

Abstract
Background
More than 1 million prostate biopsies are conducted yearly in the United States. The low specificity of prostate-specific antigen (PSA) results in diagnostic biopsies in men without prostate cancer (PCa). Additional information, such as genetic markers, could be used to avoid unnecessary biopsies.
Objective
To determine whether single nucleotide polymorphisms (SNPs) associated with PCa can be used to determine whether biopsy of the prostate is necessary.
Design, settings, and participants
The Stockholm-1 cohort (n = 5241) consisted of men who underwent a prostate biopsy during 2005 to 2007. PSA levels were retrieved from databases and family histories were obtained using a questionnaire. Thirty-five validated SNPs were analysed and converted into a genetic risk score that was implemented in a risk-prediction model.
Results and limitations
When comparing the nongenetic model (based on age, PSA, free-to-total PSA, and family history) with the genetic model and using a fixed number of detected PCa cases, it was found that the genetic model required significantly fewer biopsies than the nongenetic model, with 480 biopsies (22.7%) avoided, at a cost of missing a PCa diagnosis in 3% of patients characterised as having an aggressive disease. However, the overall genetic model does not discriminate between aggressive and nonaggressive cases.
Conclusion
Although the genetic model reduced the number of biopsies more than the nongenetic model, the clinical significance of this finding requires further evaluation.
Keywords: Diagnosis, Prostate cancer, Polygenic, Prediction model, SNP
1. Introduction
One major concern in the management of prostate cancer (PCa) is related to the overdiagnosis and subsequent overtreatment of the disease. Approximately 1 million biopsies are performed yearly in the United States, resulting in 192 000 new cases being diagnosed [1]. To decrease the number of unnecessary biopsies, prediction models with better specificity are needed to guide the clinician whether or not to recommend a biopsy of the prostate.
The majority of newly diagnosed patients will have undergone an ultrasound-guided prostate biopsy based on the presence of a prostate-specific antigen (PSA) level >3–4 ng/ml [2]. For values between 3 and 10 ng/ml, men have an estimated 20–25% risk of being diagnosed with PCa, although the majority of these men will not be diagnosed with the disease following an invasive examination [3]. New markers and predictive models, which can increase the test specificity, are therefore needed to reduce the number of unnecessary biopsies and eventually the number of men diagnosed with low-grade cancers.
In recent years, genome-wide association studies have been instrumental in identifying common genetic variants associated with complex diseases. PCa is no exception, with 36 validated genetic variants in 29 different chromosomal loci associated with disease risk. In PCa, the odds ratio (OR) associated with risk alleles varies from 1.1 to 1.6, with the majority at the lower end of the spectrum [4–15]. Due to these small increases in risk, several researchers have questioned the clinical utility of the genetic markers. However, in PCa, we have shown that the risk of disease accumulates with the increasing number of inherited risk alleles [16].
2. Patients and methods
2.1. Study population
From patient registries in two out of three pathology departments in Stockholm, Sweden, we identified 8088 men who had undergone at least one prostate biopsy between 1 January 2005 and 31 December 2007, representing 75% of all prostate biopsies conducted during this time period in Stockholm. We excluded men older than 80 yr at the time of biopsy (n = 468), men with no valid Swedish personal identification number [17] or with no valid address (n = 78), and men deceased at the time of study invitation (n = 343). Men with known PCa before 2005 were also excluded (n = 154), as were men with cancer other than adenocarcinoma detected upon prostate biopsy (n = 10).
In total, we invited 7035 men to participate in the study. The invitation included a questionnaire regarding family history of PCa. We received complete questionnaire data, written consent, and a blood sample from 5241 men (response rate: 75%) (Fig. 1). By linking the study subjects to the laboratory databases in Stockholm, we retrieved historical PSA values. We chose the PSA value closest in time to the date of biopsy, excluding values taken >130 d before. The different laboratories had different routines regarding saving results, which explains why there were more missing data on free PSA levels.
Fig. 1.
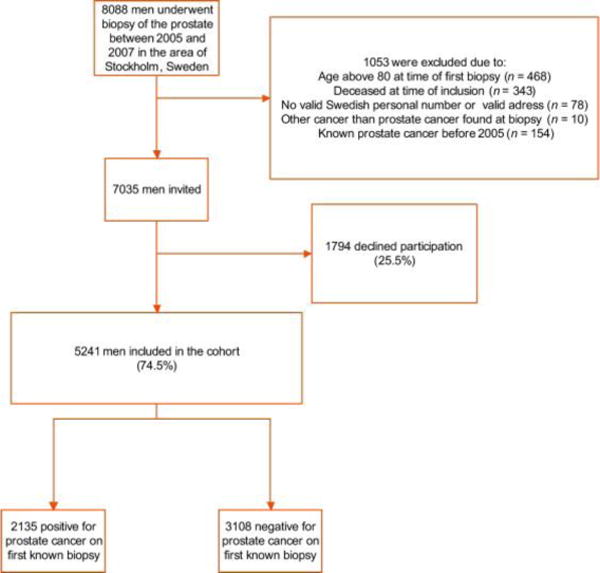
Enrolment and exclusion of participants of the Stockholm-1 study.
For information on cancer status, we linked the study participants to the Regional Cancer Registry and to the Stockholm part of the National Prostate Cancer Registry (NPCR); information on tumour stage (TNM) and grade (Gleason score) were obtained. All men diagnosed with PCa during 2005 and 2007 at the first known biopsy were classified as biopsy positive (n = 2135), and those with a noncancer diagnosis at the first known biopsy were defined as biopsy negative (n = 3106). Aggressive PCa was defined as T3-4 N1M1 or Gleason 4 + 3 and higher; nonaggressive PCa was defined as T0-2 N0/XM0/X or Gleason 3 + 4 and lower. Men with PSA >10 ng/ml were not included in the analysis concerning avoided prostate biopsies because they most likely would undergo biopsy anyway. The local ethics committee in Stockholm approved the study.
2.2. Single nucleotide polymorphism selection
We selected all published PCa-associated single nucleotide polymorphisms (SNPs; up to October 2009) reported and validated in at least one independent study population. We identified 36 SNPs in 29 chromosomal loci. Multiple SNPs have been reported in the chromosomal region 8q24. We selected eight independent SNPs from this region based on the publication of Al Olama et al [4]. The SNP rs10090154 in the 8q24 region was not included in the genotype design and was thus substituted for the proxy SNP rs1447295 [11].
2.3. Genotyping
Markers were genotyped using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry based on allele-specific primer extension with the iPLEX chemistry (SEQUENOM Inc, San Diego, CA, USA). Hardy-Weinberg calculations were performed to verify that each marker was within an allelic equilibrium in the control population. Thirty-six SNPs were genotyped in the entire sample with a 98.6% average success rate, excluding rs2660753 on chromosome 3p12, which failed completely.
2.4. Statistical analysis
All SNPs were tested for deviation from Hardy-Weinberg’s equilibrium using a permutation-based chi-square test. We assessed associations between each SNP and PCa at biopsy using a Cochran-Armitage trend test. Allelic ORs with 95% confidence intervals were computed using logistic regression models. For each man, a genetic risk score was created by summing the number of risk alleles (0,1, or 2) at each of the 35 SNPs multiplied by the logarithm of that SNP’s OR. Associations between PCa diagnosis and evaluated risk factors were explored in logistic regression analysis. Two prediction models were built to determine whether the genetic risk score could improve the prediction of PCa diagnosis: The nongenetic model included logarithmically transformed total PSA, the logarithmically transformed free-to-total PSA ratio, age at biopsy, and family history of PCa (yes or no); the genetic model also included the genetic risk score. A repeated 10-fold cross-validation was used to estimate the predicted probabilities of PCa at biopsy [18]. The genetic risk score was recalculated for each iteration of the cross-validation to preserve the independence between training and test sets, thus avoiding introducing a bias into the models’ predictive performance. The genetic model was compared with the nongenetic model in terms of (1) model fit, by likelihood ratio testing; (2) specificity, by the nonevents term of the net reclassification improvement (NRI) [19]; and (3) predictive performance, by using the cross-validated predicted probabilities to calculate the area under the receiver operating characteristic curves (AUC). Ninety-five percent confidence intervals for the AUC values were constructed using a normal approximation [20]. All reported p values are based on two-sided hypotheses.
By assuming a polygenic model and that the sibling relative risk of PCa is 2.5 [21], and by retracing the calculations in Pharoah et al [22], the predictive performance of a hypothetic genetic model can be estimated. This optimal genetic model includes a score variable constructed from SNPs explaining 100% of the population genetic risk and thus represents the optimal discrimination that can be obtained by adding genetic information to the prediction model.
3. Results
The basic characteristics of the study participants are summarised in Table 1. We successfully genotyped 5239 of the 5241 men initially included in the study. The overall results from the 35 SNPs are summarised in Table 2. In 21 of 35 SNPs, the initial reported finding was confirmed (p < 0.05). The strongest associations were observed on chromosome 8q24 rs1447295 (OR: 1.40; p < 0.0001); chromosome 19q13 rs2735839 (OR: 1.31; p < 0.0001); chromosome 22q13 rs5759167 (OR: 0.84; p < 0.0001); and chromosome 2q31 rs12621278 (OR: 0.68; p < 0.0001).
Table 1.
Number of biopsy-positive and -negative subjects and their clinical characteristics in the Stockholm-1 study
| Characteristics | Positive first biopsy (n = 2135)
|
Negative first biopsy (n = 3106)
|
|||
|---|---|---|---|---|---|
| PSA ≤10 ng/ml | All | PSA ≤10 ng/ml | All | ||
| Subjects, No. | 1359 | 2135 | 2432 | 3106 | |
| Mean age, yr (sd) | 65.0 (6.7) | 66.0 (6.9) | 63.7 (6.7) | 64.2 (6.8) | |
| Mean PSA, ng/ml (sd) | 5.9 (2.1) | 21.9 (141.1) | 5.49 (2.2) | 7.40 (6.6) | |
| Missing data (%) | 1 (0.07) | 109 (5.1) | 1 (0.04) | 230 (7.4) | |
| Mean f/t PSA (sd) | 0.15 (0.07) | 0.14 (0.07) | 0.18 (0.09) | 0.18 (0.09) | |
| Missing f/t PSA data (%) | 348 (25.6) | 696 (32.6) | 637 (26.2) | 1014 (32.6) | |
| PSA level, ng/ml (%) | |||||
| ≤4.0 | 263 (19.4) | 263 (12.3) | 639 (26.3) | 639 (20.6) | |
| >4.0–≤10.0 | 1095 (80.6) | 1095 (51.3) | 1792 (73.7) | 1792 (57.7) | |
| >10.0–≤20.0 | – | 383 (17.9) | – | 342 (11.0) | |
| >20.0 | – | 285 (13.3) | – | 103 (3.3) | |
| Missing data (%) | 1 (0.07) | 109 (5.1) | 1 (0.04) | 230 (7.4) | |
| F/t PSA level, No. (%) | |||||
| ≤0.09 | 179 (13.2) | 364 (17.0) | 180 (7.4) | 238 (7.7) | |
| 0.10–0.18 | 597 (43.9) | 799 (37.4) | 991 (40.7) | 1144 (36.8) | |
| >0.18 | 235 (173) | 276 (12.9) | 624 (25.7) | 710 (22.9) | |
| Missing data (%) | 348 (25.6) | 696 (32.6) | 637 (26.2) | 1014 (32.5) | |
| Family history of prostate cancer (%) | Yes | 399 (29.4) | 620 (29.0) | 545 (22.4) | 679 (21.9) |
| No | 958 (70.5) | 1511 (70.8) | 1884 (77.5) | 2422 (78.0) | |
| Missing data (%) | 2 (0.1) | 4 (0.2) | 3 (0.1) | 5 (0.2) | |
| Tumor stage (%) | T0, T1, Tx | 683 (50.3) | 935 (43.8) | NA | |
| T2 | 356 (26.2) | 567 (26.6) | NA | ||
| T3 | 95 (7.0) | 245 (11.5) | NA | ||
| T4 | 0 (0) | 6 (0.2) | NA | ||
| Missing data | 225 (16.5) | 382 (17.9) | NA | ||
| Nodal stage (%) | N0/X | 1103 (81.2) | 1685 (78.9) | NA | |
| N1 | 4 (0.3) | 30 (1.4) | NA | ||
| Missing data | 252 (18.4) | 420 (19.7) | NA | ||
| Metastasis stage (%) | M0/X | 1103 (81.2) | 1688 (79.1) | NA | |
| M1 | 3 (0.2) | 28 (1.3) | NA | ||
| Missing data | 253 (18.6) | 419 (19.6) | NA | ||
| Gleason score (%) | ≤5 | 87 (6.4) | 118 (5.5) | NA | |
| 6 | 631 (46.4) | 822 (38.5) | NA | ||
| 3 + 4 = 7 | 264 (19.4) | 440 (20.6) | NA | ||
| 4 + 3 = 7 | 112 (8.2) | 208 (9.7) | NA | ||
| 8 | 40 (2.9) | 118 (5.5) | NA | ||
| ≥9 | 18 (1.3) | 76 (3.6) | NA | ||
| Missing data | 207 (15.2) | 353 (16.5) | NA | ||
PSA = prostate-specific antigen; sd = standard deviation; f/t = free-to-total; NA = not applicable.
Table 2.
Genotype results from the Stockholm-1 study on established, common, prostate-cancer-susceptibility alleles
| dbSNP No. | Chromosome | Gene* | Risk allele† | Association results in the Stockholm-1 study
|
|||
|---|---|---|---|---|---|---|---|
| Risk allele frequency | Relative risk per allele | p value | Study | ||||
| rs1465618 | 2p21 | THADA | A | 0.24 | 1.08 | 8.5E-02 | Eeles et al [5] |
| rs721048 | 2p15 | EHBP1 | A | 0.19 | 1.14 | 9.9E-03 | Gudmundsson et al [8] |
| rs12621278 | 2q31.1 | ITGA6 | A | 0.94 | 1.47 | 5.1E-05 | Eeles et al [5] |
| rs4857841 | 3q21.3 | EEFSEC | A | 0.31 | 1.03 | 4.6E-01 | Gudmundsson et al [7] |
| rs12500426 | 4q22.3 | PDLIM5 | A | 0.47 | 1.06 | 1.5E-01 | Eeles et al [5] |
| rs17021918 | 4q22.3 | PDLIM5 | C | 0.65 | 1.06 | 1.6E-01 | Eeles et al [5] |
| rs7679673 | 4q24 | FLJ20032 | C | 0.59 | 1.12 | 5.3E-03 | Eeles et al [5] |
| rs9364554 | 6q25.3 | SLC22A3 | T | 0.30 | 1.02 | 7.0E-01 | Eeles et al [6] |
| rs10486567 | 7p15.2 | JAZF1 | G | 0.77 | 1.09 | 8.5E-02 | Thomas et al [9] |
| rs6465657 | 7q21.3 | LMTK2 | C | 0.49 | 0.98 | 6.8E-01 | Eeles et al [6] |
| rs1512268 | 8p21.2 | NKX3-1 | T | 0.45 | 1.01 | 8.5E-01 | Eeles et al [5] |
| rs12543663 | 8q24.21 | C | 0.30 | 1.00 | 9.8E-01 | Al Olama et al [4] | |
| rs10086908 | 8q24.21 | T | 0.68 | 1.09 | 4.4E-02 | Al Olama et al [4] | |
| rs1016343 | 8q24.21 | T | 0.25 | 1.11 | 1.9E-02 | Al Olama et al [4] | |
| rs13252298 | 8q24.21 | A | 0.72 | 1.11 | 2.3E-02 | Al Olama et al [4] | |
| rs6983561 | 8q24.21 | C | 0.04 | 1.27 | 9.9E-03 | Al Olama et al [4] | |
| rs16901979 | 8q24.21 | A | 0.04 | 1.29 | 5.2E-03 | Gudmundsson et al [15] | |
| rs16902094 | 8q24.21 | G | 0.16 | 1.12 | 3.3E-02 | Gudmundsson et al [7] | |
| rs445114 | 8q24.21 | T | 0.67 | 1.05 | 2.5E-01 | Gudmundsson et al [7] | |
| rs620861 | 8q24.21 | C | 0.67 | 1.04 | 4.1E-01 | Al Olama et al [4] | |
| rs6983267 | 8q24.21 | G | 0.53 | 1.19 | 9.8E-06 | Al Olama et al [4] | |
| rs1447295 | 8q24.21 | A | 0.12 | 1.40 | 2.8E-09 | Amundadottir et al [11] | |
| rs10993994 | 10q11.23 | MSMB | T | 0.38 | 1.15 | 4.0E-04 | Eeles et al [6] |
| rs4962416 | 10q26.13 | CTBP2 | C | 0.24 | 1.04 | 3.9E-01 | Thomas et al [9] |
| rs7127900 | 11p15.5 | A | 0.22 | 1.16 | 1.3E-03 | Eeles et al [5] | |
| rs12418451 | 11q13.2 | A | 0.29 | 1.03 | 5.6E-01 | Zheng et al [10] | |
| rs11228565 | 11q13.2 | A | 0.20 | 1.07 | 1.5E-01 | Gudmundsson et al [7] | |
| rs10896449 | 11q13.2 | G | 0.48 | 1.11 | 1.2E-02 | Thomas et al [9] | |
| rs11649743 | 17q12 | HNF1B | G | 0.79 | 1.11 | 3.2E-02 | Sun et al [13] |
| rs4430796 | 17q12 | HNF1B | A | 0.57 | 1.17 | 1.3E-04 | Gudmundsson et al [12] |
| rs1859962 | 17q24.3 | G | 0.49 | 1.13 | 1.5E-03 | Gudmundsson et al [12] | |
| rs8102476 | 19q13.2 | PPP1R14A | C | 0.52 | 1.12 | 3.8E-03 | Gudmundsson et al [7] |
| rs2735839 | 19q13.33 | KLK3 | A | 0.09 | 1.31 | 1.9E-05 | Eeles et al [6] |
| rs9623117 | 22q13.1 | TNRC6B | C | 0.20 | 1.08 | 1.1E-01 | Sun et al [14] |
| rs5759167 | 22q13.2 | BIK | G | 0.51 | 1.19 | 1.7E-05 | Eeles et al [5] |
| rs5945619 | Xp11.22 | NUDT11 | C | 0.38 | 1.27 | 3.1E-05 | Eeles et al [6] |
THADA = thyroid adenoma-associated isoform 1; EHBP1 = EH domain-binding protein 1; ITGA6 = integrin alpha chain 6; EEFSEC = elongation factor for selenoprotein translation; PDLIM5 = PDZ and LIM domain 5 isoform d; FLJ20032 = hypothetic protein LOC54790; SLC22A3 = solute-carrier family 22 member 3; JAZF1 = juxtaposed with another zinc finger gene 1; LMTK2 = lemur tyrosine kinase 2; SLC25A37 = mitochondrial solute-carrier protein; NKX3-1 = NK3 transcription factor-related locus 1; MSMB = beta-microseminoprotein isoform a precursor; CTBP2 = C-terminal binding protein 2 isoform 2; HFN1B = hepatocyte nuclear factor 1 homeobox B; PPP1R14A = protein phosphatase 1 regulatory inhibitor; KLK3 = prostate-specific antigen isoform 6; TNRC6B = trinucleotide repeat containing 6B isoform 2; BIK = BCL2-interacting killer; NUDT11 = nudix-type motif 11.
These genes are within the linkage-disequilibrium block defined by the associated variant.
Risk alleles as defined from published data cited in the table.
In further tests we restricted our analysis to men with PSA ≤10 ng/ml since there is less debate over recommending a prostate biopsy in men with a PSA >10 ng/ml, at least in cases where curative treatment is an option. We analysed the 2542 men with PSA ≤10 ng/ml and completed the data on all variables (PSA, free-to-total PSA, age, family history, and genetic score). Although all risk factors were significantly associated with a positive biopsy (Table 3), the strongest association was recorded for the combined genetic score (p < 0.0001). The predictive performance of the individual variables varied from an AUC for a positive family history of 52.0%, to 61.3% for the combined genetic score. When using the nongenetic model (ie, when combining all variables except the genetic score), the AUC increased significantly from 55.0% for PSA only, to 64.2% (Fig. 2a). By using the genetic model, the AUC was significantly improved to 67.4% (p = 0.014). The genetic model had a significantly better model fit compared with the nongenetic model (likelihood ratio test, p < 0.0001). Moreover, and most important, the genetic model had a significantly higher specificity (nonevents term of NRI, p < 0.0001); that is, it avoided significantly more biopsies than the nongenetic model for a given percentage of detected cancer. For comparison purposes, Fig. 2a also shows the results for the hypothetic genetic models (ie, if all risk SNPs were known). Fig. 2c shows the relationship between percentage of detected cancer and percentage of saved biopsies for the nongenetic and the genetic models. So far, no validated SNPs have been shown to be associated with aggressive disease rather than indolent disease. This also was the case in this study, as shown in Fig. 2b, which illustrates that the discrimination between indolent and aggressive disease is not significantly better for the genetic model compared with the nongenetic model (p = 0.83 when testing for AUC improvement).
Table 3.
Univariate and multivariate logistic regression of the risk factors for all cancers among men (n = 2542) with prostate-specific antigen (PSA) =10 ng/ml and complete information on the risk factors (age, PSA, free-to-total PSA, family history of prostate cancer, and genetic score)
| Univariate
|
Multivariate
|
|||||
|---|---|---|---|---|---|---|
| OR (95% CI) | p value | AUC (95% CI) | OR (95% CI) | p value | AUC cumulative (95% CI) | |
| PSA | 1.19 (1.14–1.24) | <0.0001 | 0.55 (0.53–0.57) | 1.17 (1.12–1.22) | 0.017 | 0.55 (0.53–0.57) |
| Free-to-total PSA | 0.68 (0.65–0.72) | <0.0001 | 0.59 (0.57–0.62) | 0.69 (0.66–0.73) | <0.0001 | 0.60 (0.58–0.62) |
| Age | 1.23 (1.18–1.29) | <0.0001 | 0.56 (0.54–0.58) | 1.17 (1.12–1.22 | <0.0001 | 0.63 (0.60–0.65) |
| Family history | 1.20 (1.16–1.25) | <0.0001 | 0.52 (0.50–0.54) | 1.41 (1.29–1.54) | 0.0001 | 0.64 (0.62–0.66) |
| Genetic score | 1.93 (1.85–2.01) | <0.0001 | 0.61 (0.59–0.63) | 1.52 (1.45–1.59) | <0.0001 | 0.67 (0.65–0.70) |
OR = odds ratio; CI = confidence interval; AUC = area under the cross-validated receiver operating characteristics curve.
Predictive performance was assessed using the AUC for individual risk factors and for the genetic model including all risk factors. AUC cumulative denotes AUC values obtained when one risk factor at the time was added to the model from PSA only to a model including all risk factors.
Fig. 2.
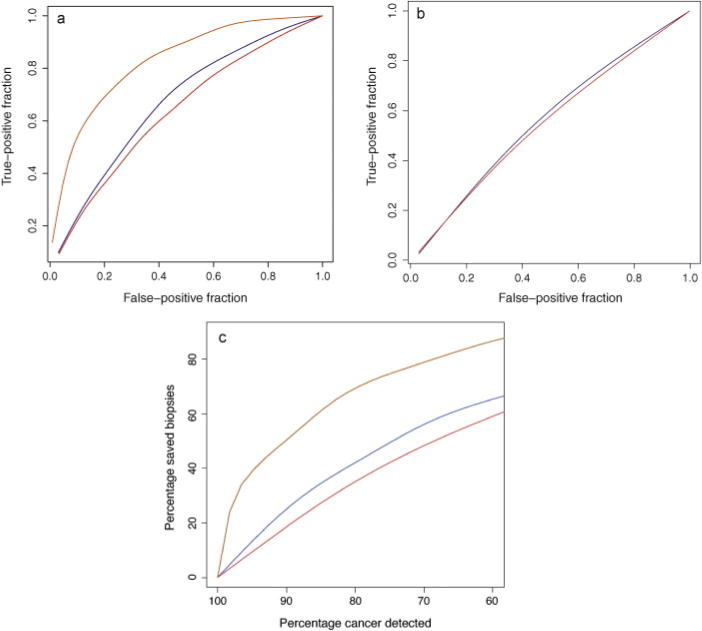
(a) Receiver operating characteristics (ROC) curve for the nongenetic (red), genetic (blue), and hypothetic (orange) model. (b) ROC curve for the nongenetic (red) and genetic (blue) models for discriminating between aggressive and indolent disease. A hypothetic model is not represented here since the heritability of aggressive cancer is not known. The genetic model does not discriminate between indolent and aggressive disease. (c) Relationship between percentage-saved biopsies and percentage-detected cancer when using the nongenetic model (red), the genetic model (blue), and the hypothetic genetic model (green). The results are based on men in the Stockholm-1 cohort study with prostate-specific antigen =10 ng/ml and complete information on the risk factors (n = 2542).
In a clinical setting it is relevant to study the models’ performance at different risk cut-off points. Using the nongenetic model and a 25% estimated risk cut-off point, 256 biopsies (10.0%) would be saved. Of these 256 biopsies, 185 (72%) were negative and 71 (28%) were positive. However, using this strategy, 8 (5%) of the aggressive cases would be missed and the men who would not be recommended a biopsy would have a 3% risk (8 of 256 cases) of having an aggressive cancer at biopsy. Using a 20% cut-off point, the number of missed aggressive cases would decrease to 2 of 143 (1%), but at the cost of doing an additional 166 biopsies.
Adding the genetic score substantially improved the model, and the number of avoided biopsies increased. Using the 25% cut-off point, 480 biopsies were avoided (22.7%), 392 men were biopsy negative (82%), and 88 were biopsy positive (18%). The problem with missing aggressive cancer cases was similar to the situation without the genetic score, with 14 missed cases (9%) and a 14 in 480 cases (3%) risk of having an aggressive cancer. A 20% cut-off point would decrease the number of missed aggressive cancers substantially from 14 to 3 cases at a cost of 267 additional biopsies.
If all PCa-risk SNPs were known, the hypothetic genetic model would, at a 20% risk cut-off point, save 25.5% of the biopsies. The cost of this would be that roughly 2% of the aggressive cancers would be missed (Table 4).
Table 4.
Biopsies conducted and cancer detected per 1000 men with a clinical prostate biopsy based on the results from the Stockholm-1 study using a cross-validated predicted risk cut-off point of 20% or 25% as the threshold for biopsy for the nongenetic model, the genetic model, and the optimal genetic model
| Model, % | Performed | Not performed | % | Cancer detected | Cancer missed | % | Aggressive cancer detected | Aggressive cancer missed | % |
|---|---|---|---|---|---|---|---|---|---|
| Nongenetic | 1000 | 0 | 0 | 365 | 0 | 0 | 60 | 0 | 0 |
| 20 | 949 | 51 | 5.10 | 352 | 13 | 3.60 | 59 | 1 | 1.70 |
| 25 | 871 | 129 | 12.90 | 338 | 27 | 7.40 | 56 | 4 | 6.70 |
| Genetic | |||||||||
| 20 | 878 | 122 | 12.20 | 344 | 21 | 5.80 | 58 | 2 | 3.30 |
| 25 | 773 | 227 | 22.70 | 321 | 44 | 12 | 55 | 5 | 8.30 |
| Optimal genetic | |||||||||
| 20 | 745 | 255 | 25.50 | 348 | 17 | 4.70 | 59* | 1* | 1.70 |
| 25 | 686 | 314 | 31.40 | 340 | 25 | 6.80 | 57* | 3* | 5 |
4. Discussion
In this large population-based study, we showed that adding a genetic score based on 35 SNPs significantly improved the specificity of the prediction model for prostate biopsies compared to models based on clinical markers used today for men with a PSA level ≤10 ng/ml. The AUC modestly, but significantly, increased from 0.64 to 0.67 when the genetic markers were added to the model. The clinical interpretation of this increment in the AUC is not obvious, thus we need to translate it into the number of biopsies that could be avoided without missing aggressive PCa at different predicted levels of individual risk. Using an estimated 20% risk of PCa as the cut-off point for conducting a biopsy, approximately 12% of the biopsies would not be performed using our genetic model compared with 5% without the nongenetic model (Table 4). This improvement could be made without missing >1% of the aggressive cancers in the population. The exact cut-off point of the risk estimate can be discussed. In our study, we observed that with a risk cut-off point of 25%, the number of saved biopsies increased to 23%, but at a cost of missing almost 10% of the aggressive cases, which in most circumstances is not acceptable.
In this prediction model we incorporated a genetic score based on the first 35 SNPs associated with PCa. In the near future we estimate that approximately 100 new PCa SNPs will be discovered by different international consortia and these probably will improve the model. In addition, hitherto-reported PCa-susceptibility SNPs show limited ability to distinguish between aggressive and indolent disease [23]. Markers of aggressive disease will be important to incorporate in future prediction models.
Our model is based on a population-based cohort of men who underwent biopsies for suspected PCa in the Stockholm area during 2005–2007. However, the final analysis was only based on approximately 70% of all men who underwent biopsies in Stockholm during these years due to the fact that one major health care provider was not included in the study. No significant differences were found on comparing the clinical data between the biopsy-positive cases included in our analysis to the NPCR in the Stockholm area. Most likely this means that our results are representative and referable to current biopsy populations in Sweden. However, in populations where the prevalence of a disease (percent positive biopsies) is lower, the specificity of the model will most likely improve. This is a likely scenario in countries where PSA is used more extensively than in Sweden (eg, the United States) and in PSA screening trials. Our models were based on a Caucasian population, and these models need to be validated in other ethnic groups.
The introduction of personalised medicine based on SNPs has been widely discussed, and there are a number of requirements that need to be met before taking these markers into the clinic [24]. We present evidence to show that SNPs can be used in a specific and important clinical situation: the decision to perform a prostate biopsy or not. There are several advantages with using SNP markers: They are easy to analyse, stable (need to be analysed only once), and affordable.
5. Conclusions
Genetic profiles based on the common genetic risk variants established to date improve the predictive accuracy of PCa diagnosis. As many more genetic risk variants are expected to be identified in the near future, the usefulness of genetic profiles is expected to increase. Large prospective clinical studies exploring the optimal use of genetic risk profiles in PCa diagnostics are highly warranted.
Acknowledgments
We thank and acknowledge all of the participants in the Stockholm-1 study. We thank Carin Cavalli-Björkman and Ami Rönnberg Karlsson for their dedicated work in the collection of data. Michael Broms is acknowledged for his skilful work with the databases. KI Biobank is acknowledged for handling the samples and for DNA extraction. Hans Wallinder at Aleris Medilab and Sven Gustafsson at Karolinska University Laboratory are thanked for their good cooperation in providing historical laboratory results. We thank the Stockholm Prostate Cancer Group and its representaitves in the NPCR, Magnus Törnblom and Stefan Carlsson, for the acquisition of clinical data.
Funding/Support and role of the sponsor: Swedish Research Council (grant no. K2010-70X-20430-04-3, and 70867901); the Swedish Cancer Foundation (grant no. 09-0677); the Hedlund Foundation; the Söderberg Foundation; the Enqvist Foundation; ALF funds from the Stockholm County Council; Stiftelsen Johanna Hagstrand och Sigfrid Linnér’s Minne; and Karlsson’s Fund for urologic and surgical research. This study was supported by the Cancer Risk Prediction Center (CRisP; http://www.crispcenter.org), a Linneus Centre (Contract ID 70867902) financed by the Swedish research Council.
Footnotes
Author contributions: Henrik Grönberg had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Aly, Grönberg, Adolfsson, Wiklund, Eklund, D’amato, Isaacs, Xu.
Acquisition of data: Aly, Adolfsson, Grönberg.
Analysis and interpretation of data: Aly, Eklund, Wiklund, Grönberg, Adolfsson, Xu, Isaacs, D’amato.
Drafting of the manuscript: Aly, Wiklund, Grönberg, Eklund.
Critical revision of the manuscript for important intellectual content: Aly, Adolfsson, Wiklund, Grönberg, Eklund, Xu, Isaacs, D’amato.
Statistical analysis: Aly, Wiklund, Eklund.
Obtaining funding: Aly, Grönberg.
Administrative, technical, or material support: Grönberg, Adolfsson, Aly, D’amato.
Supervision: Grönberg.
Other (specify): None.
Financial disclosures: I certify that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: None.
Please visit, www.eu-acme.org/europeanurology, to read and answer questions on-line. The EU-ACME credits will then be attributed automatically.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Adolfsson J, Garmo H, Varenhorst E, et al. Clinical characteristics and primary treatment of prostate cancer in Sweden between 1996 and 2005. Scand J Urol Nephrol. 2007;41:456–77. doi: 10.1080/00365590701673625. [DOI] [PubMed] [Google Scholar]
- 3.Morote J, Trilla E, Esquena S, et al. The percentage of free prostatic-specific antigen is also useful in men with normal digital rectal examination and serum prostatic-specific antigen between 10.1 and 20 ng/ml. Eur Urol. 2002;42:333–7. doi: 10.1016/s0302-2838(02)00318-4. [DOI] [PubMed] [Google Scholar]
- 4.Al Olama AA, Kote-Jarai Z, Giles GG, et al. Multiple loci on 8q24 associated with prostate cancer susceptibility. Nat Genet. 2009;41:1058–60. doi: 10.1038/ng.452. [DOI] [PubMed] [Google Scholar]
- 5.Eeles RA, Kote-Jarai Z, Al Olama AA, et al. Identification of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat Genet. 2009;41:1116–21. doi: 10.1038/ng.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eeles RA, Kote-Jarai Z, Giles GG, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008;40:316–21. doi: 10.1038/ng.90. [DOI] [PubMed] [Google Scholar]
- 7.Gudmundsson J, Sulem P, Gudbjartsson DF, et al. Genome-wide association and replication studies identify four variants associated with prostate cancer susceptibility. Nat Genet. 2009;41:1122–6. doi: 10.1038/ng.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gudmundsson J, Sulem P, Rafnar T, et al. Common sequence variants on 2p15 and Xp11.22 confer susceptibility to prostate cancer. Nat Genet. 2008;40:281–3. doi: 10.1038/ng.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas G, Jacobs KB, Yeager M, et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet. 2008;40:310–5. doi: 10.1038/ng.91. [DOI] [PubMed] [Google Scholar]
- 10.Zheng SL, Stevens VL, Wiklund F, et al. Two independent prostate cancer risk-associated loci at 11q13. Cancer Epidemiol Biomarkers Prev. 2009;18:1815–20. doi: 10.1158/1055-9965.EPI-08-0983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amundadottir LT, Sulem P, Gudmundsson J, et al. A common variant associated with prostate cancer in European and African populations. Nat Genet. 2006;38:652–8. doi: 10.1038/ng1808. [DOI] [PubMed] [Google Scholar]
- 12.Gudmundsson J, Sulem P, Steinthorsdottir V, et al. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat Genet. 2007;39:977–83. doi: 10.1038/ng2062. [DOI] [PubMed] [Google Scholar]
- 13.Sun J, Zheng SL, Wiklund F, et al. Evidence for two independent prostate cancer risk-associated loci in the HNF1B gene at 17q12. Nat Genet. 2008;40:1153–5. doi: 10.1038/ng.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun J, Zheng SL, Wiklund F, et al. Sequence variants at 22q13 are associated with prostate cancer risk. Cancer Res. 2009;69:10–5. doi: 10.1158/0008-5472.CAN-08-3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gudmundsson J, Sulem P, Manolescu A, et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007;39:631–7. doi: 10.1038/ng1999. [DOI] [PubMed] [Google Scholar]
- 16.Zheng SL, Sun J, Wiklund F, et al. Cumulative association of five genetic variants with prostate cancer. N Engl J Med. 2008;358:910–9. doi: 10.1056/NEJMoa075819. [DOI] [PubMed] [Google Scholar]
- 17.Ludvigsson JF, Otterblad-Olausson P, Pettersson BU, Ekbom A. The Swedish personal identity number: possibilities and pitfalls in healthcare and medical research. Eur J Epidemiol. 2009;24:659–67. doi: 10.1007/s10654-009-9350-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hastie T, Tibshirani R, Friedman J. The elements of statistical learning. 2. New York, NY: Springer; 2009. [Google Scholar]
- 19.Pencina MJ, D’Agostino RB, Sr, D’Agostino RB, Jr, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27:157–72. doi: 10.1002/sim.2929. discussion 207–12. [DOI] [PubMed] [Google Scholar]
- 20.Newson R. Confidence intervals for rank statistics: Somers’ D and extensions. Stata J. 2006;6:309–34. [Google Scholar]
- 21.Johns LE, Houlston RS. A systematic review and meta-analysis of familial prostate cancer risk. BJU Int. 2003;91:789–94. doi: 10.1046/j.1464-410x.2003.04232.x. [DOI] [PubMed] [Google Scholar]
- 22.Pharoah PD, Antoniou A, Bobrow M, Zimmern RL, Easton DF, Ponder BA. Polygenic susceptibility to breast cancer and implications for prevention. Nat Genet. 2002;31:33–6. doi: 10.1038/ng853. [DOI] [PubMed] [Google Scholar]
- 23.Wiklund F. Prostate cancer genomics: can we distinguish between indolent and fatal disease using genetic markers? Genome Med. 2010;2:45. doi: 10.1186/gm166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guttmacher AE, McGuire AL, Ponder B, Stefansson K. Personalized genomic information: preparing for the future of genetic medicine. Nat Rev Genet. 2010;11:161–5. doi: 10.1038/nrg2735. [DOI] [PubMed] [Google Scholar]