase activity.
INTRODUCTION
During DNA replication, dedicated replicative helicases unwind double-stranded (ds) DNA while cognate primases generate short DNA/RNA heteroduplexes on the lagging strand. These heteroduplexes prime 5′→3′ DNA synthesis by a DNA polymerase. Characterization of the gene linked to late-onset autosomal dominant progressive external ophthalmoplegia (adPEO) indicated that its product, Twinkle, was the mitochondrial DNA (mtDNA) helicase (1). The C-terminal region of Twinkle shares high similarity with the helicase C-terminal domain (CTD) of the bacteriophage T7 gene 4 (T7 gp4), a bifunctional primase-helicase protein (1–3). This CTD comprises a catalytic hexamerization domain reminiscent of the archetypal helicase RecA, and includes the Walker A and Walker B signatures (also termed H1 and H2, respectively) (4). In helicases and translocases, these motifs provide the catalytic residues for the hydrolysis of nucleoside triphosphates (NTPs), thus supplying the energy required for protein activity on DNA (4). Additional specific signatures within the helicase (H) domain—H1a, H3 and H4—assign Twinkle to the SF4 family of replicative DnaB-like helicases (3,5), whose members have a ring-shaped hexameric structure (4). In general, a cleft at the interface between two neighboring RecA-like domains of the hexamer binds the NTP, the hydrolysis of which triggers a movement between subunits. This, in turn, leads to 5′->3′ DNA unwinding (6). Further studies confirmed the assignment of Twinkle to the SF4 helicase family: functional studies with recombinant protein in vitro revealed a nucleotide hydrolysis-dependent 5′→3′ DNA unwinding activity (7–9). In addition, Twinkle, the mitochondrial DNA polymerase γ (Polγ), and the mitochondrial single-stranded (ss) DNA-binding protein (mtSSB) form a minimal replisome in vitro (10).
Previous biophysical and EM analyses showed that Twinkle forms hexamers that convert to heptamers at low salt concentrations and in the presence of Mg2+ and the ATP-analog ATPγS (5,11). Oligomeric transitions between heptamers and hexamers have also been described for T7 gp4 (6,12), as well as for SF6 helicases, which include the mini chromosome maintenance (MCM) protein complex (present in archaea and eukarya) and bacterial HB8 RuvB, involved in branch migration in Holliday junctions (4). Furthermore, studies in vitro demonstrated that Twinkle is capable of loading itself onto circular ssDNA (7,9) or onto dsDNA bubbles (7). However, Twinkle also differs from T7 gp4. It binds to dsDNA in the absence of nucleotides, and with higher affinity than when binding to ssDNA (9,11,13,14). It also displays two binding sites for ssDNA, which have been related to its annealing ability and suggests regulatory functions beyond replication (9).
The N-terminal domain (NTD) of Twinkle shows only limited sequence similarity with that of T7 gp4, but displays a similar predicted overall fold (1,3) and is classified as a prokaryotic DnaG-type primase (15). Primases of this type contain an N-terminal zinc-binding domain (ZBD), which binds ssDNA and detects trinucleotide patterns from priming-sites, and an RNA polymerase domain (RPD), which receives the priming site from the ZBD and initiates RNA primer synthesis (16). Bacterial and phage primase ZBDs typically contain a four- or five-stranded antiparallel β-sheet with two loops at one edge, which provide four cysteines (sometimes replaced by histidines) that coordinate a zinc ion (17–19). RPDs vary among primases: in E. coli DnaG (but not in T7 gp4) the RPD includes an N-terminal subdomain that binds ssDNA, orienting it toward an additional catalytic TOPRIM sub-domain (20). The TOPRIM subdomain is present in DnaG, T7 gp4 as well as in topoisomerases, nucleases and DNA repair proteins (21), and it contains an active site with metal-coordinating acidic residues that catalyze primer chain elongation (17,18,22). In addition, functional studies with either T7 gp4 (23) or DnaG (24) showed that the ZBD can interact with the RPD from its own subunit (in cis) or with that from another subunit (in trans), in either case efficiently regulating primer synthesis.
Currently, the three-dimensional (3D) organization of a bifunctional SF4 primase-helicase is only exemplified by the crystal structure of a T7 gp4 natural fragment containing only the RPD and the CTD, thus lacking the ZBD (12) (Figure 1A). In the T7gp4 hexamer, the RPD and RPD-CTD linker of one subunit interacts with the CTD of the adjacent subunit, suggesting that the interaction of each subunit with its neighbor coordinates the helicase and primase activities thus the mechanism of translocation along ssDNA (6). However, despite the predicted structural similarity between T7 gp4 and Twinkle, the bifunctional helicase-primase mechanism described for the former cannot be extrapolated to the latter because most of the activity-related metal-coordinating residues in both ZBD and RPD are absent in human Twinkle (1,3), thus precluding primase activity (13). However, there is a functional requirement for ZBD and RPD domains in Twinkle, since their partial or complete ablation results in a dramatic decrease in helicase activity (13,25). At the physiological level, Twinkle is essential for mtDNA copy number maintenance (25,26), which depends on DNA replication. Furthermore, around 50 mutations (and one in-frame 39-nucleotide duplication) within the Twinkle gene are related with the aforementioned rare diseases adPEO (1), infantile-onset spinocerebellar ataxia (IOSCA) (27), infantile onset mtDNA depletion syndromes (MDS) (28) and epileptic seizures (29), among others (see Figure 1B for domain localization of disease-related mutations and Supplementary Table S1 for references).
Figure 1.
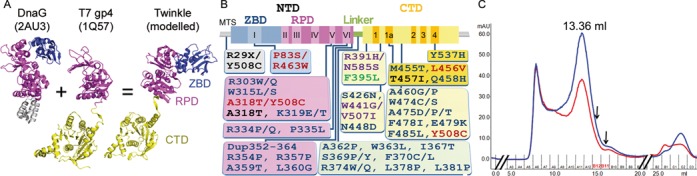
Model, mutants and purification of Twinkle. (A) Ribbon representation of DnaG (PDB code 2AU3), the natural short form of T7 gp4 (PDB 1Q57) and the modeled Twinkle monomer. ZBD is shown in blue, RPD in magenta and CTD in gold. The DnaB interaction domain of DnaG is in gray. (B) Representation of disease-causing mutations of human c10orf2 gene on a scheme showing the Twinkle domains (IOSCA, red; adPEO, blue; MDS, black; Perrault Syndrome, purple; Renal Tubulopathy, green) (references in Supplementary Table S1). Domains are represented with same colors as in panel A. MTS stands for mitochondrial targeting sequence; motifs I to VI of the primase domain and motifs 1 to 4 in the helicase domain are indicated. (C) Size exclusion chromatography profile of Twinkle produced in SF9 insect cells (see experimental procedures). The blue and red curves correspond to the absorption at 280 and 260 nm, respectively. The peak of Twinkle is labeled with the elution volume. The fractions used for GraFix analysis are indicated by arrows.
In order to examine the structural basis of Twinkle's function and gain further insight into its dysfunction in disease, we developed an efficient recombinant protein expression procedure and performed electron microscopy (EM) and small angle X-ray scattering (SAXS) studies. Our results reveal that the hexameric organization of Twinkle is clearly asymmetric at the level of ZBDs and RPDs, which allows for a network of diverse interactions between domains along the oligomer, and provides the structural framework for the polar and asymmetric processing of nucleic acids.
MATERIALS AND METHODS
Cloning, E. coli heterologous expression and purification of recombinant Twinkle
A gene construct coding for mature Twinkle (Twinkle full-length, aa 43–684, UniProt Q96RR1.1) was cloned into vector pHART1201 with a C-terminal 6 histidines tag (30). Bases encoding the 5′ and 3′ SfiI restriction sites were added to the genetic construct by PCR with oligonucleotides (oligo) complementary to Twinkle 5′ end TACCATGGGCCACCTCGGCCGAAGGAGATATACATATGGAGACTCTCCAAGCCTTGG and 3′ end ACTTAGTGGCCGAGGCGGCCGCTTTGAACGCTTGGAGGTGTC (SIGMA). Large-scale cultures of E. coli BL21-DE3 containing a pG-KJE8 plasmid (TaKaRa) were induced with 1 mg/ml L-arabinose and 6 ng/ml tetracycline, and grown at 37°C until the culture reached an A595 of 0.6. Twinkle protein expression was induced with 0.25 mM IPTG o/n at 18°C. Cells were lysed by sonication in buffer A (50m M sodium citrate pH 6.5, 1.5M NaCl, 50 mM L-arginine, 50 mM L-glutamate) containing 50 mM imidazole and EDTA-free proteinase inhibitor cocktail (Roche), and centrifuged at 20 000 rpm (JA20 Beckman rotor) for 20min at 4°C. The supernatant was loaded onto a Ni-NTA-affinity chromatography column (HisTrap HP, GE HealthCare) connected to an ÄKTA FPLC system at room temperature and washed in buffer A containing 50 mM imidazole. The protein eluted at 35% of a linear gradient of buffer B (buffer A plus 0.5M imidazole). The elution fractions were subsequently analyzed by 10% SDS-PAGE gels: the purest fractions were pooled and concentrated in a 30KDa cutoff ultrafiltrator (VIVAspin), and loaded onto a size exclusion Superose6 10/300 column (GE-HealthCare) previously equilibrated with buffer A. Eluting fractions were analyzed by 10% SDS-PAGE.
Recombinant production of Twinkle in insect cells and purification
To produce Twinkle in SF9 insect cells, a gene construct coding for mature Twinkle was cloned into vector pHAR1201 as described above, except that it spanned from aa 30 to 684. BacMagic generated Twinkle cDNA-containing virus was first tested for protein production by testing various volumes of virus stock in 10 ml SF9 cultures and assaying Twinkle expression by western-blot analysis. Based on these results all subsequent infections used 10 μl virus per 10 ml SF9 culture. Cells were harvested 72h after infection. Cells were lysed for 15min at 4°C in buffer C (100 mM Tris-HCl pH 7.8, 1.5 M NaCl, 50 mM L-Arginine, 50 mM L-Glutamate, 30 mM imidazole, 1% TX-100, 2 mM β-MeOH) containing EDTA free protease inhibitor tablets, final pH adjusted to 8.0. The lysate was sonicated (30 cycles, 1”on – 3” off) and centrifuged (3000xg) for 5min at 4°C. The cleared lysate was bound to Ni-NTA beads overnight at 4°C under gentle agitation and subsequently pelleted by centrifugation (1500xg) for 5min. The pellet was washed three times in 20–30 bed volumes with buffer D (50 mMTris-HCl pH 7.8, 1 M NaCl, 50 mM L-Arginine, 50 mM L-glutamate, 60 mM imidazole, 1 mM β-MeOH, adjusted to pH 8.0), containing 60 mM imidazole. The protein was eluted in a single step with buffer D containing 250 mM imidazole. The concentration of the eluted protein was estimated by comparison to a standard in 7.5% SDS-PAGE gels, concentrated with a 100KDa cutoff ultrafiltrator (VIVAspin 6, GE-HealthCare), and loaded onto a size-exclusion chromatography (SEC) Superose6 10/300 column (GE-HealthCare) previously equilibrated with buffer E (25 mMTris- HCl pH 7.8, 1M NaCl, 50 mM L-Arginine, 50 mM L-Glutamate), final pH adjusted to 8.0. Peak fractions were analyzed by 7.5% SDS-PAGE and stored in 10% glycerol.
Helicase activity assays
The helicase activity was assayed by radiolabeling with 32P the 5′ end of a 60nt oligo of sequence 5′ACATGATAAGATACATGGATGAGTTTGGACAAACCACAACGTAAAACGACGGCCAGTGCC-3′, whose 3′ end was complementary over 20 bases to plasmid M13(+), thus leaving a free 5′ end of 40nt. The helicase reaction was performed at 37°C for 1h at increasing concentrations of Twinkle (10, 20, 30, 60 and 100ng), and 5 μM of plasmid M13(+), in buffer G (Tris-HCl 25 mM pH 7.5, MgCl2 4.5 mM, UTP 3 mM, NaCl 27 mM, glycerol 10%, DTT 1 mM, L-arginine 0.1 M, BSA 0.1 mg/ml), and free oligonucleotides to prevent non-specific interactions. The reaction was stopped by adding buffer H (SDS 6%, EDTA 90 mM, glycerol 30%, xylene cyanol 0.25% and bromphenol blue 0.25%). Samples were loaded onto 25% acrylamide gels, which were run at 80V for 60min at RT with TBE 1x as running buffer. Gels were dried and revealed using a photosensitive autoradiographic lamina and scanned.
Gradient fixation (GraFix)
Twinkle oligomers were stabilized by using glutaraldehyde cross-linking combined with density gradient centrifugation following the GraFix method (31). A 4.5 ml gradient was formed with buffers I (HEPES 50 mM pH 7.5, NaCl 1 M, 10% glycerol) and J (same as I but with 30% glycerol and 0.15% glutaraldehyde) by using a gradient mixer and allowed to settle for 1h at 4°C. Thereafter, 300 μg of protein (200–300 μl) was applied on top of the gradient and centrifuged at 95 000x g in an MLS-50 swing-out rotor (Beckman Coulter) for 16h at 4°C. Subsequently, 300 μl fractions were collected and 80 mM glycine pH 8.0 was added to quench the cross-linking.
EM sample preparation and image processing
For cross-linked and non-crosslinked Twinkle samples, negative stained grids were prepared with 2% uranyl acetate and visualized on a JEM-1230 transmission electron microscope (JEOL Europe) at an acceleration voltage of 80 KV. The images were taken in low dose conditions at a magnification of 30 000X in a Gatan CCD camera, resulting in 2.3 Å/pixel sampling. For cryo-EM data collection, the cross-linked Twinkle oligomer was dialyzed with Millipore membrane filters to remove the glycerol and decrease the salt concentration to 100 mM. Vitrification was performed using Quantifoil holey grids (with a thin carbon film floated on) rapidly plunged into liquid ethane in a FEI Vitrobot. The images were taken in a JEM-2200FS/CR electron microscope working at 200 KV at a magnification of 50 000X, and recorded at low-dose conditions on Kodak SO-163 films. Micrographs were scanned with a Z/I Photoscan scanner (Zeiss) with a step size of 7 μm, resulting in a final pixel size of 1.4 Å.
The single particles were extracted using EMAN (32) and Spire-SPIDER package (33,34), and 2D classifications were performed in a reference-free manner based on maximum-likelihood methods implemented in XMIPP (35). The initial models were built based on the top (6-fold rotational symmetry) and side (two-fold symmetry) views of the class averages. The references for the two techniques, negative staining and cryo-EM, were calculated independently and converged on similar 3D density maps. The particles were subjected to iterative refinements following the projection-matching scheme in Spire-SPIDER. During image processing several rotational symmetries were used, from C6 up to C1, starting with 6-fold symmetry and relaxing it in a sequential manner. Given that one of the two domains lining the axial channel departed from strict six-fold symmetry, during the reconstruction step of each iteration the generated asymmetric 3D volume was sliced along the z axis and a C1 symmetry pattern was imposed in layers that construct the N-terminal region whereas a 6-fold symmetry was imposed in layers that construct the CTD region, using XMIPP tools. This approach allowed us to maximize the signal-to-noise ratio in the final reconstructed map at the level of the CTD region and to prevent the smearing-out of ZBD and RPD domains. The resolution of the final cryoEM maps was estimated at 0.15 threshold in the Fourier Shell Correlation (FSC) calculated between maps reconstructed from two halves of the image data sets (36).
Monomeric and oligomeric homology model building
A sequence similarity search with BLAST against the entire Protein Data Bank (PDB) yielded the 3D structures from T7 gp4 helicase as closest structural relatives (ZBD-RPD primase domain, PDB code 1NUI; RPD and helicase domain, 1Q57; helicase domain, 1E0J and 1E0K, see Results for references). However, none of these structures covers the longest form of T7 gp4 protein. In addition, the natural NTD of T7 gp4, especially the ZBD, is shorter than that of Twinkle. Additional searches with the Twinkle NTD (aa 43–346) alone pointed to E. coli DnaG primase (PDB code 2AU3) as closest structural relative. Thus, we built a continuous homology model of Twinkle by superposing the RPD domains of coordinates 2AU3 and 1Q57, thus connecting the Twinkle-like ZBD from DnaG with T7 gp4 helicase domain via the common RPDs. Twinkle sequence alignment to the 2AU3 and 1Q57 templates was guided by secondary structure prediction of the former, with PSIPREP (37). With these structures and the alignment, a Twinkle monomer was generated with MODELLER (version 9.13, (38)), and this was fitted six times into the cryo-EM density. The Twinkle heptamer for SAXS was generated by “fusing” seven structures of DnaG (2AU3) to the seven chains of T7 gp4 1Q57, threading the sequence with MODELLER. The Twinkle hexamer was likewise generated by using T7 gp4 1E0J CTD hexamer.
Molecular dynamics flexible fitting
The hexamer was initially fitted manually into the cryo-EM map with Chimera (39) keeping the orientation for the monomers as in the crystal structure (6,12). This was appropriate for the CTD, whereas for the NTDs we carried out molecular dynamics simulations with NAMD 2.9 (40) through the MDFF plug-in (41). The protonation state of histidine residues in the initial model were predicted with PROPKA software from the PDB2PQR package (42). Simulations of 9ns were run with the CHARMM27 force field with CMAP corrections in generalized Born implicit solvent (43,44). We performed intrinsic solvent simulations with dielectric constant 1; cutoff 16Å; switchlist 15Å; pairlistdist 18Å; GIBS on; ion concentration 0.3M; alpha cutoff 14Å; sasa on and surface tension 0.006Kcal/ml/Å2. The first minimization step was performed with a grid scaling of 0 to stabilize the initial model. Non-hydrogen atoms were coupled to the UEM potential derived from the corresponding cryo-EM density maps with a grid scaling of 0.3 kcal/mol. Simulations used restraints for secondary structure, chirality and cis-peptides derived from the initially assembled atomic model. During the initial steps of the simulation, symmetrical restraints was applied between monomers, a symmetry restraint force constant k was applied, linearly increasing from 0 to 10 (Kcal/mol)/Å2. Essentially, the overall structure of globular domains (their secondary structure elements and the relationship between them) was conserved to avoid overfitting, the motions were only allowed in linker regions, and by applying tmdk 500, a constant which scales the harmonic force applied by the restraint on a domain.
Small angle x-ray scattering (SAXS)
SAXS data of Twinkle in size exclusion chromatography (SEC) buffer were recorded at 20°C at 0.3, 0.6 and 0.9 mg ml−1 in buffer A at beamline BM29 at ESRF (Grenoble, France), covering a momentum transfer range of 0.0282 < s < 4.525 Å−1. Ten 1s frames were collected for each sample. Frames with radiation damage were discarded automatically, and buffer scattering profiles measured before and after the sample were averaged and subtracted from the protein scattering profiles by using standard protocols (45). The forward scattering, I(0), and the radius of gyration, Rg, were calculated with the Guinier approximation assuming that, at very small angles (s< 1.3/Rg), intensity is represented as I(s) = I(0)exp(-(sRg)2/3). The maximum particle dimension, Dmax, and the distance distribution were calculated from the scattering pattern with program GNOM (46). The molecular weight of the particle was calculated by comparison of the forward scattering I(0) with that from a reference solution of BSA (at 3.6 mg ml−1). In order to characterize the conformational variability of Twinkle detected in our SAXS studies (see Results), we first generated a model of a monomer with program MODELLER (38), with which we generated hexamers and heptamers (see above). Subsequently, by applying the structural model generation module of the ensemble optimization method (EOM v2.0 (47)) we defined the ZBD, RPD and the helicase domain as rigid bodies, and the connecting linkers and N- and C-terminal segments as flexible regions, and generated 5000 models with different conformations for the hexamers and heptamers. For all these models the theoretical SAXS profile was computed with the program CRYSOL (48) using standard parameters. From all these models/curves EOM selected a sub-ensemble of five conformations that optimally described the experimental SAXS curve. In order to have a better description of the conformational flexibility of Twinkle, the optimization was performed 100 times. The same procedure was repeated for sub-ensembles of 2, 10, 15, 20 and 50 conformations. Importantly, equivalent Rg and Dmax distributions were obtained independently of the size of the sub-ensemble, suggesting no overfitting (see Results).
RESULTS
In order to find optimal conditions to stabilize Twinkle in a homogeneous population with no nucleic acid bound, we screened several chemical solutions by differential scanning fluorimetry (49). Twinkle was consistently more stable at high concentrations of NaCl (typically ∼1M). Therefore, by using high salt we ensured that the sample did not bind DNA, as assessed by monitoring the ratio between the ODs at 260 and 280 nm during all steps of purification, while the particle maintained its oligomeric structure, as monitored by gel filtration (Figure 1C). Activity analysis of Twinkle in E. coli and in insect cells showed that the DNA binding and helicase activities were indistinguishable from those of Twinkle expressed by mammalian cells (Figure 2A).
Figure 2.

Activity and oligomers of Twinkle. (A) DNA unwinding activity of the three forms of Twinkle, expressed in SF9 insect cells (SF9, lanes 3 and 4), in E. coli (lanes 5 and 6) and in HEK mammalian cells (HEK, lanes 7 to 9, produced as previously reported) (25), was analyzed by monitoring the unwound ssDNA radiolabeled product of 60 bases from the M13(+) ssDNA plasmid. Lane 1 contains substrate; lane 2, boiled substrate; and the remaining lanes contain the indicated amounts of Twinkle (from 30ng to 100ng). Helicase activity of Twinkle was tested in the presence of UTP, except lane 8 for the HEK cells (lane 60ng -UTP). (B) Samples of Twinkle produced in insect cells purified by size exclusion chromatography were analyzed by negative staining EM. The two-dimensional class averages obtained by classification show the presence of hexamers, heptamers and open rings.
Twinkle forms flexible hexameric and heptameric rings
To elucidate the structural organization of Twinkle, we first carried out single-particle negative staining EM studies with Twinkle produced in either insect cells or in E. coli. In both cases the two-dimensional analysis of recombinant Twinkle revealed distinct averages that display either six or seven radial densities (Figure 2B and Supplementary Figure S1), indicating the coexistence of heptameric and hexameric ring-like species. Each monomer showed one region connected to neighboring subunits and contributing to a ring with a central channel, while another region was exposed to the solvent with variable orientations in radial arrangement (Figure 2B), thus suggesting that, in the conditions tested, Twinkle oligomers have flexible regions. This flexibility is reminiscent of the T7 gp4 crystal structure, in which the C-terminal segment of the RPD-CTD linker performs strong contacts with the CTD of the neighboring subunit, whereas the N-terminal linker segment allows variable orientations of the RPDs over adjacent CTDs. This results in a loose arrangement of all N-terminal domains with respect to the helicase ring (12).
Cryo-EM studies show asymmetry in the N-terminal ring
Despite the initial successful classification by negative staining EM, further analysis by 3D averaging techniques such as cryo-electron microscopy (cryo-EM) required a more homogeneous sample. SEC revealed species with different hydrodynamic radius, and thus heterogeneous. Fractions collected at the end of the SEC peak (Figure 1C), which were expected to contain mostly hexamers, were selected for stabilization by the GraFix method (31). This technique rigidifies the macromolecules without inducing major structural rearrangements, by fractionating different conformations or oligomerization states in a density gradient of both glycerol and cross-linker (31,50). An initial analysis by negative staining of distinct GraFix-generated fractions showed that only few contained homogeneous molecular populations. These were mostly hexamers, and suitable for cryo-EM. No heptamers were detected. The initial two-dimensional characterization of cross-linked Twinkle showed a more uniform diameter of the oligomers (Figure 3A) when compared with the negatively stained free sample (Figure 2B). After processing 12 790 particles, two types of electron density map (at 11.6 Å, cut-off of 0.15 in the FSC, Supplementary Figure S2) were calculated. One map was obtained by applying six-fold rotational symmetry to the entire particles (map C6-C6), which, consistent with the negative-staining EM, showed two stacked rings of different overall diameter encircling a central channel that widened when going from the upper narrow ring to the lower open lobules (Figure 3B). The upper ring was assigned to the helicase CTDs (‘TOP’ in Figure 3C; ∼130 Å in diameter in panel B) and the lower to the NTDs (‘BOTTOM’; ∼160 Å in diameter). The NTDs appeared in the form of six symmetric densities with no visible connections between them (Figure 3C). In addition, fostered by the T7 gp4 crystal structure, which showed variable orientations of the N-terminal domains (12), we further broke the six-fold symmetry of the widest NTDs ring while keeping the six-fold symmetry for the CTDs. This resulted in a 3D map (map C1-C6, at estimated resolution of 12 Å, Supplementary Figure S2) that showed the smaller ring with continuous density in a six-tip star shape, which was similar to the CTDs ring of map C6-C6 (compare Figure 3C and 3D, left panel). In contrast, in the ring attributed to the NTDs, four extra density blobs arose connecting five N-terminal lobules, while one NTD remained isolated (Figure 3D, central and right panels; compare with Figure 3C, same panels). Fully asymmetric image processing for both NTD and CTD yielded map C1 (Figure 3E) with features similar to map C1-C6, but in the last the signal-to-noise ratios were better. The sequence of slices from the N-terminal to the C-terminal ring of the reconstructed volume further evidenced the asymmetry of the particle (Figure 3E).
Figure 3.
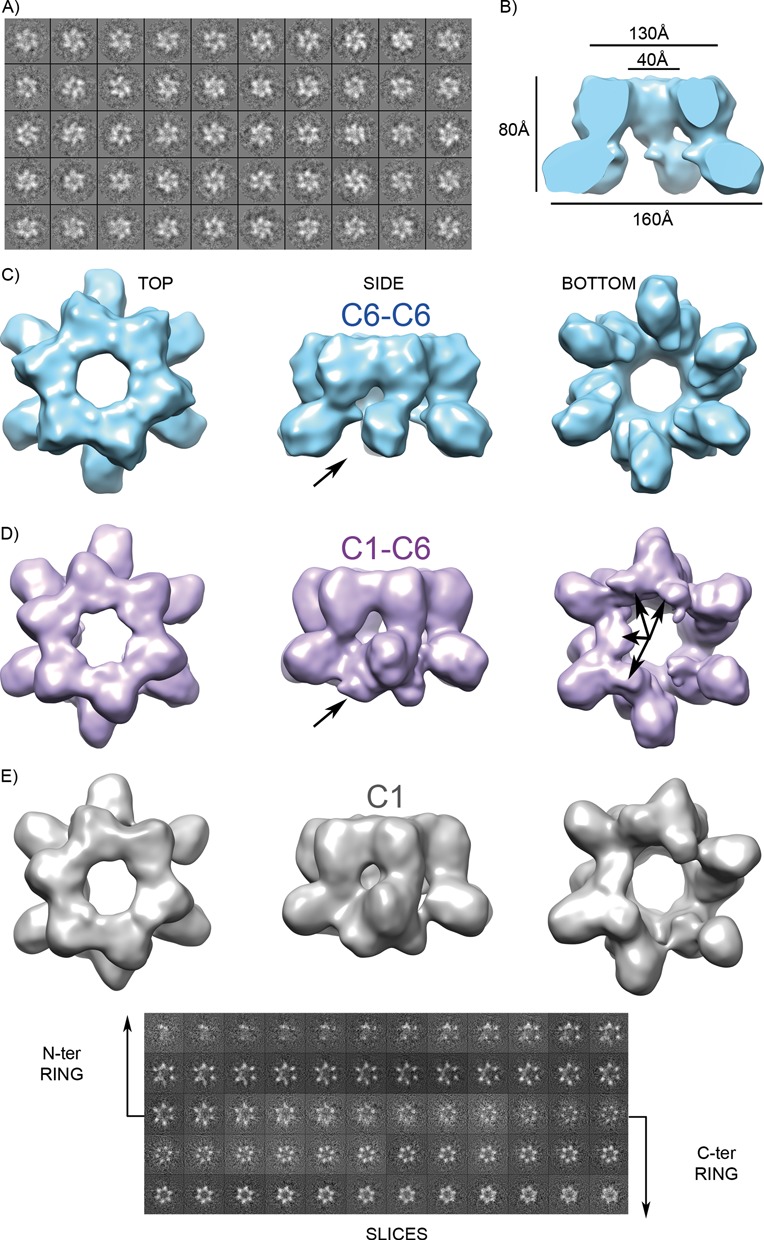
Mitochondrial helicase 3D reconstruction. (A) Cryo-EM 2D classes of a crosslinked GraFix fraction (see Results) showed hexamers. (B) The 3D reconstruction applying C6 symmetry to the whole particle (C6-C6 map) shows two stacked rings, one with continuous density and the other with no density between lobules. (C) Three views of the electron density map after applying 6-fold symmetry to the whole particle. (D) Relaxation of the symmetry at the discontinuous ring (C1-C6 map) resulted in extra density between lobules, indicated by arrows. (E) Rendering of the 3D map without imposed symmetry (C1) is displayed together with slices of the reconstructed volume. The sequence of slices reveals 6-fold symmetry in the C-terminal ring but open asymmetric rings at the N-terminal region.
Cryo-EM-based homology modeling
In order to analyze the molecular architecture of Twinkle oligomers within the cryo-EM maps, a homology-model of each of the constituting domains was built. Among the SF4 family of helicases, only partial crystal structures of T7 gp4 have been solved by X-ray crystallography to date: the CTD in complex with NTPs, which forms a helical filament (PDB codes 1CR0 to 1CR4) (51); the CTD and the linker in complex with a non-hydrolysable ATP analog forming a hexameric ring (1E0J and 1E0K) (6); a 56-kDa natural short form with excised ZBD, which crystallized as a heptameric ring (1Q57, Figure 1A) (12); and the isolated primase domain showing ZBD-RPD domain swapping between two crystallographic partners (1NUI) (17). Separately, these structures did not enable us to build a full-length model of Twinkle because the NTD of the latter contains insertions, in particular at the ZBD (see Supplementary Figure S2). Searches with BLAST against the Protein Data Bank (PDB; www.pdb.org) using only the sequence of the Twinkle N-terminal region pointed to the crystal structure of DnaG primase from A. aeolicus (PDB code 2AU3) (24), which includes both a ZBD and an RPD, as a potential structural relative. Therefore, taking into account the secondary structure prediction of Twinkle, its sequence was threaded into a chimera of the DnaG (PDB 2AU3) and T7 gp4 (PDB 1Q57) models, which were superimposed on their respective RPDs, using MODELLER (38) (Figure 1A, see Experimental Procedures).
Rigid-body fitting was performed against both C6-C6 and C1-C6 maps. In T7 gp4 oligomers, the NTDs are placed on top of the CTDs from the neighboring subunit by virtue of an extended interconnecting linker, and the same arrangement was compatible with our cryo-EM maps of Twinkle (Figure 4A). Since in both C6-C6 and C1-C6 maps a six-fold symmetry had been applied to the C-terminal ring, the CTD fit was identical in both cases and, indeed, similar to the hexameric and heptameric structures of T7 gp4, which show similar CTD interfaces (12). However, for the NTDs, fitting into the C6-C6 map was hampered by the small size of the averaged reconstructed volumes. In contrast, NTD fitting into the C1-C6 map was better but required the introduction of inter-domain flexibility by molecular dynamics (see Experimental Procedures and Supplementary Figure S4). In particular, this latter fitting required rotation of the NTDs to place four ZBDs of consecutive subunits (ZBD1 to ZBD4) into the four extra densities. Following this arrangement, the four ZBDs contact the surface facing the channel of a neighboring RPD (contact in trans) while also contacting the RPD of their own subunit (contact in cis), thus showing a head-to-tail interaction between RPDs and ZBDs (Figure 4B and C). However, whereas the RPDs fit into well-defined density and are arranged similarly within the helicase ring, the intervening ZBDs show variable orientations (Figure 4B, left panel) and protrude differently from the particle (Figure 4B, central panel): ZBD2 is closest to the RPD ring, while ZBD1, ZBD4 and ZBD3 are moved outward by, respectively, 8 Å, 9 Å and 12 Å along the vertical axis. Therefore, the RPD-ZBD contacts are not constant. No suitable density was observed for ZBD5 or ZBD6 (Figure 4B and C, left panel), thus indicating that in two neighboring subunits the ZBDs are highly flexible. Overall, the NTD has a stable part provided by the RPDs and a highly flexible part afforded by the ZBDs.
Figure 4.
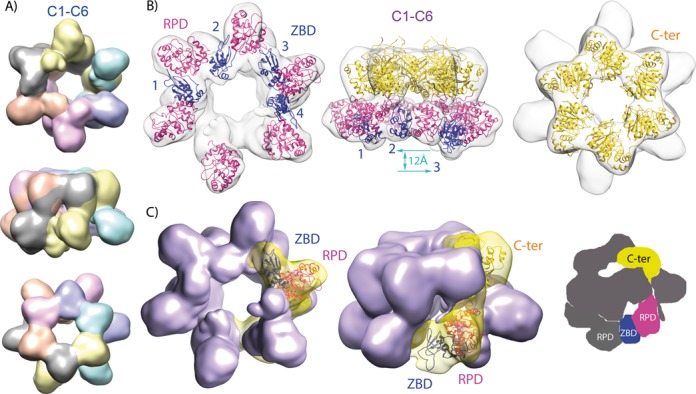
Model fitting. (A) Segmentation of the C1-C6 map into monomers upon structural fitting based on the T7 gp4 short form structural arrangement (12), in which each RPD contacts the C-terminal domain of the next subunit. (B) Organization of the molecular model upon flexible fitting into the C1-C6 3D map. The ZBD domains are numbered and represented in blue, RPDs in magenta and CTDs in gold. The C- (or N)-terminal domains are not depicted in the left (or right) volume, for clarity. The vertical blue arrow symbolizes the level difference between ZBD2 and ZBD3 (12 Å). (C) Left and central volumes are segmented around one subunit with the homology model fitted in, with same colors as in B. Right, scheme showing a ZBD contacting the RPD from its own subunit and from its neighbor simultaneously.
SAXS studies reveal high flexibility at the inter-domain connectors
To characterize the conformational dynamics of Twinkle in solution, we conducted SAXS studies with Twinkle produced in E. coli, whose gel filtration elution profile and negative-staining analysis were equivalent to those of protein produced in insect cells. The molecular mass estimated by SAXS was 454KDa, which suggested the presence of a mixture of hexamers (442 KDa) and heptamers (515 KDa). The Rg of Twinkle in solution was 69.8 Å and the pairwise distance distribution function of the curve, ρ(r) (Supplementary Figure S5), which reflects the distribution of the intra-molecular distances, showed a smooth decrease toward a maximum distance (Dmax) of 240 Å. In addition, the Kratky plot displayed a pronounced peak that did not drop to zero at high s values, which is consistent with a globular protein with flexible regions (Supplementary Figure S5). In order to explain the SAXS curve at the molecular level, hexamers (based on T7 gp4 PDB 1E0J) and heptamers (PDB 1Q57) of the aforementioned homology model were constructed, but they did not fit the experimental SAXS curves (the associated χ2 values were >10 in all cases). Considering the Kratky plot and previous reports suggesting flexibility at the interdomain loops (5), we attributed this disagreement to the absence of flexibility in our atomic models. Therefore, we applied the ensemble optimization method (EOM, (47)) to generate hexamers and heptamers with flexible inter-domain arrangements. Two pools of 5000 hexamers (based on the aforementioned C1-C6 map homology model) and heptamers (based on a chimera model of T7 gp4 heptamer 1Q57 and DnaG primase 2AU3) were generated using RanCh (47). In these models the ZBD-RPD linker (residues 157–161) and the N-terminal segment of the RPD-CTD linker (residues 361–365), as well as the N-terminal and the C-terminal tails, were defined as flexible regions with complete conformational freedom. The EOM genetic algorithm selected a sub-ensemble of five conformations with a hexamer/heptamer ratio of 3/2, which, collectively, was in agreement (χ2 = 0.71) with the SAXS curve in the complete momentum transfer range (Figure 5A). These results substantiate equilibrium between hexamers and heptamers as observed in EM. The wide Rg distribution of the EOM-selected models reflected the heterogeneity of conformations present in the sample (Supplementary Figure S5). In addition, calculation of EOM sub-ensembles using a variable number of conformations (from 2 to 50) presented similar quality of description of the experimental SAXS curve and, importantly, showed similar Rg and Dmax distributions, suggesting no overfitting of the data (Supplementary Figure S5). The representation of the five conformations sub-ensemble reveals that the helicase ring domains are connected to highly flexible linkers that orientate the NTDs mostly on one side of the Twinkle ring (Figure 5B). In addition, ZBDs and RPDs also showed multiple relative orientations toward each other. Both Twinkle models, that from the T7 gp4 heptameric ring and that obtained by EM, have a similar overall diameter (127 and 124 Å, respectively, Figure 5C). Both have a channel of more than 40 Å that could putatively accommodate either ss- or dsDNA, which would be compatible with Twinkle's function. In contrast, EOM trials using a T7 gp4 structure-based flexible hexamer did not fit the SAXS data (χ2 = 1.80); note that this structure has a ring width of 108 Å, and the central channel is 18 Å wide, so it could only thread an ssDNA strand (Figure 5C). Considering the ability of EOM to detect and quantify the flexibility of macromolecules in solution (52), this result strongly suggested that our particles in solution displayed a wide central channel. Overall, these results indicate compatibility of the flexible structure derived from the cryo-EM hexamer with a fraction of the population in solution, the rest being compatible with flexible T7 gp4-based heptamers. In addition, unbound Twinkle oligomer dimensions are consistent with binding of either ss- or dsDNA through the central channel.
Figure 5.
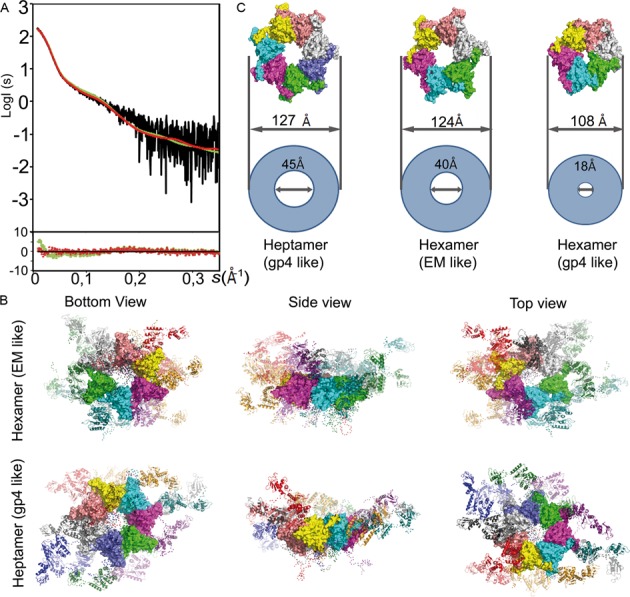
Analysis of Twinkle in solution. (A) Left panel: the experimental scattering-intensity curve (in black) is represented on a logarithmic scale as a function of the momentum transfer, s = 4πsin(θ) λ−1 (2θ, scattering angle; λ = 0.9919 Å, X-ray wavelength). The fitted EOM (see Experimental Procedures) curve for the mixture of flexible hexamers and heptamers (in red, χ2 = 0.71) and for the flexible gp4-like hexamer (in green, χ2 = 1.80) is shown. Below the panel, residuals show respective quality of fit. (B) Top and lateral molecular representation of the sub-ensemble of two models of T7 gp4 structure-based (1Q57, gp4 like) heptamers (right column) and three of EM-based hexamers (EM like, left) that collectively fit the data, superimposed by their CTD. ZBDs and RPDs are represented in ribbons while CTD rings are represented as a surface; each subunit has a different color. Red dots represent the position of Cα atoms of the flexible linkers and N- and C-terminal tails. (C) Surface representations of Twinkle CTD rings based on the T7 gp4 heptamer (PDB 1Q57) and hexamer (1E0J) and the cryo EM structure described here. Relative internal and external diameters values are represented by circles.
DISCUSSION
Our structural studies show that Twinkle forms hexamers and heptamers of variable conformation in the presence of high salt but in the absence of NTPs, Mg2+ or DNA. These findings are consistent with previous reports showing that both stability and flexibility of Twinkle increased with salt concentration (11). In general, DNA-binding proteins are stabilized by solutions containing high concentrations of NaCl that compensate the protein electropositive surfaces involved in contacts with the negatively charged phosphate backbone of nucleic acids. On the other hand, high ionic strength may disrupt electrostatic interactions between domains, resulting in greater flexibility. This has been illustrated for A. aeolicus DnaG primase, in which the ZBD and RPD, connected by a flexible linker, crystallized at low salt conditions in a compact conformation with the two domains docked through a hydrophilic interface (24) (PDB 2AU3). The same authors used FRET at physiological salt concentrations to reveal a compact form that, at high salt, extended thanks to the flexible linker. Similarly, the crystal structure of the T7 gp4 primase domain (PDB 1NUI) showed two protomers that swap their ZBDs, which perform electrostatic contacts with the symmetric RPDs by virtue of a flexible extended inter-domain linker (17). For Twinkle, flexibility of the ZBD-RPD linker was previously suggested (5) and is hereby confirmed. On the other hand, our combination of SEC with GraFix sub-fractionation allowed isolation of stabilized hexamers with a compact arrangement. This contrasts with the description of Twinkle in solution as an ensemble of more or less extended conformations. We hypothesize that a subpopulation of hexamers was locked in their compact state by the GraFix method. Overall, these results suggest that the inter-domain contacts between ZBD and RPD, but also those between RPD and CTD, are predominantly electrostatic and putatively tunable during the activity cycle of Twinkle, potentially undergoing structural rearrangements triggered by flexible linkers between domains.
Structural variability in ZBDs
For an SF4 protein displaying a structural organization homologous to a bifunctional primase-helicase, the hexameric model we present here includes a complete protomer comprising ZBD, RPD and CTD. Interestingly, the ZBDs were visible only in the absence of symmetry restraints and in four out of six protomers, thus indicating different positions and orientations of ZBDs in the stable hexamer. Since partial ZBD excision (construct Δ1–121) particularly reduces ssDNA binding (13), our cryo-EM model suggests that during translocation the ZBDs may bind ssDNA from different positions, two of them highly flexible. On the other hand, the clustering of delocalized ZBDs at one region of the NTDs ring or the open rings detected by negative staining suggest particles broken on one side. Notably, ring opening is triggered by the NTD in T7 gp4 (53) and is required for DNA loading (54).
Within the cryo-EM Twinkle hexamer, RPDs and ZBDs contact in a head-to-tail manner. Contacts between RPDs and ZBDs from different subunits are part of the transfer mechanism of the ssDNA priming site from the ZBD to the RPD in active primases (16). E. coli and A. aeolicus DnaGs and phage T7 gp4 perform efficient primer synthesis by cis and trans crosstalk between ZBDs and RPDs from different subunits (23,24). Cis and trans ZBD-RPD contacts have been shown by crystal structure analysis (17,24), yet they do not involve the active sites. In our cryo-EM model, the ZBDs contact neighboring subunit RPDs in a region facing the channel. At the back of this region, exposed to the solvent, are the amino acids that would be involved in primer synthesis in an active primase (compare Figure 4C and Supplementary Figure S6). If this contact occurred in an active SF4 primase, ZBD and RPD active sites could interact only by means of a small linker extension and a slight RPDs and ZBDs reorientation.
Contacts between N-terminal and helicase domains
Progressively longer deletion of the NTD—i.e. removal of ZBD, or ZBD and part of RPD (truncation Δ1–314)—increasingly affects ATPase and helicase activities of Twinkle, suggesting a functional or structural role of the NTD that influences the CTD folding and function (13). In our hands, a Twinkle construct that contained the CTD and the preceding inter-domain linker (Δ1–357) showed high heterogeneity both in size-exclusion chromatography and in florescence-based thermal shift assays (data not shown). This contrasted with the homogeneity of the full-length protein and suggested a weak contribution of the linker to the hexamer assembly. It also contrasts with T7 gp4, in which the linker alone stabilizes multimerization of the CTDs (6,51). Instead, the N-terminal truncation Δ1–314, which keeps the C-terminal region of the RPD, can form stable hexamers, indicating that the RPD fragment contains residues that contribute to oligomer stability (13). This is consistent with the Twinkle EM-based oligomeric model, which shows all RPD C-terminal regions facing the CTDs in rather similar orientations. In particular, vicinal RPD/CTD surfaces include motif V and VI from the RPD and motif 1a and following residues to motif 2 from the CTD (Supplementary Figure S6), which, together with the linker, are hot-spots for disease-linked mutations (Supplementary Table 1). According to previous reports (14), most of these mutants show structural instability that could have deleterious consequences in the long term. However, due to the limitation of our resolution, these detailed structural observations should be taken with caution. Nonetheless, in T7 gp4, the contact between RPD and CTD, despite being far from the active site, has shown to modulate the activity of the phage helicase (55), pin-pointing its functional importance. The functions of Twinkle include DNA translocation, which may involve structurally relevant subunit-specific CTD rotations to configure the central channel for ssDNA binding and pulling. This is the case in T7 gp4 (6), DnaB (56) and in papilloma-virus E1 helicase (from SF3) (57). Based on the Twinkle EM structure, a rotation of a CTD would affect the contact with the adjacent subunit RPD. Whether the RPD/CTD interface is disrupted or, by means of a flexible RPD-CTD linker remains intact during translocation, needs to be addressed by further research.
Heptamers, hexamers and variable central channel diameter
Studies describing hexamers and heptamers show disparate results, possibly attributable to differences in the experimental conditions (9,11). We found both hexamers and heptamers in the absence of ligands at high salt. T7 gp4 forms hexamers when bound to nucleoside di- or triphosphate and ssDNA, while a mixture of hexa- and heptamers appears in the absence of DNA. An ssDNA loading mechanism was proposed, according to which a heptamer ejects one subunit when it contacts ssDNA. This creates a gap in the ring through which the hexamer loads the ssDNA to the internal channel (54). However, because T7 gp4 binds dsDNA, other authors have proposed that heptamers translocate on it (12). Our unliganded heptamer selected by SAXS shows a wide central channel compatible with either functional model, i.e. subunit ejection upon ssDNA detection or dsDNA translocation. The EM hexamer also shows a wide channel, and could thus allocate dsDNA. However, structural studies of hexameric helicases systematically show that, upon ssDNA binding, the ring converts to a right-handed spiral with a dramatic narrowing of the central channel, which harbors ssDNA (54,56). Unliganded Twinkle hexamers could also undergo similar reshaping upon binding of cofactors or ssDNA.
Twinkle is particular in binding both dsDNA and ssDNA but, in contrast to T7 gp4, it has higher affinity for the former (9,11,13,14). In addition, a study analyzing the DNA re-annealing ability of Twinkle detected a total of three DNA-binding sites, two for ssDNA (putatively in the central channel and on the external ring surface) and one for dsDNA (9). Accordingly, sequence alignment (Supplementary Figure S3) suggests that Twinkle's central channel may also contain the CTD ssDNA-binding loops found in other SF4 helicases, and our EM model shows the RPDs oriented so that the regions involved in ssDNA binding in active primases (e.g. DnaG (20)) are on the outer surface (Supplementary Figure S6). Therefore, the cryo-EM model of Twinkle described here is also compatible with internal and external binding sites for ssDNA reannealing.
A number of mutations in Twinkle have been found to be associated with various pathologies, and insights into the molecular mechanisms underlying its function in health and disease are provided by examination of its 3D structure. In summary, the hexamer structure revealed by EM shows a compact arrangement, in which a ZBD and an RPD from one subunit contact, respectively, the RPD and CTD of a neighboring subunit. RPDs and CTDs show broadly similar arrangements, suggesting a stable, functionally relevant contact. In contrast, the ZBDs show independent orientations, which is compatible with the asymmetric, differential ZBD positioning required for DNA pulling. Interdomain flexible loops allow for multiple conformations, and in solution ZBD/RPD/CTD contacts are disrupted while the hexameric ring remains intact. Taken together, our results show that Twinkle's 3D structure supports the high plasticity needed for DNA loading and helicase activity.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank the European Synchrotron Radiation Facility (ESRF) in Grenoble (France) for access to the BioSAXS beamline and for excellent support.
Footnotes
Present addresses:
Pablo Fernández-Millán, UPR ‘Architecture et Réactivité de l'ARN’, Université de Strasbourg, CNRS, Institut de Biologie Moléculaire et Cellulaire, Strasbourg, F-67084 Cedex, France.
Joachim M. Gerhold, University of Tartu, Dept. of Biophysics and Plant Physiology, Institute of Molecular and Cell Biology, Tartu, 51010, Estonia.
Nina Rajala, Cancer Genomics, BioMediTech, University of Tampere, Tampere, FI-33014, Finland.
FUNDING
MINECO [BFU2009-07134, BFU2012-33516 to M.S., BFU2012-34873 to M.V.]; Generalitat de Catalunya [SGR2009-1366, 2014 SGR 997 to M.S.]; EU [FP7-HEALTH-2010-261460, FP7-PEOPLE-2011-290246, FP7-HEALTH-2012-306029-2 to M.S.]. P.F.-M. held an FPI fellowship from MINECO [BES-2008-005384REF]. Academy of Finland [CoE funding to J.N.S.]; Tampere University Hospital Medical Research Fund [9J119, 9K126, 9L097 to J.N.S.]; Netherlands Organization for Scientific Research [NWO: VICI grant 865.10.004 to J.N.S.]; European Molecular Biology Organization [EMBO LTF co-funded by Marie Curie Actions, EMBO LTF 1066_2011 (including MCA-EMBOCOFUND2010,GA-2010-267146) to J.M.G.]; Agence Nationale de la Recherche SPIN-HD [CHEX-2011]; ATIP-Avenir. Funding for open access charge: MINECO [BFU2012-33516 to M.S.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Spelbrink J.N., Li F.Y., Tiranti V., Nikali K., Yuan Q.P., Tariq M., Wanrooij S., Garrido N., Comi G., Morandi L., et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat. Genet. 2001;28:223–231. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- 2.Leipe D.D., Aravind L., Grishin N.V., Koonin E.V. The bacterial replicative helicase DnaB evolved from a RecA duplication. Genome Res. 2000;10:5–16. [PubMed] [Google Scholar]
- 3.Shutt T.E., Gray M.W. Twinkle, the mitochondrial replicative DNA helicase, is widespread in the eukaryotic radiation and may also be the mitochondrial DNA primase in most eukaryotes. J. Mol. Evol. 2006;62:588–599. doi: 10.1007/s00239-005-0162-8. [DOI] [PubMed] [Google Scholar]
- 4.Singleton M.R., Dillingham M.S., Wigley D.B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 5.Ziebarth T.D., Farr C.L., Kaguni L.S. Modular architecture of the hexameric human mitochondrial DNA helicase. J. Mol. Biol. 2007;367:1382–1391. doi: 10.1016/j.jmb.2007.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singleton M.R., Sawaya M.R., Ellenberger T., Wigley D.B. Crystal structure of T7 gene 4 ring helicase indicates a mechanism for sequential hydrolysis of nucleotides. Cell. 2000;101:589–600. doi: 10.1016/s0092-8674(00)80871-5. [DOI] [PubMed] [Google Scholar]
- 7.Jemt E., Farge G., Backstrom S., Holmlund T., Gustafsson C.M., Falkenberg M. The mitochondrial DNA helicase TWINKLE can assemble on a closed circular template and support initiation of DNA synthesis. Nucleic Acids Res. 2011;39:9238–9249. doi: 10.1093/nar/gkr653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Korhonen J.A., Gaspari M., Falkenberg M. TWINKLE Has 5’ -> 3’ DNA helicase activity and is specifically stimulated by mitochondrial single-stranded DNA-binding protein. J. Biol. Chem. 2003;278:48627–48632. doi: 10.1074/jbc.M306981200. [DOI] [PubMed] [Google Scholar]
- 9.Sen D., Nandakumar D., Tang G.Q., Patel S.S. Human mitochondrial DNA helicase TWINKLE is both an unwinding and annealing helicase. J. Biol. Chem. 2012;287:14545–14556. doi: 10.1074/jbc.M111.309468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korhonen J.A., Pham X.H., Pellegrini M., Falkenberg M. Reconstitution of a minimal mtDNA replisome in vitro. EMBO J. 2004;23:2423–2429. doi: 10.1038/sj.emboj.7600257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ziebarth T.D., Gonzalez-Soltero R., Makowska-Grzyska M.M., Nunez-Ramirez R., Carazo J.M., Kaguni L.S. Dynamic effects of cofactors and DNA on the oligomeric state of human mitochondrial DNA helicase. J. Biol. Chem. 2010;285:14639–14647. doi: 10.1074/jbc.M109.099663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toth E.A., Li Y., Sawaya M.R., Cheng Y., Ellenberger T. The crystal structure of the bifunctional primase-helicase of bacteriophage T7. Mol. Cell. 2003;12:1113–1123. doi: 10.1016/s1097-2765(03)00442-8. [DOI] [PubMed] [Google Scholar]
- 13.Farge G., Holmlund T., Khvorostova J., Rofougaran R., Hofer A., Falkenberg M. The N-terminal domain of TWINKLE contributes to single-stranded DNA binding and DNA helicase activities. Nucleic Acids Res. 2008;36:393–403. doi: 10.1093/nar/gkm1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Longley M.J., Humble M.M., Sharief F.S., Copeland W.C. Disease variants of the human mitochondrial DNA helicase encoded by C10orf2 differentially alter protein stability, nucleotide hydrolysis, and helicase activity. J. Biol. Chem. 2010;285:29690–29702. doi: 10.1074/jbc.M110.151795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ilyina T.V., Gorbalenya A.E., Koonin E.V. Organization and evolution of bacterial and bacteriophage primase-helicase systems. J. Mol. Evol. 1992;34:351–357. doi: 10.1007/BF00160243. [DOI] [PubMed] [Google Scholar]
- 16.Kuchta R.D., Stengel G. Mechanism and evolution of DNA primases. Biochim. Biophys. Acta. 2010;1804:1180–1189. doi: 10.1016/j.bbapap.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kato M., Ito T., Wagner G., Richardson C.C., Ellenberger T. Modular architecture of the bacteriophage T7 primase couples RNA primer synthesis to DNA synthesis. Mol. Cell. 2003;11:1349–1360. doi: 10.1016/s1097-2765(03)00195-3. [DOI] [PubMed] [Google Scholar]
- 18.Keck J.L., Roche D.D., Lynch A.S., Berger J.M. Structure of the RNA polymerase domain of E. coli primase. Science. 2000;287:2482–2486. doi: 10.1126/science.287.5462.2482. [DOI] [PubMed] [Google Scholar]
- 19.Pan H., Wigley D.B. Structure of the zinc-binding domain of Bacillus stearothermophilus DNA primase. Structure. 2000;8:231–239. doi: 10.1016/s0969-2126(00)00101-5. [DOI] [PubMed] [Google Scholar]
- 20.Corn J.E., Pelton J.G., Berger J.M. Identification of a DNA primase template tracking site redefines the geometry of primer synthesis. Nat. Struct. Mol. Biol. 2008;15:163–169. doi: 10.1038/nsmb.1373. [DOI] [PubMed] [Google Scholar]
- 21.Aravind L., Leipe D.D., Koonin E.V. Toprim–a conserved catalytic domain in type IA and II topoisomerases, DnaG-type primases, OLD family nucleases and RecR proteins. Nucleic Acids Res. 1998;26:4205–4213. doi: 10.1093/nar/26.18.4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Podobnik M., McInerney P., O'Donnell M., Kuriyan J. A TOPRIM domain in the crystal structure of the catalytic core of Escherichia coli primase confirms a structural link to DNA topoisomerases. J. Mol. Biol. 2000;300:353–362. doi: 10.1006/jmbi.2000.3844. [DOI] [PubMed] [Google Scholar]
- 23.Lee S.J., Richardson C.C. Interaction of adjacent primase domains within the hexameric gene 4 helicase-primase of bacteriophage T7. Proc. Natl. Acad. Sci. U.S.A. 2002;99:12703–12708. doi: 10.1073/pnas.202471499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corn J.E., Pease P.J., Hura G.L., Berger J.M. Crosstalk between primase subunits can act to regulate primer synthesis in trans. Mol. Cell. 2005;20:391–401. doi: 10.1016/j.molcel.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 25.Wanrooij S., Goffart S., Pohjoismaki J.L., Yasukawa T., Spelbrink J.N. Expression of catalytic mutants of the mtDNA helicase Twinkle and polymerase POLG causes distinct replication stalling phenotypes. Nucleic Acids Res. 2007;35:3238–3251. doi: 10.1093/nar/gkm215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tyynismaa H., Sembongi H., Bokori-Brown M., Granycome C., Ashley N., Poulton J., Jalanko A., Spelbrink J.N., Holt I.J., Suomalainen A. Twinkle helicase is essential for mtDNA maintenance and regulates mtDNA copy number. Human Mol. Genet. 2004;13:3219–3227. doi: 10.1093/hmg/ddh342. [DOI] [PubMed] [Google Scholar]
- 27.Nikali K., Suomalainen A., Saharinen J., Kuokkanen M., Spelbrink J.N., Lonnqvist T., Peltonen L. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Human Mol. Genet. 2005;14:2981–2990. doi: 10.1093/hmg/ddi328. [DOI] [PubMed] [Google Scholar]
- 28.Nogueira C., Almeida L.S., Nesti C., Pezzini I., Videira A., Vilarinho L., Santorelli F.M. Syndromes associated with mitochondrial DNA depletion. Ital. J. Pediatr. 2014;40:34–43. doi: 10.1186/1824-7288-40-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lonnqvist T., Paetau A., Valanne L., Pihko H. Recessive twinkle mutations cause severe epileptic encephalopathy. Brain. 2009;132:1553–1562. doi: 10.1093/brain/awp045. [DOI] [PubMed] [Google Scholar]
- 30.Moriniere J., Rousseaux S., Steuerwald U., Soler-Lopez M., Curtet S., Vitte A.L., Govin J., Gaucher J., Sadoul K., Hart D.J., et al. Cooperative binding of two acetylation marks on a histone tail by a single bromodomain. Nature. 2009;461:664–668. doi: 10.1038/nature08397. [DOI] [PubMed] [Google Scholar]
- 31.Kastner B., Fischer N., Golas M.M., Sander B., Dube P., Boehringer D., Hartmuth K., Deckert J., Hauer F., Wolf E., et al. GraFix: sample preparation for single-particle electron cryomicroscopy. Nat. Methods. 2008;5:53–55. doi: 10.1038/nmeth1139. [DOI] [PubMed] [Google Scholar]
- 32.Ludtke S.J. 3-D structures of macromolecules using single-particle analysis in EMAN. Methods Mol. Biol. 2010;673:157–173. doi: 10.1007/978-1-60761-842-3_9. [DOI] [PubMed] [Google Scholar]
- 33.Baxter W.T., Leith A., Frank J. SPIRE: the SPIDER reconstruction engine. J. Struct. Biol. 2007;157:56–63. doi: 10.1016/j.jsb.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 34.Frank J., Radermacher M., Penczek P., Zhu J., Li Y., Ladjadj M., Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 35.Scheres S.H., Nunez-Ramirez R., Sorzano C.O., Carazo J.M., Marabini R. Image processing for electron microscopy single-particle analysis using XMIPP. Nat. Protoc. 2008;3:977–990. doi: 10.1038/nprot.2008.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenthal P.B., Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 37.Buchan D.W., Minneci F., Nugent T.C., Bryson K., Jones D.T. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 2013;41:W349–W357. doi: 10.1093/nar/gkt381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eswar N., Webb B., Marti-Renom M.A., Madhusudhan M.S., Eramian D., Shen M.Y., Pieper U., Sali A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinform. 2006;2006:1–30. doi: 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 40.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kale L., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trabuco L.G., Villa E., Mitra K., Frank J., Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16:673–683. doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H., Robertson A.D., Jensen J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 43.Mackerell A.D. Jr, Feig M., Brooks C.L. 3rd. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 44.Tanner D.E., Chan K.Y., Phillips J.C., Schulten K. Parallel Generalized Born Implicit Solvent Calculations with NAMD. J. Chem. Theory Comput. 2011;7:3635–3642. doi: 10.1021/ct200563j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petoukhov M.V., Franke D., Shkumatov A.V., Tria G., Kikhney A.G., Gajda M., Gorba C., Mertens H.D.T., Konarev P.V., Svergun D.I. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Cryst. 2012;45:342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Svergun D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992;25:495–503. [Google Scholar]
- 47.Bernadó P., Mylonas E., Petoukhov M.V., Blackledge M., Svergun D.I. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 48.Svergun D.I., Barberato C., Koch M.H.J. CRYSOL - a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 1995;28:768–773. [Google Scholar]
- 49.Ericsson U.B., Hallberg B.M., Detitta G.T., Dekker N., Nordlund P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem. 2006;357:289–298. doi: 10.1016/j.ab.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 50.Lee S., Sielaff B., Lee J., Tsai F.T. CryoEM structure of Hsp104 and its mechanistic implication for protein disaggregation. Proc. Natl. Acad. Sci. U.S.A. 2010;107:8135–8140. doi: 10.1073/pnas.1003572107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sawaya M.R., Guo S., Tabor S., Richardson C.C., Ellenberger T. Crystal structure of the helicase domain from the replicative helicase-primase of bacteriophage T7. Cell. 1999;99:167–177. doi: 10.1016/s0092-8674(00)81648-7. [DOI] [PubMed] [Google Scholar]
- 52.Bernadó P. Effect of interdomain dynamics on the structure determination of modular proteins by small-angle scattering. Eur. Biophys. J. 2010;39:769–780. doi: 10.1007/s00249-009-0549-3. [DOI] [PubMed] [Google Scholar]
- 53.Ahnert P., Picha K.M., Patel S.S. A ring-opening mechanism for DNA binding in the central channel of the T7 helicase-primase protein. EMBO J. 2000;19:3418–3427. doi: 10.1093/emboj/19.13.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O'Shea V.L., Berger J.M. Loading strategies of ring-shaped nucleic acid translocases and helicases. Curr. Opin. Struct. Biol. 2014;25:16–24. doi: 10.1016/j.sbi.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu B., Lee S.J., Richardson C.C. An in trans interaction at the interface of the helicase and primase domains of the hexameric gene 4 protein of bacteriophage T7 modulates their activities. J. Biol. Chem. 2009;284:23842–23851. doi: 10.1074/jbc.M109.026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Itsathitphaisarn O., Wing R.A., Eliason W.K., Wang J., Steitz T.A. The hexameric helicase DnaB adopts a nonplanar conformation during translocation. Cell. 2012;151:267–277. doi: 10.1016/j.cell.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Enemark E.J., Joshua-Tor L. Mechanism of DNA translocation in a replicative hexameric helicase. Nature. 2006;442:270–275. doi: 10.1038/nature04943. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.