

Abstract
Background
KLF5 is a basic transcriptional factor that regulates multiple physiopathological processes. Our recent study showed that deletion of Klf5 in mouse prostate promotes tumorigenesis initiated by the deletion of Pten. While molecular characterization of Klf5-null tumors suggested that angiogenesis was partially responsible for tumor promotion, the precise function and mechanism of KLF5 deletion in prostate tumor angiogenesis remain unclear.
Results
Applying histological staining to Pten-null mouse prostates, we observed that deletion of Klf5 significantly increased the number of microvessels, accompanied by the upregulation of multiple angiogenesis-related genes based on microarray analysis with MetaCore software. In human umbilical vein endothelial cells (HuVECs), tube formation and migration, both of which are indicators of angiogenic activities, were decreased by conditioned media from PC-3 and DU 145 human prostate cancer cells with KLF5 overexpression, but increased by media from cells with KLF5 knockdown. HIF1α, a key angiogenesis inducer, was upregulated by KLF5 loss at the protein but not the mRNA level in both mouse tissues and human cell lines, as determined by immunohistochemical staining, real-time RT-PCR and Western blotting. Consistently, KLF5 loss also upregulated VEGF and PDGF, two pro-angiogenic mediators of HIF1α function, as analyzed by immunohistochemical staining in mouse tissues and ELISA in conditioned media. Mechanistically, AKT activity, which caused the accumulation of HIF1α, was increased by KLF5 knockout or knockdown but decreased by KLF5 overexpression. PI3K/AKT inhibitors consistently abolished the effects of KLF5 knockdown on angiogenic activity, HIF1α accumulation, and VEGF and PDGF expression.
Conclusion
KLF5 loss enhances tumor angiogenesis by attenuating PI3K/AKT signaling and subsequent accumulation of HIF1α in PTEN deficient prostate tumors.
Electronic supplementary material
The online version of this article (doi:10.1186/s12943-015-0365-6) contains supplementary material, which is available to authorized users.
Keywords: KLF5, Angiogenesis, HIF1α, AKT, PTEN, Prostate cancer
Introduction
Angiogenesis, the process of forming new blood vessels from pre-existing vessels, is vital for development, tissue wound healing, and tumor initiation and progression [1]. Tumor angiogenesis is activated by multiple pro-angiogenic secretory factors including VEGF, PDGF-B, bFGF and TNF-α, which are all transcriptionally activated by hypoxia inducible factor 1 (HIF1) [2], composed of HIF1α and HIF1β subunits. HIF1α is mainly regulated at the protein level by the ubiquitin proteasome pathway, whose activation depends on the oxygen level, or by the activation of PI3K/AKT and MAPK signaling in a tumor, which is independent of oxygen level [2,3]. Targeting angiogenesis has been proposed as a therapeutic approach in prostate and other types of solid tumors for decades, and a number of candidate drugs have been tested in clinical trials. However, few of them have been officially approved for cancer treatment and their success has been limited [4]. A better understanding of the molecular basis of angiogenesis should improve the development of anti-angiogenesis-based cancer therapy.
KLF5 (Krüppel-like factor 5, also known as BTEB2) encodes a basic transcription factor expressed in different tissues to regulate diverse cellular processes [5]. In tumorigenesis, KLF5 has been shown to play a context-dependent role. On one hand, its genetic locus is frequently deleted in human cancers [5,6]; a tumor suppressor function has been established in mouse models where KLF5 expression suppresses tumor growth [7,8] and deletion of Klf5 in mouse prostates promotes tumorigenesis initiated by the deletion of Pten [9]. On the other hand, interruption of KLF5 acetylation converts its function from that of a tumor suppressor to a tumor promoter, and multiple oncogenic pathways appear to be involved [8]. Recent studies also suggest that KLF5 could regulate angiogenesis. KLF5 is markedly induced in activated vascular smooth muscle cells and fibroblasts, deletion of Klf5 in mice compromises vascular remodeling involving Pdgf-a [10], and loss of Klf5 in mouse cornea epithelial cells results in abnormal neovascularization with elevated expression of angiogenesis-related genes [11,12]. In our recent study [9], we found that deletion of Klf5 in Pten-null mouse prostates resulted in larger tumors without lumens and necrosis, which were present in tumors induced by Pten deletion alone [13] but with enhanced activation of AKT and ERK, which are reported to promote tumor angiogenesis [14,15]. It is thus possible that KLF5 modulates angiogenesis during prostatic tumorigenesis.
In this study, we tested whether and how KLF5 regulates angiogenesis in the context of PTEN loss in prostate cancer. We found that in Pten-null mouse prostate tumors, deletion of Klf5 significantly promoted angiogenesis, and conditioned media from human prostate cancer cells with modulated KLF5 expression affected tube formation and migration of human umbilical vein endothelial cells (HuVECs). Expression profiling and other analyses demonstrated that Klf5 deletion led to the upregulation of multiple pro-angiogenic genes and accumulation of HIF1α, a transcription factor that stimulates angiogenesis. Additional experiments showed that accumulation of HIF1α induced by KLF5 loss depended on the activation of the PI3K/AKT signaling pathway. These findings suggest that KLF5 loss promotes tumor angiogenesis by enhancing PI3K/AKT signaling and the subsequent accumulation of HIF1α in PTEN deficient prostate cancer.
Results
Klf5 deletion promotes angiogenesis initiated by Pten deletion in mouse prostate tumors
To test whether Klf5 deletion plays a role in tumor angiogenesis, we first examined H&E stained tissue sections for the number of intraepithelial blood vessels, indicated by histological appearance and the presence of red blood cells between wildtype tissues and those with Klf5 deletion. We used dorsal prostate tumors of 8 month old mice for the Pten-null group, and whole prostate with high grade mPINs of 12 to 18 month old mice for the Pten hemizygous group. The number of microvessels was clearly increased by homozygous deletion of Klf5 in both Pten groups (Figure 1A, 1B upper panels). To more accurately determine the number of microvessels in mPINs and prostate tumors, we performed immunohistochemical staining for the Cd31 endothelial cell marker and counted the number of microvessels indicated by positive Cd31 staining (Figure 1A, B, lower panels). The density of microvessels, determined by dividing the number of Cd31-marked microvessels by the total area of epithelial cells, was significantly increased by both hemizygous and homozygous deletions of Klf5 in the Pten-null tumors and by Klf5 homozygous deletion in the Pten-hemizygous group (Figure 1, bar figures). The increase in angiogenesis correlated with the increase in tumor mass and the decrease in Klf5 expression caused by Klf5 deletion in the same mice [9]. This result indicates that Klf5 deletion promotes angiogenesis in both prostate tumors and mPINs induced by Pten deletion.
Figure 1.
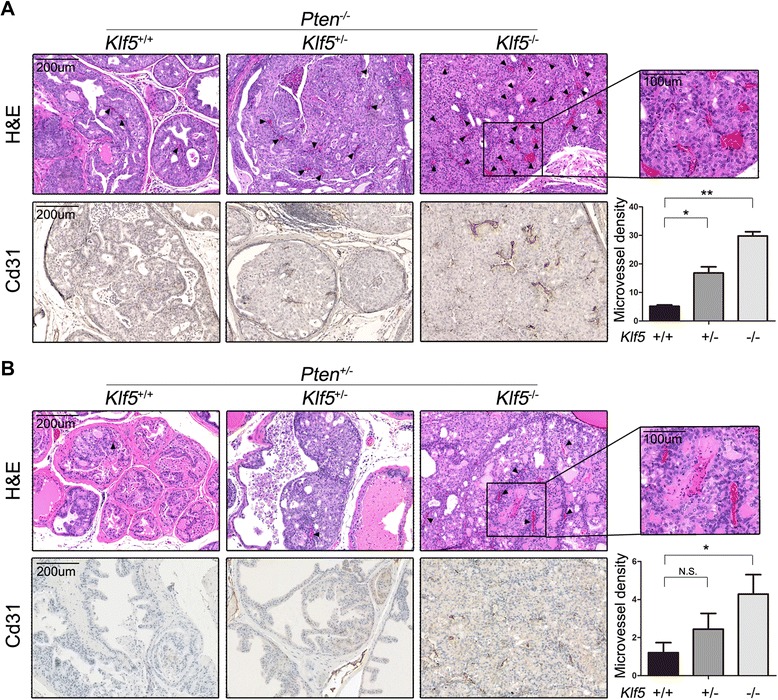
Klf5 deletion increases blood microvessels in mouse prostate tumors. H&E staining and IHC staining of Cd31 for blood microvessels from prostates at 8 months for Pten −/− (A) and 12 to 18 months for Pten +/− (B). Black arrows point to microvessels inside prostatic acini. Images at the right upper corner are higher magnifications of boxed areas. Microvessels were counted in dorsal lobes for Pten −/− prostates and the entire lobes for Pten +/− prostates. The total area of prostatic epithelia excluding the lumen space inside the acini was measured with ImageJ software. Gene status is indicated at the top of each panel, and + and – indicate wildtype and deletion of Klf5 or Pten, respectively. Three to 6 mice were used for each group. Microvessel density was defined by the number of microvessels per mm2. *, P < 0.05; **, P < 0.01; N.S., not statistically significant.
Klf5 deletion activates the angiogenesis transcriptional network and HIF1α in Pten-null mouse prostates
In our recent study, expression profiling and MetaCore analysis of mouse dorsal prostates at 6 months in the Pten-null background indicated that the process of blood morphogenesis is the most enriched upon the deletion of Klf5 [9], composed of 39 differentially expressed genes. We searched the PubMed database for articles on each of the 39 genes, and reviewed these articles to determine whether the genes are pro- or anti-angiogenic. Among the 39 genes, 33 (84.6%) were reported to promote, 3 (7.7%) to suppress, and 3 (7.7%) to have unclear effects on angiogenesis. Twenty six (79%) of the 33 pro-angiogenic genes were significantly upregulated by Klf5 deletion. The 36 genes with a promoting or repressing effect are listed in Additional file 1: Table S1, along with their expression fold changes caused by Klf5 deletion, and a heatmap of these genes is shown in Figure 2A. Dysregulation of these genes indicates an enhanced angiogenesis transcription network resulting from Klf5 deletion.
Figure 2.
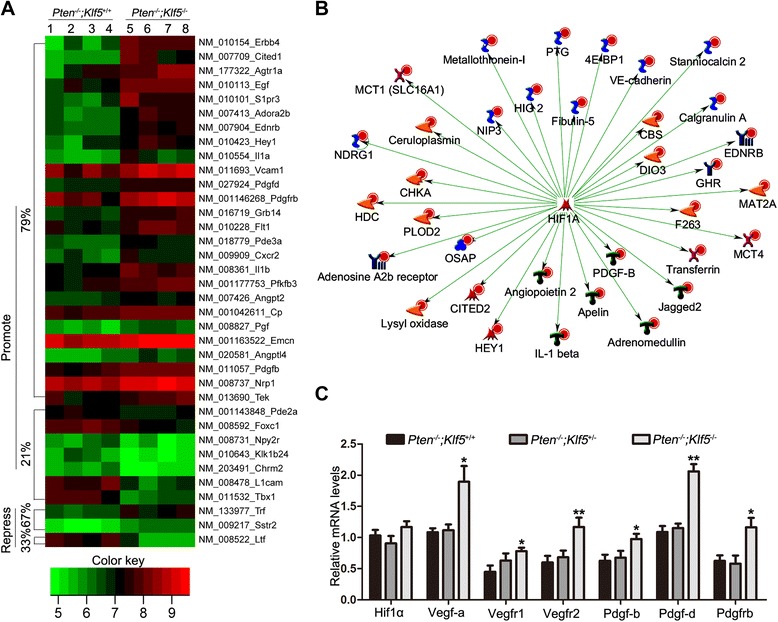
Klf5 deletion dysregulates angiogenesis-related genes and enhances HIF1α transcriptional activity in Pten-null mouse prostates. (A) Heatmap of genes involved in angiogenesis based on the MetaCore analysis and PubMed publications and that had >1.5 fold change in expression between wildtype and Klf5-null mouse prostates (4 samples per group, indicated at the top). Genes are clustered by their effects on angiogenesis, with those promoting and repressing angiogenesis marked as “promote” and “repress”, respectively. Color intensities in the heat map represent log2 values of expression intensities. The percentage of upregulated or downregulated genes is shown. (B) HIF1α is activated by Klf5 deletion, as indicated by the upregulation of its direct transcriptional target genes based on MetaCore’s interactome analysis for transcription factors. Different shapes of the nodes represent functional classifications of genes. Red dots on the upper right corner of each symbol indicate increased expression level, and green lines indicate positive regulation. (C) Detection of Hif1α/Vegf and Pdgf signaling molecules by real-time RT-PCR in 6-month-old Pten-null mouse dorsal prostates with indicated Klf5 deletion status. Data for each genotype were from 8 mice.* and **indicate P < 0.05 and P < 0.01, respectively.
Based on the process indicative of active angiogenesis, we attempted to determine whether a specific pathway or a key transcription factor was activated by Klf5 deletion to drive the pro-angiogenic phenotype, using the MetaCore program. Interestingly, HIF1α, an important mediator of tumor angiogenesis [16], was identified as a key transcription factor (Figure 2B) with p-value 4.2 × 10−147, as it affected the expression of 115 of the differentially expressed genes. Among the 115 genes, 49 were identified as direct transcriptional targets activated by HIF1α, and 34 of the 49 genes (69%) were upregulated by Klf5 deletion (Figure 2B), suggesting an activated HIF1α transcription activity. VEGF and some of its related molecules are direct targets and functional mediators of HIF1α in angiogenesis [17,18], so we analyzed mRNA expression for several molecules of the HIF1α/VEGF signaling pathway, including Hif1α, Vegf-a, Vegfr1 and Vegfr2, by real-time RT-PCR using 8 mouse dorsal prostates for each genotype of Klf5 deletion in the Pten-null background. As shown in Figure 2C, the Hif1α mRNA level was unchanged, while Vegf-a, Vegfr1 and Vegfr2 mRNA levels were all increased by Klf5 homozygous deletion. Of these, Vegf-a was not identified by the microarray analysis. Klf5 deletion upregulated Pdgf-b, Pdgf-d and Pdgfrb mRNA expression in the microarray analysis (Figure 2A), suggesting an activated PDGF signaling pathway that could play an important role in angiogenesis [19]. Pdgf-b is also a transcriptional target of HIF1α [20] and could also be a functional mediator of HIF1α. Therefore, we examined mRNA expression of these PDGF signaling molecules by real-time RT-PCR, and confirmed the microarray analysis findings that Pdgf-b, Pdgf-d and Pdgfrb were upregulated by Klf5 deletion (Figure 2C). Hemizygous deletion of Klf5 had a mild effect on the expression of these molecules. These results indicate that Klf5 deletion activates multiple key regulators of angiogenesis.
We also examined protein expression of Hif1α, Vegf, Pdgf-b and Pdgf-d in prostates with different Klf5 deletion status by IHC staining. In Pten-null prostates, although the mRNA expression of Hif1α was not affected by Klf5 deletion (Figure 2), its protein expression was increased in both the nucleus and the cytoplasm of prostatic cells by both hemizygous and homozygous deletions of Klf5 (Figure 3A). In addition, almost all cells with Klf5 deletion were positive for Hif1α expression in the nucleus (Figure 3A), which indicates an active Hif1α status [3]. In the context of Pten hemizygous deletion, accumulation of Hif1α was increased upon Klf5 deletion in prostates of one year old mice, and homozygous deletion of Klf5 also significantly increased the ratio of cells with positive Hif1α nuclear staining (Figure 3B). For pro-angiogenic factors Vegf, Pdgf-b and Pdgf-d, whose mRNA expression was upregulated by Klf5 deletion (Figure 2A, C), an increase in their protein expression was also detected in Pten-null prostates with both hemizygous and homozygous deletions of Klf5 (Figure 3C). In prostates with Pten hemizygous deletion, only homozygous Klf5 deletion increased protein expression of these factors (Figure 3D). These results further indicate that Klf5 loss induces the enhanced expression and activity of Hif1α accompanied with augmented expression of pro-angiogenic factors.
Figure 3.
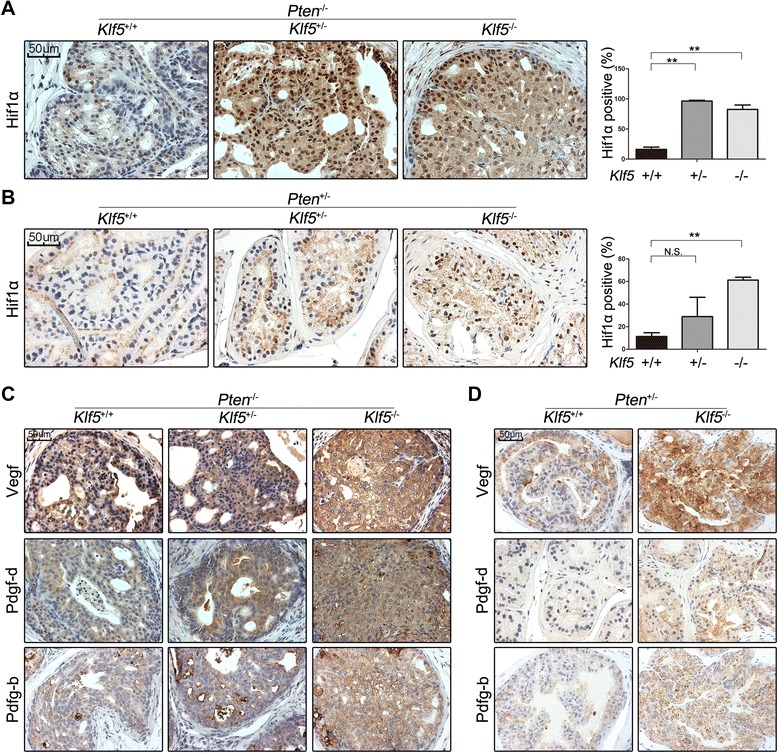
Klf5 deletion increases protein expression of Hif1α and pro-angiogenic factors. (A, B) IHC staining of Hif1α in mouse prostates at 4 months for Pten −/− and 12 months for Pten +/−. The numbers of Hif1α-positive cells and total prostatic epithelial cells were counted with ImageJ software for each mouse. (C, D) Protein expression of pro-angiogenic factors Vegf, Pdgf-d and Pdgf-b as detected by IHC staining with the same mouse tissues used in panels A and B. Three or 4 mice were used for each group. *, P < 0.05; **, P < 0.01; N.S., not statistically significant.
Dysregulation of KLF5 in prostate cancer cells impacts tube formation and migration of HuVECs
To further test the role of KLF5 in angiogenesis, we determined whether changes in KLF5 expression in human prostate cancer cells directly modulate endothelial cell behaviors such as tube formation and migration, both of which are indicative of angiogenesis. We used PC-3 (PTEN null) and DU 145 (PTEN haploinsufficient) human prostate cancer cell lines [21] and human umbilical vein endothelial cells (HuVECs) for the analysis. In vitro cultured cells eliminate any effects of tumor growth in vivo, thus providing evidence for a direct effect of KLF5 on angiogenesis. We first knocked down KLF5 using lentiviruses expressing shRNAs in PC-3 and DU 145 cells to mimic the KLF5 and PTEN status in the mouse model. Using stable cell populations with efficient knockdown of KLF5, we collected conditioned media and applied them to HuVECs for tube formation assay, in which HuVECs formed a tubular network of interconnecting branches after 5–7 hours. Knockdown of KLF5 in both PC-3 and DU 145 cell lines significantly increased the cumulative tube length and the number of tube nodes in HuVECs (Figure 4A). Considering that both PC-3 and DU 145 cell lines express a relatively lower level of KLF5 [22,23], we also overexpressed KLF5 by lentiviral infection in these two cell lines and performed HuVEC tube formation assay with conditioned media. Consistent with the knockdown result, ectopic expression of KLF5 significantly inhibited tube formation in vitro, as indicated by the cumulative tube length and the number of tube nodes (Figure 4B).
Figure 4.
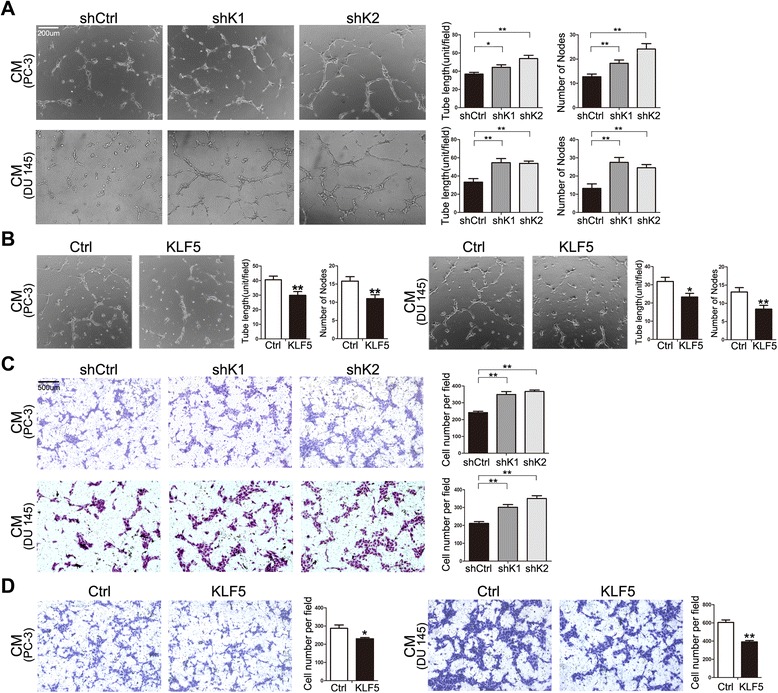
Expression changes in KLF5 in human prostate cancer cell lines modulate the angiogenic activities of HuVECs. Tube formation assay (A, B) and transwell assay (C, D) of human umbilical vein endothelial cells (HuVECs) with conditioned media (CM) from PC-3 and DU 145 cells with KLF5 knockdown (A) and ectopic expression (B) mediated by lentiviral infection. For tube formation assay, tube length and node number from ten fields were measured with ImageJ software, and each unit is 100 pixels. For transwell assay, HuVECs were seeded in the upper chamber with serum-free medium. Conditioned media was added to the lower wells, and cells migrating to the bottom side of the upper chamber were stained with crystal violet and counted from 20 fields at 4× with ImageJ software. shK1 and shK2 indicate two shRNAs targeting KLF5(sh36 and sh37) (A, C), and Ctrl and KLF5 indicate the pSin vector control and pSin-KLF5 expression construct, respectively (B, D). *, P < 0.05; **, P < 0.01.
Vascular endothelial cell migration is a critical step and another indicator of angiogenic ability [24], so we also performed transwell and wound healing assays to evaluate the effect of KLF5 dysregulation on the migration of HuVECs. Conditioned media from both PC-3 and DU 145 cell lines with KLF5 knockdown significantly promoted, whereas those from these cells with ectopic expression of KLF5 significantly inhibited, the migration of HuVECs, as indicated by the number of cells migrated through the membrane in the transwell assay (Figure 4C, D) and the extent of gap in the wound healing assay (Additional file 2: Figure S1). These in vitro assays provide another line of evidence for the role of KLF5 in angiogenesis in PTEN-deficient prostate cancer cells.
KLF5 also regulates the expression of HIF1α and other pro-angiogenic factors in human prostate cancer cells
Based on our finding that manipulation of KLF5 expression in human prostate cancer cells affected tube formation and migration of HuVECs (Figure 4), we further tested whether manipulation of KLF5 expression also affects the expression of HIF1α and other pro-angiogenic factors as in mouse tissues. We performed Western blotting to detect HIF1α expression and ELISA to detect the secretion of VEGF and PDGF-B in prostate cancer cells, whereas PDGF-D is undetectable by ELISA due to a low level of expression. As shown in Figure 5A, knockdown of KLF5 in PC-3 and DU 145 cells by lentiviral shRNA clearly increased the expression of HIF1α and PDGF-B, although the expression of VEGF was increased in PC-3 cells but slightly decreased in DU 145 cells (not statistically significant). Consistently, ectopic expression of KLF5 by lentiviral infection in the same two cell lines decreased the expression of HIF1α, VEGF and PDGF-B (Figure 5B). We also used transient transfection of siRNAs to knock down KLF5 (Figure 5C) and adenoviral infection to transiently express KLF5 (Figure 5D) in the two prostate cancer cell lines, and confirmed that knockdown and overexpression of KLF5 respectively increased and decreased the expression of HIF1α (Figure 5C, D).
Figure 5.
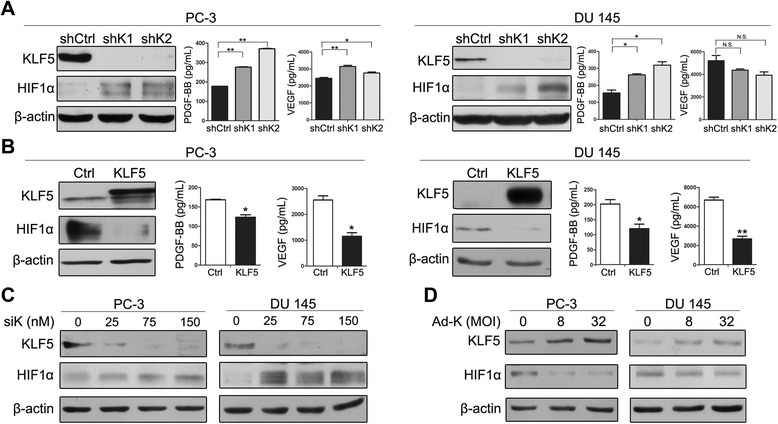
KLF5 decreases protein expression of HIF1α and secretion of pro-angiogenic factors. In PC-3 and DU 145 cells with stable knockdown (A), ectopic expression (B), RNAi-mediated transient knockdown (C), or adenovirus-mediated overexpression (D) of KLF5, HIF1α protein was detected by Western blotting whereas secretory pro-angiogenic factors PDGF-BB and VEGF in the conditioned media were detected by ELISA. shK1 and shK2 indicate 2 lentiviral shRNAs targeting KLF5 (sh36 and sh37), siK represents siRNA targeting KLF5 (siKLF5), and Ad-K represents adenovirus expressing KLF5. *, P < 0.05; **, P < 0.01; N.S., not statistically significant.
We also detected mRNA expression of HIF1α, VEGF and PDGF-B in human prostate cancer cells with altered KLF5 expression. PDGF-B and VEGF-A mRNA showed the same trend of change as their protein expression (Additional file 3: Figure S2), consistent with the results from mouse tissues (Figures 2 and 3). HIF1α mRNA expression also responded to KLF5 manipulation and showed the same trend of expression change as its protein expression in PC-3 cells (Additional file 3: Figure S2 left panel), in contrast to the result from mouse tissues where no expression change of HIF1α mRNA was detectable (Figure 2C).
KLF5 loss induces HIF1α protein accumulation by activating AKT signaling
Following the detection of the angiogenic phenotype and the activation of the HIF1α-driven pro-angiogenic molecular network upon KLF5 loss in both mouse tissues and cultured human prostate cancer cells, the fundamental question became how KLF5 loss activates the HIF1α network to promote angiogenesis. Considering that the expression change of HIF1α upon KLF5 loss mainly occurred at the protein level but not the mRNA level (Figures 2, 3 and 5) and that post-transcriptional regulation is the major mechanism of HIF1α regulation [3], we focused on the hypothesis that KLF5 loss activates HIF1α post-transcriptionally.
In our recent study, we found that Klf5 deletion activates multiple oncogenic signaling pathways including the PI3K/AKT, ERK and EGF/EGFR pathways [9]. On the other hand, it has been well established that AKT and ERK activation leads to the accumulation of HIF1α mainly by either translational activation or stabilization of HIF1α [25-29]. We therefore tested whether KLF5 loss induces HIF1α protein accumulation by activating AKT and ERK signaling. Consistent with the finding from mouse prostates [9], Western blotting demonstrated that both stable and transient knockdown of KLF5 in PC-3 and DU 145 cells increased, and ectopic expression of KLF5 decreased, the expression of activated AKT (p-AKT) (Figure 6A, B). For both EGFR and ERK, activity changes were inconsistent between human cancer cell lines and mouse tissues. Specifically, EGFR activation was slightly downregulated by both KLF5 knockdown and ectopic expression in the two cell lines, while ERK activation was downregulated upon KLF5 ectopic expression only in PC-3 cells but not in DU 145 cells. ERK activity was unchanged when KLF5 was knocked down in both the PC-3 and DU 145 cell lines. This result suggests that EGFR and ERK are less crucial than AKT in mediating the effect of KLF5 on HIF1α (Additional file 4: Figure S3). Expression changes of p-AKT (Figure 6A, B) corresponded nicely to those of HIF1α in the same cells (Figure 5).
Figure 6.
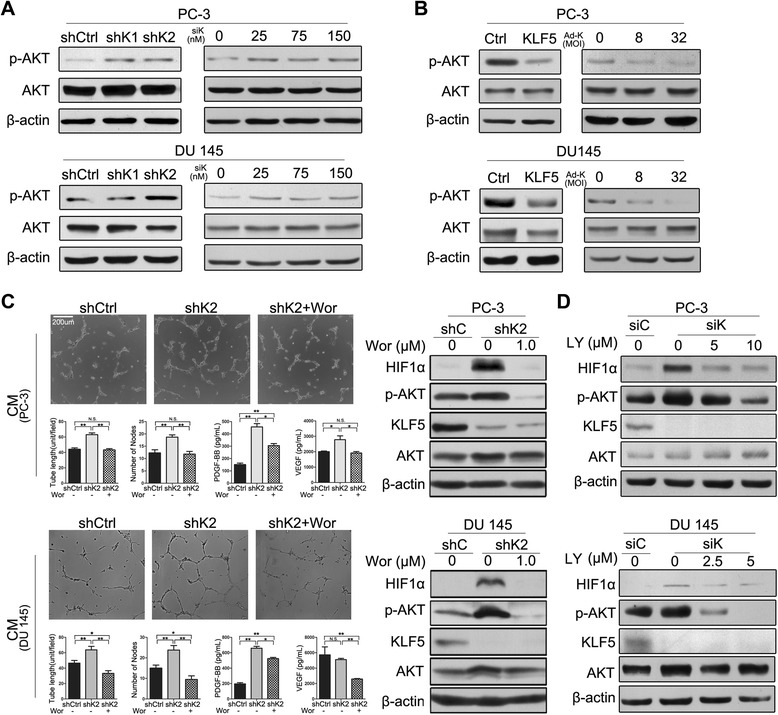
KLF5 regulates HIF1α protein via AKT1 kinase. (A, B) Phospho-AKT (S473) is upregulated by the knockdown of KLF5 (A) and downregulated by ectopic expression of KLF5 (B) in PC-3 and DU 145 human prostate cancer cells, as detected by Western blotting. (C) Wortmannin, a PI3K inhibitor, rescued the elevation of HuVEC tube formation and the expression of p-AKT, HIF1α, PDGF-B and VEGF induced by KLF5 knockdown. Conditioned media, collected after 24 hours of 1 μM Wortmannin treatment or solvent control from PC-3 and DU 145 cells with KLF5 knockdown (shK2), were used for HuVEC tube formation assay and detection of PDGF-BB and VEGF expression by ELISA. Expression of HIF1α and p-AKT was detected by Western blotting. (D) Detection of HIF1α and phospho-AKT (S473) by Western blotting in PC-3 and DU 145 cells with transient knockdown of KLF5 and treatment of PI3K inhibitor LY294002 for 3 hours. Solvent control for Wortmannin and LY294002 was DMSO. shK1 and shK2 indicate 2 lentiviral shRNAs targeting KLF5 (sh36 and sh37), siK represents siRNA targeting KLF5 (siKLF5), and Ad-K represents adenovirus expressing KLF5. The concentration of siRNA was at 100 nM for panel D. *, P < 0.05; **, P < 0.01; N.S., not statistically significant.
To further test whether AKT activation mediates HIF1α accumulation induced by KLF5 loss, we treated cells with Wortmannin, an irreversible PI3K/AKT inhibitor. We found in both PC-3 and DU 145 cells that inhibition of AKT activity by Wortmannin prevented the enhancement of tube formation and the upregulation of HIF1α, PDGF-B and VEGF by KLF5 knockdown (Figure 6C). Similarly, treatment of PC-3 and DU 145 cells with the LY294002 PI3K inhibitor upon KLF5 transient knockdown also abolished the upregulation of HIF1α protein (Figure 6D).
Discussion
Klf5 loss enhances angiogenesis in prostatic tumorigenesis
In this study, we performed histological and molecular analyses in mouse prostate tumors and human prostate cancer cells with the deletion or knockdown of KLF5 and PTEN, and obtained multiple lines of evidence suggesting the role of KLF5 loss in tumor angiogenesis. First of all, Klf5 loss enhanced angiogenesis not only during prostatic tumorigenesis initiated by homozygous deletion of Pten (Figure 1A) but also in mPINs induced by hemizygous deletion of Pten (Figure 1B), and Klf5’s homozygous deletion was more potent than its hemizygous deletion in the increase of microvessels (Figure 1). Secondly, conditioned culture media from human prostate cancer cells with deficient or insufficient PTEN and KLF5 knockdown increased tube forming capacity and migration of HuVECs (Figure 4), which are indicative of angiogenic potential. Overexpression of KLF5 in human prostate cancer cells showed a consistent effect on HuVECs (Figure 4). Thirdly, at the molecular level Klf5 deletion significantly elevated the expression of HIF1α and its pro-angiogenic functional effectors such as VEGF and PDGF (Figures 2, 3 and 5), which represent the most potent inducers of tumor angiogenesis. Our findings are consistent with a previous study in which Klf5 deletion in mouse corneas enhanced neovascularization [12], indicating that KLF5 loss in some epithelial tissues such as prostate and cornea promotes angiogenesis.
While KLF5 loss enhances angiogenesis, the extent of enhancement may depend on tumorigenesis including the development of cancer precursors such as mPIN, because deletion of Klf5 alone, which does not induce neoplastic alterations in mouse prostates [30], did not affect angiogenesis and HIF1α expression (data not shown). On the other hand, other genetic alterations combined with Pten deletion more often do not affect angiogenesis in mouse prostate tumors. For example, overexpression of COUP-TFII in Pten–null prostates results in aggressive metastasis-prone prostate tumors but does not alter angiogenesis [31]. In addition, there is no evidence for alterations in angiogenesis when other tumor suppressor genes such as p53, Smad4, Nkx3-1 and p27 are deleted in Pten-null mouse prostates [32-35].
Our recent study showed that interruption of KLF5 acetylation also promotes prostate cancer growth [8]. However, tumor promotion by unacetylated KLF5 does not appear to affect angiogenesis, as the number of microvessels and the expression of HIF1α and VEGF were not affected by changes in the acetylation status of KLF5 (data not shown), and angiogenesis was not among the affected processes based on bioinformatic analysis [8].
KLF5 loss enhances angiogenesis partially by upregulating HIF1α and its downstream pro-angiogenic factors including VEGF and PDGF
In understanding how KLF5 loss enhances tumor angiogenesis, our Gene Ontology (GO) enrichment analysis of differentially expressed genes upon Klf5 deletion identified blood morphogenesis as the most affected biological process, involving a molecular signature of activated pro-angiogenic genes (Figure 2A). In addition, network analysis of the differentially expressed genes led to the identification of HIF1α, a crucial inducer of angiogenesis, as a key transcription factor activated by Klf5 deletion, which transcriptionally activated 34 genes upon Klf5 deletion (Figure 2B). Among the 34 genes, secretory protein Pdgf-b is a well-established transcriptional target of HIF1α and a crucial regulator of angiogenesis [2,20] (Figure 2C). Although VEGF, another key angiogenesis regulator and secretory protein induced by HIF1α [17], was not identified by the microarray analysis as being upregulated by Klf5 deletion, our expression analyses demonstrated that KLF5 loss indeed upregulated HIF1α at the protein level and VEGF and PDGF at both the mRNA and protein levels in both mouse prostate tumors and human prostate cancer cells with deficient or insufficient PTEN (Figure 2, 3 and 5).
In addition to the well-established angiogenesis regulators HIF1α, VEGF and PDGF [36], other molecules could also be involved in enhanced angiogenesis after KLF5 loss. For example, Apelin and Adora2b, both of which are transcriptional targets of HIF1α, were upregulated by Klf5 deletion (Figure 2B) and have been shown to promote angiogenesis in certain types of cells and animal models [37,38]. Another group among the 36 genes that play a role in angiogenesis and were upregulated by Klf5 deletion in mouse prostates (Figure 2A, Additional file 1: Table S1) included Il1 and Ang, both encoding secretory proteins in prostatic epithelial cells that could also mediate the role of KLF5 loss in angiogenesis.
Upregulation of HIF1α by KLF5 loss is likely mediated by the activation of PI3K/AKT signaling
HIF1 consists of two subunits, the α unit, which is induced by hypoxia, and the β unit, which is expressed constitutively [16]. Regulation of HIF1α mainly occurs at the protein level, since it is degraded by the ubiquitin proteasome pathway (UPP) under normoxia conditions but is stabilized under hypoxia conditions in a tumor environment [2,3]. HIF1α can also be upregulated independent of hypoxia upon the activation of growth factor-dependent signaling pathways [3]. For example, forced PTEN overexpression reduces HIF1α expression, whereas activated PI3K/AKT signaling upregulates HIF1α expression [28,29]. Our recent study established that Klf5 deletion leads to the activation of PI3K/AKT and ERK signaling in mouse prostate tumors [9]. Using cultured human prostate cancer cell lines with deficient or insufficient PTEN, not only did we confirm the activation of AKT by KLF5 loss (Figure 6A), we also found that activated AKT indeed mediated the upregulation of HIF1α by KLF5 loss, since treatment with PI3K inhibitors prevented both the activation of AKT and the accumulation of HIF1α (Figure 6C, D). Treatment with PI3K inhibitors also prevented the enhancement of tube formation by KLF5 loss (Figure 6C). Therefore, one mechanism for the upregulation of HIF1α by KLF5 loss in PTEN deficient or insufficient prostate cancer cells appears to be the activation of AKT. Previous studies have demonstrated that PI3K/AKT signaling promotes HIF1α protein accumulation by either increasing its translation or by inhibiting its degradation or both [25,39,40]. Currently it is unknown which process is most involved in HIF1α accumulation induced by KLF5 loss and subsequent PI3K/AKT activation.
It is well known that PTEN is a lipid phosphatase whose loss activates PI3K/AKT signaling [41], which is related to HIF1α upregulation and enhanced angiogenesis in some human cancer cells [42,43]. Our recent study demonstrated that Klf5 loss in Pten-null prostate tumors also activates PI3K/AKT signaling by upregulating multiple extracellular growth factors (e.g. EGF), cytokines and their receptors [9]. Deletion of Klf5 alone in mouse prostates did not affect angiogenesis (data not shown), and neither did Pten deletion alone in mouse prostates cause apparent angiogenesis [13]. Therefore, KLF5 loss and PTEN loss had an additive effect on the activation of PI3K/AKT signaling [9], HIF1α accumulation (Figure 6), and angiogenesis (Figure 1). The precise function of KLF5 in mediating AKT activation is still under investigation.
While two other oncogenic signaling pathways, EGF/EGFR and ERK, were activated in mouse prostate tumors upon Klf5 deletion [9], and KLF5 could positively regulate EGFR expression and ERK activation [44,45], their changes in activity in response to KLF5 modulation were inconsistent between human prostate cancer cell lines PC-3 and DU 145 and mouse prostate tissues (Additional file 4: Figure S3). Therefore, whereas EGFR and ERK activation may still play a role in the upregulation of HIF1α by Klf5 deletion in mouse prostate tumors, they are less crucial for KLF5 to regulate HIF1α in human cancer cell lines PC-3 and DU 145.
There are other mechanisms that could account for how KLF5 regulates HIF1α protein, including transcriptional regulation, miRNA and hypoxia. In colon and lung cancer cells, KLF5 was shown to directly regulate the transcription of HIF1α [46,47]. However, our results (Figure 2 & Additional file 3: Figure S2) showed that KLF5 did not alter the transcription of HIF1α either in mouse prostates or in human prostate cancer cells. HIF1α can also be regulated by miRNAs including miR-138 [48]. While miR-138 appeared to be downregulated by Klf5 deletion in mouse prostate tumors, it was unaltered by KLF5 loss in human prostate cancer cells (data not shown). Hypoxia is a potent inducer of HIF1α, but it did not appear to be necessary for KLF5 loss to induce HIF1α, at least in cancer cell lines, because the morphological and molecular alterations in PC-3 and DU 145 cells were detected under normoxic conditions (Figures 4, 5 and 6). Nevertheless, it remains to be clarified whether these additional mechanisms are involved in the HIF1α accumulation induced by KLF5 loss.
Enhanced angiogenesis by KLF5 loss may partially account for its tumor promoting function and have implications in human prostate cancer
Our recent study demonstrated that Klf5 deletion promotes mouse prostate tumorigenesis initiated by Pten deletion, as indicated by the accelerated emergence and progression of mPIN caused by Pten insufficiency and the more severe morphological and molecular abnormalities of prostate tumors caused by Pten deficiency [9]. In that study, enhanced cell proliferation was identified as a mechanism for the tumor promoting effect of Klf5 deletion [9]. Our current study suggests that enhanced angiogenesis by KLF5 loss is likely another mechanism for the tumor promoting effect of KLF5 loss during prostatic tumorigenesis initiated by other oncogenic factors, which is in addition to its previously reported promoting function in tumor cell proliferation [9].
During the development of human prostate cancer, chromosome 13q is one of the most frequently deleted chromosomes, and the deletion involves multiple regions and genetic foci in 13q14 and 13q21, including RB1 and FOXO1 at 13q14 and KLF5 at 13q21 [22,49-52]. Whereas RB1 is a well-established tumor suppressor gene at 13q14, the tumor suppressor function of KLF5 has also been demonstrated in both xenograft and knockout models [7,9]. In addition, conventional comparative genomic hybridization (CGH) studies indicate that deletion spanning the KLF5 locus could occur in as much as 39% of prostate cancers [6], and our earlier deletion analyses suggest that hemizygous deletion is more common than homozygous deletion in prostate cancer [22,50]. In the latest genome-wide copy number change studies, KLF5’s deep deletion, which likely indicates homozygous deletion, has been detected in 10% of prostate cancers in The Cancer Genome Atlas (TCGA) database (tcga-data.nci.nih.gov) and in 20% of metastatic prostate cancers in another database [53], as analyzed by the cBioPortal program [54,55]. Hemizygous deletion induces haploinsufficiency of KLF5 [9,10], and our recent publication demonstrated that hemizygous deletion of Klf5 in mouse prostates indeed promotes tumor development [30]. Therefore, frequent hemizygous and homozygous deletions induce frequent insufficiency and deficiency, respectively, of KLF5 during prostatic carcinogenesis. Genetic amplification and overexpression of the WWP1 E3 ubiquitin ligase, which degrades KLF5 protein, also frequently occur in human prostate cancer [56,57], causing excess protein degradation and insufficiency of KLF5. Therefore, enhanced tumor angiogenesis represents another mechanism for how KLF5 insufficiency and deficiency could promote the development and progression of human prostate cancer.
As in many other types of cancers, prostatic carcinogenesis depends on the accumulation of multiple genetic and epigenetic alterations, and deletions of KLF5 and PTEN can occur in the same tumors [22,58-61]. For example, 7 of the 25 (28%) prostate cancers with KLF5 deep deletion in the TCGA database and 4 of the 11 (36%) metastatic prostate cancers in another database also have PTEN deep deletion or truncating mutation [53], as revealed by the cBioPortal analysis [54,55]. Further related to our current findings, it has been reported that PTEN inactivation is positively associated with higher microvessel densities in clinically localized prostate cancer [43], and PTEN expression is negatively associated with VEGF expression in gastric carcinomas [62]. Another study demonstrated that in high-grade prostate adenocarcinomas, 46% of them showed higher nuclear HIF1α immunostaining, and 89% expressed higher levels of VEGF [63]. In the context of concurrent deletions of KLF5 and PTEN in human prostate cancer, our findings further suggest that during the development of human prostate cancer, KLF5 loss and PTEN loss cooperate to enhance tumor angiogenesis by upregulating HIF1α and VEGF, which remains to be examined.
In summary, we found that deletion of Klf5 promoted angiogenesis in Pten deletion-initiated mouse prostate tumors. Promotion of angiogenesis by Klf5 loss involved the upregulation of pro-angiogenic molecules including HIF1α and its downstream targets VEGF and PDGF. Upregulation of HIF1α was mediated at least in part by the activation of PI3K/AKT oncogenic signaling. Consistently, knockdown of KLF5 in PTEN-insufficient human prostate cancer cells increased tube formation and migration in HuVECs, enhanced PI3K/AKT activity, and upregulated HIF1α, VEGF and PDGF. These findings indicate that KLF5 loss enhances angiogenesis during prostatic carcinogenesis initiated by other oncogenic factors, and that enhanced angiogenesis could be a mechanism for the tumor promoting function of KLF5 loss.
Materials and methods
Cell lines, siRNAs, inhibitors and antibodies
Human umbilical vein endothelial cells (HuVECs) were purchased from Life Technologies and cultured in Medium 200 with low serum growth supplement (LSGS) (Carlsbad, CA). PC-3 and DU 145 human prostate cancer cell lines and the 293 T human embryonic kidney cell line were purchased from American Type Culture Collection (ATCC) (Manassas, VA), and propagated following ATCC’s instructions.
SiRNA against KLF5 (SiKLF5), developed in a previous study [64] with the sequence of AAGCUCACCUGAGGACUCATT, was used to knock down KLF5 in human cells at a concentration of 150 nM or as indicated in related figures. The control siRNA (siCtrl), AAUUCUCCGAACGUGUCACGUTT, was purchased from Thermo Fisher (Waltham, MA) used to bring the siRNA concentration to the same in concentration gradient RNAi assay.
PI3K inhibitors Wortmannin and LY294002 were purchased from Cell Signaling Technology (Danvers, MA), and dissolved in dimethyl sulfoxide (DMSO).
Antibodies used in this study included the following: anti-HIF1α and anti-PDGF-B from Novus Biologicals (Littleton, CO), anti-PDGF-D from Santa Cruz (Santa Cruz, CA), anti-phospho-AKT (S473), anti-AKT, anti-phospho-EGFR (Y1068), anti-EGFR, anti-phospho-ERK1/2 and anti-ERK1/2 from Cell Signaling Technology, anti-VEGF and anti-CD31 from Abcam (Cambridge, MA), and anti-β-actin from Sigma. The antibody against KLF5 has been described previously [23].
Mouse prostate tissues with different Klf5 and Pten deletion status
All mouse prostate tissues with different Klf5 and/or Pten deletion status, including prostate tumors, mouse prostatic intraepithelial neoplasia (mPIN) and control tissues of different ages, were from our recently published study [9].
Production of lentiviruses and adenoviruses and their infection of cells
Lentiviruses expressing KLF5 and shRNA targeting KLF5 were produced by transfecting respective plasmids in 293 T cells following the protocols described on the Addgene website (http://www.addgene.org/lentiviral/protocols-resources/). The PLKO.1 expression vector was used for shRNA expression, and the 2 shRNAs targeting KLF5, sh36 and sh37, were described in our previous study [65]. The pSin expression vector was used for KLF5 expression (pSin-KLF5) as in our previous study [66]. Following our previously described procedures [65], viruses were added to culture media to infect PC-3 and DU 145 cells, and medium containing 1 μg/ml puromycin was used to select infected cells for at least 96 hours to obtain populations for analysis.
Adenoviruses expressing KLF5 (Ad-KLF5) and GFP (Ad-GFP) were prepared and used following our established procedures [64]. MOI (multiplicity of infection) was defined as the average number of virus particles infecting a cell.
RNA isolation, real-time RT-PCR and microarray expression analysis
Mouse dorsal prostates or tumor tissues were dissected freshly and stored in liquid nitrogen, and total RNA was isolated by using the RNeasy Mini Kit (Qiagen, Valencia, CA) following the manufacturer’s instructions. TRIzol reagent (Life technologies) was used for isolating total RNA from cultured cellsfollowing previously established procedures [67]. First-strand cDNA was synthesized from total RNA using a Promega reverse transcription kit (Madison, WI).
Real-time RT-PCR was performed using the SYBR Premix reagent (Takara, Tokyo, Japan) and ABI Fast 7500 Real-time PCR System (Applied Biosystems, South San Francisco, CA). The 2(−△△Ct) method was used to calculate relative fold changes with GAPDH as the internal control. Primers used for real-time RT-PCR are listed in Additional file 5: Table S2.
Genes with significantly different expression between Klf5 wildtype and Klf5-null 6-month old prostate tumors in the context of Pten deletion, as described in our recent study [9], were uploaded into the web-based MetaCore platform (http://thomsonreuters.com/metacore/), and angiogenesis-related genes were enriched using Gene Ontology (GO)-based functional process enrichment. Whether a gene functions in angiogenesis was further evaluated by literature review among PubMed publications with “angiogenesis” and a gene’s name as key words. The algorithm of “activated transcription factor regulating genes in the experimental data” was used to build a HIF1α-centered network with those that were not only direct transcriptionally activated targets of HIF1α but were also upregulated by Klf5 deletion.
Immunohistochemistry (IHC), Western blotting and enzyme-linked immunosorbent assay (ELISA)
IHC staining was performed following our established protocols [30] and nuclei were stained with hematoxylin. Mounted slides were scanned with the Hamamatsu NanoZoomer scanner (Hamamatsu Corporation, Bridgewater, NJ) or photographed with a microscope. When needed, cell numbers were counted by using the ImageJ program to calculate the ratio of positive cells for a protein. Western blotting was performed following our previously established procedures [68]. For secretory factors, conditioned culture media were collected from 100% confluent cells in 100-mm dishes, and commercial ELISA kits against VEGF and PDGF-BB from Abcam were used to determine the expression level of VEGF and PDGF-BB following the manufacturer’s instructions.
Tube formation assay in HUVECs
Prostatic epithelial cells were seeded in 100-mm dishes, grown to 90% confluence at 48 hours, washed with PBS twice, and then cultured in 3 ml of medium containing 1% serum for another 24 hours to reach 100% confluence. Conditioned media were then collected, centrifuged at 1500 rpm/min, and the supernatant was used for experiments as previously described [69]. A total of 3 × 104 HuVECs were seeded into each well of a 48-well plate coated with growth-factor reduced Matrigel (BD Biosciences, San Jose, CA). After culturing for 5 and 8 hours with conditioned media or the HuVEC culture medium, the capillary-like structures were captured with the Olympus IX51 microscope, and the extent of tube formation was assessed by measuring the cumulative tube length and counting the number of tube nodes (the intersection among 3 or more tubes) in a 10× field using ImageJ software. At least 10 fields were examined to determine cell tube formation ability [70].
Wound healing assay in HuVECs
Confluent monolayer HuVECs cultured in 24-well plates were scratched with the tip of a 10-μl pipette tip to generate a cross wound, washed with PBS twice, and cultured with conditioned media for another 13–15 hours to allow the gap to close. Photographs of gaps at 0 and 13–15 hours were taken with an Olympus IX51 inverted microscope at 10× magnification, and the extent of wound closure was assessed by calculating the ratio of the initial to the final wound area with Image J software. At least 4 fields were examined to determine cell migration ability [71].
Transwell migration assay in HuVECs
Five x 104 HuVECs in 200 μl serum-free medium were seeded in the upper chamber of an insert (BD Biosciences) in a 24-well plate, and 800 μl of conditioned medium was added to the lower chamber. After 15–17 hours, cells in the insert were fixed in 4% paraformaldehyde (Sigma) and stained with 0.1% crystal violet (Sigma). Cells inside the transwell chamber were scraped with a cotton swap, while cells outside the chamber were photographed with an inverted microscope. The number of cells per field at 4× magnification was counted with Image J software, and at least 10 fields were examined to determine cell migration ability [65].
Statistical analysis
Results from real-time RT-PCR and ELISA were expressed as means ± standard derivation, while readings from other experiments were expressed as means ± standard error. The statistical significance for a difference between two groups was determined by unpaired Student t test. A P-value of 0.05 or smaller was considered statistically significant.
Acknowledgements
The authors thank Dr. Ganji Nagaraju Purnachandra and Dr. Xiaoou Sun of Emory University for technical assistance during this study, Ms. Rini Pauly and Mr. Gregory Doho of the Biostatistics and Bioinformatics Shared Resource at Emory Winship Cancer Institute for assistance in microarray data analysis, Mr. Yudong Xia of E-GENE, Shenzhen, China for assistance in the generation of the heatmap, and Dr. Anthea Hammond of Emory for manuscript editing. Xinpei Ci was supported by the graduate student program of the China Scholarship Council (No. 201206200049). This work was supported in part by grants R01CA87921 and R01CA171189 from the National Cancer Institute, National Institutes of Health and grant 81130044 from the National Natural Science Foundation of China. Research reported in this publication was supported in part by the Integrated Cellular Imaging Shared Resource, the Emory Integrated Genomics Core, and the Biostatistics and Bioinformatics Shared Resource of Winship Cancer Institute of Emory University and NIH/NCI under award number P30CA138292. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- KLF5
Krüppel-like factor 5
- HIF1α
Hypoxia-inducible factor 1-alpha
- PTEN
Phosphatase and tensin homolog
- PI3K
Phosphatidylinositol-4,5-bisphosphate 3-kinase
- ERK
Extracellular signal-regulated kinases
- MAPK
Mitogen-activated protein kinase
- EGF
Epidermal growth factor
- VEGF
Vascular endothelial growth factor
- PDGF
Platelet-derived growth factor
- HuVEC
Human umbilical vein endothelial cells
- IHC
Immunohistochemistry
- ELISA
Enzyme-linked immunosorbent assay
- GO
Gene ontology
Additional files
Differentially expressed genes involved in angiogenesis, as identified by the MetaCore program. Fold change between Klf5-wildtype and Klf5-null groups are shown for each of the genes. P and R indicate promoting and repressive functions, respectively, of a gene in angiogenesis.
Expression changes in KLF5 in PC-3 and DU 145 prostate cancer cells modulate wound healing in HuVECs. Confluent HuVECs cultured in conditioned media were scratched with a pipette tip and photographed from 4 fields at 0 and 13-15 hours for each sample. shK1 and shK2 indicate two shRNAs targeting KLF5 (sh36 and sh37) (A), Ctrl and KLF5 indicate lentiviruses expressing the pSin vector control and KLF5, respectively (B). Areas of wound were measured by using ImageJ software to indicate the extent of wound closure. *, P<0.05; **, P<0.01.
Modulation of KLF5 expression affects mRNA expression of pro-angiogenic factors. Real-time RT-PCR was used to detect mRNA expression of HIF1α, VEGF-A, PDGF-B and PDGF-D mRNA in PC-3 and DU 145 prostate cancer cells with stable knockdown (A) or ectopic expression (B) of KLF5. shK1 and shK2 indicate two shRNAs targeting KLF5 (sh36 and sh37) (A), Ctrl and KLF5 indicate pSin vector control and pSin-KLF5 (B), respectively. *, P<0.05; **, P<0.01. (TIFF 334 kb)
EGFR and ERK activation do not appear to mediate the upregulation of HIF1α by KLF5 knockdown in human prostate cancer cell lines. Protein expression of p-EGFR (Y1068), EGFR, p-ERK and ERK were determined by Western blotting in PC-3 and DU 145 cells with stable knockdown or ectopic expression of KLF5. shK1 and shK2 indicate two shRNAs targeting KLF5 (sh36 and sh37), and Ctrl and KLF5 indicate the pSin vector control and pSin-KLF5 expression construct, respectively.
Primer sequences used in this study.
Footnotes
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
JD, XC and WZ designed the experiments; XC performed most of the experiments; CX provided the mouse tissue samples and contributed to the first figure; BZ, ZZ and JN assisted with experiments and provided discussion; JD and XC wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Xinpei Ci, Email: cixinpei@126.com.
Changsheng Xing, Email: hunterxcs85@gmail.com.
Baotong Zhang, Email: baotong.zhang@emory.edu.
Zhiqian Zhang, Email: zzhang8@mdanderson.org.
Jenny Jianping Ni, Email: jianping.jenny.ni@emory.edu.
Wei Zhou, Email: wzhou2@emory.edu.
Jin-Tang Dong, Email: j.dong@emory.edu.
References
- 1.Risau W, Flamme I. Vasculogenesis. Annu Rev Cell Dev Biol. 1995;11:73–91. doi: 10.1146/annurev.cb.11.110195.000445. [DOI] [PubMed] [Google Scholar]
- 2.Martinez-Jabaloyas JM, March-Villalba JA, Navarro-Garcia MM, Dasi F. Anti-angiogenic therapies in prostate cancer. Expert Opin Biol Ther. 2013;13:1–5. doi: 10.1517/14712598.2013.733366. [DOI] [PubMed] [Google Scholar]
- 3.Bardos JI, Ashcroft M. Negative and positive regulation of HIF-1: a complex network. Biochim Biophys Acta. 2005;1755:107–20. doi: 10.1016/j.bbcan.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Schweizer MT, Carducci MA. From bevacizumab to tasquinimod: angiogenesis as a therapeutic target in prostate cancer. Cancer J. 2013;19:99–106. doi: 10.1097/PPO.0b013e31827e0b86. [DOI] [PubMed] [Google Scholar]
- 5.Dong JT, Chen C. Essential role of KLF5 transcription factor in cell proliferation and differentiation and its implications for human diseases. Cell Mol Life Sci. 2009;66:2691–706. doi: 10.1007/s00018-009-0045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dong JT. Chromosomal deletions and tumor suppressor genes in prostate cancer. Cancer Metastasis Rev. 2001;20:173–93. doi: 10.1023/A:1015575125780. [DOI] [PubMed] [Google Scholar]
- 7.Nakajima Y, Akaogi K, Suzuki T, Osakabe A, Yamaguchi C, Sunahara N, et al. Estrogen regulates tumor growth through a nonclassical pathway that includes the transcription factors ERbeta and KLF5. Sci Signal. 2011;4:ra22. [DOI] [PubMed]
- 8.Li X, Zhang B, Wu Q, Ci X, Zhao R, Zhang Z, et al. Interruption of KLF5 acetylation converts its function from tumor suppressor to tumor promoter in prostate cancer cells. Int J Cancer. 2014;136:536–46. doi: 10.1002/ijc.29028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xing C, Ci X, Sun X, Fu X, Zhang Z, Dong EN, et al. Klf5 deletion promotes Pten deletion-initiated luminal-type mouse prostate tumors through multiple oncogenic signaling pathways. Neoplasia. 2014;16:883–99. doi: 10.1016/j.neo.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shindo T, Manabe I, Fukushima Y, Tobe K, Aizawa K, Miyamoto S, et al. Kruppel-like zinc-finger transcription factor KLF5/BTEB2 is a target for angiotensin II signaling and an essential regulator of cardiovascular remodeling. Nat Med. 2002;8:856–63. doi: 10.1038/nm738. [DOI] [PubMed] [Google Scholar]
- 11.Kenchegowda D, Swamynathan S, Gupta D, Wan H, Whitsett J, Swamynathan SK. Conditional disruption of mouse Klf5 results in defective eyelids with malformed meibomian glands, abnormal cornea and loss of conjunctival goblet cells. Dev Biol. 2011;356:5–18. doi: 10.1016/j.ydbio.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kenchegowda D, Harvey SA, Swamynathan S, Lathrop KL, Swamynathan SK. Critical role of Klf5 in regulating gene expression during post-eyelid opening maturation of mouse corneas. PLoS One. 2012;7 doi: 10.1371/journal.pone.0044771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209–21. doi: 10.1016/S1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 14.Jiang BH, Liu LZ. AKT signaling in regulating angiogenesis. Curr Cancer Drug Targets. 2008;8:19–26. doi: 10.2174/156800908783497122. [DOI] [PubMed] [Google Scholar]
- 15.Dimova EY, Michiels C, Kietzmann T. Kinases as upstream regulators of the HIF system: their emerging potential as anti-cancer drug targets. Curr Pharm Des. 2009;15:3867–77. doi: 10.2174/138161209789649358. [DOI] [PubMed] [Google Scholar]
- 16.Mabjeesh NJ, Amir S. Hypoxia-inducible factor (HIF) in human tumorigenesis. Histol Histopathol. 2007;22:559–72. doi: 10.14670/HH-22.559. [DOI] [PubMed] [Google Scholar]
- 17.Ahluwalia A, Tarnawski AS. Critical role of hypoxia sensor–HIF-1alpha in VEGF gene activation. Implications for angiogenesis and tissue injury healing. Curr Med Chem. 2012;19:90–7. doi: 10.2174/092986712803413944. [DOI] [PubMed] [Google Scholar]
- 18.Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–13. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshida D, Kim K, Noha M, Teramoto A. Hypoxia inducible factor 1-alpha regulates of platelet derived growth factor-B in human glioblastoma cells. J Neurooncol. 2006;76:13–21. doi: 10.1007/s11060-005-3279-0. [DOI] [PubMed] [Google Scholar]
- 21.Bastola DR, Pahwa GS, Lin MF, Cheng PW. Downregulation of PTEN/MMAC/TEP1 expression in human prostate cancer cell line DU145 by growth stimuli. Mol Cell Biochem. 2002;236:75–81. doi: 10.1023/A:1016191913274. [DOI] [PubMed] [Google Scholar]
- 22.Chen C, Bhalala HV, Vessella RL, Dong JT. KLF5 is frequently deleted and down-regulated but rarely mutated in prostate cancer. Prostate. 2003;55:81–8. doi: 10.1002/pros.10205. [DOI] [PubMed] [Google Scholar]
- 23.Chen C, Sun X, Ran Q, Wilkinson KD, Murphy TJ, Simons JW, et al. Ubiquitin-proteasome degradation of KLF5 transcription factor in cancer and untransformed epithelial cells. Oncogene. 2005;24:3319–27. doi: 10.1038/sj.onc.1208497. [DOI] [PubMed] [Google Scholar]
- 24.Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circ Res. 2007;100:782–94. doi: 10.1161/01.RES.0000259593.07661.1e. [DOI] [PubMed] [Google Scholar]
- 25.Pore N, Jiang Z, Shu HK, Bernhard E, Kao GD, Maity A. Akt1 activation can augment hypoxia-inducible factor-1alpha expression by increasing protein translation through a mammalian target of rapamycin-independent pathway. Mol Cancer Res. 2006;4:471–9. doi: 10.1158/1541-7786.MCR-05-0234. [DOI] [PubMed] [Google Scholar]
- 26.Liu LZ, Li C, Chen Q, Jing Y, Carpenter R, Jiang Y, et al. MiR-21 induced angiogenesis through AKT and ERK activation and HIF-1alpha expression. PLoS One. 2011;6 doi: 10.1371/journal.pone.0019139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mottet D, Dumont V, Deccache Y, Demazy C, Ninane N, Raes M, et al. Regulation of hypoxia-inducible factor-1alpha protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta pathway in HepG2 cells. J Biol Chem. 2003;278:31277–85. doi: 10.1074/jbc.M300763200. [DOI] [PubMed] [Google Scholar]
- 28.Fang J, Ding M, Yang L, Liu LZ, Jiang BH. PI3K/PTEN/AKT signaling regulates prostate tumor angiogenesis. Cell Signal. 2007;19:2487–97. doi: 10.1016/j.cellsig.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zundel W, Schindler C, Haas-Kogan D, Koong A, Kaper F, Chen E, et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000;14:391–6. [PMC free article] [PubMed] [Google Scholar]
- 30.Xing C, Fu X, Sun X, Guo P, Li M, Dong JT. Different expression patterns and functions of acetylated and unacetylated Klf5 in the proliferation and differentiation of prostatic epithelial cells. PLoS One. 2013;8 doi: 10.1371/journal.pone.0065538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qin J, Wu SP, Creighton CJ, Dai F, Xie X, Cheng CM, et al. COUP-TFII inhibits TGF-beta-induced growth barrier to promote prostate tumorigenesis. Nature. 2013;493:236–40. doi: 10.1038/nature11674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–30. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding Z, Wu CJ, Chu GC, Xiao Y, Ho D, Zhang J, et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature. 2011;470:269–73. doi: 10.1038/nature09677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abate-Shen C, Banach-Petrosky WA, Sun X, Economides KD, Desai N, Gregg JP, et al. Nkx3.1; Pten mutant mice develop invasive prostate adenocarcinoma and lymph node metastases. Cancer Res. 2003;63:3886–90. [PubMed] [Google Scholar]
- 35.Di Cristofano A, De Acetis M, Koff A, Cordon-Cardo C, Pandolfi PP. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat Genet. 2001;27:222–4. doi: 10.1038/84879. [DOI] [PubMed] [Google Scholar]
- 36.Chung AS, Lee J, Ferrara N. Targeting the tumour vasculature: insights from physiological angiogenesis. Nat Rev Cancer. 2010;10:505–14. doi: 10.1038/nrc2868. [DOI] [PubMed] [Google Scholar]
- 37.Kalin RE, Kretz MP, Meyer AM, Kispert A, Heppner FL, Brandli AW. Paracrine and autocrine mechanisms of apelin signaling govern embryonic and tumor angiogenesis. Dev Biol. 2007;305:599–614. doi: 10.1016/j.ydbio.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 38.Desai A, Victor-Vega C, Gadangi S, Montesinos MC, Chu CC, Cronstein BN. Adenosine A2A receptor stimulation increases angiogenesis by down-regulating production of the antiangiogenic matrix protein thrombospondin 1. Mol Pharmacol. 2005;67:1406–13. doi: 10.1124/mol.104.007807. [DOI] [PubMed] [Google Scholar]
- 39.Choi D, Han J, Lee Y, Choi J, Han S, Hong S, et al. Caffeic acid phenethyl ester is a potent inhibitor of HIF prolyl hydroxylase: structural analysis and pharmacological implication. J Nutr Biochem. 2010;21:809–17. doi: 10.1016/j.jnutbio.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 40.Joshi S, Singh AR, Zulcic M, Durden DL. A macrophage-dominant PI3K isoform controls hypoxia-induced HIF1alpha and HIF2alpha stability and tumor growth, angiogenesis, and metastasis. Mol Cancer Res. 2014;12:1520–31. doi: 10.1158/1541-7786.MCR-13-0682. [DOI] [PubMed] [Google Scholar]
- 41.Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. 2012;4:a011189. doi: 10.1101/cshperspect.a011189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60:1541–5. [PubMed] [Google Scholar]
- 43.Giri D, Ittmann M. Inactivation of the PTEN tumor suppressor gene is associated with increased angiogenesis in clinically localized prostate carcinoma. Hum Pathol. 1999;30:419–24. doi: 10.1016/S0046-8177(99)90117-X. [DOI] [PubMed] [Google Scholar]
- 44.Yang Y, Goldstein BG, Nakagawa H, Katz JP. Kruppel-like factor 5 activates MEK/ERK signaling via EGFR in primary squamous epithelial cells. FASEB J. 2007;21:543–50. doi: 10.1096/fj.06-6694com. [DOI] [PubMed] [Google Scholar]
- 45.Liu R, Zheng HQ, Zhou Z, Dong JT, Chen C. KLF5 promotes breast cell survival partially through fibroblast growth factor-binding protein 1-pERK-mediated dual specificity MKP-1 protein phosphorylation and stabilization. J Biol Chem. 2009;284:16791–8. doi: 10.1074/jbc.M808919200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee SJ, No YR, Dang DT, Dang LH, Yang VW, Shim H, et al. Regulation of hypoxia-inducible factor 1alpha (HIF-1alpha) by lysophosphatidic acid is dependent on interplay between p53 and Kruppel-like factor 5. J Biol Chem. 2013;288(35):25244–53. doi: 10.1074/jbc.M113.489708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li X, Liu X, Xu Y, Liu J, Xie M, Ni W, et al. KLF5 promotes hypoxia-induced survival and inhibits apoptosis in non-small cell lung cancer cells via HIF-1alpha. Int J Oncol. 2014;45:1507–14. doi: 10.3892/ijo.2014.2544. [DOI] [PubMed] [Google Scholar]
- 48.Yeh YM, Chuang CM, Chao KC, Wang LH. MicroRNA-138 suppresses ovarian cancer cell invasion and metastasis by targeting SOX4 and HIF-1alpha. Int J Cancer. 2013;133:867–78. doi: 10.1002/ijc.28086. [DOI] [PubMed] [Google Scholar]
- 49.Hyytinen ER, Frierson HF, Boyd JC, Chung LW, Dong JT. Three distinct regions of allelic loss at 13q14, 13q21-22, and 13q33 in prostate cancer. Genes Chromosomes Cancer. 1999;25:108–14. doi: 10.1002/(SICI)1098-2264(199906)25:2<108::AID-GCC6>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 50.Dong JT, Chen C, Stultz BG, Isaacs JT, Frierson HF., Jr Deletion at 13q21 is associated with aggressive prostate cancers. Cancer Res. 2000;60:3880–3. [PubMed] [Google Scholar]
- 51.Dong XY, Chen C, Sun X, Guo P, Vessella RL, Wang RX, et al. FOXO1A is a candidate for the 13q14 tumor suppressor gene inhibiting androgen receptor signaling in prostate cancer. Cancer Res. 2006;66:6998–7006. doi: 10.1158/0008-5472.CAN-06-0411. [DOI] [PubMed] [Google Scholar]
- 52.Williams JL, Greer PA, Squire JA. Recurrent copy number alterations in prostate cancer: an in silico meta-analysis of publicly available genomic data. Cancer Genet. 2014;207:474–88. doi: 10.1016/j.cancergen.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 53.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. [DOI] [PMC free article] [PubMed]
- 56.Chen C, Sun X, Guo P, Dong XY, Sethi P, Cheng X, et al. Human Kruppel-like factor 5 is a target of the E3 ubiquitin ligase WWP1 for proteolysis in epithelial cells. J Biol Chem. 2005;280:41553–61. doi: 10.1074/jbc.M506183200. [DOI] [PubMed] [Google Scholar]
- 57.Chen C, Sun X, Guo P, Dong XY, Sethi P, Zhou W, et al. Ubiquitin E3 ligase WWP1 as an oncogenic factor in human prostate cancer. Oncogene. 2007;26:2386–94. doi: 10.1038/sj.onc.1210021. [DOI] [PubMed] [Google Scholar]
- 58.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 59.Vlietstra RJ, van Alewijk DC, Hermans KG, van Steenbrugge GJ, Trapman J. Frequent inactivation of PTEN in prostate cancer cell lines and xenografts. Cancer Res. 1998;58:2720–3. [PubMed] [Google Scholar]
- 60.Hermans KG, van Alewijk DC, Veltman JA, van Weerden W, van Kessel AG, Trapman J. Loss of a small region around the PTEN locus is a major chromosome 10 alteration in prostate cancer xenografts and cell lines. Genes Chromosomes Cancer. 2004;39:171–84. doi: 10.1002/gcc.10311. [DOI] [PubMed] [Google Scholar]
- 61.Saramaki OR, Porkka KP, Vessella RL, Visakorpi T. Genetic aberrations in prostate cancer by microarray analysis. Int J Cancer. 2006;119:1322–9. doi: 10.1002/ijc.21976. [DOI] [PubMed] [Google Scholar]
- 62.Zheng H, Takahashi H, Murai Y, Cui Z, Nomoto K, Niwa H, et al. Expressions of MMP-2, MMP-9 and VEGF are closely linked to growth, invasion, metastasis and angiogenesis of gastric carcinoma. Anticancer Res. 2006;26:3579–83. [PubMed] [Google Scholar]
- 63.Lekas A, Lazaris AC, Deliveliotis C, Chrisofos M, Zoubouli C, Lapas D, et al. The expression of hypoxia-inducible factor-1alpha (HIF-1alpha) and angiogenesis markers in hyperplastic and malignant prostate tissue. Anticancer Res. 2006;26:2989–93. [PubMed] [Google Scholar]
- 64.Guo P, Dong XY, Zhang X, Zhao KW, Sun X, Li Q, et al. Pro-proliferative factor KLF5 becomes anti-proliferative in epithelial homeostasis upon signaling-mediated modification. J Biol Chem. 2009;284:6071–8. doi: 10.1074/jbc.M806270200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang B, Zhang Z, Xia S, Xing C, Ci X, Li X, et al. KLF5 activates microRNA 200 transcription to maintain epithelial characteristics and prevent induced epithelial-mesenchymal transition in epithelial cells. Mol Cell Biol. 2013;33:4919–35. doi: 10.1128/MCB.00787-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martin-Garrido A, Williams HC, Lee M, Seidel-Rogol B, Ci X, Dong JT, et al. Transforming growth factor beta inhibits platelet derived growth factor-induced vascular smooth muscle cell proliferation via Akt-independent, smad-mediated cyclin D1 downregulation. PLoS One. 2013;8 doi: 10.1371/journal.pone.0079657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu X, Zhu Z, Li W, Fu X, Su D, Fu L, et al. Chromodomain helicase DNA binding protein 5 plays a tumor suppressor role in human breast cancer. Breast Cancer Res. 2012;14:R73. doi: 10.1186/bcr3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Z, Zhang B, Li W, Fu L, Fu L, Zhu Z, et al. Epigenetic silencing of miR-203 upregulates SNAI2 and contributes to the invasiveness of malignant breast cancer cells. Genes Cancer. 2011;2:782–91. doi: 10.1177/1947601911429743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nagaraju GP, Park W, Wen J, Mahaseth H, Landry J, Farris AB, et al. Antiangiogenic effects of ganetespib in colorectal cancer mediated through inhibition of HIF-1alpha and STAT-3. Angiogenesis. 2013;16:903–17. doi: 10.1007/s10456-013-9373-6. [DOI] [PubMed] [Google Scholar]
- 70.Shi X, Liu M, Li D, Wang J, Aneja R, Zhou J. Cep70 contributes to angiogenesis by modulating microtubule rearrangement and stimulating cell polarization and migration. Cell Cycle. 2012;11:1554–63. doi: 10.4161/cc.19954. [DOI] [PubMed] [Google Scholar]
- 71.Sun X, Shi X, Liu M, Li D, Zhang L, Liu X, et al. Mdp3 is a novel microtubule-binding protein that regulates microtubule assembly and stability. Cell Cycle. 2011;10:3929–37. doi: 10.4161/cc.10.22.18106. [DOI] [PubMed] [Google Scholar]