


Abstract
Mutation rates are used to calibrate molecular clocks and to link genetic variants with human disease. However, mutation rates are not uniform across each eukaryotic genome. Rates for insertion/deletion (indel) mutations have been found to vary widely when examined in vitro and at specific loci in vivo. Here, we report the genome-wide rates of formation and repair of indels made during replication of yeast nuclear DNA. Using over 6000 indels accumulated in four mismatch repair (MMR) defective strains, and statistical corrections for false negatives, we find that indel rates increase by 100 000-fold with increasing homonucleotide run length, representing the greatest effect on replication fidelity of any known genomic parameter. Nonetheless, long genomic homopolymer runs are overrepresented relative to random chance, implying positive selection. Proofreading defects in the replicative polymerases selectively increase indel rates in short repetitive tracts, likely reflecting the distance over which Pols δ and ϵ interact with duplex DNA upstream of the polymerase active site. In contrast, MMR defects hugely increase indel mutagenesis in long repetitive sequences. Because repetitive sequences are not uniformly distributed among genomic functional elements, the quantitatively different consequences on genome-wide repeat sequence instability conferred by defects in proofreading and MMR have important biological implications.
INTRODUCTION
Insertions and deletions of bases into DNA (indels) are on average more deleterious than base pair substitutions. Studies performed over many years have provided important insights into the mechanisms by which indels are generated (reviewed in (1)). One particularly well-known mechanism involves slippage of the two DNA strands during replication of repetitive sequences (2). This slippage can generate mismatches with one or more extra bases in the primer strand that can result in insertions, or one or more uncopied bases in the template strand that can result in deletions. The rate at which indels result from replication infidelity depends on the initial probability of forming misaligned intermediates, on the efficiency of proofreading of indel mismatches during replication, and on the efficiency of mismatch repair (MMR) of misaligned intermediates that escape proofreading (3).
Our understanding of the contributions of each of these processes to indel rates has largely been derived from studies that monitor indels in small DNA targets that do not capture the sequence diversity of whole genomes. A broader picture is now being obtained by mutation accumulation studies that examine whole genomes by deep sequencing. For example, studies of haploid Saccharomyces cerevisiae strains with defects in MMR have described the locations of about 100 single base mutations in the 12 000 000 base pair nuclear genome (4–6). We too examined mutation accumulation in a MMR-defective haploid yeast strain, in this case encoding a mutator variant DNA Polymerase δ (Pol δ) (7). The pattern of mutations in that strain, in combination with earlier work at specific loci, supported the idea that Pol δ and DNA Polymerase α (Pol α) primarily synthesize the nascent lagging strand (8), whereas DNA Polymerase ϵ (Pol ϵ) primarily synthesizes the nascent leading strand (9).
In our initial whole genome study (7), and in a more recent study of haploid yeast that accumulated mutations during a greater number of generations (10), there was clear evidence for purifying selection against deleterious mutations. Because purifying selection could potentially bias interpretations of genome-wide instability, perhaps especially for highly deleterious indels, we have switched to the use of diploid yeast strains to examine replication fidelity across the yeast nuclear genome. Using this approach, we identified more than 35 000 base substitutions that accumulated in diploid yeast strains encoding wild type or mutator variants of Pols α, δ or ϵ, each of which was either proficient or deficient in MMR (11). That analysis provided a broad view of replication fidelity and the efficiency of MMR for single base-base mismatches that result in base substitutions. The same study also identified >6000 indel mutations. Here, we analyze the rates, distributions and sequence contexts of these indels, in order to better understand the mechanisms of formation of indel mismatches during replication of the yeast nuclear genome and the consequences of defects in their removal by proofreading and MMR.
MATERIALS AND METHODS
Yeast strains, strain construction, mutation accumulation experiments, genomic DNA preparation, Illumina library preparation, genome sequencing, reference genome assembly, genomic feature selection, variant base pair calling, nucleosome mapping, determining the magnitude of selective pressure, mutable motif detection and multiple hypothesis testing all proceeded as previously described (11). Indel rates are calculated as described (11), but with target size set by counts of repeat tracts of specific length and adjusted for estimated false negative rates (see below).
Calculating the number of expected repeats across the genome
In percolation theory, K(p) is the mean run count per site or mean run density (12), where a run of length s (an s-run) is an isolated group of s consecutive characters that individually occur with probability p within a larger character string of length n. The mean run density is calculated from the total number of runs, Rn, which, ignoring boundaries, satisfies
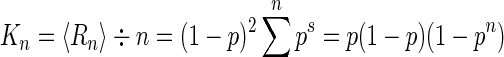 |
and
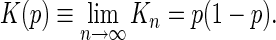 |
The mean expected run count, Rs, for a given run length is
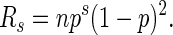 |
For ‘runs’ where the repeated unit is a set of m characters, if n >> ms, then this method may be generalized by redefining p and Rs as
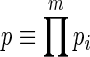 |
and
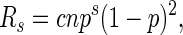 |
where c adjusts the probability for the number of patterns within a repeat class after accounting for circular permutations and complementary sequences.
For example, the expected number of Class 201 repeats of exactly 2 units (AT2 or TA2; c = 2) across the yeast genome (given 38% GC content, thus 31% each for A and T) is
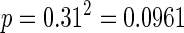 |
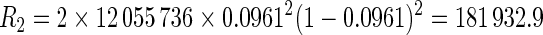 |
Estimating the rate of false negative insertion and deletion calls
Sequencing reads that map to the reference genome such that they overlap but do not cross all of a given homopolymer sequence (HP) are included in local coverage calculations but provide no information on changes in HP length. The longer an HP and the shorter a read, the more likely it is that the read will push the apparent allelic fraction of an insertion or deletion event (indel) within the HP below filter thresholds. One solution to this is to lower filter thresholds, but this would also result in a higher false positive rate. An alternate solution is to accept the false negative rate while accounting for it in indel rate calculation for HPs of particular lengths.
If L is the HP length, D is the average coverage depth, R is the length of a read, and C is the lower allelic fraction cutoff, then the false negative rate, B(L;D,p), may be calculated from a binomial cumulative distribution function
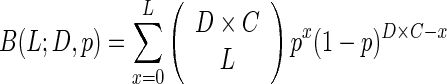 |
where p is the fraction of reads that cross the entire HP,
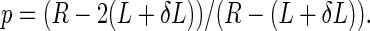 |
Indel rates are corrected by simply dividing by 1 − B(L;D,p). For example, the estimated false negative rate for 14 bp tracts is 0.797, suggesting that 79.7% of deletions in A14 tracts will not be detected due to random chance (Figure 1E). Deletion rates in A14 tracts are thus divided by 0.203 (or multiplied by 4.92) to correct for the missing population.
Figure 1.
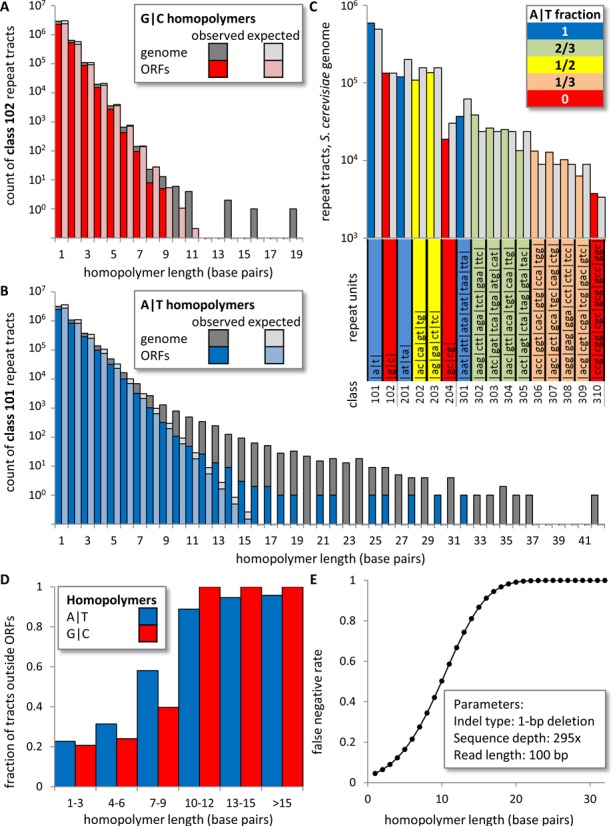
Repeat classes and counts. (A) Observed and expected counts of G/C homopolymers within the L03 reference genome, relative to open reading frames (ORFs) Observed (dark gray) and expected (light gray) counts in the genome are overlaid with observed (red) and expected (pink) counts in ORFs. (B) As per (A), but for A/T homopolymers with observed and expected counts in ORFs in dark and light blue, respectively. (C) Observed (colored) and expected (light gray) counts for the sixteen classes of homopolymers and di- and trinucleotide repeats in the reference genome. Class numbers and repeated units are tabulated beneath. Expected counts were derived from the overall GC-content of the genome via percolation theory (s-runs; see ‘Materials and Methods’ section). (D) The fraction of A/T (dark blue) and G/C (red) homopolymers (by length in bp) found outside of ORFs. Most short homopolymers are located within ORFs, while nearly all long homopolymers are found outside of ORFs. (E) Estimated false negative rates for single-base deletions in homopolymers at a mean sequencing depth of 295× with 100 bp reads (see ‘Materials and Methods’ section).
RESULTS
Collecting mutations and determining mutation rates
The indels analyzed here were identified in an earlier study (11) of MMR+ and MMR− yeast strains encoding either wild-type replicases or variant alleles of Pol α (pol1-L868M), Pol δ (pol3-L612M) or Pol ϵ (pol2-M644G). Multiple clonal isolates of these eight strains were passaged on solid media, their genomes were sequenced, and mutations were identified by comparison to a reference genome for each strain. Sequencing of MMR− pol2-M644G genomes at different passage numbers confirmed that mutation counts increased linearly with passage number, indicating minimal purifying selection against deleterious mutations. Based on the total number of generations over which mutations accumulated (Table 1), indel counts were used to calculate mutation rates per base per generation. Rates in MMR− strains define the rate at which the DNA replication fork generates indel mismatches, and the ratio of rates in MMR− and MMR+ strains estimates MMR efficiency.
Table 1. Insertion and deletion counts.
| Polymerase variant | Wild type | L868M Pol α | L612M Pol δ | M644G Pol ϵ | ||||
|---|---|---|---|---|---|---|---|---|
| MMR status | + | − | + | − | + | − | + | − |
| Isolates | 8 | 5 | 6 | 7 | 8 | 4 | 8 | 6 |
| Passages | 240 | 142 | 180 | 178 | 240 | 90 | 240 | 158 |
| Generations | 7200 | 4260 | 5400 | 5340 | 7200 | 2700 | 7200 | 4740 |
| Mutations | Count | Count | Count | Count | Count | Count | Count | Count |
| −A or –T | 0 | 955 | 0 | 934 | 2 | 1382 | 3 | 1375 |
| −C or –G | 0 | 11 | 0 | 56 | 0 | 195 | 2 | 60 |
| +A or +T | 0 | 46 | 0 | 112 | 1 | 244 | 1 | 168 |
| +C or +G | 0 | 5 | 0 | 6 | 0 | 49 | 0 | 17 |
| >1-bp delete | 0 | 135 | 0 | 55 | 0 | 42 | 0 | 142 |
| >1-bp Insert | 0 | 8 | 0 | 10 | 0 | 3 | 0 | 11 |
Adapted from (11).
Target size analysis
We began by tallying the number of A/T and G/C base pairs in the yeast genome that are present as non-iterated bases and in repetitive sequences of different composition and length (Figure 1, Supplementary Table S1). This information was used to calculate indel rates per base pair per generation for the different types of indels identified by whole genome sequencing.
Types and number of indels
Among 30 genomes sequenced from four MMR+ strains, only nine indels were identified, seven single base deletions and two single base insertions, all from strains with Pol δ and Pol ϵ mutator variants (11). This result contrasts sharply with the 6020 indels identified in the four polymerase backgrounds among 22 genomes sequenced from MMR− strains (Table 1, adapted from (11)). The vast majority of those indels were the loss or gain of a single base pair (Table 1). The number of deletions far exceeds the number of insertions, and the number of deletions and insertions is much higher for A/T as compared to G/C base pairs. Deletions and insertions of two or more base pairs were also observed (listed in Supplementary Table S2 and complied by class in Supplementary Table S3). Here again, deletions exceeded insertions and more events involved A/T as compared to G/C base pairs.
Single-base indel rates
Among 5615 total single-base indels in the genomes of MMR− strains (Table 1), all but 79 occurred in homonucleotide runs. From their locations and the target size information (Figure 1), we calculate that indel rates increase by more than 100 000-fold as the number of consecutive base pairs in a homopolymeric run increases (Figure 2A–D). Interestingly, many more indels involve A/T pairs as compared to G/C pairs (Table 1). However, the number of runs of A/T pairs in the genome (Figure 1B and C) greatly exceeds the number of runs of G/C pairs (Figure 1A and C). As but one example, the yeast genome contains 788 runs of 10 A/T base pairs but only six runs of 10 G/C base pairs. Thus, after correcting for differences in target size at each run length, single base indel rates per G/C base pair replicated per generation (Figure 2B) are as high, and occasionally exceed, indel rates for A/T base pairs (Figure 2A). For A/T pairs, rates for deletions (Figure 2A) generally exceed rates for insertions (Figure 2C), in some cases by up to 100-fold. For G/C base pairs, rates for deletions (Figure 2B) and insertions (Figure 2D) also vary, but are generally more similar to each other. Single base indel rates increase with homonucleotide run length in all four replicase backgrounds (colored lines). Compared to the strain with wild type replicases, strains with variant replicases have indel rates that are selectively elevated in shorter as compared to longer runs (Figure 2A–D). This pattern is evident in Figure 2E and F, which display the ratios of rates for the variant versus wild type replicases for deletion (panels A/B) or insertion (panels C/D) of A/T and G/C base pairs as a function of run length in the mismatch repair defective strains.
Figure 2.
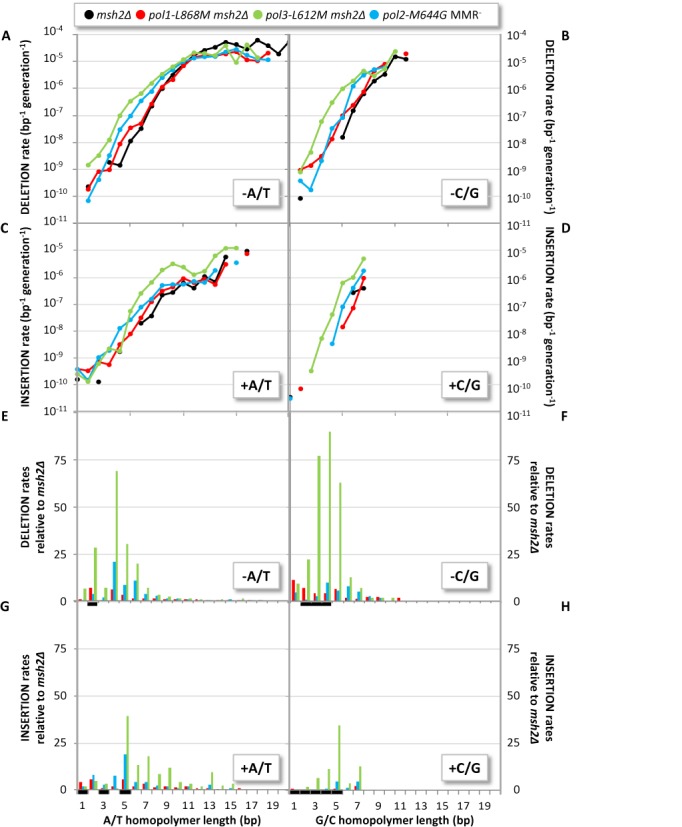
Effects of run length and polymerase variant on single base indel rates. (A–D) Single-base deletion (A and B) and insertion (C and D) rates within A/T (A and C) and G/C (B and D) homopolymers (by length in bp), colored by genotype: msh2Δ (black); pol1-L868M msh2Δ (red); pol3-L612M msh2Δ (green); and pol2-M644G MMR− (blue). All rates take into account the number of homopolymers of each type and length (Figure 1) and are adjusted to account for false negative rates (‘Materials and Methods’ section). Note some pol2-M644G MMR− isolates are pol2-M644G msh3/6Δ rather than pol2-M644G msh2Δ, but that the rates and spectra of these two genotyped are indistinguishable. (E–H) As per (A–D), but with the vertical axis indicating fold increase relative to the msh2Δ strain. Black bars indicate homopolymer types where no events were observed in the msh2Δ strain. There the vertical bars in (E–H) represent the lower bounds of fold rate increases, estimated as the ratio given one hypothetical msh2Δ event.
Multi-base indel rates
In the four MMR− strains, a total of 406 mutations involved deleting or inserting multiple base pairs (Additional files 2 and 3). Among these, 304 mutations involved deleting multiple A/T pairs from homonucleotide runs, 276 of which were two-base deletions. Based on target size analysis (Figure 1), we calculated rates per base pair per generation as a function of run length for these two-base deletions (Figure 3A). At most run lengths, rates are substantially lower for loss of two A/T base pairs (Figure 3A) as compared to loss of one A/T base pair (Figure 2A). The genome also contains many runs of dinucleotide repeats (Figure 1C). Among the four types of dinucleotide repeats, indels occurred in only two types, TA/AT and CA/TG (Additional files 2 and 3). Among these were 56 single-repeat-unit deletions, 47 of which were in TA/AT. When rates for these events were calculated as a function of increasing numbers of nucleotides in repeat units, the rates (Figure 3B) were substantially lower than rates for deleting one (Figure 2A) or even two A/T base pairs (Figure 3A) from homonucleotide runs containing an equivalent number of nucleotides. A large difference in rates is not limited to deletions from repeats containing A/T pairs. No dinucleotide deletions were observed in GC/CG repeats, despite their frequent presence in the yeast genome (Figure 1C) and despite the fact that rates for deleting single G/C base pairs (Figure 2B) are as high as for deleting single A/T base pairs (Figure 2A). Slippage of three nucleotides during replication of trinucleotide repeats is even less likely because, despite the fact that trinucleotide repeats make up >10% of the yeast genome (Figure 1C), deletions from trinucleotide repeat units were observed only three times (Supplementary Table S2) among more than 6000 indels. In comparison to multi-base deletions, far fewer multi-base insertions were observed. Nonetheless, two base pair insertions into homonucleotide and di-nucleotide runs of A/T base pairs were observed in numbers sufficient to estimate some rates (Figure 3C and D).
Figure 3.
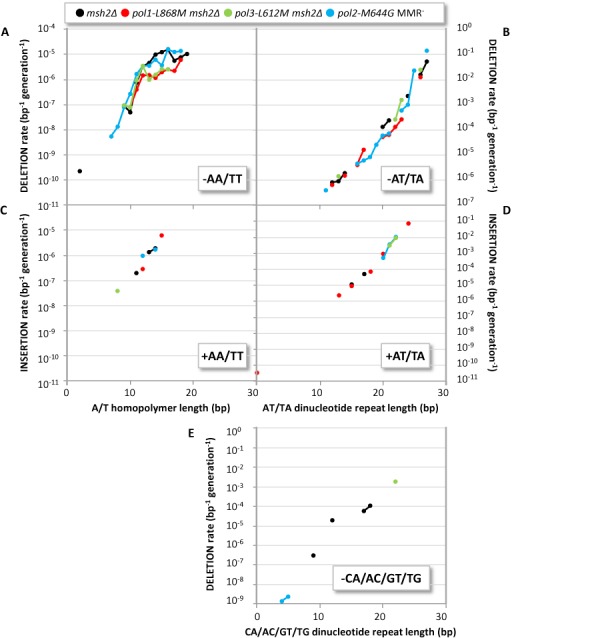
Multi-base indels rates versus repeat length. Multi-base deletion (A, B and E) and insertion (C and D) rates within A/T homopolymers (A and C) and in dinucleotide repeat tracts (AT/TA in B and D, CA/AC/GT/TG in E), colored by genotype: msh2Δ (black); pol1-L868M msh2Δ (red); pol3-L612M msh2Δ (green); and pol2-M644G MMR− (blue). All rates take into account the number of repeat tracts of each type and length (Figure 1 and Supplementary Figure S1) and are adjusted to account for false negative rates (‘Materials and Methods’ section).
DISCUSSION
The observation that replication error rates for single base indels strongly increase across the whole genome as the number of consecutive base pairs in a homonucleotide run increases supports earlier results at a single locus (13) or in a few repeat tracts (14), and previous mutation accumulation studies involving much smaller numbers of indels (4–6). The underlying mechanism for this relationship is undoubtedly DNA strand slippage during replication, as originally proposed (2) and supported by substantial evidence since then (reviewed in (1) and see (6)). In the absence of MMR, single base deletion rates per base replicated are 10−5 in long runs but only 10−10 for non-iterated base pairs (Figure 2A–D). This 100 000-fold difference in the rate of formation of a single base mutation depending on the local sequence in which it resides is the greatest DNA sequence context effect on replication fidelity of any known genomic parameter.
Single base deletion rates are higher than insertion rates, with a generally greater differential between deletions and insertions seen for runs of A/T as compared to G/C base pairs (Figure 2A–D). A high deletion to insertion ratio is also observed during synthesis in vitro by DNA polymerases (reviewed in (3)). This bias has been rationalized by the idea that, for a run of any particular length, base–base hydrogen bonding must be disrupted for one more base pair in order to create an insertion mismatch with an unpaired base in the primer strand as compared to a deletion mismatch with an unpaired base in the template strand (see Figure 2 in (15)). The difference in the amount of energy required would be more than sufficient to explain the single base deletion to insertion ratios observed here (Figure 2A and B versus Figure 2C and D), which vary from a few-fold to ∼100-fold (e.g. in runs of 12 A/T base pairs). Differences in polymerase interactions with unpaired bases in the template versus primer strand (reviewed in (16), and see Figure 2E–H) may also contribute to a deletion bias.
Many more single base indels involved A/T as compared to G/C base pairs in the genome as a whole (Table 1). However, the yeast genome contains far more (Figure 1A), and on average longer runs of A/T (Figure 1B) as compared to G/C base pairs (Figure 1C). When these different target sizes are taken into account, indel rates per base pair per generation are similar and sometimes slightly higher in runs of G/C base pairs than in runs of A/T base pairs of corresponding lengths (Figure 2A and B versus Figure 2C and D). The similarity in rates in G/C versus A/T runs is not necessarily anticipated, because strand slippage to create misaligned intermediates requires disruption of hydrogen bonds between correct base pairs, which logically should be more difficult for G/C base pairs as compared to A/T base pairs. The similarities in rates thus imply that strand slippage errors during replication is not only limited thermodynamically, but also kinetically and/or structurally. Kinetic and/or structural effects are also suggested by numerous differences in indel rates in the replicase variant strains (Figures 2 and 3). Overall, these observations reveal that DNA sequence context effects on single base indel rates during replication in vivo vary by polymerase, by the strand containing the unpaired base, and by the location of the unpaired base within a run relative to the polymerase active site (Figure 4, discussed below).
Figure 4.
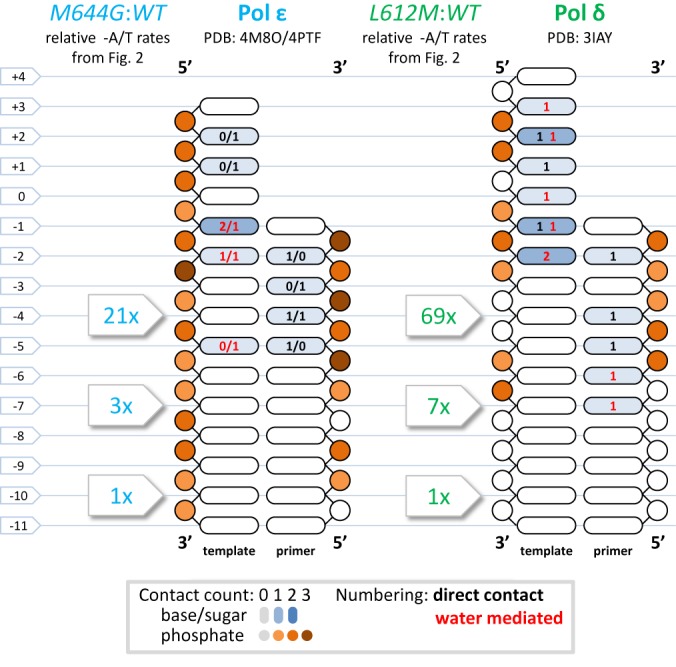
DNA footprints of Polymerases δ and ϵ active sites. Schematic plots comparing fold increases in A/T deletion rates, relative to the msh2Δ strain (Figure 2E), to DNA oligonucleotides bound to the catalytic sites of DNA Polymerases δ and ϵ in extant crystal structures (color coded by protein–DNA contact count, location, and type; Protein Data Bank codes are noted on the figure). Fold rate increases peak in 4 bp homopolymers and then fall to approximately msh2Δ levels as homopolymer lengths increase to 10 bp or more, where slippage loops pass beyond the range of proofreading exonuclease domains.
Rates for deleting two A/T pairs from homonucleotide runs are also high (Figure 3A). Interestingly, Monte Carlo simulations suggest that the rates for these two-base deletions are not significantly different from what would be expected for sequential accumulation of two, single-base deletions over multiple generations. This suggests that the two-base deletions in A/T runs result from two independent, sequential slippage events rather than from slippage of two bases in a single polymerization cycle. This differs from the situation for two-base deletions in TA/AT dinucleotide repeats (Figure 3B), which are most simply explained by slippage of a single, two-base repeat unit in one replication cycle. Interestingly, unlike the higher rates of single base deletions as compared to additions discussed above, rates for deleting an TA/AT dinucleotide repeat (Figure 3B) are similar to rates of inserting an TA/AT dinucleotide repeat (Figure 3D). This observation may be related to the fact that indels involving TA/AT dinucleotides primarily occur in longer repeats. Two consecutive misaligned bases may be able to reside about equally well in either the template or primer strand if they are far removed from the polymerase active site, as is possible in runs of 10 or more dinucleotide repeats (Figure 3B and D). It is also interesting that, although the yeast genome contains many other runs of di- and trinucleotide repeats (Figure 1), indels were predominantly detected in TA/AT repeats, with only a few occurrences in CA/TG repeats (Additional files 2 and 3), none in GC/CG repeats, and only three occurrences in trinucleotide repeats. Thus, for repetitive sequences containing equivalent numbers of repeat units, the rate of slippage of two or three consecutive template strand bases in one replication cycle is much lower than the rate of slippage of a single nucleotide, and rates differ for repeat units of different base composition.
To place the likelihood of slippage of multiple bases during replication into perspective, consider that in the msh2Δ strain, the average deletion rate per base per generation in TA/AT dinucleotide repeats is 2.6 × 10−9. This rate is similar to the rate (2.4 × 10−9) at which the very common T-dG mismatch is generated (11)). Remarkably, when MMR is defective, indel rates per base per generation exceed 10−2 in sequences containing many consecutive TA/AT repeats (Figure 3B and D), far higher than the rates of any and all mutation classes measured in the URA3 reporter gene in similar strains (17). This extraordinary rate in long repeats in MMR-defective strains, and the fact that only nine total single base indels were observed in the MMR+ strains (Table 1), illustrates the critical role of MMR in protecting the genome against indel mismatches generated during replication in a wide range of different repetitive sequences. Our data in yeast quantitatively illustrate why microsatellite instability, which is clinically monitored in very long repetitive sequences (reviewed in (18); revised Bethesda guidelines (19)), is such a strong signature of MMR-defective tumors (discussed in (1)).
Interestingly, two of the five A/T deletions that were observed in MMR+ strains were of non-iterated base pairs. This contrasts sharply with the fact that in MMR− strains, >99.9% of A/T deletions were in homonucleotide runs. Thus, the genome-wide average apparent MMR efficiency for removing A/T base pair deletion mismatches from runs is 24 000-fold, while the genome-wide average for non-iterated A/T base pairs is only 40-fold, a difference of 600-fold. This large difference may reflect intrinsically more efficient MMR of single base deletion mismatches in runs. A non-exclusive hypothesis is that some single base deletions of non-iterated sequences may arise via an alternative mechanism, such as during bypass of DNA lesions. If that occurred during replication, the presence of the lesion could reduce MMR efficiency. If the mismatch arose outside the context of normal replication, e.g. during post-replicative gap-filling, MMR might be unavailable for repair.
Compared to the strain with wild type replicases, the strains encoding variant replicases have indel rates that are selectively elevated in shorter runs (Figure 2E–H). The differences are most evident in the pol3-L612M strain (Figure 2E–H), and they gradually decrease as the run length increases to 10 pairs. A similar trend is also apparent in the pol2-M644G strain (Figure 2E–H). We suggest that the preferentially higher rates in short runs are due to defective proofreading by L612M Pol δ and M644G Pol ϵ, despite the fact that their exonuclease active sites have not been perturbed. This is because proofreading efficiency depends on the balance between excising and extending a mismatch (reviewed in (3)), and compared to their wild-type parents, L612M Pol δ and M644G Pol ϵ are promiscuous for mismatch extension (9,20) the M644G Pol ϵ strain (but not the L612M Pol δ strain (21)), has slightly elevated dNTP pools (22) that will drive mismatch extension, has been shown to have reduced proofreading (23). Moreover, we previously showed that proofreading of single-base deletion mismatches in vitro decreases with increasing homonucleotide run length (see (24) and reviewed in (1)), and this relationship holds in vivo for runs of A/T base pairs at a specific location in yeast (13,25). This makes sense in light of the DNA ‘footprints’ seen in the crystal structures of Pols δ and ϵ (26–28). Both replicases make numerous contacts with the DNA backbone and the bases within five to seven base pairs of the active site (Figure 4). Beyond those base pairs, DNA contacts are fewer and only occur with the backbone. Thus an unpaired base within the first 5–7 bp of the active site should interfere with extension and tip the balance to excision more so than if the unpaired base is present further upstream. This logic rationalizes why proofreading provides little protection against indels in long runs (13,25), whereas MMR is hugely important. This logic also nicely explains why all four replicase backgrounds exhibit similar indel rates in long runs (Figure 2A–D), and it accounts for the preferentially higher indels rates in short runs in the pol3-L612M and pol2-M644G strains.
The preferential increases in indel rates in short runs conferred by defective proofreading implies that defective proofreading will selectively render coding sequences at risk of indel mutagenesis. This is because, as indicated by our target size analysis (Figure 1D and E), coding sequences contain a higher proportion of short repetitive sequences as compared to non-coding sequences. The prediction that defective proofreading will selectively render coding sequences at risk of indel mutagenesis can be examined in future studies of indel mutagenesis in tumors known to harbor mutations in Pols δ and ϵ ((29–35), reviewed in (36–38)). Our data predict that, just as indels in long repetitive sequences (microsatellite instability) are diagnostic for MMR defective tumors, indels in short repetitive sequences should be diagnostic of proofreading defects. In support of this prediction, the mutational footprint of a yeast strain encoding a mutator allele of ribonucleotide reductase (39) with elevated pyrimidine triphosphate concentrations is characterized by deletion hotspots in short runs, and these hotspots are nicely explained by enhanced extension of indel mismatches at the expense of proofreading. Elevating dNTP pools, either through mutations or following DNA damage (40), is but one of several mechanisms to reduce proofreading efficiency. Others include mutations that inactivate the 3′-exonuclease of a replicative DNA polymerase, mutations in polymerase domains that promote mismatch extension (as here) and mutations that prevent switching from the polymerase to the exonuclease active site (23,41).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Monica Frazier and Jordan St Charles for helpful comments on the manuscript.
FUNDING
Division of Intramural Research of the National Institutes of Health, National Institute of Environmental Health Sciences [Z01 ES065070 to T.K.]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1.Garcia-Diaz M., Kunkel T.A. Mechanism of a genetic glissando: structural biology of indel mutations. Trends Biochem. Sci. 2006;31:206–214. doi: 10.1016/j.tibs.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 2.Streisinger G., Okada Y., Emrich J., Newton J., Tsugita A., Terzaghi E., Inouye M. Frameshift mutations and the genetic code. This paper is dedicated to Professor Theodosius Dobzhansky on the occasion of his 66th birthday. Cold Spring Harb. Symp. Quant. Biol. 1966;31:77–84. doi: 10.1101/sqb.1966.031.01.014. [DOI] [PubMed] [Google Scholar]
- 3.Kunkel T.A. Evolving views of DNA replication (in)fidelity. Cold Spring Harb. Symp. Quant. Biol. 2009;74:91–101. doi: 10.1101/sqb.2009.74.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zanders S., Ma X., Roychoudhury A., Hernandez R.D., Demogines A., Barker B., Gu Z., Bustamante C.D., Alani E. Detection of heterozygous mutations in the genome of mismatch repair defective diploid yeast using a Bayesian approach. Genetics. 2010;186:493–503. doi: 10.1534/genetics.110.120105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma X., Rogacheva M.V., Nishant K.T., Zanders S., Bustamante C.D., Alani E. Mutation hot spots in yeast caused by long-range clustering of homopolymeric sequences. Cell Rep. 2012;1:36–42. doi: 10.1016/j.celrep.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lang G.I., Parsons L., Gammie A.E. Mutation rates, spectra, and genome-wide distribution of spontaneous mutations in mismatch repair deficient yeast. G3 (Bethesda) 2013;3:1453–1465. doi: 10.1534/g3.113.006429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Larrea A.A., Lujan S.A., Nick McElhinny S.A., Mieczkowski P.A., Resnick M.A., Gordenin D.A., Kunkel T.A. Genome-wide model for the normal eukaryotic DNA replication fork. Proc. Natl. Acad. Sci. U.S.A. 2010;107:17674–17679. doi: 10.1073/pnas.1010178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nick McElhinny S.A., Gordenin D.A., Stith C.M., Burgers P.M., Kunkel T.A. Division of labor at the eukaryotic replication fork. Mol. Cell. 2008;30:137–144. doi: 10.1016/j.molcel.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pursell Z.F., Isoz I., Lundstrom E.B., Johansson E., Kunkel T.A. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317:127–130. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serero A., Jubin C., Loeillet S., Legoix-Ne P., Nicolas A.G. Mutational landscape of yeast mutator strains. Proc. Natl. Acad. Sci. U.S.A. 2014;111:1897–1902. doi: 10.1073/pnas.1314423111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lujan S.A., Clausen A.R., Clark A.B., MacAlpine H.K., MacAlpine D.M., Malc E.P., Mieczkowski P.A., Burkholder A.B., Fargo D.C., Gordenin D.A., et al. Heterogeneous polymerase fidelity and mismatch repair bias genome variation and composition. Genome Res. 2014;24:1751–1764. doi: 10.1101/gr.178335.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finch S.R. Mathematical Constants. NY: Cambridge University Press; 2003. [Google Scholar]
- 13.Tran H.T., Keen J.D., Kricker M., Resnick M.A., Gordenin D.A. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol. Cell. Biol. 1997;17:2859–2865. doi: 10.1128/mcb.17.5.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wierdl M., Dominska M., Petes T.D. Microsatellite instability in yeast: dependence on the length of the microsatellite. Genetics. 1997;146:769–779. doi: 10.1093/genetics/146.3.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kunkel T.A. Misalignment-mediated DNA synthesis errors. Biochemistry. 1990;29:8003–8011. doi: 10.1021/bi00487a001. [DOI] [PubMed] [Google Scholar]
- 16.Bebenek K., Kunkel T.A. Streisinger revisited: DNA synthesis errors mediated by substrate misalignments. Cold Spring Harb. Symp. Quant. Biol. 2000;65:81–91. doi: 10.1101/sqb.2000.65.81. [DOI] [PubMed] [Google Scholar]
- 17.Lujan S.A., Williams J.S., Pursell Z.F., Abdulovic-Cui A.A., Clark A.B., Nick McElhinny S.A., Kunkel T.A. Mismatch repair balances leading and lagging strand DNA replication fidelity. PLoS Genet. 2012;8:e1003016. doi: 10.1371/journal.pgen.1003016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heinimann K. Toward a molecular classification of colorectal cancer: the role of microsatellite instability status. Front. Oncol. 2013;3:272. doi: 10.3389/fonc.2013.00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Umar A., Boland C.R., Terdiman J.P., Syngal S., de la Chapelle A., Ruschoff J., Fishel R., Lindor N.M., Burgart L.J., Hamelin R., et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst. 2004;96:261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nick McElhinny S.A., Stith C.M., Burgers P.M., Kunkel T.A. Inefficient proofreading and biased error rates during inaccurate DNA synthesis by a mutant derivative of Saccharomyces cerevisiae DNA polymerase delta. J. Biol. Chem. 2007;282:2324–2332. doi: 10.1074/jbc.M609591200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams J.S., Clausen A.R., Lujan S.A., Marjavaara L., Clark A.B., Burgers P.M., Chabes A., Kunkel T.A. Evidence that processing of ribonucleotides in DNA by topoisomerase 1 is leading-strand specific. Nat. Struct. Mol. Biol. 2015 doi: 10.1038/nsmb.2989. doi:10.1038/nsmb.2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nick McElhinny S.A., Kumar D., Clark A.B., Watt D.L., Watts B.E., Lundstrom E.B., Johansson E., Chabes A., Kunkel T.A. Genome instability due to ribonucleotide incorporation into DNA. Nat. Chem. Biol. 2010;6:774–781. doi: 10.1038/nchembio.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reha-Krantz L.J. DNA polymerase proofreading: multiple roles maintain genome stability. Biochim. Biophys. Acta. 2010;1804:1049–1063. doi: 10.1016/j.bbapap.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 24.Kroutil L.C., Register K., Bebenek K., Kunkel T.A. Exonucleolytic proofreading during replication of repetitive DNA. Biochemistry. 1996;35:1046–1053. doi: 10.1021/bi952178h. [DOI] [PubMed] [Google Scholar]
- 25.Roberts J.D., Nguyen D., Kunkel T.A. Frameshift fidelity during replication of double-stranded DNA in HeLa cell extracts. Biochemistry. 1993;32:4083–4089. doi: 10.1021/bi00066a033. [DOI] [PubMed] [Google Scholar]
- 26.Hogg M., Osterman P., Bylund G.O., Ganai R.A., Lundstrom E.B., Sauer-Eriksson A.E., Johansson E. Structural basis for processive DNA synthesis by yeast DNA polymerase varepsilon. Nat. Struct. Mol. Biol. 2014;21:49–55. doi: 10.1038/nsmb.2712. [DOI] [PubMed] [Google Scholar]
- 27.Jain R., Rajashankar K.R., Buku A., Johnson R.E., Prakash L., Prakash S., Aggarwal A.K. Crystal structure of yeast DNA polymerase epsilon catalytic domain. PLoS One. 2014;9:e94835. doi: 10.1371/journal.pone.0094835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swan M.K., Johnson R.E., Prakash L., Prakash S., Aggarwal A.K. Structural basis of high-fidelity DNA synthesis by yeast DNA polymerase delta. Nat. Struct. Mol. Biol. 2009;16:979–986. doi: 10.1038/nsmb.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Briggs S., Tomlinson I. Germline and somatic polymerase epsilon and delta mutations define a new class of hypermutated colorectal and endometrial cancers. J. Pathol. 2013;230:148–153. doi: 10.1002/path.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Church D.N., Briggs S.E., Palles C., Domingo E., Kearsey S.J., Grimes J.M., Gorman M., Martin L., Howarth K.M., Hodgson S.V., et al. DNA polymerase epsilon and delta exonuclease domain mutations in endometrial cancer. Hum. Mol. Genet. 2013;22:2820–2828. doi: 10.1093/hmg/ddt131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rohlin A., Zagoras T., Nilsson S., Lundstam U., Wahlstrom J., Hulten L., Martinsson T., Karlsson G.B., Nordling M. A mutation in POLE predisposing to a multi-tumour phenotype. Int. J. Oncol. 2014;45:77–81. doi: 10.3892/ijo.2014.2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshida R., Miyashita K., Inoue M., Shimamoto A., Yan Z., Egashira A., Oki E., Kakeji Y., Oda S., Maehara Y. Concurrent genetic alterations in DNA polymerase proofreading and mismatch repair in human colorectal cancer. Eur. J. Hum. Genet. 2011;19:320–325. doi: 10.1038/ejhg.2010.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.TCGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kandoth C., Schultz N., Cherniack A.D., Akbani R., Liu Y., Shen H., Robertson A.G., Pashtan I., Shen R., Benz C.C., et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palles C., Cazier J.B., Howarth K.M., Domingo E., Jones A.M., Broderick P., Kemp Z., Spain S.L., Guarino E., Salguero I., et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013;45:136–144. doi: 10.1038/ng.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heitzer E., Tomlinson I. Replicative DNA polymerase mutations in cancer. Curr. Opin. Genet. Dev. 2014;24C:107–113. doi: 10.1016/j.gde.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henninger E.E., Pursell Z.F. DNA polymerase epsilon and its roles in genome stability. IUBMB Life. 2014;66:339–351. doi: 10.1002/iub.1276. [DOI] [PubMed] [Google Scholar]
- 38.Wheeler D.A., Wang L. From human genome to cancer genome: the first decade. Genome Res. 2013;23:1054–1062. doi: 10.1101/gr.157602.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar D., Abdulovic A.L., Viberg J., Nilsson A.K., Kunkel T.A., Chabes A. Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res. 2011;39:1360–1371. doi: 10.1093/nar/gkq829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chabes A., Georgieva B., Domkin V., Zhao X., Rothstein R., Thelander L. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell. 2003;112:391–401. doi: 10.1016/s0092-8674(03)00075-8. [DOI] [PubMed] [Google Scholar]
- 41.Jin Y.H., Garg P., Stith C.M., Al-Refai H., Sterling J.F., Murray L.J., Kunkel T.A., Resnick M.A., Burgers P.M., Gordenin D.A. The multiple biological roles of the 3′–>5′ exonuclease of Saccharomyces cerevisiae DNA polymerase delta require switching between the polymerase and exonuclease domains. Mol. Cell. Biol. 2005;25:461–471. doi: 10.1128/MCB.25.1.461-471.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.