


Abstract
To effectively modulate the gene expression within an infected mammalian cell, the pathogen Mycobacterium tuberculosis would need to bring about epigenetic modifications at appropriate genomic loci. Working on this hypothesis, we show in this study that the mycobacterial protein Rv2966c is a 5-methylcytosine-specific DNA methyltransferase that is secreted out from the mycobacterium and gets localized to the nucleus in addition to the cytoplasm inside the host cell. Importantly, Rv2966c binds to specific DNA sequences, methylates cytosines predominantly in a non-CpG context and its methylation activity is positively influenced by phosphorylation. Interestingly, like the mammalian DNA methyltransferase, DNMT3L, Rv2966c can also interact with histone proteins. Ours is the first study that identifies a protein from a pathogenic bacteria with potential to influence host DNA methylation in a non-canonical manner providing the pathogen with a novel mechanism to alter the host epigenetic machinery. This contention is supported by repression of host genes upon M. tuberculosis infection correlated with Rv2966c binding and non-CpG methylation.
INTRODUCTION
The response of a host cell to infection by a pathogenic bacteria exemplifies the ability of a cell to mount a coordinated and calibrated response to environmental cues. Studies that have examined the interaction of a mammalian host cell with pathogenic bacteria show that this interaction is not limited to eliciting an immune response. It also manifests as changes in specific cell-signalling cascades and expression of multiple genes amongst others (1,2).
As modulation of gene expression in a cell is achieved by changes in epigenetic modifications like DNA methylation and histone modifications (3–5), it is possible that epigenetic modifications themselves change upon infection. The epigenetic changes observed during host–pathogen interaction could be a result of either direct or indirect interaction of pathogen-specific factors (protein or RNA) with the cellular machinery. Studies that have examined pathogen factor-specific changes in the host cell epigenome are very few and the underlying mechanisms are not very well understood. Only recently a few reports have shown that pathogens exploit host epigenome plasticity to ensure their successful survival (6,7). Proteins like RomA in Legionella pneumophila and NuE in Chlamydia trachomatis have been shown to modify the host histones at H3 (at Lysine 14) and H3, H4 respectively (8,9). Listeria monocytogens protein LLO is known to induce a signalling cascade that eventually alters the chromatin (10). Existence of histone mimics (influenza viral protein NS1) has also been reported (11). While there have been a few studies that have examined the role of bacterial proteins affecting histone modifications in the host, we were unable to find any report in the literature that had looked at bacterial proteins that affect the host DNA methylation.
Mycobacterial genome have been found to be rich in G + C content (12). Mycobacterial species contains methyltransferases that are not canonical dam or dcm DNA methyltransferases as is evident from the presence of 5-methylcytosine in addition to 6-methyladenine in their genome (13). While a DNA methyltransferase (Rv3263) that can methylate adenine in the mycobacterial genome has been recently identified (14), no mycobacterial DNA methyltransferase has been characterized that can methylate DNA in cytosine context. As cytosine methylation is observed extensively in mammalian cells, the aim of the present study was to identify and characterize a mycobacterial protein that could perform cytosine methylation in the host cell upon infection.
To identify such a DNA methyltransferases from mycobacteria the following biochemical criterion were set: (i) ability to methylate DNA in cytosine context; (ii) ability to be secreted out by mycobacterium; and (iii) ability to localize to mammalian host cell nucleus. Here, we report that the mycobacterial protein Rv2966c is a 5-methylcytosine-specific DNA methyltransferase that satisfies all these criteria and has the potential to alter the DNA methylation patterns in the host cell. Rv2966c was found to be secreted out from the mycobacterium and showed ability to localize to the host mammalian cell nucleus. Importantly, Rv2966c methylated specific DNA sequences predominantly in a non-CG context DNA, interacted with histone proteins and was functionally correlated with repression of specific host cell genes. Interestingly, the DNA binding and methylation activity of Rv2966c was dependent on its phosphorylation status, it being phosphorylated by multiple Mycobacterium tuberculosis kinases.
MATERIALS AND METHODS
Cloning, expression and purification of Rv2966c and its mutants
Full length and deletion mutants of Rv2966c were generated by PCR amplification of Rv2966c from M. tuberculosis H37Rv BAC clone library (15). For kinase assay, Rv2966c was cloned into the HindIII–NotI sites of pET-DUET-MBP and pET-DUET-MBP-kinase vectors. For mycobacterial expression, the full length Rv2966c was cloned in NdeI–HindIII sites of pVV16 mycobacterial shuttle vector. Full length Rv2966c was also cloned into the HindIII–EcoRI sites of pEGFPc3 mammalian transfection vector. Deletion mutants of Rv2966c were cloned in HindIII–EcoRI site in pEGFPc3 vector. 6x-HIS tagged Rv2966c protein was purified from Escherichia coli BL21-DE3 using 0.2 mM IPTG induction for 16 h at 18°C following established protocols (16).
DNA methyltransferase assay and DNA methylation analysis
DNA methylation assay was performed using 25 ng protein, poly(dI-dC) (100 ng, Sigma) or poly(CG) (50 pmol), poly(CA) (100 pmol), or poly(CT) (100 pmol), poly(CATG) (50 pmol) or poly(CACC) (100 pmol) DNA oligos and 2 μM of tritiated S-adenosyl methionine (SAM) for 5 min (or as otherwise indicated) in assay buffer (10 mM Tris pH7.4, 1 mM dithiothreitol (DTT), 400 μg/ml bovine serum albumin (BSA), 50 mM KCl) at 30°C. All the oligos were 60 bp in length. The reaction was stopped by addition of 1 ml of ice-cold 10% TCA followed by incubation on ice for 30 min. GF/C filters were pre-soaked with 50 μM cold SAM prior to spotting the precipitated DNA. The filters were washed with 10% TCA, water and 95% ethanol (in the given order), dried at 70°C, soaked in 4 ml of scintillation fluid and counts (DPMs) were taken in the Tri-Carb Scintillation Counter (Perkin Elmer).
When the DNA methyltransferase assay was to be followed by bisulfite sequencing, 200 μM cold SAM and 100 ng of the PCR product was used per reaction. The PCR product was generated from the DNA fragments excised from EMSA gel and cloned in pBluescript SK + vector using T7F and SKR primers. DNA, after the methyltransferase assay, was converted for bisulfite sequencing using the Epitect Bisulfite Kit (Qiagen) following manufacturer's instructions. Bisulfite PCR product was generated using the following primers:
5′ TGAAATAATGTTTAGTTGGGATTTG 3′
5′ ATTACTACTCTAAAAAACTACAAAAC 3′
For bisulfite analysis of the endogenous H2AFY2 intronic region from uninfected and M. tuberculosis H37Rv infected PMA treated THP1 cells (THP1 macrophages), the following bisulfite primers were used:
5′ AGGTTTAGGAGGGGTTTGTTAATAG 3′
5′ CACCCAAAAAAATAAAAATCTATCC 3′
pETDuet-based kinase assay in E. coli
Escherichia coli BL21-DE3 transformed with individual pET DUET constructs containing 6xHis-tagged Rv2966c and maltose-binding protein (MBP)-tagged mycobacterial kinases (either full length or kinase domains) were induced and processed as described before (17). Input samples were probed for expression of kinases by western using MBP antibody (Millipore). Fraction eluted from the Ni-NTA column was probed for enrichment using Rv2966c antibody (Imgenex, India) and phosphorylation of 6xHis-tagged Rv2966c using phospho-Thr antibody (CST).
Rv2966c antibody was raised against the E. coli purified Rv2966c protein in rabbit and the serum was purified by Protein-A agarose affinity chromatography (Imgenex, India). The specificity of the antibody was confirmed by western blotting (Supplementary Figure S1).
Affinity pull down assay
For Ni-NTA affinity pull down, protein lysate of HEK293 cells transfected with 6xHis-Rv2966c construct was prepared 24 h post-transfection in a 10 mM 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) buffer (pH7.0) containing 50 mM KCl, 1% Triton-X-100, 0.25% sodium deoxycholate, 20 mM imidazole and 10 mM NaF, incubated for 2 h with equilibrated Ni-NTA beads followed by three washes in the same buffer and analysed for bound proteins on a sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE).
For ConA affinity pull down, 400 μg protein lysate (Mycobacterium bovis BCG lysate or culture filtrate) in a buffer containing 20 mM Tris–HCl pH 7.4, 0.5 M NaCl, 1 mM MnCl2 and 1 mM CaCl2 was incubated with 15 μl of equilibrated ConA Sepharose beads (Amersham) overnight. The beads were washed thrice with the same buffer and bound protein was analysed on a SDS-PAGE.
Mycobacterium culture filtrate preparation
Electrocompetent M. bovis BCG were prepared and transformed using 500 ng of plasmid as described previously (18). For preparing culture filtrate from M. bovis BCG, M. tuberculosis H37Rv, Rv-pptr-B (19,20) and RvΔpknG (21) strains, the following methodology was used. Strains were revived from glycerol stocks and grown in 10 ml 7H9-ADC medium. Large scale cultures were inoculated with 1% primary cultures in modified Sauton's media and harvested once cultures reached OD600 of 0.8–1.0 (22). For Rv-pptr-B, culture was revived in 7H9-ADC medium in presence of inducer pristinamycin. Thereafter, the cultures were washed twice in phosphate-buffered saline (PBS) containing 0.05% Tween 80 and were used to seed fresh cultures in Sauton medium at an initial A600 of 0.1 in the absence of inducer. Cultures were centrifuged at 4000g for 30 min, the supernatant was passed through 0.22 μM filter to remove any debris and concentrated to 1/100th of the original volume using Amicon 10 kDa centrifugal filter units and stored as culture filtrates. The culture filtrate thus concentrated was estimated by Bradford's method and protein concentrations determined. Cell lysates were prepared by sonicating the mycobacterial pellet with glass beads. GroEL1 antibody was a kind gift from Dr Shekhar Mande, NCCS, Pune, India.
FITC labelling
For FITC labelling of proteins we followed an established protocol provided by FITC manufacturer (Sigma). Briefly, 10 mg/ml FITC dissolved in sodium bicarbonate buffer (pH 9.5) was used. One milligram Rv2966c protein was added to 4 mg/ml of FITC solution and allowed to bind for 6 h at 4°C with continuous agitation. Labelled-protein was dialysed overnight against PBS at 4°C. After 24 h of recovery in fresh Roswell Park Memorial Institute (RPMI) medium, Phorbol 12-myristate 13-acetate (PMA)-treated THP1 cells were incubated with 20 μg of labelled protein for 30–45 minutes. All the steps were performed in the dark. Cells were fixed and examined in a confocal microscope.
Transient transfection and infection of mammalian cells
Vector alone or GFP-tagged Rv2966c (full length and deletion mutants) were transfected in HEK293 cells (a kind gift from Dr Gayatri Ramakrishna, who obtained it from the Cell Culture Stock Centre at National Centre for Cell Science (NCCS), Pune) using Lipofectamine 2000 (Invitrogen) and in THP1 cells using Amaxa Nucleofection Kit (Lonza) followed by 12 h treatment with PMA (Sigma).
Infection of PMA treated THP1 cells with GFP::M. bovis BCG was done at an approximate MOI of 10. For immunofluorescence, 24 h after transfection or 24 h after infection, cells were washed with PBS, fixed using 4% para-formaldehyde and permeabilized with 0.1% Triton X-100. For immunostaining, Rv2966c antibody and Alexa568-conjugated secondary antibody was used. Cells were then mounted using diamidino-2-phenylindole dye (DAPI)-containing Vectashield (Vector Laboratories) and examined by confocal microscopy.
EMSA
THP1 genomic DNA was sonicated and fragments between 100 and 300 bp were excised from the agarose gel. The DNA fragments were then end-repaired using T4 DNA polymerase, ligated with 32γP-labelled adapter and used as probe. EMSA was performed as described before (23) with the exception of binding reaction at 30°C for 20 min. Poly(dI-dC) was added in the binding buffer wherever it is mentioned. The band corresponding to the DNA–protein complex was excised, DNA was eluted by boiling the gel slice in nuclease-free water followed by PCR using the adaptor oligos (LK102 and LK103, NimbleGen System). After two rounds of PCR, the amplified DNA was resolved on agarose gel, individual bands were cut, cloned into the pBluescript vector and Rv2966c-bound DNA sequences were identified by sequencing.
For validation, individual probes for DNA fragments obtained as above and listed in Supplementary Table S2 were prepared by PCR using 32αP-dCTP in the nucleotide mix. The primers used for obtaining the PCR probes are listed below.
LK102 5′ GCGGTGACCCGGGAGATCTGAATTC 3′
LK103 5′ GAATTCAGATC 3′
T7F 5′ GTAATACGACTCACTATAGGGC 3′
SKR 5′ CCGCTCTAGAACTAGTGGATC 3′
The diffuse nature of the radioactive signal for the probe could suggest DNA degradation.
Expression analysis by real time PCR
Mycobacterium tuberculosis infected THP1 macrophages were harvested 48 h post-infection and RNA was isolated using All Prep Kit (Qiagen). One microgram of RNA was converted to cDNA using Superscript III (Invitogen). The change in expression upon infection was evaluated by real time PCR using Mesa Green qPCR Mastermix Plus (Eurogentec) in ABI Prism SDS 7500 system. GAPDH was used as an internal control. Ct values were normalized for uninfected and infected (48 h) samples against 0 h (uninfected and infected) samples. Ratio of infected versus uninfected was then plotted.
PET112LRTF 5′ GCTGTCAGTGAGAGTCCTGT 3′
PET112LRTR 5′ AGTCTTGCCTTCCCTCTTCC 3′
GRK5RTF 5′ TGCTCACGAAAGATGCGAAG 3′
GRK5RTR 5′ ATGTCCAGCACGTCCTTACA 3′
H2AFY2RTF 5′ AATGACGAGGAGCTCAACCA 3′
H2AFY2RTR 5′ TCCTGCCTCTTTTCTCTGGG 3′
ZNF64RTF 5′ ATCAGAGGAGGTTTCAGCCC 3′
ZNF64RTR 5′ GGTGTGTCCCTGGGTCATAA 3′
GAPDHRTF 5′ TGCTGGCGCTGAGTACGTCG 3′
GAPDHRTR 5′ GGGTGGCAGTGATGGCATGG 3′
Mass spectrometry analysis
For mass spectrometry analysis, purified protein (phosphorylated) was run on SDS-PAGE, excised as gel plug and MALDI analyses was performed on a mass spectrometer (Bruker) in the NGTF facility of CDFD.
For LC–MS, the culture filtrate from M. bovis BCG expressing Rv2966c was resolved on SDS-PAGE. Gel plug for proteins between 34 and 50 kDa (with reference to pre-stained marker, Fermentas) was excised and sent to Taplin Mass Spectrometry Facility, Harvard Medical School, for analysis.
Chromatin immunoprecipitation
PMA-treated THP1 cells were infected with 6xHis-Rv2966c::M. bovis BCG at an MOI of 20:1for 24 h, followed by 24 h of rest. Following infection, cells were washed with PBS and cross-linked with 2.7% formaldehyde for 10 min at room temperature. The reaction was quenched with 125 mM glycine for 5 min. Cells were harvested and resuspended in 0.1% SDS. Chromatin was prepared by sonicating the cells in Bioruptor (Diagenode) to obtain DNA fragments in the range of 100–300 bp. For each ChIP, chromatin from 10 million of infected or uninfected cells was used. Chromatin was incubated with 2 μg of anti-His antibody (abcam) or IgG (Rabbit, Diagenode) for 12 h, followed by incubation with Protein A beads for 4 h. All the steps were done using auto ChIP kit (Diagenode) in the IPStar Compact machine (Diagenode). Ten percent chromatin was used as input.
Source of antibodies used
GroEL1 antibody (raised in mice) was a kind gift from Dr Shekhar C. Mande, NCCS, Pune. Antibodies against H3K36me3 (ab9050), H3K4me3 (ab8580), 6xHis (ab9108), CFP10 ab45074, H4 (ab10158) and H3 (ab1791) were procured from Abcam. H3k27me3 (#07-449) antibody was purchased from Millipore.
RESULTS
Identification of Rv2966c as a candidate DNA methyltransferase
Mycobacterium tuberculosis genome has been reported to be devoid of any dam or dcm homologue and hence lacks methylation at the respective canonical sites (13). However, the existence of both of 6-methyladenosine and 5-methylcytosine has been reported (12–14). Methyltransferase for N6-MdA, mama (Rv3263), has also been recently reported (14). We initiated the identification of a mycobacterial cytosine methyltransferase by mining the Tuberculist database. Of the 29 genes that were listed as putative methyltransferase in the Tuberculist database (24), we identified 17 genes that could be putative DNA methyltransferases based on the presence of DNA-binding and SAM binding domains (Supplementary Table S1). We cloned and expressed six genes followed by a SAM-based DNA methyltransferase assay on four of these proteins (Figure 1A, see Materials and Methods). Of these four proteins, Rv2966c and Rv3366 showed DNA methylation activity (Figure 1A). Here, we report characterization of the mycobacterial protein Rv2966c. Rv2966c is a 188 amino acid protein with N-terminal target recognition domain (25) and the predicted catalytic AdoMet methyltransferase domain at the C-terminus (Figure 1B). In vitro, it can bind both to DNA and RNA (25). Rv2966c contains the SAM-binding motif 51YAGSG55 (motif I F/Y XGXG), the invariant cysteine in motif IV ‘155TC156’ (motif IV ‘PC’) of cytosine DNA methyltransferases. Rv2966c is highly conserved (100%) between M. tuberculosis and M. bovis BCG but has been reported to be non-essential by Himar-1 based transposon mutagenesis in H37Rv strain (26).
Figure 1.

Rv2966c is a DNA methyltransferase. (A) DNA methylation activity of Rv2966c and few other candidate mycobacterial proteins was examined using tritiated-SAM in a filter-based assay. DNA blank was used as negative control and nuclear extract from THP1 cells was used as positive control. The name of the candidate mycobacterial proteins are given below the X-axis. (B) Line diagram representation of the mycobacterial Rv2966c protein. Rv2966c is a 188 aa long protein with a N-terminal Target Recognition Domain (TRD) and the predicted SAM-binding AdoMet methyltransferase domain. (C) The change in Rv2966c enzyme activity with time was studied using 25 ng Rv2966c protein, 100 ng poly(dI-dC) (substrate) and 2 μM of tritiated SAM (methyl group donor) in a filter-based assay. (D) The effect of Rv2966c concentration on its enzymatic activity was studied using 100 ng poly(dI-dC) (substrate) and 2 μM of tritiated SAM (methyl group donor) in a filter-based assay. (E) The effect of SAM (methyl group donor) concentration on enzyme activity was studied using 100 ng of poly(dI-dC) (substrate) and 40 ng of protein in a filter-based assay. All reactions were done at 30°C for 5 min unless specified. Each experiment was repeated at least three times. Activity was calculated as pmoles of methyl group transferred per minute per mg of protein. Error bars represent standard deviation (SD). * indicates significant difference (Student's t-test, *P < 0.05; **P < 0.005).
Rv2966c is a functional DNA methyltransferase
Since our aim was to probe the cytosine methylation potential of the protein, poly(dI-dC), which has been well established as a DNA substrate for mammalian cytosine-DNMTs under in vitro conditions, was used (27). Tritiated S-adenosyl methionine was used as the methyl group donor to determine the in vitro enzymatic properties of Rv2966c and as shown in Figure 1A, Rv2966c does methylate poly(dI-dC) DNA substrate.
Interestingly, we found that DNA methylation activity of Rv2966c decreased with increasing incubation time as well as with increasing protein concentration (Figure 1C and D). The decrease in activity at higher concentrations of enzyme could be due to aggregation of protein molecules. That this was the case became evident from the higher light scattering that was observed with increasing concentration of Rv2966c (Supplementary Figure S2). The decrease in enzyme activity with time was similar to what has been reported for the Tet2 protein (28). On the other hand, Rv2966c DNA methyltransferase activity increased with increase in SAM concentration (Figure 1E). Taken together, these observations demonstrate the functional cytosine methyltransferase activity of Rv2966c.
Rv2966c acquires post translation modifications
We observed anomalous migration of Rv2966c protein on a SDS-PAGE. Instead of the expected molecular weight of ∼19.8 kDa, the protein band was detected at 34 kDa (Supplementary Figure S3). The observed band was confirmed to be Rv2966c by peptide competition assay (Supplementary Figure S4).
Since post translational modifications are known to influence protein activity as well as its mobility on a SDS-PAGE, we first sought to identify if Rv2966c indeed was modified. Protein phosphorylation, especially by Ser/Thr kinases (STKs), and glycosylation are the most common modifications implicated in anomalous migration of proteins (29,30). Reports indicate that mycobacterial proteins can indeed be glycosylated and phosphorylated (17,31).
To examine whether Rv2966c was glycosylated, we performed affinity pull down of glycosylated proteins from M. bovis BCG cell lysate with ConA-Sepharose, which binds specifically to α-d-mannosyl and α-d-glucosyl residues (32). The pulled down glycosylated proteins were probed with Rv2966c antibody. Rv2966c protein was detected in the ConA pull down fraction indicating that it was indeed glycosylated (Figure 2A).
Figure 2.
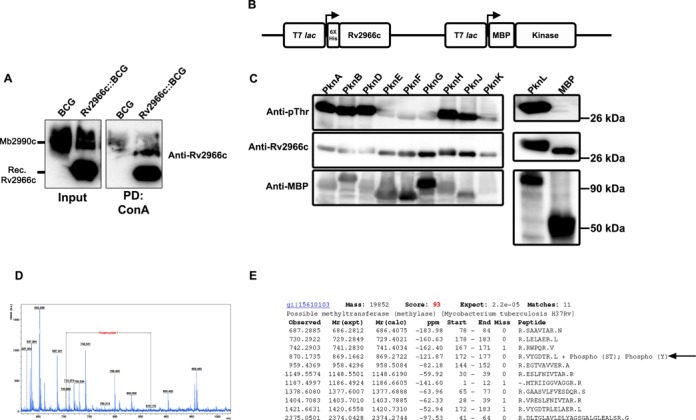
Rv2966c acquires post-translation modifications. (A) Rv2966c acquires glycosylation. ConA pull down was performed on M. bovis BCG or Rv2966c::M. bovis BCG cell lysate. The Input and pull down proteins were probed with Rv2966c antibody. PD – ConA pull down samples. Mb2990c represents the endogenous homolog of Rv2966c from M. bovis BCG. (B) A line diagram showing pET DUET construct with 6xHis-Rv2966c and either MBP or MBP-tagged mycobacterial kinases. (C) Rv2966c is phosphorylated by multiple mycobacterial Ser/Thr kinases. Upon co-expression of the mycobacterial kinase and Rv2966c in E. coli using the pET DUET system, Rv2966c was pulled down using Ni-NTA beads and probed with Rv2966c and phosphothreonine antibodies. Input lysates were also probed with anti-MBP antibody to check for the levels of kinases. The names of the mycobacterial kinases tested in our assay are given on top of the panels. (D and E) Mass spectrometric analysis of Rv2966c. (D) MS spectra of the recombinant 6xHis-Rv2966c from M. bovis BCG lysate. Fragments of the phosphorylated Rv2966c have a mass shift of 160 (shown by horizontal line) indicating a phosphorylation modification site in the peptide. (E) Mass spectrometry data analysis by MASCOT showing the peptide 172VYGDTR177 with phospho- modifications (marked by an arrow).
To test whether Rv2966c gets phosphorylated at any of the 16 threonine present in the protein, 6xHis-tagged Rv2966c gene was cloned into pET-DUET vector that also contained MBP-tagged mycobacterium kinases (21; there are 11 eukaryotic-like Ser/Thr kinases in the mycobacterial genome; Figure 2B for representation, see ‘Materials and Methods’ for details of the kinases used). 6xHis-Rv2966c was affinity purified using Ni-NTA column from the E. coli protein lysate and probed for threonine phosphorylation. As can be seen in Figure 2C, Rv2966c was found to be a substrate for multiple kinases, namely, PknA, PknB, PknD, PknH, PknJ and PknL. This was further confirmed by mass spectrometry analysis of Rv2966c incubated with PknB protein that prefers T over S as the phospho-acceptor. Two of the threonine residues in the peptide ‘ATTCAPLTWPEGW’ corresponding to the amino acids 153–165 in the protein were found to be phosphorylated (Supplementary Figure S5A and B). Interestingly, the same peptide also contains the catalytic cysteine (C156) residue.
To examine the phosphorylation status of Rv2966c in mycobacterium, we affinity pulled down the recombinant 6xHis-Rv2966c (the 25 kDa band) from Rv2966c::M. bovis BCG lysate and examined it by mass spectrometry (MS) for phosphorylation. One of the peptides from the C-terminus of Rv2966c, 172VYGDTR177 with tyrosine at 173 and threonine at 176 position was found to be phosphorylated (Figure 2D and E). Thus, our results show glycosylation and phosphorylation of Rv2966c suggesting that mycobacterial proteins can have dual post-translational modifications.
Rv2966c phosphorylation modulates its DNA binding and methylation activity
Recent studies on Dnmt1, a mammalian cytosine DNA methyltransferase, have shown that phosphorylation by AKT1 and CDKs regulates its stability in vivo and affects its cellular pool and hence activity (33,34). In order to study whether phosphorylation had any effect on its DNA-binding and methylation activity, we tested these properties of Rv2966c in presence and absence of phosphorylation. To examine its DNA-binding activity we performed EMSA analysis for the phosphorylated and unphosphorylated Rv2966c protein with 32P-labelled and adapter-ligated THP1 genomic DNA fragments in the range of 100–300 bp (as DNA binding motif for Rv2966c is not known). In a comparison between the DNA-binding ability of unphosphorylated (expressed from the pET23 and pET-DUET MBP vectors) and phosphorylated (expressed from pET-DUET MBP PknB/PknA vectors) Rv2966c protein, the binding of the unphosphorylated protein to DNA was found to be much weaker than that observed for PknB phosphorylated Rv2966c protein (Figure 3A). For the phosphorylated protein we also observed an additional slower migrating DNA-protein complex (Figure 3A).
Figure 3.
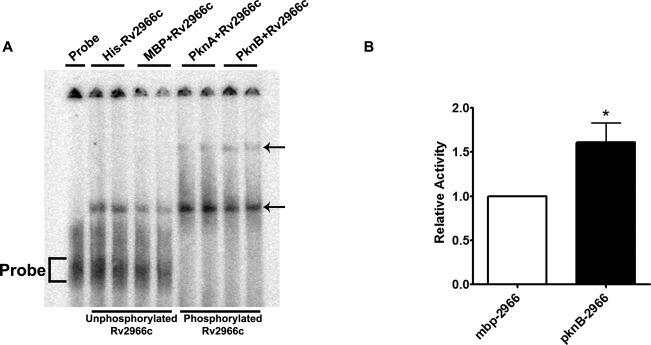
Phosphorylation influences Rv2966c DNA methylation activity. (A) DNA binding capability of Rv2966c was enhanced by phosphorylation. γ32P-labelled THP1 genomic DNA fragments were incubated with unphosphorylated and phosphorylated Rv2966c. The DNA–protein complex was resolved on a 5% acrylamide EMSA gel. Lane description is on the top of each lane. Arrows indicate the position of the DNA–protein complex. Probe is marked by a bracket symbol. (B) DNA methylation activity of the unphosphorylated and PknB-phosphorylated Rv2966c was compared using filter based assay. Twenty-five nanograms of Rv2966c was incubated with 100 ng poly(dI-dC) and 200 μM SAM (tritiated SAM was spiked with cold SAM in a ratio of 1:20) for 5 min at 30°C. Activity of the phosphorylated protein relative to that of unphosphorylated protein was calculated. The experiment was repeated thrice. Error bars represent standard seviation (SD). * indicates significant difference (Student's t-test, *P < 0.05).
Furthermore, DNA methyltransferase activity of phosphorylated and unphosphorylated Rv2966c protein was compared by filter based assay, wherein transfer of tritiated methyl group was monitored from S-adenosyl methionine to poly(dI-dC) per minute per mg of protein. For PknB phosphorylated Rv2966c protein a statistically significant increase in DNA methyltransferase activity was observed as compared to the unphosphorylated protein (Figure 3B). These observations suggest a positive effect of phosphorylation on the DNA-binding and cytosine methylation ability of Rv2966c.
Rv2966c is a secretory protein
For a protein to be able to influence the mammalian host machinery, an important criterion would be its ability to be secreted out of the bacterial cell. In order to study whether Rv2966c and its M. bovis BCG homolog Mb2990c (100% similar) were being secreted, M. tuberculosis H37Rv and M. bovis BCG were cultured in modified sauton's medium and culture filtrates were analysed for the endogenous protein using Rv2966c antibody. As can be seen in Figure 4A and B, the endogenous Rv2966c (Mb2990c) protein was detected in both the cell lysate (lanes 1 and 3) and the culture filtrate (lanes 2 and 4) indicating that the endogenous protein was indeed secreted.
Figure 4.
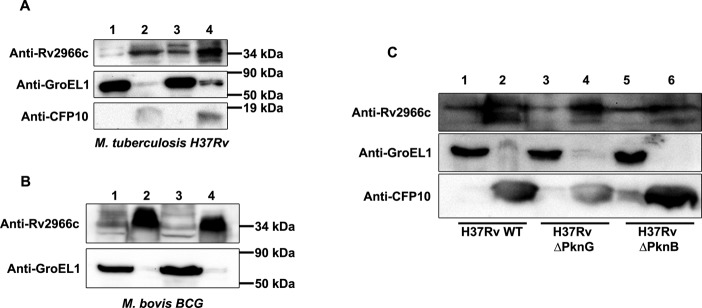
Rv2966c is secreted out by mycobacterium. Mycobacterial cell lysate and culture filtrate (in Sauton's medium) were electrophoresed on a SDS-PAGE and probed with anti-Rv2966c, anti-GroEL1 (non-secreted protein) and anti-CFP10 (secreted protein) antibodies. (A) Secretion in M. tuberculosis H37Rv. Lanes 1 and 3 are mycobacterial cell lysate; lanes 2 and 4 are culture filtrates from two different M. tuberculosis H37Rv cultures. (B) Secretion in M. bovis BCG. Rv2966c antibody detected the 100% similar M. bovis protein Mb2990c in both the cell lysates (lanes 1 and 3) and culture filtrates (lanes 2 and 4). Two independent M. bovis BCG cultures were analysed. (C) Rv2966c secretion in M. tuberculosisH37Rv is not dependent on phosphorylation by PknB, PknG. Culture filtrates were prepared from H37Rv, Rv-pptr-B and RvΔpknG strains of M. tuberculosis. Secretion of Rv2966c was compared by loading 50 μg of both lysates and culture filtrate on SDS-PAGE and probing with anti-Rv2966c. GroEL1 and CFP-10 were used as negative and positive control for secretion respectively. Lanes 1, 3 and 5: cell lysate; 2, 4 and 6: culture filtrate.
Since DNA methylation activity of Rv2966c was found to be modulated by phosphorylation, we decided to check whether the same was true for Rv2966c secretion. As shown before, Rv2966c gets phosphorylated by PknB but not by PknG (Figure 2). In order to address the role of PknB and PknG in secretion of Rv2966c, we utilized M. tuberculosis mutants Rv-pptr-B, wherein the PknB gene in the genome was replaced with a pristinamycin- inducible copy, and RvΔpknG, a gene replacement mutant of PknG (19,25). A comparison was made between M. tuberculosis H37Rv, Rv-pptr-B, grown in the absence of inducer and RvΔpknG strains for their ability to secrete Rv2966c protein into the culture filtrate. As can be seen in Figure 4C, Rv2966c was found to be secreted in both wild type and mutant H37Rv strains (compare lanes 2, 4 and 6 in the Rv2966c panel). This could either suggest that phosphorylation of Rv2966c by PknB and PknG had no role in secretion or phosphorylation by a different kinase was involved in Rv2966c secretion. That phosphorylation of Rv2966c may not have any role to play in its secretion was further established by our finding that the non-secreted recombinant Rv2966c in the M. bovis BCG cell lysate was also phosphorylated (using phospho-Thr antibody; Supplementary Figure S6, lane 1). While this may suggest that phosphorylation may not be involved in Rv2966c secretion but as the entire spectrum of Rv2966c phosphorylation is not available, the role of phosphorylation in secretion cannot be completely ruled out. The secreted Rv2996c protein was also glycosylated as ConA affinity pull down of M. bovis BCG culture filtrate showed the presence of Rv2966c (Supplementary Figure S7).
Rv2966c is secreted into the host cell and can localizes to nucleus
Many proteins are known to enter living cells and overcome endo-lysosomal entrapment. In fact, this transduction of live cells by functional proteins has been used as an alternative strategy to transient transfection of culture cells (35–37). Since mycobacterium also utilizes the endoocytic machinery to enter the macrophage and manipulates the cellular machinery to prevent phagolysosome formation (38,39), we wanted to test whether Rv2966c could enter PMA-treated THP1 cells (THP1 macrophages) and overcome endo-lysosomal entrapment. To examine this possibility, THP1 macrophages in culture were incubated with recombinant Rv2966c protein conjugated to FITC for 30–45 min and the localization of the protein was visualized by confocal microscopy. Unlike FITC-BSA that predominantly localized to the THP1 plasma membrane, FITC-Rv2966c was found to be localized to cytoplasm, nucleus and the nucleolus of the THP1 cells (Figure 5A).
Figure 5.
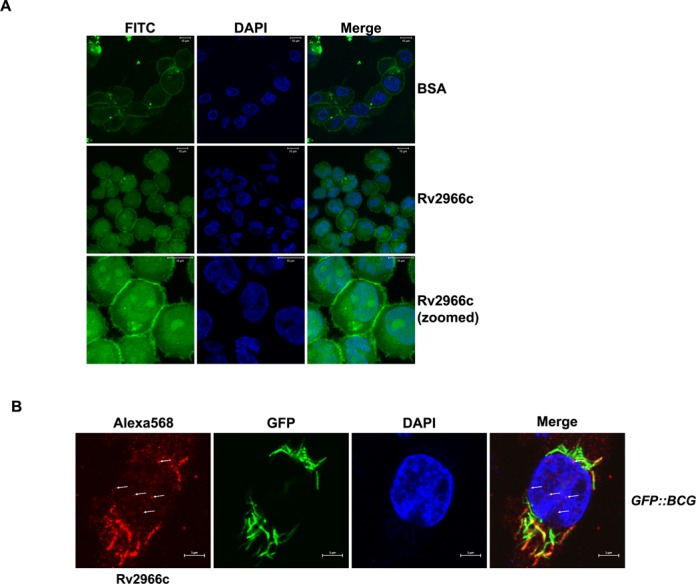
Rv2966c is secreted in to the host cell upon infection. (A) Rv2966c can overcome endo-lysosomal entrapment. FITC-conjugated Rv2966c or BSA were incubated with PMA-treated THP1 cells for 30–45 min following which the cells were fixed and analysed by confocal microscopy. Unlike FITC-BSA that stained only the outer cell membrane, FITC-Rv2966c localized to all parts of the cell including nucleus. (B) Rv2966c is secreted into the host cell upon infection. Confocal pictures of PMA-treated THP1 cells infected with M. bovis BCG overexpressing GFP (GFP::BCG). Signal for the endogenous Mb2990c (Rv2966c homolog) was detected by immunostaining with Rv2966c antibody + Alexa568 conjugated secondary antibody. Notice diffused signal for Rv2966c in contrast to GFP signal that is limited to the bacilli in GFP::M. bovis BCG. Arrow mark the Rv2966c signal in the host nucleus.
To investigate whether this was also true in vivo, THP1 macrophages were infected with M. bovis BCG overexpressing GFP (GFP::M. bovis BCG) and examined by confocal microscopy. GFP signal in THP1 cells infected with GFP::M. bovis BCG appeared restricted to within the boundary of the bacilli (Figure 5B, GFP). On the other hand, the signal for Rv2966c (immuno-stained with Alexa568-Rv2966c antibody) was diffused and localized throughout the host cell (Figure 5B). Rv2966c signal could also be detected in the nucleus (marked by arrows). This indicated once again that Rv2996c was indeed secreted into the host cell upon mycobacterial infection.
C-terminal residues dictate the nuclear localization of Rv2966c
To further test the subcellular localization potential of Rv2966c, we performed transient transfection of the GFP-tagged Rv2966c in HEK293 and THP1 cells. In both cells types (Figure 6A, B), Rv2966c was localized to nucleus in addition to cytoplasm and nucleolus.
Figure 6.
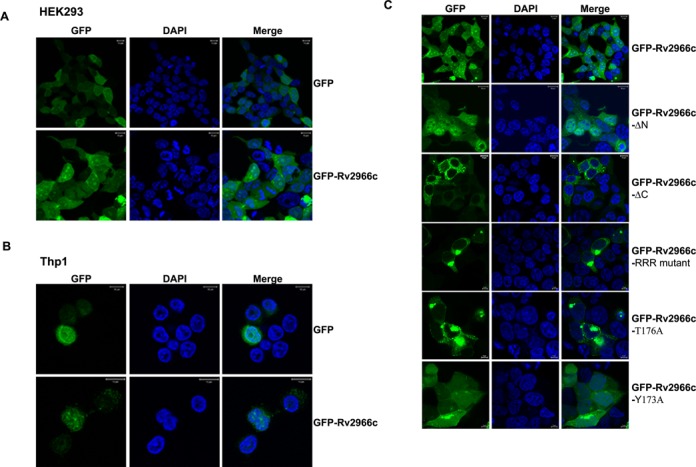
Rv2966c can localize to the host nucleus. (A and B) Localization of GFP-Rv2966c (upper panel) upon transfection into HEK293 (A) and THP1 macrophages (B). The localization of GFP alone (using pEGFPc3-GFP construct, lower panel) was used as a control. (C) Identification of NLS in Rv2966c. Localization of Rv2966c mutants where either ‘N’ and ‘C’ terminus were deleted or point mutations were made in the C-terminus of Rv2966c, was studied upon transfection of these constructs into HEK293 cells. As compared to full-length Rv2966c, Rv2966c-ΔC, Rv2966c-RRR mutant and Rv2966c-T176A showed exclusively cytoplasmic localization. Rv2966c-ΔN was absent only from the nucleolus and Y173A showed no change in localization. ΔN: N-terminus deletion; ΔC: C-terminus deletion; RRR mutant: arginine at 171, 177 and 183 position mutated to alanine. T176A: Threonine at 176 mutated to alanine. Y173A: tyrosine at 173 mutated to alanine.
Presence of Rv2966c both in the nucleus and cytoplasm suggested nucleo-cytoplasmic shuttling of the protein. Prediction tools like NucPred (40) and NLS Mapper (41) could not identify any putative Nuclear Localization Signal (NLS) in the Rv2966c protein. To examine if nuclear and/or nucleolar signals were present in the Rv2966c protein, N- and C-terminal deletion mutants were generated for the GFP-tagged Rv2966c wherein 32 amino acids from the N-terminus (comprising the target recognition domain) or 20 amino acids from C-terminal were deleted respectively. When the GFP-tagged deletion mutants were transfected into HEK293, N-terminus deletion mutants was found to have lost its capacity for nucleolar localization but was able to localize elsewhere in the cytoplasm and nucleus (Figure 6C, second panel). On the other hand, the C-terminal deletion mutant showed predominantly cytosolic retention of the mutant protein (Figure 6C, third panel). Further analysis of the deleted 20 amino acids from the C-terminal showed the presence of three arginines at 171, 177 and 183 in the pattern, RX5RX5R. Mutant Rv2966c protein with these three arginine residues mutated to alanine (Rv2966c-RRR mutant) showed reduced nuclear localization. However, this altered localization was not seen in all the cells. Since R177 was present in the peptide 172VYGDTR177 that was previously found to be phosphorylated in M. bovis BCG (Figure 2D and E), we decided to check if Rv2966c phosphorylation had any role in its localization to the nucleus. As we were transfecting Rv2966c construct in vitro into HEK293 cells, we first confirmed the phosphorylation status of the transfected GFP-Rv2966c protein. Both western (Supplementary Figure S8) and mass spectrometry (Supplementary Figure S9) analysis showed that Rv2966c was indeed phosphorylated by the host cell kinases. In fact, the peptide ‘168WPQRVYGDTR177’ similar to the one observed for M. bovis BCG was found to be phosphorylated (Supplementary Figure S9B). To examine if Y173 or T176 were involved in nuclear localization of Rv2966c, we mutated them individually to alanine. T176A mutant showed nuclear localization defect similar to that observed for the RRR mutant but Y173A had no effect (Figure 6C). This could either indicate that a novel NLS was present in the C-terminus of Rv2966c and the residues R171, R177 and R183 and T176 were part of this novel NLS or that these residues within the C-terminus regulate the interaction of Rv2966c with a host protein for a piggy-back entry into the host nucleus.
Rv2966c binds to specific DNA sequences in the THP1 genome and methylates non-CpG dinucleotides
Mammalian DNA methyltransferases are known to bind and methylate DNA non-specifically (42). To test whether the binding that we observed in Figure 3A was non-specific or specific to particular DNA sequences, the Rv2966c-bound DNA fragments in the EMSA gels were excised, cloned and sequenced (see ‘Materials and Methods’ section). Several DNA species were obtained (Supplementary Table S2) that mapped to different regions of the human genome. A few individual DNA sequences from them were PCR amplified and used in EMSA analysis. As can be seen in Figure 7A for a few representative DNA sequences, Rv2966c showed binding to all of them. As expected, phosphorylated Rv2966c bound DNA much more strongly than the unphosphorylated form (Figure 7A). However, when we used pBluescriptSK MCS as a DNA substrate in our binding assay, we found negligible binding to Rv2966c (Figure 7A, panel 4) suggesting that Rv2966c was binding to specific DNA sequences.
Figure 7.
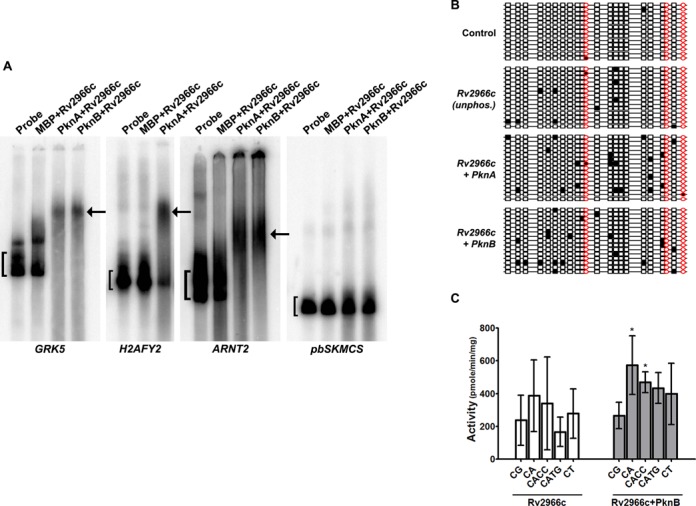
Rv2966c binds and methylates non-CpG cytosines within specific DNA sequences from the THP1 genome. (A) Rv2966c binds specific DNA sequences. α32P-labelled DNA probes were prepared for three fragment from the THP1 genomic DNA (see Supplementary Table S2) and control pBluescript vector. EMSA was done with both unphosphorylated and PknA/PknB phosphorylated proteins in the presence of poly(dI-dC) (see labels on top of the panels). Names of the genes, regions for which probes were synthesized, are indicated below the panels. pbSKMCS is the probe from the pBluescript vector. Brackets indicate the band corresponding to the probe; arrow indicates the DNA-protein complex band. (B) Bisulfite analysis of in vitro Rv2966c methylated H2AFY2 gene fragment. To examine DNA methylation activity of Rv2966c, bisulfite sequencing was performed on the DNA fragment from the H2AFY2 gene after incubation with unphosphorylated and phosphorylated Rv2966c. Thirteen or more clones were screened for each sample. Control indicates incubation with BSA. Red circles represent CpG dinucleotides. Non-CpG cytosines in CpA, CpC and CpT dinucelotides are represented by rectangles. Filled symbols indicates methylation of cytosine in the dinucleotide. (C) Comparison of in vitro DNA methylation activity of unphosphorylated and PknB phosphorylated Rv2966c with CpG and non-CpG DNA oligo substrates and using tritiated-SAM as methyl donor in a filter-based assay as indicated. The error bars represent standard deviation (SD). * indicate significant difference (Student's t-test, *P < 0.05).
To examine whether Rv2966c was capable of methylating these specific DNA sequences in the THP1 genome, DNA methylation analysis was done by performing bisulfite sequencing on in vitro Rv2966c methylated DNA fragment. Figure 7B shows the methylation profile for the intronic fragment from the H2AFY2 gene. As compared to control (incubation with BSA), incubation with both phosphorylated (PknA/PknB) and unphosphorylated Rv2966c protein showed significant methylation. While for control, only 14% clones showed methylated cytosines, for unphosphorylated and PknA/PknB phosphorylated Rv2966c 57, 67 and 73% clones showed cytosine methylation respectively. Surprisingly, we detected negligible CpG methylation (denoted by red circles) but significant amount of non-CpG methylation (denoted by black boxes), specifically in CpA and CpT dinucleotides (Figure 7B). The non-CpG was observed in both CHH and CHG context. To confirm the preference of Rv2966c for cytosine in non-CpG context, in vitro methylation assay for Rv2966c (both unphosphorylated and pknB phosphorylated forms) was performed using double stranded DNA oligos that were either poly(CG), poly(CA), poly(CACC), poly(CATG) or poly(CT). As can be seen from Figure 7C, phosphorylated Rv2966c methylated the CA and CACC oligos significantly more that the CG oligos.
This was also true in vivo upon infection of THP1 macrophages with M. tuberculosis H37Rv. In the methylation profile of the intronic region from the H2AFY2 gene analysed by bisulfite sequencing, no difference was observed in the CpG methylation between infected and uninfected THP1 cells. Similar to what we had observed in the in vitro assay, a significant increase of non-CpG methylation was observed in infected THP1 cells for the H2AFY2 intron (Figure 8A). As has been reported earlier (43), increased non-CpG methylation was present on the antisense strand (representative region shown in Figure 8A). As compared to 70% of clones that showed non-CpG methylation in infected cells, only 10% clones showed methylation in uninfected THP1 cells. Non-CpG methylation occurs at very low levels in differentiated cells (43). In uninfected cells only 0.6% of the non-CpG cytosines showed methylation. This increased to ∼2% in infected THP1 macrophages (Figure 8B). As the efficiency of THP1 infection by M. tuberculosis H37Rv was only about 50% (see ‘Materials and Methods’ section), we expect the increase in non-CpG methylation to be significantly more in infected cells.
Figure 8.
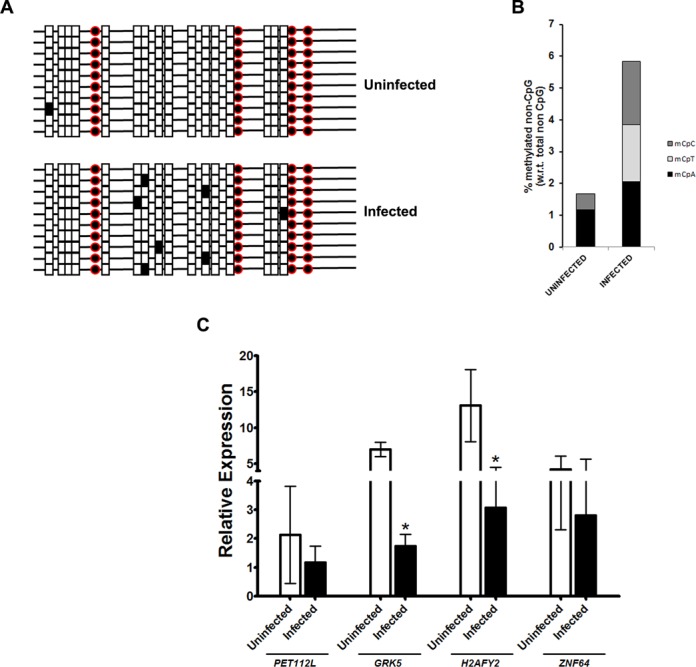
Non-CpG methylation upon M. tuberculosis H37Rv infection. (A) Bisulfite analysis of H2AFY2 intronic region in PMA treated THP1 cells after M. tuberculosis H37Rv infection. Ten clones were sequenced for both uninfected and infected THP1 cells. Red circles represent CpG dinucleotides. Non-CpG cytosines in CpA, CpC and CpT dinucelotides are represented by rectangles. Filled symbols indicates methylation of cytosine in the dinucleotide. (B) Statistical analysis of methylated cytosine dinucleotides in M. tuberculosis H37Rv infected and uninfected THP1 cells. (C) Human H2AFY2 and GRK5 genes are repressed in THP1 cells infected by M. tuberculosis H37Rv. Real-time RT-PCR was performed for the indicated genes (mentioned below the X-axis) on RNA isolated from uninfected and M. tuberculosis H37Rv infected (48 h post-infection) THP1 macrophages. To take into account the effect of culturing on expression the levels were normalized against the expression of these gene at the time of infection (0 h). Open bars indicate expression in uninfected cells while filled bars are for infected cells. The experiment was done at least thrice in duplicates. The error bars represent standard deviation (SD). * indicate significant difference (Student's t-test, *P < 0.05).
To examine the functional significance of the observed non-CpG methylation during infection, we analysed the expression of some of these genes upon M. tuberculosis H37Rv infection of THP1 macrophages. The infected cells were maintained in culture for 48 h followed by quantitative real-time RT-PCR expression analysis of some of the genes mentioned in Supplementary Table S2. To take into account the changes that could be a result of cell culture, the relative expression of each gene in cells 48 h after infection was normalized with the values for the genes at the time of infection (0 h). As can be seen in Figure 8C, significant decrease in expression was seen for the H2AFY2 and GRK5 genes but not for PET112L and ZNF64.
Rv2966c interacts with histones H3 and H4
As Rv2966c localizes to the host nucleus, we were interested in finding whether it can influences the host epigenetic circuitry only through direct interaction with the host DNA or whether there were other proteins that it interacts with. In order to identify host proteins that interact with Rv2966c, we performed affinity pull down assay on recombinant E. coli purified 6xHis-Rv2966c that had been incubated with M. bovis BCG infected THP1 protein lysate. Amongst the Rv2966c interactants identified by mass spectrometry, were the proteins histone H4 and NPM1 (nucleophosmin).
NPM1 is a multi-functional nucleo-cytoplasmic shuttling protein, a known histone chaperone and is implicated in regulation of transcription due to its association with RNA polymerase II, transcription factors like YY1 and chromatin remodelling complex SNF2 (44,45). The interaction of Rv2966c with NPM1 was validated by performing affinity pull down for NPM1 on HEK293 cells co-transfected with SFB-NPM1 and 6xHis-Rv2966c constructs and probing with Rv2966c antibody (see 'Materials and Methods' section). Rv2966c indeed interacted with NPM1 (Figure 9A). Since NPM1 is a nucleo-cytoplasmic shuttling protein it is possible that the interaction of Rv2966c with NPM1 is responsible for its nuclear localization.
Figure 9.
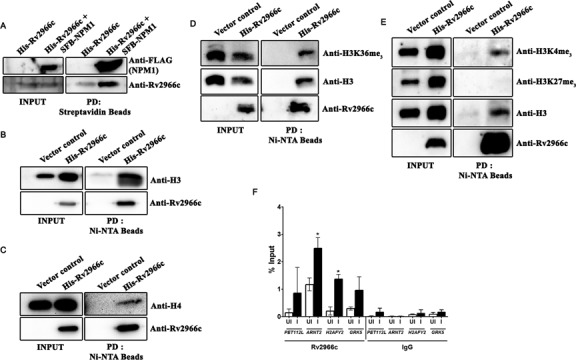
Rv2966c interacts with histone proteins. (A) Interaction with NPM1. Protein lysate from HEK293 cells transfected with 6xHis-Rv2966c alone or co-transfected with 6xHis-Rv2966c and SFB-NPM1 was subjected to pull down with streptavidin (interacts with the SFB tag) and the bound proteins were examined for the presence of NPM1 and Rv2966c. FLAG is part of SFB triple tag and FLAG antibody detected SFB-NPM1 fusion protein. (B and C) Interaction with histone H3 and H4. Protein lysate from HEK293 cells transfected with 6xHis-Rv2966c or pcDN3.1 (vector control) was subjected to pull down with Ni-NTA beads and probed for the presence of Rv2966c and H3 (B) or H4 (C). PD: pull down; Input: total protein before pull down. (D and E.) Western blot analysis to examine interaction of Rv2966c with specific modifications of histone H3 as indicated. Histone H3 and Rv2966c antibodies were used as control. (F) ChIP analysis for the indicated genomic regions (see Supplementary Table S2 for genomic coordinates) using His antibody. IgG was used as a control. UI, uninfected and I, 6xHisRv2966c::M. bovis BCG infected THP1 macrophages.
To investigate the interaction of Rv2966c with core histones, Ni-NTA affinity pull down was carried out on HEK293 cells transfected with 6xHis-Rv2966c and the western blot was probed with H3 and H4 antibody. Rv2966c was found to be associated with both histone H3 and H4 in both the pull downs (Figure 9B and C).
DNMT3L, belonging to the de novo DNMT3 family of DNA methyltransferase, has been shown to interact with histone H3 over a region that includes lysine 4 (46–48). Moreover, a recent report has indicated that targeted methylation of specific sequence within the mammalian genome can be achieved by the interaction of DNA methyltransferase, DNMT3B and SETD2 mediated H3K36me3 methylation (49). To probe whether Rv2966c was binding to a specific modification of histone H3, we again resorted to Ni-NTA affinity pull down on HEK293 cells transfected with 6xHis-Rv2966c and the western blot was probed with antibodies specific to H3K4me3, H3K27me3 and H3K36me3. As can be seen in Figure 9D and E, Rv2966c binds to H3 when H3K36 and H3K4 (active chromatin marks, 4, 5) were methylated but not when H3K27 (inactive chromatin marks, 4, 5) was methylated indicating that like DNMT3L and DNMT3B, Rv2966c has the ability to interact with specific modifications of histone H3.
As shown above, Rv2966c binds to specific DNA sequences in vitro (Figure 7). To examine whether Rv2966c was part of the chromatin at these sequences in the genome in vivo, chromatin immunoprecipitation was carried out on THP1 macrophages infected with 6xHisRv2966c::M. bovis BCG using His antibody. Genomic region encompassing the Rv2966c binding sequences within the PET112L, H2AFY2, ARNT2 and GRK5 genes were examined. As shown in Figure 9F, Rv2966c was found to be associated with the chromatin in the genomic regions of H2AFY2, ARNT2 and GRK5 and this binding was statistically significant for H2AFY2 and ARNT2 (see Supplementary Table S2 for the genomic coordinates).These interaction studies clearly indicate the multifaceted nature of the Rv2966c protein allowing it to interact with host chromatin at multiple levels.
DISCUSSION
Here we show the mycobacterial protein Rv2966c to be a DNA methyltransferase with the potential to modulate host DNA methylation in a non-CpG context. Our study adds another facet to the growing list of bacterial proteins that have the potential to modulate host epigenetic circuitry (6–11). This is in addition to the well-known interfaces of interaction between a pathogen and its host like surface receptor, phagocytosis, cytosolic factors, etc. (1,2). We also show for the first time that along with specific changes in the host histone modifications, host DNA methylation changes at specific loci could be achieved by a bacterial protein.
Rv2966c methylates cytosines predominantly in non-CpG context
The mycobacterial protein Rv2966c has been categorized as a methyltransferase based on the presence of the SAM-binding domain and the DPPY motif in the catalytic domain. It also contains the Target Recognition Domain (TRD) that is able to recognize DNA and RNA substrates (25). Rv2966c has previously been described as an RsmD-like methyltransferase that methylates 16S rRNA in vitro (25). However, our study clearly demonstrates that Rv2966c can methylate DNA at cytosine residues. While a N6A methyltransferase has been reported in mycobacteria (14), ours is the first report showing cytosine methylation by a mycobacterial DNA methyltransferase. Importantly, we show that Rv2966c can methylate cytosine predominantly in a non-CpG context particularly at CpA and CpT dinucleotides. In case of mammals, non-CpG methylation has only been detected in cancer samples, embryonic stem cells and mature human neurons but at a very low frequency when compared to CpG methylation (50–52). Since CpA/CpT non-CpG methylation is asymmetric, its maintenance would require a de novo methylation with every replication event. Like mature human neurons, M. tuberculosis infected macrophages do not undergo cell division so the established non-CpG would persist till the macrophage eventually dies. We do see a correlation between Rv2966c, non-CpG methylation and H2AYF2 gene repression in THP1 macrophages infected with M. tuberculosis H37Rv. It would now be interesting to examine the possibility that the non-CpG DNA methylation effected by Rv2966c indeed modulates the larger epigenetic framework of the macrophage genome upon infection by pathogenic mycobacterium.
Mammalian DNA methyltransferase specifically methylate CpG dinucleotides but no specific DNA sequence or motif to which they bind has been defined (42). Our study shows that Rv2966c binds only to certain DNA fragments from the THP1 genome but not the DNA sequence from the vector pBluescript (Figure 6). While the prokaryotic Dcm methyltransferases also bind to specific DNA sequence motifs (53), we could not find Dcm-binding motifs within all the DNA fragments that showed binding to Rv2966c. In addition, Rv2966c methylates CpA and CpT dinucleotides that are not within the Dcm recognition sequences. Previous studies have also found that dam and dcm recognition sites are unmethylated in mycobacterial species (13). This indicates that Rv2966c is a dual natured DNA methyltransferase as it retains the property of a dcm/dam like prokaryotic DNA methyltransferase to bind specific DNA sequences but has gained ability to methylate cytosines that are not canonical dcm/dam sites.
Rv2966c interacts with the host epigenetic circuitry
Our study shows that Rv2966c is secreted out of the mycobacterial cell. Why would a mycobacteria want to secrete a protein that is involved in methylation of DNA (this study) or 16s rRNA (25)? Not only is Rv2966c secreted out of the mycobacterium, it has the potential to overcome endo-lysosomal entrapment and be localized to the nucleus inside the host cell. In the light of the fact that Rv2966c is a DNA methyltransferase that can methylate non-canonical Dcm sites, its secretory nature would suggest that mycobacteria could be using Rv2966c to modulate the host epigenome and transcriptional machinery. This contention was strengthened by our findings that Rv2966c was a part of host chromatin, it interacts with histone H3 and H4 and has the ability to differentiate between different histone H3 modifications. One of the mammalian DNA methyltransferases, DNMT3L, has been shown to be a link between DNA methylation and histone modifications as it can regulate DNA methylation as well as bind to histone H3. DNMT3L is also involved in nuclear reprogramming (46–48). That Rv2966c shows properties similar to a mammalian epigenetic effector indicates that its interaction with the host epigenetic machinery at multiple levels could be an important event in the nuclear reprogramming of the macrophages during M. tuberculosis infection.
Rv2966c binding to specific DNA sequence would indicate that this modulation might be of a specific set of genes in the host. The observation that the transcription of the host genes that showed binding sites for Rv2966c (Supplementary Table S2) was altered upon mycobacterial infection supports this hypothesis. One of the Rv2966c binding regions was present within H2AFY2, a macro histone H2A family member and postulated to be acting as a barrier to reprogramming (54). Another region that showed binding to Rv2966c was present within the GRK5 gene. GRK5 was also found to be markedly repressed in THP1 macrophages infected with Mycobacterium tuberculosis H37Rv. As a G-protein coupled receptor kinase (subfamily Ser/Thr protein kinase), GRK5 has been reported to regulate the mobility of polymorphonuclear leukocytes that migrate to site of injury (inflammation), are effectors of innate immune system and kill the invading pathogen (55).
An interesting corollary to our result was the observation that Rv2966c is modified by dual post translational modifications and that its DNA methylation activity but not secretion was influenced by phosphorylation. Rv2966c was found to be phosphorylated by multiple mycobacterial kinases. Rv2966c was also phosphorylated by mammalian kinases in vitro. This perhaps was expected as mycobacterial kinases have been found to be similar to the Ser/Thr kinases present in mammalian cells (17,20). The modulation of DNA methyltransferase activity by phosphorylation throws up the possibility of either the host cell controlling Rv2966c activity by kinases/phospahatases or mycobacterium using the services of host kinases to regulate the host–pathogen epigenetic interaction.
In conclusion, our study explores the possibility that a mycobacterial protein could modulate DNA methylation in the mammalian host cell. While CpG methylation is the normal mode of DNA methylation observed in most mammalian cell types, instances of non-CpG methylation are seen only in specific tissues and for very specific functions (50–52). The prospect of a pathogenic bacteria using a multi-faceted protein to interact with the host epigenetic machinery, which in turn uses a non-canonical DNA methylation approach and its interaction with histones H3 and H4 to alter the host epigenetic framework and thence control transcriptional machinery provides a novel paradigm in host–pathogen interaction and needs further investigation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Imtiyaz Yaseen for his help in some of the experiments. G.S. and S.U. are recipients of Senior Research Fellowship of the Council of Scientific and Industrial Research (CSIR), India towards the pursuit of a PhD degree of the Manipal University, Manipal and JNU, New Delhi, respectively.
FUNDING
Department of Biotechnology, Government of India [BT/PR3260/BRB/10/967/2011 to S.K. and V.K.N.]. Funding for open access charge: The open access publication charge for this paper has been waived by Oxford University Press.
Conflict of interest statement. None declared.
REFERENCES
- 1.Bhavsar A.P., Guttman J.A., Finlay B.B. Manipulation of host-cell pathways by bacterial pathogens. Nature. 2007;449:827–834. doi: 10.1038/nature06247. [DOI] [PubMed] [Google Scholar]
- 2.Lamkanfi M., Dixit V.M. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe. 2010;8:44–54. doi: 10.1016/j.chom.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 3.Jaenisch R., Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 2003;33:245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 4.Berger S.L. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 5.Li B., Carey M., Workman J.L. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 6.Bobetsis Y.A., Barros S.P., Lin D.M., Weidman J.R., Dolinoy D.C., Jirtle R.L., Boggess K.A., Beck J.D., Offenbacher S. Bacterial infection promotes DNA hypermethylation. J. Dent. Res. 2007;86:169–174. doi: 10.1177/154405910708600212. [DOI] [PubMed] [Google Scholar]
- 7.Paschos K., Allday M.J. Epigenetic reprogramming of host genes in viral and microbial pathogenesis. Trends Microbiol. 2010;18:439–447. doi: 10.1016/j.tim.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pennini M.E., Perrinet S., Dautry-Varsat A., Subtil A. Histone methylation by NUE, a novel nuclear effector of the intracellular pathogen Chlamydia trachomatis. PLoS Pathogen. 2010;6:e1000995. doi: 10.1371/journal.ppat.1000995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rolando M., Sanulli S., Rusniok C., Gomez-Valero L., Bertholet C., Sahr T., Margueron R., Buchrieser C. Legionella pneumophila effector RomA uniquely modifies host chromatin to repress gene expression and promote intracellular bacterial replication. Cell Host Microbe. 2013;13:395–405. doi: 10.1016/j.chom.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Hamon M.A., Batsché E., Régnault B., Tham T.N., Seveau S., Muchardt C., Cossart P. Histone modifications induced by a family of bacterial toxins. Proc. Natl. Acad. Sci. U.S.A. 2007;104:13467–13472. doi: 10.1073/pnas.0702729104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marazzi I., Ho J.S., Kim J., Manicassamy B., Dewell S., Albrecht R.A., Seibert C.W., Schaefer U., Jeffrey K.L., Prinjha R.K., et al. Suppression of the antiviral response by an influenza histone mimic. Nature. 2012;483:428–433. doi: 10.1038/nature10892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srivastava R., Gopinathan K.P., Ramakrishnan T. Deoxyribonucleic acid methylation in mycobacteria. J. Bacteriol. 1981;148:716–719. doi: 10.1128/jb.148.2.716-719.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hemavathy K.C., Nagaraja V. DNA methylation in mycobacteria: absence of methylation at GATC (dam) and CCA/TGG (Dcm) sequences. FEMS Immunol. Med. Microbiol. 1995;11:291–296. doi: 10.1111/j.1574-695X.1995.tb00159.x. [DOI] [PubMed] [Google Scholar]
- 14.Shell S.S., Prestwich E.G., Baek S.H., Shah R.R., Sassetti C.M., Dedon P.C., Fortune S.M. DNA methylation impacts gene expression and ensures hypoxic survival of Mycobacterium tuberculosis. PLoS Pathog. 2013;9:e1003419. doi: 10.1371/journal.ppat.1003419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brosch R., Gordon S.V., Billault A., Garnier T., Eiglmeier K., Soravito C., Barrell B.G., Cole S.T. Use of a Mycobacterium tuberculosis H37Rv bacterial artificial chromosome library for genome mapping, sequencing, and comparative genomics. Infect. Immun. 1998;66:2221–2229. doi: 10.1128/iai.66.5.2221-2229.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maniatis T., Fritsch E.F., Sambrook J. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbour Laboratory; 1982. [Google Scholar]
- 17.Khan S., Nagarajan S.N., Parikh A., Samantaray S., Singh A., Kumar D., Roy R.P., Bhatt A., Nandicoori V.K. Phosphorylation of enoyl-acyl carrier protein reductase InhA impacts mycobacterial growth and survival. J. Biol. Chem. 2010;285:37860–37871. doi: 10.1074/jbc.M110.143131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parish T., Stoker N.G. Electroporation of Mycobacteria. Methods Mol. Biol. 1998;101:129–144. doi: 10.1385/0-89603-471-2:129. [DOI] [PubMed] [Google Scholar]
- 19.Forti F., Crosta A., Ghisotti D. Pristinamycin-inducible gene regulation in mycobacteria. J. Biotechnol. 2009;140:270–277. doi: 10.1016/j.jbiotec.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 20.Chawla Y., Upadhyay S.K., Khan S., Nagarajan S.N., Forti F., Nandicoori V.K. Protein Kinase B (PknB) of Mycobacterium tuberculosis is essential for growth of the pathogen in vitro as well as for survival within the host. J. Biol. Chem. 2014;289:13858–13875. doi: 10.1074/jbc.M114.563536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cowley S., Ko M., Pick N., Chow R., Downing K.J., Gordhan B.G., Betts J.C., Mizrahi V., Smith D.A., Stokes R.W., et al. The Mycobacterium tuberculosis protein serine/threonine kinase PknG is linked to cellular glutamate/glutamine levels and is important for growth in vivo. Mol. Microbiol. 2004;52:1691–1702. doi: 10.1111/j.1365-2958.2004.04085.x. [DOI] [PubMed] [Google Scholar]
- 22.Gebhard S., Cook G.M. Differential regulation of high-affinity phosphate transport systems of Mycobacterium smegmatis: identification of PhnF, a repressor of the phnDCE operon. J. Bacteriol. 2008;190:1335–1343. doi: 10.1128/JB.01764-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis S.E., Konradi C. Analysis of DNA–protein interactions in the nervous system using the electrophoretic mobility shift assay. Methods. 1996;10:301–311. doi: 10.1006/meth.1996.0107. [DOI] [PubMed] [Google Scholar]
- 24.Lew J.M., Kapopoulou A., Jones L.M., Cole S.T. TubercuList – 10 years after. Tuberculosis. 2011;91:1–7. doi: 10.1016/j.tube.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 25.Kumar A., Saigal K., Malhotra K., Sinha K.M., Taneja B. Structural and functional characterization of Rv2966c protein reveals an RsmD-like methyltransferase from Mycobacterium tuberculosis and the role of its N-terminal domain in target recognition. J. Biol. Chem. 2011;286:19652–19661. doi: 10.1074/jbc.M110.200428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sassetti C.M., Boyd D.H., Rubin E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 27.Pedrali-Noy G., Weissbach A. Mammalian DNA methyltransferases prefer poly (dI-dC) as substrate. J. Biol. Chem. 1986;261:7600–7602. [PubMed] [Google Scholar]
- 28.Ito S., Shen L., Dai Q., Wu S.C., Collins L.B., Swenberg J.A., He C., Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;33:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shirai A., Matsuyama A., Yashiroda Y., Hashimoto A., Kawamura Y., Arai R., Komatsu Y., Horinouchi S., Yoshida M. Global analysis of gel mobility of proteins and its use in target identification. J. Biol. Chem. 2008;283:10745–10752. doi: 10.1074/jbc.M709211200. [DOI] [PubMed] [Google Scholar]
- 30.Unal E.S., Zhao R., Qiu A., Goldman I.D. N-linked glycosylation and its impact on the electrophoretic mobility and function of the human proton-coupled folate transporter (HsPCFT) Biochim. Biophys. Acta. 2008;1778:1407–1414. doi: 10.1016/j.bbamem.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith G.T., Sweredoski M.J., Hess S. O-linked glycosylation sites profiling in Mycobacterium tuberculosis culture filtrate proteins. J. Proteomics. 2014;97:296–306. doi: 10.1016/j.jprot.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Satchidanandam V., Kumar N., Jumani R.S., Challu V., Elangovan S., Khan N.A. The glycosylated Rv1860 protein of Mycobacterium tuberculosis inhibits dendritic cell mediated TH1 and TH17 polarization of T cells and abrogates protective immunity conferred by BCG. PLoS Pathog. 2014;10:e1004176. doi: 10.1371/journal.ppat.1004176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Estève P.O., Chang Y., Samaranayake M., Upadhyay A.K., Horton J.R., Feehery G.R., Cheng X., Pradhan S. A methylation and phosphorylation switch between an adjacent lysineand serine determines human DNMT1 stability. Nat. Struct. Mol. Biol. 2011;18:42–48. doi: 10.1038/nsmb.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lavoie G., St-Pierre Y. Phosphorylation of human DNMT1: implication of cyclin-dependent kinases. Biochem. Biophys. Res. Commun. 2011;409:187–192. doi: 10.1016/j.bbrc.2011.04.115. [DOI] [PubMed] [Google Scholar]
- 35.Richard J.P., Melikov K., Vives E., Ramos C., Verbeure B., Gait M.J., Chernomordik L.V., Lebleu B. Cell-penetrating peptides: a reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003;278:585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 36.Nakase I., Niwa M., Takeuchi T., Sonomura K., Kawabata N., Koike Y., Takehashi M., Tanaka S., Ueda K., Simpson J.C., et al. Cellular uptake of arginine-rich peptides: roles for macropinocytosis and actin rearrangement. Mol. Ther. 2004;10:1011–1022. doi: 10.1016/j.ymthe.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 37.Sugita T., Yoshikawa T., Mukai Y., Yamanada N., Imai S., Nagano K., Yoshida Y., Shibata H., Yoshioka Y., Nakagawa S., et al. Comparative study on transduction and toxicity of protein transduction domains. Br. J. Pharmacol. 2008;153:1143–1152. doi: 10.1038/sj.bjp.0707678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen L., Pieters J. The Trojan horse: survival tactics of pathogenic mycobacteria in macrophages. Trends Cell. Biol. 2005;15:269–276. doi: 10.1016/j.tcb.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 39.de Chastellier C. The many niches and strategies used by pathogenic mycobacteria for survival within host macrophages. Immunobiology. 2009;214:526–542. doi: 10.1016/j.imbio.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 40.Brameier M., Krings A., MacCallum R.M. NucPred—predicting nuclear localization of proteins. Bioinformatics. 2007;23:1159–1160. doi: 10.1093/bioinformatics/btm066. [DOI] [PubMed] [Google Scholar]
- 41.Kosugi S., Hasebe M., Tomita M., Yanagawa H. Systematic identification of cell cycle-dependent yeast nucleo-cytoplasmic shuttling proteins by prediction of composite motifs. Proc. Natl. Acad. Sci. U.S.A. 2009;106:10171–10176. doi: 10.1073/pnas.0900604106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bestor T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000;9:2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- 43.Lister R., Pelizzola M., Dowen R.H., Hawkins R.D., Hon G., Tonti-Filippini J., Nery J.R., Lee L., Ye Z., Ngo Q.M., et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Colombo E., Alcalay M., Pelicci P.G. Nucleophosmin and its complex network: a possible therapeutic target in hematological diseases. Oncogene. 2011;30:2595–2609. doi: 10.1038/onc.2010.646. [DOI] [PubMed] [Google Scholar]
- 45.Lindström M.S. NPM1/B23: A Multifunctional Chaperone in Ribosome Biogenesis and Chromatin Remodeling. Biochem. Res. Int. 2011:195209. doi: 10.1155/2011/195209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gowher H., Liebert K., Hermann A., Xu G. Mechanism of stimulation of catalytic activity of Dnmt3A and Dnmt3B DNA-(cytosine-C5)-methyltransferases by Dnmt3L. J. Biol. Chem. 2005;280:13341–13348. doi: 10.1074/jbc.M413412200. [DOI] [PubMed] [Google Scholar]
- 47.Ooi S.K., Qiu C., Bernstein E., Li K., Jia D., Yang Z., Erdjument-Bromage H., Tempst P., Lin S.P., Allis C.D., et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gokul G., Ramakrishna G., Khosla S. Reprogramming of HeLa cells upon DNMT3L overexpression mimics carcinogenesis. Epigenetics. 2009;4:322–329. doi: 10.4161/epi.4.5.9239. [DOI] [PubMed] [Google Scholar]
- 49.Baubec T., Colombo D.F., Wirbelauer C., Schmidt J., Burger L., Krebs A.R., Akalin A., Schübeler D. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature. 2015 doi: 10.1038/nature14176. doi:10.1038/nature14176. [DOI] [PubMed] [Google Scholar]
- 50.Ziller M.J., Müller F., Liao J., Zhang Y., Gu H., Bock C., Boyle P., Epstein C.B., Bernstein B.E., Lengauer T., et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011;7:e1002389. doi: 10.1371/journal.pgen.1002389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arand J., Spieler D., Karius T., Branco M.R., Meilinger D., Meissner A., Jenuwein T., Xu G., Leonhardt H., Wolf V., et al. In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 2012;8:e1002750. doi: 10.1371/journal.pgen.1002750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lister R., Mukamel E.A., Nery J.R., Urich M., Puddifoot C.A., Johnson N.D., Lucero J., Huang Y., Dwork A.J., Schultz M.D., et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boyer H.W., Chow L.T., Dugaiczyk A., Hedgpeth J., Goodman H.M. DNA substrate site for the EcoRII restriction endonuclease and modification methylase. Nat. New Biol. 1973;244:40–43. doi: 10.1038/newbio244040a0. [DOI] [PubMed] [Google Scholar]
- 54.Gaspar-Maia A., Qadeer Z.A., Hasson D., Ratnakumar K., Leu N.A., Leroy G., Liu S., Costanzi C., Valle-Garcia D., Schaniel C., et al. MacroH2A histone variants act as a barrier upon reprogramming towards pluripotency. Nat. Commun. 2013;4:1565. doi: 10.1038/ncomms2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fan J., Malik A.B. Toll-like receptor-4 (TLR4) signaling augments chemokine-induced neutrophil migration by modulating cell surface expression of chemokine receptors. Nat. Med. 2003;9:315–321. doi: 10.1038/nm832. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.