


Abstract
DNA-dependent T7 RNA polymerase (T7 RNAP) is the most powerful tool for both gene expression and in vitro transcription. By using a Next Generation Sequencing (NGS) approach we have analyzed the polymorphism of a T7 RNAP-generated mRNA pool of the mboIIM2 gene. We find that the enzyme displays a relatively high level of template-dependent transcriptional infidelity. The nucleotide misincorporations and multiple insertions in A/T-rich tracts of homopolymers in mRNA (0.20 and 0.089%, respectively) cause epigenetic effects with significant impact on gene expression that is disproportionally high to their frequency of appearance. The sequence-dependent rescue of single and even double InDel frameshifting mutants and wild-type phenotype recovery is observed as a result. As a consequence, a heterogeneous pool of functional and non-functional proteins of almost the same molecular mass is produced where the proteins are indistinguishable from each other upon ordinary analysis. We suggest that transcriptional infidelity as a general feature of the most effective RNAPs may serve to repair and/or modify a protein function, thus increasing the repertoire of phenotypic variants, which in turn has a high evolutionary potential.
INTRODUCTION
T7 RNA polymerase (T7 RNAP) catalyzes synthesis of RNA in the presence of a DNA template containing a specific T7 phage promoter, i.e. φ10 (1–3). The specificity of the promoter recognition, simple structure of the enzyme, independence from additional protein cofactors and much higher rate of synthesis in comparison to the Escherichia coli host RNAP, makes T7 RNAP widely used. T7-based expression systems are broadly employed to prepare biologically active mRNA in vivo (3,4) and in vitro (5,6). Preparative quantities of a defined length of RNA is generated by run off transcription (7), often as labeled RNA probes (8) and also by amplification of a linear aRNA (amplified antisense) based on cDNA (9,10). T7 RNAP rifampicin-resistance allows directly for exclusive overexpression of the target gene, and indirectly, for specific protein radiolabeling (3). However, transcriptional errors introduced by this RNAP have been reported. Nucleotide misincorporation (11) and transcriptional slippage were detected on extended runs of homopolymers, at the initiation (12), elongation (13) and termination steps of transcription (14). What is more, the DNA template-independent but RNA template-directed addition of an extra nucleotide at the 3′ terminus as well as template-dependent slippage was observed (15,16). Moreover, the ability of T7 RNAP for translesion synthesis on over a dozen different types of DNA damage is well known (17).
For years overproduction of heterologous proteins in E. coli cells by the T7 phage derived expression system has been the most widely used method of choice (2,3). However, it turned out that this expression system is ‘over-efficient’ and causes both a high metabolic burden on the host cells and a severe imbalance between the T7-based transcription process, translation, and mRNA stability (18,19). In general, this leads to an accumulation of protein aggregates or deposition of inclusion bodies which are composed of both misfolded and accompanying proteins (20). To avoid this, numerous approaches were developed which would reduce the transcriptional rate mainly by lowering the abundance of T7 RNAP production (21–28).
Here, we describe another aspect of T7 RNAP that was revealed after mboIIM2 DNA methyltransferase of Moraxella bovis expression analyses (29,30). In light of the results presented here, bacteria with a target gene carrying a single or double insertion/deletion (InDel) mutation that should cause frameshift wild type reading frame are phenotypically heterogenous. Predominantly, through a transcriptional slippage mechanism in homopolymer A- and T-rich stretches, T7 RNAP infidelity can rescue such mutations and abolish the expected null phenotype. Moreover, it leads to the production of a mixture of protein variants, with partially changed internal sequences. In this work, the potential role of phenotypic mutations in the evolution of the protein and their stability is discussed.
MATERIALS AND METHODS
Bacterial strains, culture conditions
Escherichia coli DH10B and ER2566 (DE3) both from New England Biolabs and Tuner (DE3) (Novagen) were grown aerobically in Luria–Bertani (LB) broth or M9 minimal medium (31), supplemented with 0.2% Casamino Acids (Difco Laboratories) and 0.2% glucose at 37°C with shaking at 180 rpm. Where appropriate, kanamycin (Km), ampicillin (Amp), chloramphenicol (Cm) and tetracycline (Tc) were added at final concentrations of 50, 100, 15 and 15 μg/ml, respectively. In the host's transcription inhibition experiment rifampicin (Rif) was added to 200 μg/ml. When necessary, isopropyl-β-d-thiogalactopyranoside (IPTG) 1 mM/ml and l-arabinose (0.04%) was added for the induction of gene expression.
Genetic techniques
Standard protocols (31) and kits were used for purification of the plasmid DNA (A&A Biotechnology, Poland), DNA digestion with restriction endonucleases, DNA ligation with T4 DNA ligase, PCR techniques with PfuPlus DNA polymerase (all from Eurx-Gdansk, Poland), as well as for DNA sequencing of the mboIIM2 mutated derivatives (Genomed, Poland).
Single and multiple site-directed mutagenesis
InDel variants of methyltransferases mboIIM2 and mboIIM1 of M. bovis ATCC 10900 (29,30), ncuIM2 of Neisseria cuniculi ATCC 14688 (32) and chloramphenicol-resistance gene cat from transposon Tn9 (33) were constructed by a nucleotide deletion/insertion in the reverse primers. Supplemental Tables S1 and S2 include a list of the oligonucleotides used and a description of the plasmid construction. Appropriate plasmid templates were PCR-amplified with high fidelity PfuPlus DNA polymerase (Eurx-Gdansk, Poland) according to the manufacturer's instructions (50 ng of plasmid template was added to a 50-μl PCR). One microliter (10 u) of the DpnI enzyme (Fermentas) was added directly to the PCRs to eliminate the parental plasmid. Following a 1.5-h incubation at 37°C, the DNA products were resolved in agarose gels, appropriate bands were cut out and aliquots containing purified DNA were transformed into DH10B competent E. coli cells. All the plasmid modifications were confirmed by Sanger DNA sequencing using the BigDye Terminator v3.1 (Applied Biosystems, USA).
gfp reporter constructs
Plasmids pET24mboIIMB.3 containing the mboIIM2 wild type gene (30) and pET24mboIIMB.4 carrying its single nucleotide deletion mutation variant (mboIIM2ΔA356), were used as templates in PCR to obtain shorter deletion variant genes (378 and 377 nt long, respectively), both containing a putative slippage region. The resulting plasmids, obtained by cloning as described in (34), named pETmboBwtΔ378 and pETmboBΔA356Δ377, were used for the construction of translational fusions with the gfp gene from pGreenTIR (35). The proximal part of the gfp gene sequence was modified from the wild type 5′-ATG AGT AAA GGA- [MSKG] to 5′-ATG GAT CCA AAA GGA- [MDPKG, gfp0] or 5′-ATG GAT CCA AAG GAG [MDPKE, gfp-1]), and then inserted as PCR products between the BamHI and EcoRI sites of pETmboBwtΔ378 and pETmboBΔA356Δ377. The resulting series of four plasmids named pETmboBwtΔ375gfp0, pETmboBwtΔ375gfp-1, pETmboBΔA356Δ375gfp0 and pETmboBΔA356Δ375gfp-1, differ from each other by the wild type (gfp0) or frameshifted (gfp-1) variants of the gfp gene, respectively, enabling 0 or −1 frame reading of fusions with mboIIM2Δ378 and mboIIM2ΔA356Δ377 gene fragments. Plasmids pETmboBΔA356Δ562gfp0 and pETmboBΔA356Δ562gfp-1 with a longer variant of the mboIIM2 gene (562 nt)were constructed in a similar way. All the plasmid modifications were confirmed by DNA sequencing.
Western blotting
ER2566 cells harboring various pET24a-mboIIM2 plasmids were grown in LB medium at 37°C and induced with 1 mM IPTG for 2 h. Protein samples (100 μl each) were electrophoresed on 10–12.5% sodium dodecyl sulfate (SDS)-polyacrylamide gel (PAGE) and electroblotted onto nitrocellulose membrane (Bio-Rad) as follows. In cases that required a higher resolution of small peptides, 12.5% Tris-Tricine SDS-PAGE was used (36). Cell pellets were resuspended in water, an appropriate amount of 4× SDS-PAGE loading dye was added and the samples were run on a gel. Transfer to a nitrocellulose membrane was performed in the electrophoretic transfer cell at 50 V for 3 h or 15 V overnight in Towbin buffer, with or without 20% methanol, respectively (37). The membrane was blocked at 4°C for 1 h in phosphate buffered saline (PBS) buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4 pH7.4) with 5% skimmed milk. The membrane was then probed with rabbit anti-M2.MboII (30) or rabbit anti-DnaA antibodies (courtesy of Dr M. Glinkowska, University of Gdansk) diluted 1:1250 and 1:1333, respectively, in TBS-T buffer (50 mM Tris–HCl, 150 mM NaCl, 0.05% Tween 20, pH 7.6) with 5% skimmed milk for 1.5 h at room temperature. After three washes with TBS-T, the membrane was incubated for 1 h with a goat anti-rabbit secondary antibody conjugated with alkaline phosphatase (AP, 1:30 000, Sigma) for 1 h at room temperature. The membrane was washed three times and a specific protein was visualized by adding BCIP/NBT solution (Fermentas). The GFP was probed using a mouse primary antibody (Santa Cruz Biotechnology or Novous Biologicals) at 1:4000 in TBS-T with 5% skimmed milk followed by a goat anti-mouse secondary antibody conjugated with horse radish peroxidase (HRP, 1:4000, Sigma). The membrane was washed three times and a specific protein was visualized by adding chemiluminescent substrate solution (Bio-Rad) and a series of exposures on X-ray film.
Protein radiolabeling
The expression of radiolabeled plasmid-encoded methyltransferase gene products was performed by IPTG (1 mM) induction of ER2566 cells grown at 37°C in M9 medium, supplemented with appropriate antibiotics. 45 min after induction, the host's RNAP inhibitor, rifampicin, was added at a concentration of 200 μg/ml followed by pulse-labeling with 35S methionine (3). 100-μl samples of the culture were labeled for 5 min with 5 μCi 35S methionine at 30 min intervals. Samples were centrifuged, suspended in SDS/PAGE loading dye and loaded on to 10%-SDS/PAGE (38). Then, transfer to a nitrocellulose membrane (Bio-Rad) was performed, and protein visualization was done by autoradiography.
The M2.MboII MTase assays
Enzyme activity was determined by monitoring the DNA protection level (resistance to cleavage by endonuclease R.MboII, 1 h at 37°C) of plasmid isolated from the M2.MboII producing cells. The samples were analyzed by 0.8 % (w/v) agarose gel electrophoresis. The second M2.MboII specific methylation assay was based on monitoring of the enzyme-catalyzed transfer of 3H-labeled methyl groups from S-adenosyl-l-methionine ([3H]AdoMet) to DNA. The assay was performed in a 30 μl reaction mixture containing 0.3 μg pUC18 DNA (or synthetic oligoduplexes (Supplementary Table S2), C1 + C2—substrate with a single recognition site 5′-GAAGA3′/5′TCTTC-3′, or C3 + C4—substrate with a nonspecific site 5′GAACA3′/5′TGTTC3′ site), 10 mM Tris–HCl (pH 7.0), 22 nCi 3H AdoMet (1 Ci = 3.7×1010 Bq), and an aliquot of the P-11 phosphocellulose chromatography fraction (1 h, 37 °C). The reaction was stopped by adding 30 μl of 50 % trichloroacetic acid. The sample was then centrifuged (10 000 × g, 10 min) and the pellet was washed with 1 ml 70 % (v/v) ethanol, centrifuged, and dried. Scintillation counting was used to estimate the incorporated radioactivity.
Total RNA extraction
Five milliliters of E. coli ER2566 cells harboring pET24mboIIMB.4 were harvested while growing in the exponential phase after 2 h IPTG induction of expression. The bacterial pellet was resuspended in 1 ml of stayRNA reagent (A&A Biotechnology). The RNA was then extracted using the Total RNA Mini Plus kit (A&A Biotechnology) according to the manufacturer's instructions. After elution, the RNA was treated with DNase I for 30 min at 37°C (Thermo Scientific) and then concentrated using the Total RNA Mini Plus Concentrator kit (A&A Biotechnology). The final concentration (1.5 μg/μl) and purity was checked by measuring its absorbance at 260/280 nm in a spectrophotometer (Nanodrop).
Qualitative and quantitative polymorphism of mboIIM2ΔA356 mRNA by NGS approach
Preparation of a DNA library for NGS technique was performed by Genomed Ltd (Warsaw, Poland). Four microgram of total bacterial RNA served as a template in a reverse transcription reaction using 1 μl of a specific primer RT-mboB (20 μM), 1 μl Superscript III reverse transcriptase 200 u/μl (Invitrogen), and dNTPs mix (1 mM final concentration). The reaction was performed according to the procedure described by the manufacturer. In accordance with the recommendations for use of a specific primer for first strand cDNA synthesis, this step was carried out at an elevated temperature of 55°C. Given the large number of template RNA used, an RNase inhibitor was not added to the reaction. Second strand cDNA synthesis and the subsequent steps to create a library were performed with a set of NEBNext Ultra Directional RNA Library Preparation Kit for Illumina (New England Biolabs). Further reactions were performed with the buffers, following the procedure according to the manufacturer's instructions. At the stage of enrichment of the library, unilateral labeling was applied in a PCR reaction using a universal primer and a primer containing the index sequence NEBNext Multiplex Oligos for Illumina (New England Biolabs). Because the procedure does not take into account the fragmentation step required before NGS sequencing technique in the MiSeq device (Illumina), the last stage in preparation of the library was the purification of an appropriate fraction of DNA fragments using AMPureXP magnetic beads (Beckman Culture).
The libraries were sequenced using the MiSeq Reagent Kit v2 (Illumina) in the paired-end mode with a read length of 150 bases. Alignment was performed using CLC Genomics Workbench 7 and an additional analysis was performed by Piledriver software (https://github.com/arq5x/piledriver) and custom scripts. The mboIIM2 reference gene (GenBank KM192157) was used for the mapping of sequencing reads, and identification of mutations.
Analysis of the N-terminal amino acid sequence of truncated variants of M2.MboII
N-terminal protein sequence analysis was performed at BioCentrum Ltd (Krakow, Poland). Sequentially detached phenylthiohydantoin derivatives of amino acids were identified using the Procise 491 (Applied Biosystems, Foster City, USA) automatic sequence analysis system, according to the standard protocol of the manufacturer.
Identification of the domain composition of M2.MboII protein by mass spectrometry
All LC–MS-MS/MS analyses (liquid chromatography coupled to tandem mass spectrometry) were done at a Mass Spectrometry Lab (IBB PAN, Warsaw). Prior to the analysis, gel slices were subjected to a standard ‘in-gel digestion’ procedure during which the proteins were reduced with 100 mM dithiotreitol (DTT) (30 min at 56°C), alkylated with iodoacetamide (45 min in a darkroom at room temperature) and digested overnight with trypsin (sequencing Grade Modified Trypsin, Promega) or pepsin (Promega). The resulting peptides were eluted from gel with 0.1% trifluoroacetic acid (TFA) and 2% acetonitrile (ACN). The peptide mixture was applied to RP-18 precolumn (nanoACQUITY Symmetry® C18, Waters) using 0.1% TFA as a mobile phase and then applied to a nano-HPLC RP-18 column (nanoACQUITY BEH C18, Waters) using an ACN gradient (0–35% in 70 min) in the presence of 0.05% formic acid with a flow rate of 250 nl/min. The column outlet was directly coupled to the ion source of the spectrometer working in the regime of data dependent MS to MS/MS switch (Orbitrap Velos mass spectrometer—Thermo Electron Corp.). The acquired raw data were processed by Mascot Distiller followed by Mascot Search (Matrix Science, UK) against the predicted reference peptide masses of the derived M2.MboII. The search parameters for the precursor and product ions mass tolerance were 15 ppm and 0.6 Da, respectively; trypsin/pepsin specificity: one missed cleavage sites allowed; fixed modification of cysteine by carbamidomethylation and variable modification of and methionine oxidation. Peptides with a Mascot Score exceeding the threshold value corresponding to <5% False Positive Rate, calculated by the Mascot procedure, and with the Mascot Score >30 were considered to be positively identified.
Fluorescence microscopy
Samples of Tuner (DE3) or ER2566 (DE3) bacterial cultures (0.2 ml) bearing a gfp-tagged reporter were taken for fluorescence microscopy studies. Cell membranes were stained with the fluorescent dye SynaptoRed C2 (FM4-64) (Sigma) at a final concentration of 5 μg/ml for 10 min. The DNA was visualized by staining with DAPI (4,6-diamidino-2-phenylindole) at 1 μg/ml for 10 min. Samples were then immobilized on 1-mm 1.5% agarose pads dissolved in LB medium and visualized using a Leica DMI4000B microscope fitted with a DFC365FX camera (Leica). The following Leica filter sets were used: N2.1 (for FM4-64), green fluorescent protein (GFP) and A4 (for DAPI). Images were collected and processed using LAS AF 3.1software (Leica).
RESULTS
Frameshifting during expression of the mboIIM2 gene with a single-nucleotide deletion restores the wild type phenotype
In our previous work, we demonstrated that the mboIIM2 gene codes for methyltransferase, a 32 kDa protein responsible for the specific methylation of DNA sequences (30). Here, we monitored expression of its mutant derivative with the use of a T7 expression system. Expression of the mboIIM2ΔA356 gene with a single adenine deletion gave rise to an expected premature translation stop and production of a truncated (14.5 kDa) inactive protein, called M2.MboIIΔA356 (Figure 1A). However, to our surprise, the plasmid obtained from the IPTG-induced bacteria was protected against MboII endonuclease digestion demonstrating the presence of specific methylation. After the preliminary fractionation of the bacterial lysate with the use of phosphocellulose (P-11) chromatography, a full-length 32 kDa protein was detected. Both forms of protein reacted with specific rabbit polyclonal anti-M2.MboII antibody (Figure 1B), and were also visualized by 35S methionine pulse-labeling in a T7 phage-specific expression system (Figure 1C). Also, the specific activity of the full-length protein obtained here was identical to the previously reported wild type M2.MboII (30). Fractions demonstrated by western blot to contain full-length M2.MboII, exhibited specific activity toward both double- (Figure 1D) and single-stranded DNA (not shown). Similarly, we observed the production of both forms of the protein with diversified intensity irrespective of the RNAP/promoter used (T7 phage/φ10 or E. coli /ParaBAD), the number of copies of the gene (plasmid or chromosome), or the actual temperature values during expression (37 or 22°C) (data not shown). N-terminal amino acid sequencing of small and full-length proteins was carried out by automated Edman degradation. The sequences of the first 10 amino acid residues (M1-N2-T3-I4-F5-F6-K7-D8-S9-R10) were found to be identical and consistent with the nucleotide sequence of the gene (GenBank KM192157).
Figure 1.
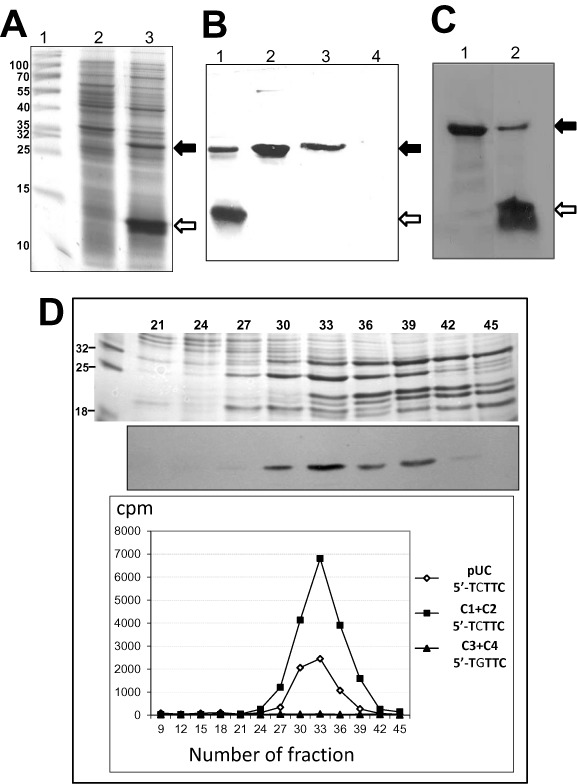
Synthesis of the wild type M2.MboII methyltransferase from the mutated gene mboIIM2ΔA356 through the frameshifting mechanism. (A) Overproduction of M2.MboII protein in T7 phage expression system. Protein lysate was resolved by 10%-SDS-PAGE and stained with Coomassie blue. Lane 1, molecular size markers. Lane 2, lysate of non-induced E. coli ER2566 with pET24mboIIMB.4 carrying mboIIM2ΔA356. Lane 3, lysate of IPTG induced ER2566. Black arrow—position of full-length M2.MboII (32 kDa), white arrow—M2.MboIIΔA356 truncated variant (14.5 kDa). (B) Immunodetection of western-blotted M2.MboII protein variants. Lane 1, lysate of IPTG induced ER2566 with pET24mboIIMB.4. Lane 2, 1.5 μg of purified WT M2.MboII. Lanes 3 and 4, eluates of fractions #33 and #45 after phosphocellulose (P-11) chromatography (see panel D). (C) Autoradiogram of protein profile after 1mM IPTG induction of expression of mboIIM2 in ER2566 bacteria. A pulse-labeling with 35S-methionine following 1.5 h rifampicin blockage of the host's transcription after 1.5 h IPTG induction was performed. Lane 1, strain with pETmboMB.3 carrying WT gene; lane 2, strain with pETmboMB.4 carrying mboIIM2ΔA356. (D) DNA methylation assay. Top: fractions after phosphocellulose chromatography separated by SDS-PAGE stained with Coomassie. Middle: western-blotting with anti-M2.MboII serum. Bottom: M2.MboII specific activity (cpm) in subsequent fractions using 3H-AdoMet cofactor and DNA substrates containing specific 5′-TCTTC sequences (plasmid pUC18 [squares] and oligoduplex DNA C1 + C2 [diamonds]) or nonspecific sequence 5′-TGTTC (oligoduplex, DNA C3 + C4, [triangles]) (Supplementary Table S2).
To assess the degree of uniqueness of this phenomenon we introduced single deletion mutations inside or close to a poly(A) tract in the isomethylomer ncuIM2 gene of N. cuniculi ATCC 14688 (32), the cognate methyltransferase mboIIM1 of M. bovis ATCC 10900 (29,30) and chloramphenicol-resistance gene cat from Tn9 (Supplementary Figure S1, Tables S1 and S2) (33). After T7-specific expression we were able to recover all the proteins as full-length specimens (which was confirmed by immunodetection), while retaining their specific activities (data not shown).
T7 RNAP generates high polymorphism of mRNA
Since programmed ribosomal frameshifting is very rare and sequence-specific (39), we hypothesized that the majority of the observed in-frame restoration in the investigated InDel mutants of mboIIM2 and other genes are likely to involve erroneous transcription. This would involve nucleotide insertion/deletion into homopolymeric tracts which are located relatively close to the previously introduced mutation site, and more importantly, they should be located upstream of the first 0 frame stop codon. To evaluate the extent of erroneous variations among the pool of mRNA sequences transcribed from the mboIIM2ΔA356 gene, we isolated total RNA from IPTG-induced ER2566 cells. Using a paired-end enriched-mRNA sequencing approach (NGS Illumina), we analyzed the proximal part of the mRNA (1–376 nt) containing the deletion mutation A356 in the 351–355 nt poly(A) region. Among 16968955 reads (mean 45130 × coverage of each nucleotide position) we detected 34635 misincorporations (0.20%), 15146 insertions (0.089%) and 2799 deletions (0.016%) (Supplementary Figure S2, Supplementary Tables S3 and S4). Of particular interest to us was the distribution of insertion mutations, as the most likely cause of a deletion mutation repair. We detected four homopolymer runs at the following positions: 13 nt (6T), 279 nt (7A), 351 nt (5A) and 364 nt (7T) which represented 1.06, 3.78, 10.47 and 1.19% of the frequency of total insertions. The percentages of the multiple insertion spectrum in these regions are shown in Table 1. The frequency of insertions in other homopolymer runs was at a much lower level (Supplementary Figure S2A [iii]). It seems that not only the length of a particular homopolymer run, but also the type of nucleotide composition and the context of the adjacent sequence is important for the incidence of transcriptional slippage. For example, for neighboring 4T (227 nt) and 4A (231 nt) repeats, the frequency is disproportionately different 0.037 and 0.306%, respectively (Supplementary Table S3). Similarly, for 3T (321nt) and 3A (324 nt) repeats, the frequencies are 0.0021 and 0.013%, respectively. Apparently, poly(A) type homopolymer, regardless of the number of repeated nucleotides, induced the strongest insertion frequencies (e.g. 3A [195 nt] versus 4T [347 nt], 0.11 and 0.06%, respectively). The most active region for transcriptional slippage is poly(5A) 351–355 nt. On the basis of data obtained from the analysis of mRNA polymorphism, we hypothesized that the insertion of a single A in this particular position is the probable cause of the A356 deletion rescue.
Table 1. Frequency of insertion at the A/T homopolymer hot spots in 376 nt mRNA of the mboIIM2ΔA356 proximal part.
| Position of the first nucleotide of the homopolymer run (length) | Fraction of total reads showing insertions (%) (number of insertion reads/total reads per base) | Homoinsertion length detected | Fraction of the insertions having a given length (%) |
|---|---|---|---|
| 13T (6) | 1.06 (316/29 898) | T | 93.7 |
| TT | 5.4 | ||
| TTT | 0.9 | ||
| 279A (7) | 3.78 (3261/86 286) | A | 88.8 |
| AA | 8.8 | ||
| AAA | 1.9 | ||
| 351A (5) | 10.47 (9603/91 678) | A | 66.9 |
| AA | 7.4 | ||
| AAA | 5.5 | ||
| AAAA | 12.8 | ||
| AAAAA | 2.1 | ||
| AAAAAA | 1.0 | ||
| AAAAAAA | 2.3 | ||
| 364T (7) | 1.19 (1049/87 793) | T | 98.7 |
| TT | 1.2 | ||
| TTT | 0.1 |
Coordinates according to the mboIIM2 gene sequence deposited in GenBank KM192157. For the purpose of this study we define a homopolymer as three or more adjacent nucleotides. Note, that in mRNA there are Us instead of Ts. A/Ts underlined restore the frame of reading for the mboIIM2ΔA356 gene.
Our analysis showed that deletion due to slippage was relatively rare (Supplementary Figure S2, Supplementary Table S3). The highest incidence of deletion (2%) was detected in the 364–370 poly(T) region.
Disruption of homopolymer continuity eliminates epigenetic mboIIM2ΔA356 gene repair
We constructed several site-specific nucleotide replacements that systematically interrupted the continuity of the homopolymeric T + A run (L-tract, 347–355 nt), the triple A (M—360–362 nt), and the R-tract, consisting of seven T (364–370 nt), upstream of the TAA stop codon of mboIIM2ΔA356 (Figure 2A and B). Transcriptional insertion of a single nucleotide into one of these tracts is the simplest way to restore the wild-type frame, preserving the wild-type amino acid composition completely (insertion in poly(A) of the L-tract) or partially (insertion in the M- or R-tract). We changed all the selected codons in such a manner as to preserve the wild type amino acid sequence (116–124 ILKNTKLFL), if the most probable frameshifting insertion in the poly(A) of the L-tract region (351–355 nt) were to take place. The only exception was the mutant (A5)mutR2, which may generate 123L instead of F, but without influence on its methylating activity (data not shown). All synonymous codons used here have a preferable codon usage index for E. coli (http://www.kazusa.or.jp/codon/). After inducing of mboIIM2ΔA356 replacement mutants expression, the M2.MboII product pattern was immunodetected by western blotting (Figure 2C). The relative amount of full-length M2.MboII in relation to the short form of the protein varies depending on the type and number of exchanges introduced. The restoration of the full-length protein was almost completely eliminated only after six nucleotide alterations ([A5]mutLMR6, Figure 2C, and corresponding 2D, lane 9). The proportion between the main short product (14.5 kDa) and its degradation derivatives, and the full-length product (32.1 kDa) was estimated densitometrically and normalized to the DnaA protein concentration in exponentially growing cells (Supplementary Figure S3). The changes in the poly(T + A) 347–355 nt of L-tract were the most effective to stop wild-type phenotype recovery. However, interruption of continuity in this homopolymeric run by nucleotide replacements only did not abolish slippage completely (up to 15% as compared to (A5)ΔA356). To assess influence of the TAATG sequence located between 370 and 374 nt (the last nucleotide of the stop codon UAA [0 frame] overlaps the first of the start codon AUG [−1 frame]) as the possible stimulatory signal in frameshift generation, we changed it to TACTC (sense codons in both reading frames). The alteration of these codons did not affect frameshift occurrence (Supplementary Table S2).
Figure 2.
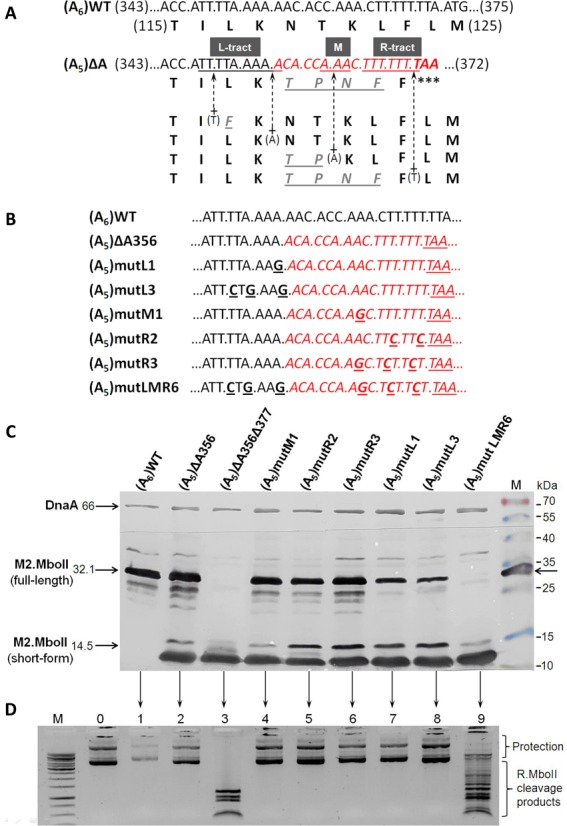
Elimination of transcriptional slippage by systematic discontinuity of T/A homopolymeric tracts in the 346–375 nt region of the mboIIM2ΔA356 gene. (A) Four of the simplest ways to restore a A356 deletion by single nucleotide insertion (plus arrow) through a transcriptional slippage mechanism and their influence on amino acid sequence changes (underlined). The T/A homopolymer runs (underlined) are named L-tract, M and R-tract, respectively. (B). A set of site-specific mutagenized mboIIM2ΔA356 genes with interrupted poly(T + A) tracts. Short names are used (see Supplementary Table S1). (C) Immunodetection of M2.MboII in total protein extracts from cultures harboring plasmids with various replacement mutations of the mboIIM2ΔA356 gene. Bacterial lysates were run on Tricine-SDS–12.5% PAGE, western blotted and immunodetected against M2.MboII protein. DnaA protein was used as an internal marker. M—molecular size markers (Fermentas) including the M2.MboII protein. (D) The relative level of plasmid methylation by the individual M2.MboII mutated variant. Plasmid DNA was digested with R.MboII, resolved by 0.8% agarose gel electrophoresis and visualized with ethidium bromide. Lanes 1–9 correspond to the variants from panel (C). M—molecular weight marker 1 kb (Fermentas). 0—undigested pET-(A5)LMR6 plasmid. Inverted image of the gel is shown.
Insertion of the non-templated A in the 351–355 nt poly(5A) region of mboIIM2 rescues the wild type phenotype
To confirm the character of changes in the mRNA of the mboIIM2ΔA356 gene involved in wild-type reading frame restoration, we created a fusion gene with the gfp reporter. We cloned the PCR product containing a 28 nt fragment of the mboIIM2ΔA356 gene, including deletion of A356 which generated stop codon TAA, fused with the GFP encoding gene (35) in a pET24a vector (Supplementary Tables S1 and S2). This allowed us to identify the amino acid sequence of the N-terminal part of the hybrid protein ‘M2.MboII’-GFP. To eliminate the possibility of employing ATG codon overlapping the stop site as an independent translational start for the gfp, we mutagenized it into a sense AGG. We assumed that the hybrid protein could arise only after nucleotide insertion in the mboIIM2 part of the gene. We expressed the mboIIM2-A5gfp0 gene and observed very strong and uniformly dispersed fluorescence under microscopy (data not shown). The preliminary purified hybrid protein (33 kDa) underwent automated Edman degradation. As expected, the sequence of the first three amino acid residues represented the part of the T7 protein 10 (M1-A2-S3), and the next seven residues (L118-K119-N120-T121-K122-L123-F124) were found to be identical to the wild type M2.MboII. This result confirmed the predominant insertion of the single A into the poly(5A) region (351–355 nt) in the wild-type phenotype restoration. It was consistent with the high frequency of A-type insertion obtained by RNA-seq analysis (Table 1).
Mass spectrometry analysis proved amino-acid heterogeneity in the population of the full-length M2.MboII
To explore the rescue character of the A356 deletion mutation in greater detail, the full-length (32 kDa) variant M2.MboII arising from mboIIM2ΔA356 gene expression was resolved by PAGE and subjected to mass spectrometry LC–MS-MS/MS analysis (40). We compared the masses of peptide fragments of the protein with a sequence database supplied by us using the MASCOT program (http://www.matrixscience.com) (41). MS/MS analysis allowed us to find two major peptides: the wild-type (122–132 LFLMDTYLWNR, Supplementary Figure S4A) and mutated (119–132 TPNFFLMDTYLWNR and TILKTPNF [115–122], trypsin or pepsin digestion derivatives, respectively) with some replaced amino acids (Supplementary Figure S4B). Their presence reflected that two modes of repair had occurred, first by insertion of an additional A into the 351–355 nt region (UUA.AAA.AAC) or U in the 364–370 nt region (UUU.UUU.UUA), respectively (Figure 2A, Table 2), both eliminating the UAA stop codon (370–372 nt). This data was in agreement with the insertion frequency in those regions determined by NGS mRNA analysis (Table 1). We were convinced that the mutated variant of the M2.MboII retained methylation activity by construction of a double mutant (deletion/insertion) gene—mboIIM2ΔA356+T371 (Table 2, Supplementary Table S1).
Table 2. List of InDel mutations assayed by the T7 RNAP in vivo transcription of the mboIIM2 gene.
| InDel location | Sequence context of InDel sites(1) | Full-length protein/MTase activity(2) | Possible restoration of gene continuity by transcriptional slippage mode(3) |
|---|---|---|---|
| None (WT) | (106)..GCC AAA GAT TGG TTA | +++/+++ | |
| (311)..AT ATT TTT GAT TTA AAT AG | |||
| (346)..ATT TTA AAA AAC ACC | |||
| (361)..AAA CTT TTT TTA ATG G | |||
| (616)..TTT GGC GTG CAT TCA GCG | |||
| ΔG112 | (106)..GCC AAA ΔGATT GGT TA | +/++ | +A113 ..GCC AAA AAT TGG TTA.. |
| ΔG112/ΔA356 | (106)..GCC AAA ΔGATT GGT../ | −/− | +A113/+A356 ..AAA AAT TGG../..AAA AAC ACC.. |
| (346)..TTT TAA AAA ΔACA | |||
| ΔT327 | (311)..AT ATT TTT GAT TTA AAΔTA GC | +/+++ | +A328 ..ATT TTT GAT TTA AAAAGC.. or |
| +T323 ..ATT TTT GAT TTT AAA AGC.. | |||
| ΔT327/ΔA328 | (311)..AT ATT TTT GAT TTA AAΔT ΔAG | ++/− | +T324/+A328 ..AT ATT TTT GAT TTT AAAAG.. or |
| +2A327 ..AT ATT TTT GAT TTA AAAAG | |||
| ΔA356 | (346)..ATT TTA AAA AΔACA CC…/ | +++/+++ | +A356 ..ATT TTA AAA AAC ACC..(4,5a) or |
| …(362)(..AAC TTT TTT TAA TGG..) | +T372 ..AAC TTT TTT TTA ATG..(5bc) | ||
| ΔA356/A372C/G374C | (346)..ATT TTA AAA ACA CC../ | +++/+++ | +A356 ..ATT TTA AAA AAC ACC.. or |
| (361)..AAC TTT TTT TA[C] T[C] | +T371 ..AAC TTT TTT TTA CTC.. | ||
| ΔA355/ΔA356 | (346)..ATT TTA AAA ΔAΔACAC C | ++/++ | +2A355 ..ATT TTA AAA AAC ACC.. or |
| +A355/+T371 ..ATT TTA AAA ACA../..TTT AAT.. | |||
| +T372 | (361)..AAA CTT TTT TTT AAT GG | +++/+++ | ΔT372 ..CTT TTT TTΔTA ATG G.. or |
| +2A 357 ..ATT TTA AAA AAAACA.. | |||
| ΔA356/ΔT371 | (346)..ATT TTA AAA AΔACA../ | ++/+ | +2A 356 ..ATT TTA AAA AAACAC.. or |
| (361)..AAC TTT TTT ΔTAAT | +A356/+T371 ..ATT TTA AAA AAC../..CTT TTT TTA.. or | ||
| +2T370 ..AAC TTT TTTTTA ATG.. | |||
| ΔA356/+T371 | (346)..ATT TTA AAA AΔACA../ | +++/+++ | +T371 insertion compensate the A356 deletion, restoring itself to the original frame from this point |
| (361)..AAC TTT TTT TTA AT | |||
| ΔA630 | (616)..TTT GGC GTG CAT TCΔAG CG.. | −/− | +T619 ..TTT TGG CGT GCA TTC GCG.. |
| C621G/ΔA630 | (616)..TTT GG[G] GTG CAT TCΔAG CG.. | −/− | +G623 ..TTT GGG GGT GCA TTC GCG.. |
(1)Coordinates according to the mboIIM2 gene sequence deposited in GenBank KM192157.
(2)Relative production of full-length M2.MboII protein (32 kDa) in comparison to the wild type/relative methylation activity level (evaluated by plasmid protection assay). (+++) high, (++) medium, (+) low, (−) none.
(3)For simplicity T nucleotide was used instead of U nucleotide.
(4)Amino acid composition LKNTKLF (117–123) confirmed by Edman degradation analysis.
(5)Amino acid composition (a) LFLMDTYLWNR (122–132), (b) TPNFFLMDTYLWNR (119–132) and (c) TILKTPNF (115–122) confirmed by LC–MS-MS/MS mass spectrometry analysis after trypsin (a,b), or pepsin (c) proteolysis of the M2.MboIIΔA356.
Legend: (Δ) nucleotide deletion; (A) nucleotide insertion; ([G]) nucleotide replacement; italization—changes affecting the reading frame causing codon(s)/(amino acid(s) alteration in comparison to the wild type.
gfp reporter fusions demonstrate the high frameshifting potential of T7 RNAP
Initially, we used the GFP reporter to determine the minimal slippage region and also to examine the possibility of −1 ribosomal frameshifting recoding (42). Since our analysis, arising from the prediction of the mboIIM2 mRNA secondary structure, showed the likely existence of a H-type RNA pseudoknot structure in the ‘slippery region’ (not shown), we fused the gfp gene in various combinations, downstream of the frameshifting site of mboIIM2. The gene fusions consisted of the proximal 377/378 nt part of mboIIM2ΔA356 or wild type mboIIM2 genes, and 0 frame or −1 frameshifted gfp gene (Figure 2A). They all eliminated pseudoknot formation. After expression of the fusion genes we observed the high level of frameshifting, which revealed that the H-type pseudoknot was indispensable as possible stimulatory signal. Moreover, to assess influence of the TAATG sequence located between 370 and 374 nt (the last nucleotide of the stop codon UAA [0 frame] overlaps the first of the start codon AUG [-1 frame]) as the possible stimulatory signal in frameshift generation, we changed the sequence to TACTC (sense codons in both reading frames) (Supplementary Table S2). The alteration of these codons did not affect frameshift occurrence. We concluded that our results do not support translational mechanism of frameshiting. Next, to find out more about the frameshifting ability of the T7 RNAP in vivo, we used a set of constructed fusion genes to analyze all possible (+) or (−) slippage events generated in most active 351–356 nt and 364–371 nt homopolymeric regions. The gfp fusions allowed us to detect all out-of-frame transcriptional signals generated by T7 RNA polymerase in the slippery region, whereas the MboII-GFP the means to hybrids provided distinguish the slippage products from each other and also from degradation products after PAGE electrophoresis. Therefore, due to the gfp reporter we could detect specific out of frame products. It is worth noting that only mboIIM2wtΔ378gfp0 should produce the hybrid protein in the 0 frame. Surprisingly, it appeared that transcriptional slippage was present not only during expression of the mboIIM2ΔA356 nucleotide deletion mutant gene, but also in case of the wild type mboIIM2 gene expression. In all four cases we observed a fluorescence signal (Figure 2B) and detected a 41.7 kDa M2.MboIIΔ377(378)–GFP hybrid protein, using anti-M2.MboII polyclonal antibody and an anti-GFP monoclonal antibody (Figure 2C and D). Analysis of the western-blotted protein products indicated ‘regular’ 0 frame truncated products of M2.MboII (14.6 and 16.8 kDa) and the hybrid protein, formed due to a frameshift in the transcription. All the hybrid proteins formed pole-deposited aggregates in the E. coli cells (43,44). In the case of M2.MboIIΔA356Δ377–GFP0 hybrid, we observed a uniformly distributed fluorescence signal which also indicated the presence of the dispersed GFP protein alone, unbound with the M2.MboIIΔ377 component. The production of GFP is possible by adapting the internal AUG codon overlapping the upstream UAA stop codon of mboIIM2 (Figure 2A). However, the pictures from phase contrast microscopy showed the presence of aggregates deposited at poles similar to M2.MboII ΔA356Δ377-GFP-1 (not shown), which was also confirmed in a western-blotting experiment (Figure 2C). Our results indicated that at least the poly(A) and poly(T) regions located between 351–355/356 nt and 364/365–371 nt, respectively, were the hot spots for high level transcript slippage, both for the wild-type and deletion mutation mboIIM2 gene. Insertion of an additional A or U in either hot spot was a likely mechanism of mutation rescue in the case of two fusion genes, mboIIM2ΔA356Δ377gfp0 and mboIIM2wtΔ378gfp-1. Contrary to this, in the case of the double deletion mutant mboIIM2ΔA356Δ377gfp-1, the deletion of a single U nucleotide caused mutation rescue. However, we cannot exclude the possibility of eliminating UAA stop codon upstream of gfp by double nucleotide insertion in A- and/or T-rich homopolymers. Interestingly, the nucleotide insertion/deletion mechanism was ineffective to rescue the two deletions located a considerable distance from each other (mboIIM2ΔA356Δ562gfp-1). Western blot analysis revealed the presence of 0 frame M2.MboII short product (14.6 kDa, 370 nt UAA stop codon) and the slightly longer +1 frameshifted 24.2 kDa incomplete M2.MboII-GFP hybrid (first possible UAG stop codon in proximal part of gfp-1 gene at 613 nt) (Supplementary Figure S5). Due to the long distance between the sites of the deletion, all possible combinations of −1 or +2 reparative frameshifting generated stop codons. The complete 49.1 kDa hybrid protein M2.MboIIΔ562-Gfp could only arise in case of the mboIIM2ΔA356Δ562gfp0 fusion gene (Supplementary Figure S5).
Various InDel frameshifting mutations in the mboIIM2 gene are restored by T7 RNA transcription in slippage-prone sequences
To address the question of whether more frameshifting mutations can be rescued by other slippery-inducing regions of the gene, we created several single and double nucleotide insertion/deletion mutants (Supplementary Tables S1 and S2). They were located mainly in the proximal part of the mboIIM2 gene (Table 2). All of them interrupted the continuity of the translational reading frame by the premature appearance of a stop codon. Indeed, there were more slippage-inducing sites in the mboIIM2 gene which could provoke RNA editing by the T7 RNAP. We observed that the production of full-length proteins and restoration of their methylation activity depended on the nucleotide context in the vicinity of the InDel mutations and also their location in the protein domain (Figure 4, Table 2). Thus, in some cases restoration of the full-length protein did not lead to recovery of its full catalytic activity. We also observed a different level of gene expression for each mutant, manifested as a reciprocal proportion in concentrations between the short variants of the protein, translated from unchanged mRNA containing InDel mutation, and full-length variants formed by translation from erroneously synthesized mRNA (Figure 4). Hence, such ‘repair’ of deletions by the insertion of a single nucleotide(s) is dependent on the type and length of the homopolymer adjacent to the mutation site, as well as the transcript slippage efficiency of RNAP. For example, the production of the full-length protein may be inefficient (Figure 4A, lanes 1 and 2) but biologically effective to sufficiently methylate the MboII recognition sites (Figure 4B, lanes 12 and 16). Interestingly, in the case of ΔG112 mutant rescue, we observed the unexpected expression of a shorter protein of 21.71 kDa, (Figure 4A, lane 1) starting from the internal ATG initiation codon (268–270 nt, as was apparent from the mass spectrometry analysis (not shown). However, an insertion by erroneous transcription is not always effective in ‘repairing’ the deletion mutation. For example, the lack of a proper nucleotide context downstream of the 5A homopolymer (601–605 nt) nearby a single deletion site (A630) means that any insertion of a single A generates UGA stop codon (606–608 nt). Thus, such a mutant seems to be ‘irreparable’, even after creating a tetra-nucleotide poly(G) region (C621G/ΔA630, Table 2), most likely because it is not effective in slippage induction. We faced a different situation in the case of +T372 single-insertion mutation. The highest frequency of nucleotide deletion mapped in that region (2%) provides a plausible explanation for the likely way to restore a +T372 frameshift mutation. Unlike what has been described so far, the deletion of a single U nucleotide from a long 8U-region is the presumable mechanism of wild-type gene restoration. This is confirmed by the efficient production of a full-length protein and its high level methylation activity (Figure 4A, lane 4 and 4B, lane 24, respectively).
Figure 4.
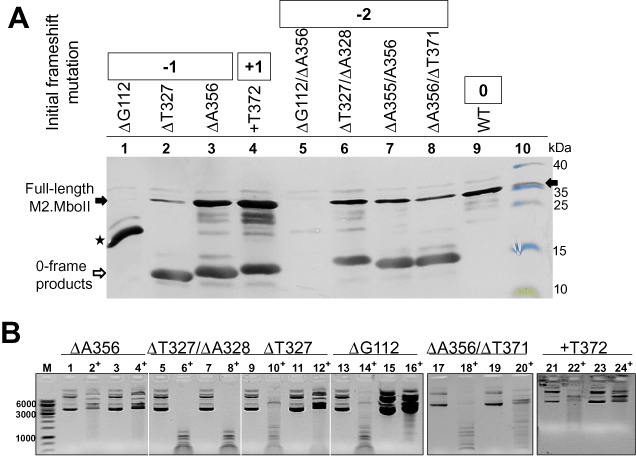
Synthesis of the full-length M2.MboII protein after epigenetic frameshift repair of some InDel mutations of the mboIIM2 gene. (A) Equal amounts of total protein extracts from cultures harboring plasmids with various InDel mutations (lanes 1–9) were analyzed after Tricine-SDS-PAGE and western blotting using anti-M2.MboII antibodies. Lane 10, molecular size markers (Fermentas) including purified M2.MboII protein. Black arrow—position of the full-length M2.MboII (32 kDa); white arrow—position of short M2.MboII variants; asterisk—shorter variant of M2.MboII product (21.71 kDa). (B) The relative protection level of the MboII sites in plasmids bearing the selected mboIIM2 InDel mutants against R.MboII digestion. Plasmid aliquots from non-induced (lanes 1–2, 5–6, 9–10, 13–14, 17–18, 21–22) and IPTG -induced cultures (3–4, 7–8, 11–12, 15–16, 19–20, 23–24) were digested with R.MboII (+). DNA was resolved by 0.8% agarose gel electrophoresis and visualized with ethidium bromide. Samples 17–20 and 21–24 were run on separate gels. Inverted image is shown.
It seems that the transcriptional repair of a double deletion is more complicated and complex in its diversity. We showed that the recovery of double deletion mutation occurred regardless of whether they were initially next to each other or at some distance. Based on several examples, we demonstrated a negative correlation between the efficiency of production of the full-length protein and the increase of distance between the mutation sites. Hence, there is no protein production and no activity restoration in the case of ΔG112/ΔA356 double mutation (Figure 4A, lane 5). Another aspect is the type of missing nucleotide of mutants and type of inserted nucleotides during erroneous transcription (the same versus different). Presumably, most codon changes are associated with negative alteration in protein activity, particularly in the domains responsible for DNA binding, catalysis, etc. The restoration of biological activity appears to be easier in the case of double deletion of nucleotides in close proximity to each other but of the same type (Δ2A355/356, Figure 4A, lane 7, positive methylation activity not shown). In contrast, the epigenetic restoration of the double deletion mutant ΔT327/ΔA328 resulted in the production of an inactive full-length protein (Figure 4A, lane 6, and Figure 4B, lane 8) or the production of a protein with a low level of activity, as was seen in the case of the mutant ΔA356/ΔT371 (Figure 4A, lane 8 and Figure 4B, lane 20).
DISCUSSION
Among the many processes observed contributing to the alteration of the genetic information at the mRNA level (RNA editing), the best reported to date is transcriptional slippage (45–47). Programmed transcriptional slippage can regulate the level and specificity of gene expression (48,49). Recently, more convincing evidence showed that transcriptional inaccuracy was able to restore the wild-type phenotype of mutant genes (50–55). It was proposed that inaccuracy in gene expression in individual living cells may be important for the generation of diversity during environmental challenge (51,53,56). In fact, epigenetic effects have been observed after slippage during initiation, elongation and termination at homopolymer runs in E. coli both in vitro (14,16) and in vivo (57–60).
We provide evidence that the intrinsic infidelity of T7 RNAP in transcription brings significant phenotypic consequences, observed as frameshift mutation restoration and the production of a heterogenous population of the full-length protein. It was reasonable to hypothesize that the transcriptional slippage mechanism at the A/T homopolymer regions of mRNA contributed to the observed restoration of the InDel disrupted genes, enabling the synthesis of full-length proteins (Figures 1 and 2). By means of NGS and genetic techniques we identified four non-uniformly distributed erroneous hot spots in the examined part of the mboIIM2 mRNA with a high frequency of InDel events (Supplementary Figure S2). Among them, the highest insertion frequency (almost 11%) was displayed by the poly(A) region 351–355 nt (Table 1). As shown in Table 1 a single insertion error is dominant over multiple insertions or deletion events. Site-specific mutagenization of the most insertion inducing A/T-rich regions (Figure 2) and the outcome of the mboIIM2-gfp fusion genes expression clearly demonstrate a significant impact of the A/T homopolymers on the quality and quantity of a given gene's expression (Figures 2–4). Based on our results, it seems that utilization of transcriptional slippage in the context of repair of accidental frameshift mutations may be more common than thought so far (50). It is likely that homopolymers have a nonrandom location within genes and can be conserved over evolutionary time (50,55,61). Recently, the occurrence of a strong correlation between the frequency of InDel frameshifting bypasses and nucleotide homopolymer repeats was reported for HaeIII methyltransferase coding gene variants (57). The significance of the phenomenon described above might seem ambiguous. A model study with error-prone T7 RNAP showed that synthesis of dysfunctional proteins based on erroneous transcripts was highly deleterious for T7 phage fitness (62). Similarly, a frequent loss of the fragment of DNA with the T7 RNAP gene was observed when wild-type T7 was grown on an E. coli host which already expressed T7 RNAP (63). The large proportion of amino acid substitutions arising during T7 RNAP-based (or any RNAPs) expression is deleterious for protein activity and function (64). However, it cannot be excluded that some of the altered phenotypes may carry the potential for adaptation under pressure of selective conditions (65). Indeed, we showed here that for some InDel mutant genes transcriptional infidelity may repair and/or modify a protein's structure/activity, increasing the repertoire of phenotypic variants. This could be achieved with restoration of the original genetic sequence or appearance of other, usually less-effective, codon alternatives incompatible with the original DNA template (Table 2, mboIIM2ΔA356). We demonstrated the presence of the mutated variant of the full-length M2.MboII with four amino acid replacements in the protein's population, manifesting a high level of a site-specific DNA methylation activity. Therefore, it seems, we are dealing with a new situation, synthesis of a heterogeneous population of mRNAs whose rescue of the gene function leads to the production of a heterogeneous full-length proteins with amino acid alterations. We have reason to believe therefore that the final biological outcome of protein recovery and its real impact on cellular function does not depend simply on the RNAP potential for erroneous changes. For example, we observed a lack of recovery for double deletion mutation located at a considerable distance from each other (217 nt) in the mboIIM2Δ356Δ563gfp-1 gene fusion (Supplementary Figure S5), and a successful rescue in the case of mboIIM2Δ356Δ377gfp-1 gene (distance of 26 nt, Figure 3). Transcriptional slippage occurred in both cases but could only be manifested phenotypically in the latter case. The probability of the stop codon generation is much higher for deletion locations separated by long distances. Obviously, as was shown recently for HaeIII methyltransferase a partial or complete wild-type phenotype return was dependent on the location of the InDel mutation with respect to the particular domain of the enzyme (nonconserved versus conserved regions (57). The mutant spectrum provides a reservoir of protein variants which might become adequate in response to changes in the molecular environment in the cell. Apparently, changes in protein structure and function are always the cost which must be paid for life support under mutagenic conditions (17).
Figure 3.
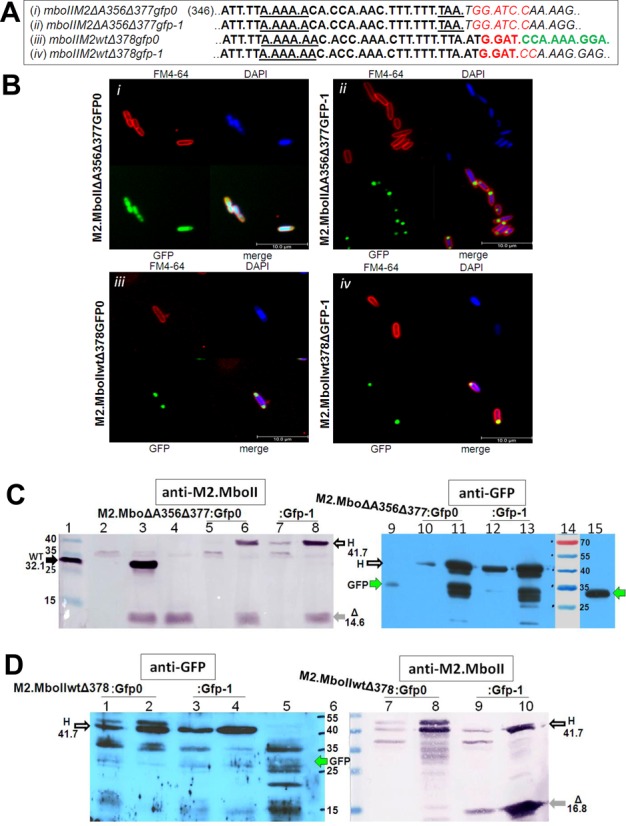
Monitoring of T7 RNAP-based frameshifting by identification of the A356 deletion mutant and wild-type M2.MboIIΔ377(378)-GFP hybrid protein products. (A) The nucleotide sequence of the junction site in four mboIIM2Δ377(378)gfp fusion genes. Note, the triplets indicate the zero frame codons and only mboIIM2wtΔ378gfp0 should produce the hybrid protein in this frame (the gfp part of the fusion gene is marked in green). BamHI recognition site is marked in red, italicized nucleotides mean failure of Gfp recovery in 0 frame. (B) Images show fluorescence microscopy of induced E. coli ER2566 containing four plasmids with an appropriate mboIIM2gfp gene. DAPI and FM4-64 were added to stain DNA and cell membrane, respectively. Scale bar ≈ 10 μm. (C) Immunodetection of M2.MboIIΔA356Δ377-GFP hybrids by anti-M2.MboII rabbit polyclonal antibody/anti-rabbit-AP and anti-GFP mouse monoclonal/anti-mouse-HRP. The positions of the hybrid (H), GFP, full-length (WT) and short-form (Δ) M2.MboII proteins are indicated by arrows. (D) Immunodetection of the M2.MboIIwtΔ378-GFP hybrids (H) by anti-M2.MboII rabbit polyclonal antibody/anti-rabbit-AP and anti-GFP mouse monoclonal/anti-mouse-HRP.
A similar phenomenon was observed in eukaryotic cells in a process referred to as molecular misreading (66,67). Frameshift mutations were generated near short simple repeat hot spots (68). Besides this, the propensity of eukaryotic RNA viruses to a very high mutation rate is generally known and recognized as ambivalent (69–71). On the one hand this allows for rapid adaptation in response to selective pressure, on the other hand, it also leads to a reduction or loss of ability to infect host cells by exceeding the safe error threshold (72,73). Study of the serial plaque-to-plaque transfers of FMDV RNA virus provided evidence of mutation accumulation and fitness decrease (74–76). Interestingly, a heterogeneity of length of the internal polyadenosine extension in leader sequence of the Lab protease gene was found (74). An increase in the length of the poly(A) tract (up to 50 nucleotides) correlated negatively with the relative fitness of the virus clones. Most probably, it had a deep impact on the initiation of translation efficiency by changing at least the secondary structure of mRNA. Hence, a shortening of the internal polyadenylate is associated with the fitness recovery process (75). Moreover, half of all the fixed substitutions in the capsid protein affected internal amino acid residues constitute a structural role (74,75).
It appears that the intrinsic properties of RNAPs can make an equally unpredictable impact on the final outcome of gene expression as well as the process of differentiation of genes. It is known that the error rate of transcripts generated by E. coli RNAP in vivo is significantly high, roughly between the values of 10−5 and 10−3 per residue, and location-dependent (77). Generally, this is true for all other types of RNAPs, both single and multisubunit. It is likely that the wild type RNAP has evolved some ‘slippage ability’, which may be regulated under certain cellular conditions (51,53,60,67,68). It would be interesting to know what the contribution of the transcriptional slippage to the mechanisms of regulation precision in the level of a gene expression is. Since the level of transcription primarily determines the overall expression level (78), we suggest that homopolymeric regions of genes susceptible for InDel mutations, especially the A/T type, may play the role of a structural determinant decreasing availability of the wild-type mRNA, and thus negatively influencing the level of wild-type protein synthesis. The hypothesis about the role of homopolymeric tracts, namely the reduction of efficiency of the expression of genes carrying poly(A) regions has already been raised by others (54,55). We intend to address this issue in our further work.
We propose that the observed high tolerance for erroneous incorporation at the level of transcription must be considered as survival strategy. Aside from the practical significance (the presence of proteins with different properties within cellular population) this phenomenon provides a new understanding of the role of the mRNA population that is inconsistent with the DNA template and deserves further investigation toward homopolymer tracts frequency and arrangement in the genome and their regulatory role.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We wish to thank Patryk Plisinski, Agata Fischer and Dawid Koscielniak for their technical assistance. We wish to express special thanks to Dariusz Nowicki (University of Gdansk, Poland) for his excellent work on the fluorescence microscopy.
FUNDING
National Science Centre (Poland) [2012/07/BNZ2/01782 to M.S.]. Funding for open access charge: National Science Centre (Poland) [2012/07/BNZ2/01782].
Conflict of interest statement. None declared.
REFERENCES
- 1.Davanloo P., Rosenberg A.H., Dunn J.J., Studier F.W. Cloning and expression of the gene for bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 1984;81:2035–2039. doi: 10.1073/pnas.81.7.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Studier F.W., Moffatt B.A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- 3.Tabor S., Richardson C.C. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc. Natl. Acad. Sci. U.S.A. 1985;82:1074–1078. doi: 10.1073/pnas.82.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenberg A.H., Lade B.N., Chui D.S., Lin S.W., Dunn J.J., Studier F.W. Vectors for selective expression of cloned DNAs by T7 RNA polymerase. Gene. 1987;56:125–135. doi: 10.1016/0378-1119(87)90165-x. [DOI] [PubMed] [Google Scholar]
- 5.Fuerst T.R., Niles E.G., Studier F.W., Moss B. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 1986;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nevin D.E., Pratt J.M. A coupled in vitro transcription-translation system for the exclusive synthesis of polypeptides expressed from the T7 promoter. FEBS Lett. 1991;291:259–263. doi: 10.1016/0014-5793(91)81297-l. [DOI] [PubMed] [Google Scholar]
- 7.Schenborn E.T., Mierendorf R.C., Jr A novel transcription property of SP6 and T7 RNA polymerases: dependence on template structure. Nucleic Acids Res. 1985;13:6223–6236. doi: 10.1093/nar/13.17.6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graifer D., Karpova G. General approach for introduction of various chemical labels in specific RNA locations based on insertion of amino linkers. Molecules. 2013;18:14455–14469. doi: 10.3390/molecules181214455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Gelder R.N., von Zastrow M.E., Yool A., Dement W.C., Barchas J.D., Eberwine J.H. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc. Natl. Acad. Sci. U.S.A. 1990;87:1663–1667. doi: 10.1073/pnas.87.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang E., Miller L.D., Ohnmacht G.A., Liu E.T., Marincola F.M. High-fidelity mRNA amplification for gene profiling. Nat. Biotechnol. 2000;18:457–459. doi: 10.1038/74546. [DOI] [PubMed] [Google Scholar]
- 11.Pomerantz R.T., Temiakov D., Anikin M., Vassylyev D.G., McAllister W.T. A mechanism of nucleotide misincorporation during transcription due to template-strand misalignment. Mol. Cell. 2006;24:245–255. doi: 10.1016/j.molcel.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin C.T., Muller D.K., Coleman J.E. Processivity in early stages of transcription by T7 RNA polymerase. Biochemistry. 1988;27:3966–3974. doi: 10.1021/bi00411a012. [DOI] [PubMed] [Google Scholar]
- 13.Volchkov V.E., Becker S., Volchkova V.A., Ternovoj V.A., Kotov A.N., Netesov S.V., Klenk H.D. GP mRNA of Ebola virus is edited by the Ebola virus polymerase and by T7 and vaccinia virus polymerases. Virology. 1995;214:421–430. doi: 10.1006/viro.1995.0052. [DOI] [PubMed] [Google Scholar]
- 14.Macdonald L.E., Zhou Y., McAllister W.T. Termination and slippage by bacteriophage T7 RNA polymerase. J. Mol. Biol. 1993;232:1030–1047. doi: 10.1006/jmbi.1993.1458. [DOI] [PubMed] [Google Scholar]
- 15.Cazenave C., Uhlenbeck O.C. RNA template-directed RNA synthesis by T7 RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 1994;91:6972–6976. doi: 10.1073/pnas.91.15.6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Groebe D.R., Uhlenbeck O.C. Characterization of RNA hairpin loop stability. Nucleic Acids Res. 1988;16:11725–11735. doi: 10.1093/nar/16.24.11725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doetsch P.W. Translesion synthesis by RNA polymerases: occurrence and biological implications for transcriptional mutagenesis. Mutat. Res. 2002;510:131–140. doi: 10.1016/s0027-5107(02)00258-0. [DOI] [PubMed] [Google Scholar]
- 18.Chevrier-Miller M., Jacques N., Raibaud O., Dreyfus M. Transcription of single-copy hybrid lacZ genes by T7 RNA polymerase in Escherichia coli: mRNA synthesis and degradation can be uncoupled from translation. Nucleic Acids Res. 1990;18:5787–5792. doi: 10.1093/nar/18.19.5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iost I., Guillerez J., Dreyfus M. Bacteriophage T7 RNA polymerase travels far ahead of ribosomes in vivo. J. Bacteriol. 1992;174:619–622. doi: 10.1128/jb.174.2.619-622.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Villaverde A., Carrio M.M. Protein aggregation in recombinant bacteria: biological role of inclusion bodies. Biotechnol. Lett. 2003;25:1385–1395. doi: 10.1023/a:1025024104862. [DOI] [PubMed] [Google Scholar]
- 21.Dubendorff J.W., Studier F.W. Controlling basal expression in an inducible T7 expression system by blocking the target T7 promoter with lac repressor. J. Mol. Biol. 1991;219:45–59. doi: 10.1016/0022-2836(91)90856-2. [DOI] [PubMed] [Google Scholar]
- 22.Mertens N., Remaut E., Fiers W. Tight transcriptional control mechanism ensures stable high-level expression from T7 promoter-based expression plasmids. Biotechnology. 1995;13:175–179. doi: 10.1038/nbt0295-175. [DOI] [PubMed] [Google Scholar]
- 23.Miroux B., Walker J.E. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 24.Narayanan A., Ridilla M., Yernool D.A. Restrained expression, a method to overproduce toxic membrane proteins by exploiting operator-repressor interactions. Protein Sci. 2011;20:51–61. doi: 10.1002/pro.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan S.H., Malcolm B.A. Reduced background expression and improved plasmid stability with pET vectors in BL21 (DE3) Biotechniques. 2000;29:1234–1238. doi: 10.2144/00296st03. [DOI] [PubMed] [Google Scholar]
- 26.Sektas M., Szybalski W. Novel single-copy pETcoco vector with dual controls for amplification and expression. InNovations. 2002;14:6–8. [Google Scholar]
- 27.Studier F.W. Use of bacteriophage T7 lysozyme to improve an inducible T7 expression system. J. Mol. Biol. 1991;219:37–44. doi: 10.1016/0022-2836(91)90855-z. [DOI] [PubMed] [Google Scholar]
- 28.Wagner S., Klepsch M.M., Schlegel S., Appel A., Draheim R., Tarry M., Hogbom M., van Wijk K.J., Slotboom D.J., Persson J.O., et al. Tuning Escherichia coli for membrane protein overexpression. Proc. Natl. Acad. Sci. U.S.A. 2008;105:14371–14376. doi: 10.1073/pnas.0804090105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bocklage H., Heeger K., Muller-Hill B. Cloning and characterization of the MboII restriction-modification system. Nucleic Acids Res. 1991;19:1007–1013. doi: 10.1093/nar/19.5.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furmanek-Blaszk B., Boratynski R., Zolcinska N., Sektas M. M1.MboII and M2.MboII type IIS methyltransferases: different specificities, the same target. Microbiology. 2009;155:1111–1121. doi: 10.1099/mic.0.025023-0. [DOI] [PubMed] [Google Scholar]
- 31.Sambrook J., Fritsch E.F., Maniatis T. Molecular Cloning. A Laboaratory Manual. 2nd edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 32.Furmanek B., Sektas M., Wons E., Kaczorowski T. Molecular characterization of the DNA methyltransferase M1.NcuI from Neisseria cuniculi ATCC 14688. Res. Microbiol. 2007;158:164–174. doi: 10.1016/j.resmic.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 33.Mruk I., Blumenthal R.M. Tuning the relative affinities for activating and repressing operators of a temporally regulated restriction-modification system. Nucleic Acids Res. 2009;37:983–998. doi: 10.1093/nar/gkn1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sektas M., Furmanek-Blaszk B. Improvement in the visual discrimination of recombinant clones by size reduction of non-recombinant colonies. J. Microbiol. Methods. 2013;95:302–303. doi: 10.1016/j.mimet.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 35.Miller W.G., Lindow S.E. An improved GFP cloning cassette designed for prokaryotic transcriptional fusions. Gene. 1997;191:149–153. doi: 10.1016/s0378-1119(97)00051-6. [DOI] [PubMed] [Google Scholar]
- 36.Schagger H., von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 37.Towbin H., Staehelin T., Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U.S.A. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 39.Gurvich O.L., Baranov P.V., Zhou J., Hammer A.W., Gesteland R.F., Atkins J.F. Sequences that direct significant levels of frameshifting are frequent in coding regions of Escherichia coli. EMBO J. 2003;22:5941–5950. doi: 10.1093/emboj/cdg561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hess D., Covey T.C., Winz R., Brownsey R.W., Aebersold R. Analytical and micropreparative peptide mapping by high performance liquid chromatography/electrospray mass spectrometry of proteins purified by gel electrophoresis. Protein Sci. 1993;2:1342–1351. doi: 10.1002/pro.5560020817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perkins D.N., Pappin D.J., Creasy D.M., Cottrell J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 42.Giedroc D.P., Cornish P.V. Frameshifting RNA pseudoknots: structure and mechanism. Virus Res. 2009;139:193–208. doi: 10.1016/j.virusres.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carrio M.M., Corchero J.L., Villaverde A. Dynamics of in vivo protein aggregation: building inclusion bodies in recombinant bacteria. FEMS Microbiol. Lett. 1998;169:9–15. doi: 10.1111/j.1574-6968.1998.tb13292.x. [DOI] [PubMed] [Google Scholar]
- 44.Scheu K., Gill R., Saberi S., Meyer P., Emberly E. Localization of aggregating proteins in bacteria depends on the rate of addition. Front. Microbiol. 2014;5:418. doi: 10.3389/fmicb.2014.00418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anikin M., Molodtsov V., Temiakov D., McAllister W.T. Transcript slippage and recoding. In: Atkins JF, Gesteland RF, Bujnicki JM, editors. Recoding: Expansion of Decoding Rules Enriches Gene Expression. 24th edn. Springer, New York, NY: 2010. pp. 409–432. [Google Scholar]
- 46.Sharma V., Firth A.E., Antonov I., Fayet O., Atkins J.F., Borodovsky M., Baranov P.V. A pilot study of bacterial genes with disrupted ORFs reveals a surprising profusion of protein sequence recoding mediated by ribosomal frameshifting and transcriptional realignment. Mol. Biol. Evol. 2011;28:3195–3211. doi: 10.1093/molbev/msr155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turnbough C.L., Jr Regulation of gene expression by reiterative transcription. Curr. Opin. Microbiol. 2011;14:142–147. doi: 10.1016/j.mib.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Larsen B., Wills N.M., Nelson C., Atkins J.F., Gesteland R.F. Nonlinearity in genetic decoding: homologous DNA replicase genes use alternatives of transcriptional slippage or translational frameshifting. Proc. Natl. Acad. Sci. U.S.A. 2000;97:1683–1688. doi: 10.1073/pnas.97.4.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turnbough C.L., Jr, Switzer R.L. Regulation of pyrimidine biosynthetic gene expression in bacteria: repression without repressors. Microbiol. Mol. Biol. Rev. 2008;72:266–300. doi: 10.1128/MMBR.00001-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baranov P.V., Hammer A.W., Zhou J., Gesteland R.F., Atkins J.F. Transcriptional slippage in bacteria: distribution in sequenced genomes and utilization in IS element gene expression. Genome Biol. 2005;6:R25. doi: 10.1186/gb-2005-6-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gordon A.J., Halliday J.A., Blankschien M.D., Burns P.A., Yatagai F., Herman C. Transcriptional infidelity promotes heritable phenotypic change in a bistable gene network. PLoS Biol. 2009;7:e44. doi: 10.1371/journal.pbio.1000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gordon A.J., Satory D., Halliday J.A., Herman C. Heritable change caused by transient transcription errors. PLoS Genet. 2013;9:e1003595. doi: 10.1371/journal.pgen.1003595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meyerovich M., Mamou G., Ben-Yehuda S. Visualizing high error levels during gene expression in living bacterial cells. Proc. Natl. Acad. Sci. U.S.A. 2010;107:11543–11548. doi: 10.1073/pnas.0912989107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tamas I., Wernegreen J.J., Nystedt B., Kauppinen S.N., Darby A.C., Gomez-Valero L., Lundin D., Poole A.M., Andersson S.G. Endosymbiont gene functions impaired and rescued by polymerase infidelity at poly(A) tracts. Proc. Natl. Acad. Sci. U.S.A. 2008;105:14934–14939. doi: 10.1073/pnas.0806554105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wernegreen J.J., Kauppinen S.N., Degnan P.H. Slip into something more functional: selection maintains ancient frameshifts in homopolymeric sequences. Mol. Biol. Evol. 2010;27:833–839. doi: 10.1093/molbev/msp290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Atkins J.F., Elseviers D., Gorini L. Low activity of -galactosidase in frameshift mutants of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1972;69:1192–1195. doi: 10.1073/pnas.69.5.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rockah-Shmuel L., Toth-Petroczy A., Sela A., Wurtzel O., Sorek R., Tawfik D.S. Correlated occurrence and bypass of frame-shifting insertion-deletions (InDels) to give functional proteins. PLoS Genet. 2013;9:e1003882. doi: 10.1371/journal.pgen.1003882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wagner L.A., Weiss R.B., Driscoll R., Dunn D.S., Gesteland R.F. Transcriptional slippage occurs during elongation at runs of adenine or thymine in Escherichia coli. Nucleic Acids Res. 1990;18:3529–3535. doi: 10.1093/nar/18.12.3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xiong X.F., Reznikoff W.S. Transcriptional slippage during the transcription initiation process at a mutant lac promoter in vivo. J. Mol. Biol. 1993;231:569–580. doi: 10.1006/jmbi.1993.1310. [DOI] [PubMed] [Google Scholar]
- 60.Zhou Y.N., Lubkowska L., Hui M., Court C., Chen S., Court D.L., Strathern J., Jin D.J., Kashlev M. Isolation and characterization of RNA polymerase rpoB mutations that alter transcription slippage during elongation in Escherichia coli. J. Biol. Chem. 2013;288:2700–2710. doi: 10.1074/jbc.M112.429464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Passel M.W., Ochman H. Selection on the genic location of disruptive elements. Trends Genet. 2007;23:601–604. doi: 10.1016/j.tig.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 62.Brakmann S., Grzeszik S. An error-prone T7 RNA polymerase mutant generated by directed evolution. Chembiochem. 2001;2:212–219. doi: 10.1002/1439-7633(20010302)2:3<212::AID-CBIC212>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 63.Yin J. Evolution of bacteriophage T7 in a growing plaque. J. Bacteriol. 1993;175:1272–1277. doi: 10.1128/jb.175.5.1272-1277.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Goldsmith M., Tawfik D.S. Potential role of phenotypic mutations in the evolution of protein expression and stability. Proc. Natl. Acad. Sci. U.S.A. 2009;106:6197–6202. doi: 10.1073/pnas.0809506106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aharoni A., Gaidukov L., Khersonsky O., Mc Q.G.S., Roodveldt C., Tawfik D.S. The ‘evolvability’ of promiscuous protein functions. Nat. Genet. 2005;37:73–76. doi: 10.1038/ng1482. [DOI] [PubMed] [Google Scholar]
- 66.Evans D.A., van der Kleij A.A., Sonnemans M.A., Burbach J.P., van Leeuwen F.W. Frameshift mutations at two hotspots in vasopressin transcripts in post-mitotic neurons. Proc. Natl. Acad. Sci. U.S.A. 1994;91:6059–6063. doi: 10.1073/pnas.91.13.6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van Leeuwen F., van der Beek E., Seger M., Burbach P., Ivell R. Age-related development of a heterozygous phenotype in solitary neurons of the homozygous Brattleboro rat. Proc. Natl. Acad. Sci. U.S.A. 1989;86:6417–6420. doi: 10.1073/pnas.86.16.6417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Den Hurk W.H., Willems H.J., Bloemen M., Martens G.J. Novel frameshift mutations near short simple repeats. J. Biol. Chem. 2001;276:11496–11498. doi: 10.1074/jbc.M011040200. [DOI] [PubMed] [Google Scholar]
- 69.Drake J.W. Rates of spontaneous mutation among RNA viruses. Proc. Natl. Acad. Sci. U.S.A. 1993;90:4171–4175. doi: 10.1073/pnas.90.9.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Holland J., Spindler K., Horodyski F., Grabau E., Nichol S., VandePol S. Rapid evolution of RNA genomes. Science. 1982;215:1577–1585. doi: 10.1126/science.7041255. [DOI] [PubMed] [Google Scholar]
- 71.Lauring A.S., Andino R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010;6:e1001005. doi: 10.1371/journal.ppat.1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Domingo E., Sabo D., Taniguchi T., Weissmann C. Nucleotide sequence heterogeneity of an RNA phage population. Cell. 1978;13:735–744. doi: 10.1016/0092-8674(78)90223-4. [DOI] [PubMed] [Google Scholar]
- 73.Drake J.W., Holland J.J. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. U.S.A. 1999;96:13910–13913. doi: 10.1073/pnas.96.24.13910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Escarmis C., Davila M., Charpentier N., Bracho A., Moya A., Domingo E. Genetic lesions associated with Muller's ratchet in an RNA virus. J. Mol. Biol. 1996;264:255–267. doi: 10.1006/jmbi.1996.0639. [DOI] [PubMed] [Google Scholar]
- 75.Escarmis C., Davila M., Domingo E. Multiple molecular pathways for fitness recovery of an RNA virus debilitated by operation of Muller's ratchet. J. Mol. Biol. 1999;285:495–505. doi: 10.1006/jmbi.1998.2366. [DOI] [PubMed] [Google Scholar]
- 76.Escarmis C., Gomez-Mariano G., Davila M., Lazaro E., Domingo E. Resistance to extinction of low fitness virus subjected to plaque-to-plaque transfers: diversification by mutation clustering. J. Mol. Biol. 2002;315:647–661. doi: 10.1006/jmbi.2001.5259. [DOI] [PubMed] [Google Scholar]
- 77.Rosenberger R.F., Hilton J. The frequency of transcriptional and translational errors at nonsense codons in the lacZ gene of Escherichia coli. Mol. Gen. Genet. 1983;191:207–212. doi: 10.1007/BF00334815. [DOI] [PubMed] [Google Scholar]
- 78.Guimaraes J.C., Rocha M., Arkin A.P. Transcript level and sequence determinants of protein abundance and noise in Escherichia coli. Nucleic Acids Res. 2014;42:4791–4799. doi: 10.1093/nar/gku126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.