

Abstract
Purpose
Recurrent gene mutations, chromosomal translocations and acquired genomic copy number aberrations (aCNA) have been variously associated with AML patient outcome. However, knowledge of the co-occurrence of such lesions and the relative influence of different types of genomic alterations on clinical outcomes in AML is still evolving.
Experimental Design
We performed SNP 6.0 array-based genomic profiling of aCNA/cnLOH along with sequence analysis of 13 commonly mutated genes on purified leukemic blast DNA from 156 prospectively enrolled non-FAB-M3 AML patients across the clinical spectrum of de novo, secondary, and therapy-related AML.
Results
TP53 and RUNX1 mutations are strongly associated with the presence of SNP-A-based aCNA/cnLOH, while FLT3 and NPM1 mutations are strongly associated with the absence of aCNA/cnLOH. The presence of mutations in RUNX1, ASXL1 and TP53, elevated SNP-A-based genomic complexity, and specific recurrent aCNAs predicted failure to achieve a complete response to induction chemotherapy. The presence of ≥1 aCNA/cnLOH and higher thresholds predicted for poor long-term survival irrespective of TP53 status, and the presence of ≥1 aCNA/cnLOH added negative prognostic information to knowledge of mutations in TET2, IDH1, NPM1, DNMT3A and RUNX1. Results of multivariate analyses support a dominant role for TP53 mutations and a role for elevated genomic complexity as predictors of short survival in AML.
Conclusions
Integrated genomic profiling of a clinically relevant adult AML cohort identified genomic aberrations most associated with SNP-A-based genomic complexity, resistance to intensive induction therapies and shortened overall survival. Identifying SNP-A-based lesions adds prognostic value to the status of several recurrently mutated genes.
Keywords: AML, genomic copy number aberrations, gene mutations, refractory disease, survival
INTRODUCTION
Substantial progress has been made in the characterization of recurrent genomic aberrations in acute myelogenous leukemia (AML) and their association with clinical outcomes. In particular, risk groups of AML based on karyotype-based cytogenetics remain important in daily clinical decision-making (1–3). Over the last few years, several groups have extended the discovery of multiple recurrently mutated protein-coding genes in AML by reporting the prognostic and therapeutically predictive significance of individual mutations (including NPM1 (4–6), FLT3 (7–9), CEBPA (10,11), DNMT3A (12–14), RUNX1 (15–17), TET2 (18,19), ASXL1 (20,21), IDH1 (22,23), IDH2 (24,25), NRAS/KRAS (26,27), TP53 (28–30) and others (31–34)) as well as combinations of these mutations (35–38) in AML patient cohorts. In addition, several groups have published studies demonstrating the correlation of specific SNP array-detected aCNA or the total number of aCNA/cnLOH (genomic complexity) with clinical outcomes (29,39–43). Importantly, however, the relative influence on clinical AML outcomes of different types of genomic alterations, including gene mutations, structural genomic changes or aCNA/cnLOH, remains unclear as lesion types rarely occur in isolation. While a few integrated analyses of the patterns of co-occurrence of various genomic alterations have been published (44), these studies have not extended findings to the study of chemoresistance and survival.
The majority of studies assessing the prognostic value of mutated genes in AML have been restricted to AML with normal karyotype (NK-AML) since this category is clinically heterogeneous, lacks a prognostic structural genomic marker and because some mutated genes are enriched in NK-AML. The published cohorts studied often are selected based on clinically-derived classifications of AML subgroups, including secondary AML (sAML) that arises in the setting of an antecedent myeloid neoplasm, and de novo AML (dnAML) that presents without a history of either previous therapy or a pre-existing myeloid neoplasm. Since many of these studies are based on cohorts of patients enrolled on therapeutic trials, they are subject to selection based on age and other inclusion and exclusion criteria, which adds further restrictions on the population analyzed.
To further refine basic biological knowledge of patterns of occurrence of genomic lesion types in AML and of the effects of various genomic aberration types on AML outcome, we profiled a consecutively enrolled prospective AML cohort for aCNA/cnLOH using SNP 6.0 array profiling and determined the mutation status of 13 recurrently mutated genes using Sanger sequencing. This approach has allowed us to integrate multiple genomic features across all major clinical subgroups of non-M3 AML and across the full age range of adult patients. Here we identify genomic traits that associate with clinical AML subtypes, gene mutations that associate with genomic complexity or stability, and genomic traits that associate with sensitivity or resistance to standard induction therapy and ultimately survival.
METHODS
Patients
Between March 2005 and June 2011, 173 patients with previously untreated non-M3 AML were enrolled into this study at the University of Michigan Comprehensive Cancer Center. The study was approved by the University of Michigan Institutional Review Board (IRBMED #2004-1022) and written informed consent was obtained from all patients prior to enrollment. Of the samples from 173 patients, 156 resulted in both array-based data for paired samples (blasts versus buccal DNA [N=154] or remission marrow DNA [N=2]) and complete mutational profiling. Cytogenetic risk stratification was based on SWOG criteria (1). Induction chemotherapy was categorized as “high-intensity” if it included an anthracyline combined with cytarabine or other cytotoxic agents or if it included high-dose cytarabine or clofarabine alone or combined with other agents. Regimens including only DNA hypomethylating agents, lenolidamide or small molecule inhibitors were defined as “low-intensity” (Supplementary Tables 1 and 2).
Cell Isolation and Preparation of Sample DNA
Peripheral blood or bone marrow mononuclear cells from AML patients were isolated using Ficoll gradient separation followed by cryopreservation as previously described (45). Briefly, AML blast DNA used for SNP 6.0 profiling and DNA sequencing was extracted from thawed AML cell samples that were negatively column enriched for blasts using anti-human CD3 (Miltenyi Biotec [MB] 130-050-101), anti-human CD14 (if blasts were negative for CD14 expression; MB 130-050-201), anti-human CD19 (if blasts were negative for CD19 expression; MB 130-050-301), and anti-human CD235a (MB 130-050-301) microbeads to remove lymphocytes, mature monocytes, and nucleated red blood cells. The enriched samples were washed and stained with fluorescein isothiocyanate-conjugated anti-CD33, phycoerythrin-conjugated anti-CD13, and allophycocyanin-conjugated anti-CD45 (all antibodies: eBioscience). Live cells (PI-negative) were gated for blasts by identifying cells with intermediate-intensity staining for CD45 and low- to moderate-intensity side scatter. CD33 and CD13 were used to further discriminate blasts versus erythroid and mature myeloid lineage cells. This strategy resulted in a ≥90% pure blast population (45) that was confirmed with morphological inspection of cytospins. DNA was extracted as previously described (46).
Exon resequencing of 13 recurrently mutated genes in AML
All coding exons and adjacent intronic sequences of CEBPA, ASXL1, TET2, DNMT3A, RUNX1, and BCORL1 along with exon 12 of NPM1, exons 13–15 and 20 of FLT3, exons 2–10 of TP53, exons 2–3 of KRAS and NRAS, exon 2 of IDH1, and exon 4 of IDH2 with adjacent intronic sequences were resequenced using PCR and sequencing primers designed with the primer3 tool; sequence information was generated as previously described (45). All mutations were confirmed using unamplified tumor DNA and paired patient buccal DNA as templates.
Array Data Analysis
Affymetrix CEL files for each blast and buccal sample underwent initial quality control screening with the Genotyping Console program afterwhich Affymetrix “Birdseed” algorithm generated SNP call files with call rates for the entire sample group between 93.64% and 99.45%, with a mean call rate of 98.17%; none gave out-of-bounds results.
Genomic copy number heatmaps were generated as previously described (45). Two independent observers visually inspected parallel heatmap copy number images of AML blast and paired normal DNA samples generated through dChip. Only those copy number changes detected in blast DNA but not in paired normal DNA were called somatic, and lesions had to be ≥30 SNP positions in length to be scored positive. All visually determined aCNA were corroborated with an algorithmic lesion calling method as previously described (47). LOH analysis was performed using LOH tool v2 as previously described (29). All resulting aCNA/cnLOH are listed in Supplementary Table 3 and SNP 6.0 array data files for all 156 patient samples analyzed were deposited in the GEO public database (accession number GSE61323).
TCGA Data Analysis
The recently published AML cohort of The Cancer Genome Atlas (TCGA) provided additional clinically-annotated gene mutation and SNP-A patient data (44). To facilitate a meaningful comparison of aCNA/cnLOH from TCGA with the Michigan cohort, SNP 6.0 Affymetrix CEL files from paired normal and tumor TCGA AML specimens were downloaded from dbGaP after obtaining data access approval (dbGaP approval #29753-3). These files were analyzed with the identical informatics pre-processing pipeline and visual and algorithmic lesion calling methods used for the Michigan SNP 6.0 arrays above.
Of the 200 published matched pairs, twenty were excluded due to a diagnosis of APL (AML-M3), two for out-of-bounds results (TCGA-2855 and -2883), two for failure to pass the “Birdseed” algorithm (TCGA-2848 and -2981), and six for high visual and algorithmic background noise that precluded accurate lesion calling (TCGA-2802, -2833, -2843, -2847, -2876 and -2891). This resulted in 170 matched pairs with a complete catalog of aCNA/cnLOH, which is included in Supplementary Table 3. Gene mutation status for the thirteen genes of interest in this study and clinical characteristics including survival data based on the 5/13/2013 update were obtained from published supplementary data (44). Notable differences in the genomic profiling of TCGA specimens compared with the Michigan cohort include the use of whole exome or genome sequencing followed by non-Sanger based confirmation of gene mutation status and the use of tumor samples that were not enriched for leukemic cells.
Statistical Methods
Overall survival was defined as time (in months) between AML diagnosis and the patient’s death. For patients still alive the censoring date was September 1, 2013. Univariate and bivariate analyses were based on Kaplan-Meier estimates of survivor functions. Median survival times were estimated directly from the survivor function estimates. Significance levels for group-wise comparisons in the univariate analyses assess whether the hazard ratio between groups differs from 1 in a Cox proportional hazards model. For bivariate analyses, Cox models were fit to sub-samples defined by the levels of one of the two factors being analyzed. The reported p-value assesses whether the univariate hazard ratio for the other factor differs from 1 within the sub-sample. Multivariate analyses were based on Cox proportional hazards models with additive effects for the factors as reported in the results. The reported significance levels assess whether the hazard for a given factor differs from 1 when the other factors in the model are held fixed.
Comparison of clinical features of the various groups studied was performed using two-sided Chi-squared, Mann-Whitney, and Kruskal-Wallis tests. Pairwise associations between gene mutations, recurrent aCNA/cnLOH, etiological AML subtypes, and type of response to induction chemotherapy were calculated using a two-sided Fisher’s exact test corrected for multiple hypotheses testing with the Benjamini-Hochberg FDR algorithm and denoted using q-values. The standard error of the mean (SEM) was calculated for the mean number of aCNA/cnLOH associated with each mutated gene.
RESULTS
Patient Characteristics
Characteristics of the 156 prospectively enrolled, previously untreated, non-M3 FAB subtype AML patients (referred to hereafter as the “MI-All-AML” group) in this study are summarized in Supplementary Tables 1 and 2. Of the 156 patients, 114 underwent SNP-A analysis and limited gene sequencing in a previously published study (29). Sixty-six percent, 22%, and 12% of cases were dnAML, sAML, or tAML, respectively and 35% (54/156) of cases had normal cytogenetics. Within the dnAML group 17% had a recurrent genomic abnormality, 19% had myelodysplasia-related changes without a history of an antecedent myeloid neoplasm, and 64% were diagnosed as AML-not-otherwise-specified. Forty-two percent (43/103) of dnAML cases had normal cytogenetics. Regarding therapy, 93%, 66%, and 78% of dnAML, sAML, and tAML cases, respectively, received high-intensity induction chemotherapy, and 30% (47/156) underwent allogeneic stem cell transplantation while in complete remission, 45% (21/47) of whom were alive >3 years after diagnosis. Seventy-six percent (119/156) of patients had died at the time of data analysis, and median follow-up for surviving patients was 57.4 months.
Clinical characteristics of the AML TCGA cohort are published elsewhere (44). Of note, the TCGA data set comprises only dnAML cases and is compared in this study with the dnAML subgroup of Michigan cases (hereafter referred to as the “TCGA-dnAML” and “MI-dnAML” groups, respectively). The clinical features of the TCGA-dnAML and MI-dnAML groups are well matched with the exception of the fractions receiving high-intensity treatment (79% vs 93%, respectively; p<0.01). The comparison of these two groups is summarized in Supplementary Table 1.
Mutational spectrum of thirteen genes across clinical AML subsets
Mutation frequency of 13 recurrently mutated genes is summarized in Figure 1A. Eighty percent (125/156) of cases had a mutation in at least one of these genes. IDH2 (19% vs. 3%), NPM1 (24% vs. 3%), and FLT3 (21% vs. 11%) mutations were enriched in dnAML compared with sAML (p≤0.05 for all), while IDH1 (17% vs. 8%), ASXL1 (26% vs. 13%), and TP53 (26% vs. 8%) mutations were overrepresented in sAML compared with dnAML (p≤0.05 for all). For tAML, 50% (9/18) of cases harbored mutant TP53 (Figure 1B). ASXL1 occurred more frequently in patients ≥60 years old (p=0.05) due to the increased proportion of sAML in that population; however, comparing age ≥60 to age <60 in the dnAML group alone showed enrichment of RUNX1 and TET2 in older patients (both 22% vs. 7%, respectively; p<0.05), while the remaining eleven genes showed no significant difference in mutation frequency. Co-occurrence analysis of mutations in all AML cases demonstrated mutual exclusivity of TP53 and RUNX1, NPM1 and RUNX1, and IDH1 and IDH2 (Figure 1A). A detailed catalog of gene mutations for each case is included in Supplementary Table 4. Mutation frequency in these subgroups was consistent with published data with the exception of ASXL1 in dnAML (13% vs 3%)(36,44) and TP53 in tAML (50% vs 18–21%)(48,49).
Figure 1. Landscape of gene mutations and aCNA/cnLOH in AML.
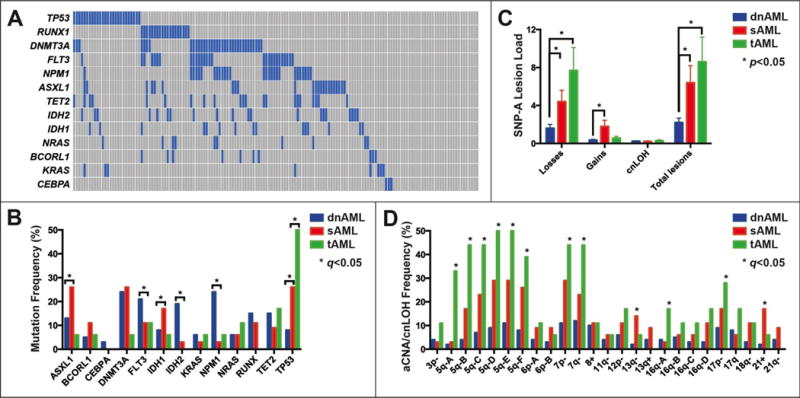
(A) Mutational spectrum for all AML cases. Demonstration of mutual exclusivity between TP53 and RUNX1: blue boxes indicate a mutation, gray boxes indicate wildtype. (B) Gene mutation frequency in clinical subtypes (dnAML, sAML and tAML) of AML. (C) Average number of losses, gains, or cnLOH in clinical subtypes of AML. (D) Frequency of minimally amplified or deleted (MADR) regions in clinical subtypes of AML. Genomic boundaries of MADRs are listed in Supplementary Table 4.
Topography of aCNA and cnLOH
A total of 455 copy number losses, 106 copy number gains, and 38 instances of cnLOH were identified, with 20% <1 Mb and 47% <5 Mb (Supplementary Table 3). aCNA/cnLOH were common in the MI-All-AML group with 62% (97/156) of cases harboring ≥1 lesion and 38% (59/156) of cases with ≥2 lesions (median 2, range 1–39); similar findings were noted in the dnAML subgroup with ≥1 lesion seen in 56% (58/103) of cases and ≥2 lesions in 29% (30/103) of cases (median 2, range 1–25). In addition, aCNA/cnLOH were detected in 31% (17/55) of cases with a normal karyotype. In cases with abnormal karyotype, additional SNP-A lesions not detected by karyotype were found in 51% of cases (53/102). The average number of copy number losses (1.6 vs. 4.4 vs. 7.7) as well as total aCNA/cnLOH (2.2 vs. 6.4 vs. 8.6) increased substantially from dnAML to sAML and tAML (p<0.05), while the average number of gains (0.3 vs. 1.8) increased between dnAML and sAML (p<0.05) (Figure 1C).
Recurrent minimally amplified or deleted regions (MADR) were determined by first identifying genomic regions in which aCNA/cnLOH occurred in ≥5% of cases. For each of these overlapping aCNA/cnLOH, the boundaries of the MADR were defined by the genomic interval containing the peak number of involved cases. For large regions multiple MADR were permitted if the peak number of involved cases decreased by ≥2. MADR were identified on chromosomes 3p, 5q, 6p, 7, 8, 11q, 12p, 13q, 16q, 17p, 17q, 18q, and 21 and are listed in Supplementary Table 4. Deletions of the distal half of 13q14.2 as well as trisomy 21 were enriched in sAML, while deletions of 5q, 7p, 7q, 16q21, monosomy 7, and deletions and cnLOH of 17p were enriched in tAML (Figure 1D).
Associations of gene mutations with aCNA/cnLOH
Mutation status and aCNA/cnLOH were analyzed for each case. Notably, co-occurrence of a gene mutation with an aCNA/cnLOH than produced a hemizygous or homozygous mutant state at that gene’s locus was uncommon. Occasional cases with mutated RUNX1, TET2, and FLT3 demonstrated aCNA/cnLOH encompassing their respective loci (16% [4/24], 8% [2/25], and 11% [4/37], respectively). The remaining nine genes had ≤1 cases with co-occurring mutations and aCNA/cnLOH. The exception was the presence of 17p deletions and cnLOH involving the TP53 locus (27% [7/26] of TP53 mutant cases with 17p deletions and 35% [9/26] of TP53 mutant cases with 17p- cnLOH).
Examining the association of gene mutations with each MADR revealed significant association between KRAS and trisomy 8 (q=0.04), BCORL1 and trisomy 21 (q=0.01), and TP53 with all recurrent aCNA/cnLOH except for 6p-, 11q-, 13q+, 21+, and 21q-.
Associations of gene mutations with SNP-A-based genomic complexity
Comparison of SNP-A-based genomic lesion loads in the MI-All-AML, MI-dnAML or a combined dnAML (MI-dnAML and TCGA-dnAML) group further subdivided by individual gene mutations and organized by fraction of cases with 0, 1, 2, 3, 4, 5–10 and >10 aCNAs showed multiple interesting findings: i) Distinctly high genomic complexity in TP53 mutated cases (mean 15.5, SEM 2.1); ii) low or no complexity in cases carrying IDH1, FLT3, NPM1 or CEBPA mutations; iii) >50% of cases with mutations in BCORL1, KRAS, RUNX1, NRAS, TET2, DNMT3A, ASXL1 or IDH2 harbored ≥1 aCNA/cnLOH (Figure 2A), and, iv) AML cases without mutations in the 13 genes analyzed here were quite complex. In the MI-All-AML group, TP53 mutations were strongly associated with the presence of aCNA/cnLOH and NPM1 mutations with their absence (q<0.01 for both), while in the pooled dnAML group TP53 and RUNX1 mutations were strongly associated with the presence of aCNA/cnLOH and NPM1 and FLT3 mutations with their absence (q<0.01 for all).
Figure 2. Spectrum of the association of aCNA/cnLOH load with gene mutations.
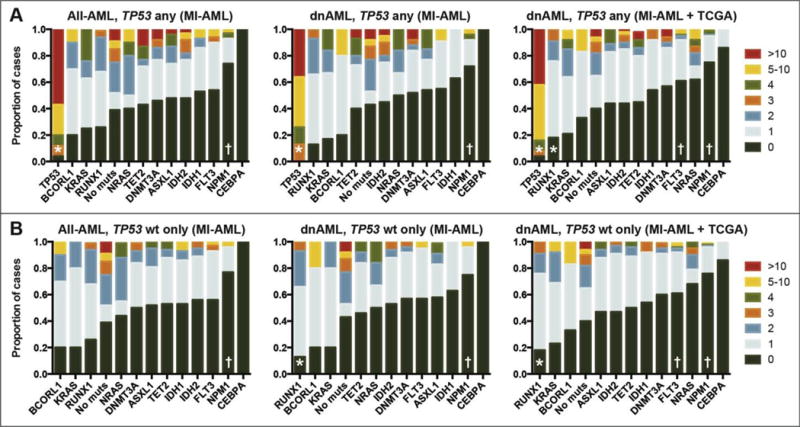
Proportion of cases with various numbers of aCNA/cnLOH for each gene mutation in the MI-All-AML, MI-dnAML and TCGA-dnAML groups, respectively, in cases with any TP53 mutation status (A) or in TP53 wildtype only (B). “*” and “†” denote significant (q<0.01) association with the presence or absence of aCNA/cnLOH, respectively.
A parallel analysis was performed on TP53 wildtype cases; in this group, a subset of cases within each mutation-carrying subgroup still carried high lesion loads (Figure 2B) and substantial differences existed in the fraction of cases that carried no lesions. Specifically, in the pooled dnAML group, RUNX1 mutations remained strongly associated with the presence of aCNA/cnLOH and NPM1 and FLT3 mutations remained strongly associated with their absence (q<0.01 for all). In both the TP53 any and TP53 wildtype analyses, the majority of cases with BCORL1 and KRAS mutations also contained aCNA/cnLOH and almost all cases of CEBPA lacked aCNA/cnLOH; however, the lower number of cases with these mutations precluded statistically significant associations.
Association of gene mutations, aCNA/cnLOH and karyotype with refractory AML
Within the total cohort (MI-All-AML), 132 patients received full-intensity induction chemotherapy; of these, 13 patients died during cycle one of induction chemotherapy and one declined further treatment and these patients were excluded from subsequent analysis. Of the remaining 118 patients, 65% (76/118) achieved a morphological complete remission (CR) after one cycle of therapy (referred to hereafter as the “sensitive” group), 15% (18/118) achieved a CR after two cycles of therapy (the “intermediate” group), and 20% (24/118) failed to achieve a CR after at least two cycles of therapy (the “refractory” group). Univariate analysis of the 118 patients examining the effect of response to induction therapy on median OS demonstrated a significant and profound difference in outcome between the sensitive, intermediate, and refractory groups (median OS of 25.8, 12.5, and 4.2 months, respectively; p<0.03 for all) (Figure 3A).
Figure 3. Genomic lesion associations with response to induction chemotherapy.
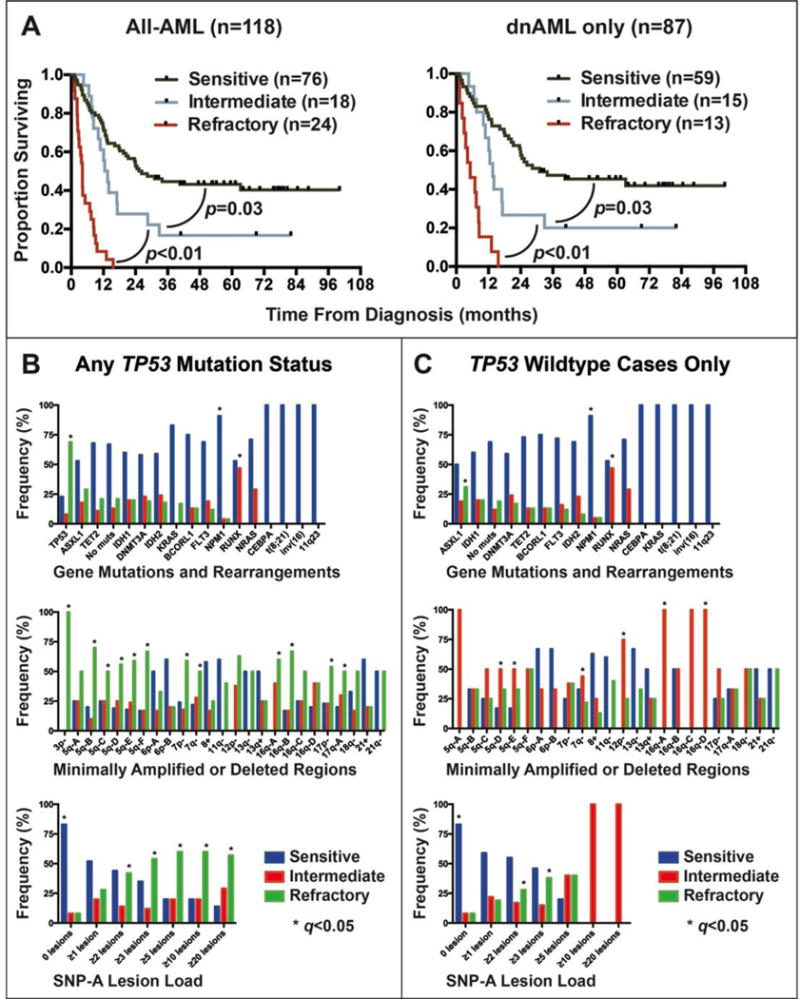
Univariate analysis of median overall survival in the MI-All-AML (N=156) and MI-dnAML (N=103) subgroups based on type of response to therapy (A). Frequency of gene mutations and structural genomic aberrations, minimally amplified or deleted regions, and aCNA/cnLOH load stratified by response to treatment in (B) all cases or in (C) TP53 wildtype cases only.
This profound survival difference prompted evaluation of genomic features associated with these distinct responses to therapy. All cases with CEBPA biallelic mutations, t(8;21), inv(16), and any 11q23 abnormality (N=3, 5, 6, and 4, respectively) were sensitive to treatment. NPM1 mutations and the absence of any aCNA/cnLOH were highly associated with the sensitive group (q<0.001 for both). RUNX1 mutations, on the other hand, were the only genomic feature highly associated with the intermediate group (q<0.001). TP53 mutations (q<0.001); SNP-A detected deletions of 3p, 5q, 7p, 7q, 12q, 16q, 17p, and 17q (q<0.05 for all); the presence of ≥2 or ≥3 aCNA/cnLOH (q=0.01 and q<0.001, respectively); and monosomal (MK) or complex karyotype (CK) (q<0.001 for both) were all associated with the refractory group. The frequency of all measured genomic features in each response group is displayed in Figure 3B.
Given the dominant correlation of TP53 mutations with refractory disease, MK, CK, and aCNA/cnLOH lesion load, the TP53 wildtype fraction (N=105) was evaluated in a similar fashion. NPM1 mutations and the absence of aCNA/cnLOH remained highly associated with the sensitive group (q<0.02) and RUNX1 mutations with the intermediate group (q<0.001). SNP-A-based deletions of 5q, 7q, 12p, and 16q still correlated with a decreased response to therapy but were associated with the intermediate group (q≤0.05 for all) rather than the refractory group. ASXL1 mutations emerged as significantly associated with the refractory group (q=0.05) as well as cases with ≥2 aCNA/cnLOH (q=0.02). The frequency of all measured genomic features in the TP53 wildtype group is shown in Figure 3C. Additional representations of genomic lesion frequency within specific types of therapy response are shown in Supplementary Figure 1.
Age, cytogenetics and gene mutations and survival in the AML cohorts studied
In the MI-All-AML group, median overall survival (OS) for age ≥60 (6.5 months) versus age <60 (24.0 months) and for favorable (not reached) versus intermediate (14.0 months) versus unfavorable (4.3 months) cytogenetics were significantly different (p<0.01 for all). Likewise, in the MI-dnAML-only subgroup the median OS for age ≥60 (8.4 months) versus age <60 (26.3 months) and median OS for favorable (not reached) versus intermediate (18.0 months) versus unfavorable (4.5 months) were significantly different (p<0.04 for all).
In the MI-All-AML cohort, a statistically significant increase in median survival was observed for NPM1 mutated (26.9 months) versus NPM1 wildtype (9.0 months) cases (p=0.01), and a significant and profound decrease in median survival was seen with TP53 mutated (2.9 months) versus TP53 wildtype (13.4 months) cases (p<0.001). The remaining eleven genes showed no significant difference in median survival.
In the MI-dnAML subgroup alone (N=103), a significant decrease in median survival was seen with TET2 mutated (6.8 months) versus TET2 wildtype (17.8 months) cases (p=0.03) and TP53 mutated (2.9 months) versus TP53 wildtype (18.0 months) cases (p<0.01). A trend toward decreased median survival was noted with RUNX1 mutated (11.5 months) versus RUNX1 wildtype (18.1 months) cases (p=0.06). The remaining ten genes showed no significant difference in median survival.
In the TCGA-dnAML group (N=170), a significant decrease in survival was again seen in TP53 mutated (7.0 months) versus TP53 wildtype (16.4 months) cases (p<0.01), RUNX1 mutated (12.5 months) and RUNX1 wildtype (14.6 months) cases (p=0.04), and DNMT3A mutated (9.9 months) versus DNMT3A wildtype (15.5 months) cases (p=0.05). The remaining ten genes showed no significant difference in median survival. Kaplan-Meier curves of univariate analyses based on gene mutation status for the MI-All-AML, MI-dnAML, and TCGA-dnAML cohorts are shown in Supplementary Figure 2.
Kaplan-Meier analysis of SNP 6.0 array-based genomic complexity and effects on survival in AML
Univariate analysis of the MI-All-AML group demonstrated a significant decrease in median survival for ≥1 lesions (7.9 months) versus 0 lesions (23.9 months) (p<0.001), ≥2 lesions (6.6 months) versus <2 lesions (15.9 months) (p<0.001), ≥3 lesions (4.2 months) versus <3 lesions (13.6 months) (p<0.001), and ≥4 lesions (4.0 months) versus <4 lesions (13.5 months) (p<0.001) (Figure 4A).
Figure 4. Univariate analyses of increasing aCNA/cnLOH burden.
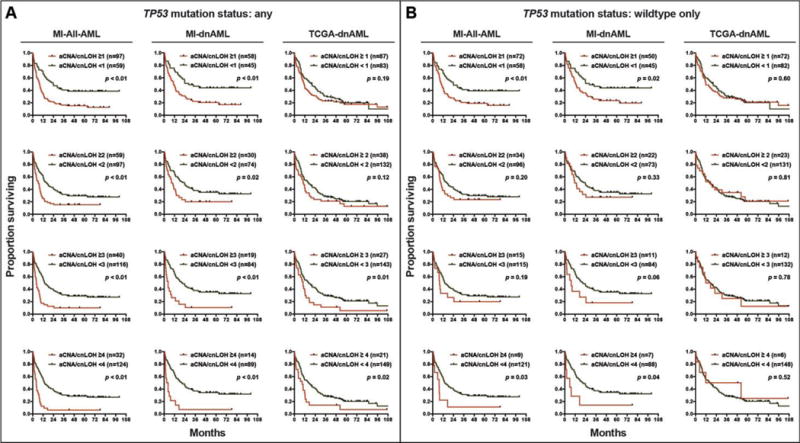
Univariate analysis of the effect of increasing aCNA/cnLOH lesion count on median overall survival in the MI-All-AML, MI-dnAML and TCGA-dnAML groups, respectively, in cases with any TP53 mutation status (A) or in TP53 wildtype only (B).
Corresponding results for the MI-dnAML group are: SNP-A-derived aCNA/cnLOH lesions ≥1 lesion (11.0 months) versus 0 lesions (28.4 months) (p=0.01), ≥2 lesions (8.5 months) versus <2 lesions (22.4 months) (p=0.03), ≥3 lesions (3.8 months) versus <3 lesions (20.5 months) (p<0.001), and ≥4 lesions (2.9 months) versus <4 lesions (20.0 months) (p<0.001) (Figure 4A). This effect was also seen in the TCGA-dnAML group for ≥3 lesions (9.9 months) versus <3 lesions (16.4 months) (p=0.01) and ≥4 lesions (8.9 months) versus <4 lesions (16.4 months) (p=0.02) (Figure 4A).
In MI-dnAML cases with wildtype TP53 (N=95), aCNA/cnLOH burden remained associated with a decrease in median survival, with a trend toward significance for ≥3 lesions (6.6 months) versus <3 lesions (20.5 months) (p=0.06) and a significant difference for ≥4 lesions (4.0 months) versus <4 lesions (20.6 months) (p=0.04) (Figure 4B).
A combined analysis of gene mutations and SNP-A-detected aCNA/cnLOH on survival in AML
Since mutational profiling is increasingly employed for clinical risk stratification, we assessed the prognostic value of knowledge of aCNA/cnLOH counts in AML patients stratified by individual gene mutations. To increase power of this analysis, the MI-dnAML and the TCGA-dnAML cohorts were combined.
The presence of ≥1 aCNA/cnLOH in patients with mutations in TET2, IDH1, NPM1 or DNMT3A significantly worsened survival as compared with lack of aCNA/cnLOH, with similar trends detected in patients carrying RUNX1 or FLT3 mutations (Figure 5). For example, median survival was 6.6 months versus 29.4 months in patients with TET2 mutations and presence or absence of aCNA/cnLOH, respectively (p<0.01), and 12.9 months versus 56.3 months for patients with IDH1 mutations and presence or absence of aCNA/cnLOH, respectively (p=0.01).
Figure 5. Bivariate analyses of gene mutations and aCNA/cnLOH.
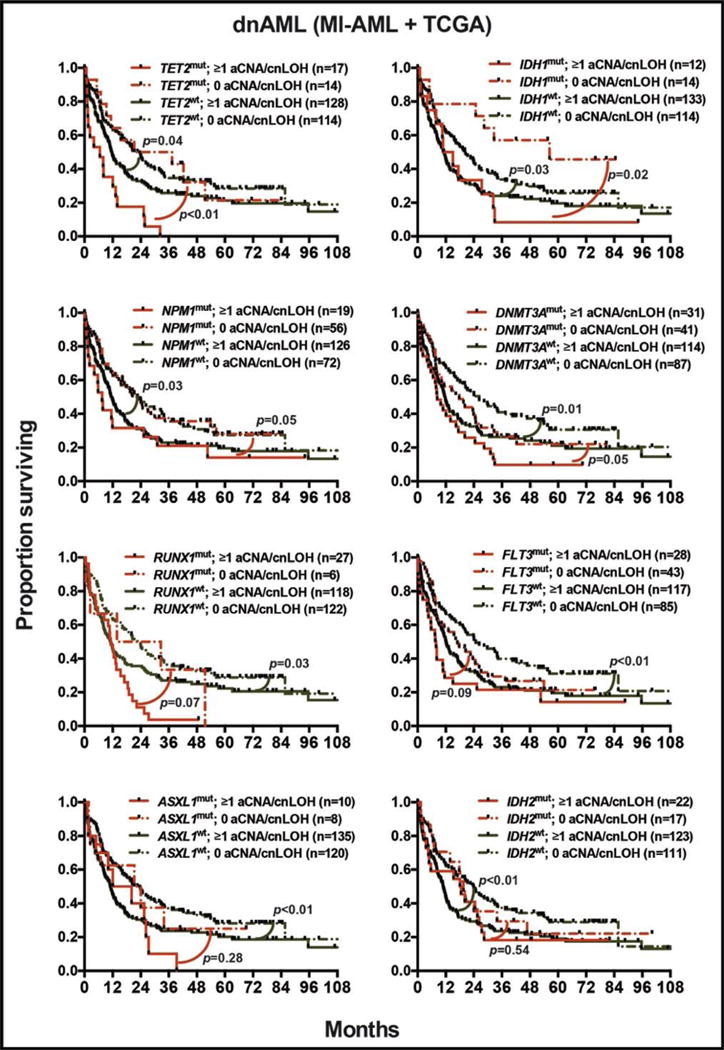
Bivariate analyses of the effect of gene mutation status combined with the presence or absence of aCNA/cnLOH on median overall survival in the pooled MI-dnAML and TCGA-dnAML groups (N=273).
Multivariate survival analyses of age, cytogenetics, SNP 6.0 array-based genomic complexity, and gene mutations in AML
Multivariate analysis was performed using a Cox proportional hazards model of single variables in combination with age and cytogenetic risk group. For the MI-All-AML group, increased genomic complexity defined as ≥4 SNP-A lesions demonstrated a strong, independent negative impact on overall survival (HR=1.98 [95% CI 1.20–3.28], p <0.01) with ≥3 SNP-A lesions trending toward an independent negative effect as well (HR=1.57 [95% CI 0.95–2.57], p=0.07). In separate models, TP53 mutations showed a strong, independent negative prognostic effect (HR=2.65 [95%CI 1.47–4.81], p=0.001) (Table 1). No other gene mutations showed an independent effect on overall survival in this model.
Table 1. Results of multivariate survival analyses.
Multivariate analysis of overall survival in the MI-All-AML cohort (middle column) and MI-dnAML cohort (right column). Models using age- and aCNA/cnLOH-based risk factors along with gene mutation status are shown in the upper section; models using age- and cytogenetics-based risk factors along with number of aCNA/cnLOH or gene mutation status are shown in the lower section.
| MI-All-AML Group (N=156) | MI-dnAML Group (N=103) | ||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Variables | Hazard Ratio | 95% CI | p | Hazard Ratio | 95% CI | p | |
| Age + aCNA/cnLOH load + additional variable | Age ≥60 vs. <60 | 2.74 | 1.85–4.05 | <0.001 | 2.16 | 1.32–3.55 | <0.01 |
| aCNA/cnLOH ≥3 vs. <3 | 2.30 | 1.52–3.48 | <0.001 | 2.61 | 1.45–4.68 | <0.01 | |
| TET2 mutated vs. wildtype | 1.10 | 0.64–1.89 | 0.73 | 1.85 | 0.97–3.51 | 0.06 | |
|
| |||||||
| Age ≥60 vs. <60 | 2.62 | 1.76–3.89 | <0.001 | 2.29 | 1.40–3.74 | <0.01 | |
| aCNA/cnLOH ≥3 vs. <3 | 2.42 | 1.59–3.68 | <0.001 | 2.45 | 1.37–4.38 | <0.01 | |
| ASXL1 mutated vs. wildtype | 1.43 | 0.87–2.36 | 0.15 | 1.22 | 0.62–2.39 | 0.56 | |
|
| |||||||
| Age ≥60 vs. <60 | 2.73 | 1.85–4.04 | <0.001 | 2.35 | 1.45–3.82 | <0.001 | |
| aCNA/cnLOH ≥3 vs. <3 | 2.29 | 1.52–3.45 | <0.001 | 2.37 | 1.34–4.19 | <0.01 | |
| DNMT3A mutated vs. wildtype | 1.00 | 0.65–1.55 | 0.99 | 1.11 | 0.64–1.93 | 0.69 | |
|
| |||||||
| Age ≥60 vs. <60 | 2.72 | 1.84–4.03 | <0.001 | 2.35 | 1.44–3.81 | <0.001 | |
| aCNA/cnLOH ≥3 vs. <3 | 2.27 | 1.50–3.43 | <0.001 | 2.40 | 1.34–4.32 | <0.01 | |
| FLT3-ITD mutated vs. wildtype | 0.92 | 0.52–1.64 | 0.77 | 1.07 | 0.55–2.11 | 0.83 | |
|
| |||||||
| Age ≥60 vs. <60 | 2.72 | 1.84–4.02 | <0.001 | 2.34 | 1.44–3.81 | <0.001 | |
| aCNA/cnLOH ≥3 vs. <3 | 2.08 | 1.37–3.17 | <0.001 | 2.23 | 1.24–4.02 | <0.01 | |
| NPM1 mutated vs. wildtype | 0.62 | 0.36–1.09 | 0.09 | 0.79 | 0.43–1.43 | 0.42 | |
|
| |||||||
| Age ≥60 vs. <60 | 2.82 | 1.90–4.19 | <0.001 | 2.46 | 1.52–4.00 | <0.001 | |
| aCNA/cnLOH ≥3 vs. <3 | 1.23 | 0.69–2.34 | 0.52 | 1.60 | 0.77–3.31 | 0.19 | |
| TP53 mutated vs. wildtype | 3.23 | 1.54–6.77 | <0.01 | 3.65 | 1.31–10.2 | 0.01 | |
|
| |||||||
| Age + cytogenetics + additional variable | Age ≥60 vs. <60 | 2.85 | 1.93–4.21 | <0.001 | 2.40 | 1.48–3.90 | <0.001 |
| Unfavorable cytogenetics vs. not | 1.84 | 1.19–2.85 | <0.01 | 1.66 | 0.96–2.85 | 0.06 | |
| aCNA/cnLOH ≥4 vs. <4 | 1.98 | 1.20–3.28 | <0.01 | 2.50 | 1.29–4.84 | <0.01 | |
|
| |||||||
| Age ≥60 vs. <60 | 2.83 | 1.91–4.19 | <0.001 | 2.32 | 1.43–3.76 | <0.001 | |
| Unfavorable cytogenetics vs. not | 1.89 | 1.19–2.99 | <0.01 | 1.53 | 0.85–2.77 | 0.15 | |
| aCNA/cnLOH ≥3 vs. <3 | 1.57 | 0.95–2.57 | 0.07 | 1.92 | 1.02–3.63 | 0.04 | |
|
| |||||||
| Age ≥60 vs. <60 | 2.87 | 1.94–4.25 | <0.001 | 2.51 | 1.55–4.06 | <0.001 | |
| Unfavorable cytogenetics vs. not | 1.64 | 1.02–2.64 | 0.04 | 1.43 | 0.77–2.68 | 0.25 | |
| TP53 mutated vs. wildtype | 2.65 | 1.47–4.81 | <0.001 | 4.08 | 1.56–10.7 | <0.01 | |
|
| |||||||
| Age ≥60 vs. <60 | 2.90 | 1.96–4.29 | <0.001 | 2.25 | 1.38–3.68 | <0.01 | |
| Unfavorable cytogenetics vs. not | 2.35 | 1.61–3.45 | <0.001 | 2.20 | 1.28–3.79 | <0.01 | |
| TET2 mutated vs. wildtype | 1.12 | 0.65–1.92 | 0.68 | 1.90 | 1.00–3.63 | 0.05 | |
|
| |||||||
| Age ≥60 vs. <60 | 2.84 | 1.91–4.22 | <0.001 | 2.43 | 1.50–3.96 | <0.001 | |
| Unfavorable cytogenetics vs. not | 2.32 | 1.59–3.40 | <0.001 | 1.97 | 1.17–3.34 | 0.01 | |
| ASXL1 mutated vs. wildtype | 1.14 | 0.70–1.87 | 0.59 | 1.01 | 0.52–1.95 | 0.99 | |
|
| |||||||
| Age ≥60 vs. <60 | 2.89 | 1.96–4.28 | <0.001 | 2.45 | 1.51–3.98 | <0.01 | |
| Unfavorable cytogenetics vs. not | 2.37 | 1.61–3.48 | <0.001 | 2.00 | 1.18–3.38 | <0.01 | |
| DNMT3A mutated vs. wildtype | 1.12 | 0.72–1.74 | 0.62 | 1.19 | 0.68–2.05 | 0.54 | |
Similarly, in multivariate analysis of the MI-dnAML subgroup, the presence of ≥3 SNP-A lesions had an independent negative effect on overall survival (HR=1.92 [95%CI 1.01–3.68], p=0.04), as did the presence of ≥4 lesions (HR=2.50 [95%CI 1.29–4.84], p<0.01). In separate models, TP53 mutations likewise showed a strong, independent negative impact on prognosis (HR 4.08 [95% CI 1.56–10.66], p<0.01) as did TET2 (HR=1.90 [95% CI 1.00–3.63], p=0.05) (Table 1). No other gene mutations showed an independent effect on overall survival in multivariate analysis.
Models of MI-dnAML that included age, TP53 mutations and the presence of ≥3 or ≥4 SNP-A lesions showed elevated HR of 1.60 [95% CI 0.77–3.31] and 1.80 [95% CI 0.74–4.37] for ≥3 and ≥4 SNP-A lesions, respectively, that were NS (p=0.20 and 0.19); however, conclusions are limited by the small number of cases (N=11) that both had an aCNA/cnLOH count ≥3 and were TP53 wildtype.
Multivariate analyses were also performed on the TCGA-dnAML group. In models that included age and cytogenetics, DNMT3A and FLT3–ITD mutations showed an independent adverse prognostic effect (HR 1.67 [95% CI 1.12–2.48], p=0.01; and HR 1.60 [95% CI 1.00–2.55], p=0.04, respectively) with a trend toward an independent adverse effect of TP53 mutations (HR 1.81 [95% CI 0.92–3.56], p=0.08) (Supplementary Table 5). An independent adverse effect of aCNA/cnLOH count ≥3 in TP53 wildtype or mutant cases was not detected. Analysis of the clinical features of the TP53 mutant and wildtype subgroups within the TCGA and MI-AML groups shows that while the TP53 mutant groups are well matched (median age 67 vs 68 and BMT as consolidation in 27% vs 25%, respectively), the TCGA TP53 wildtype subgroup with aCNA/cnLOH count ≥3 harbored substantial differences compared with MI-AML (median age 60 vs 69 and BMT as consolidation in 67% vs 18% [p=0.03]), which may explain the lack of observed effect of elevated genomic complexity on outcomes in the TCGA TP53 wildtype subgroup.
DISCUSSION
Recent publications in AML have underscored the importance of simultaneous analysis of competing genomic variables when determining an individual mutated gene’s association with survival (35–37,50). While these studies have focused on the relative influence of a given mutant gene compared with a pool of other mutant genes, our study integrates the majority of known recurrently mutated genes with SNP-A-based genomic complexity. We characterized the DNA of leukemic blasts from 156 consecutive prospectively enrolled AML patients using SNP 6.0 arrays and Sanger-based mutational analysis of TP53, RUNX1, DNMT3A, NPM1, FLT3, ASXL1, TET2, IDH1, IDH2, NRAS, KRAS, BCORL1 and CEBPA to evaluate the relationship between chromosomal translocations, aCNA/cnLOH and gene mutations and their association with response to induction chemotherapy and overall survival. The cohort studied comprised consecutively evaluated patients without a priori selection for clinically defined subgroups, such as dnAML, sAML, and tAML, or normal karyotype versus aberrant karyotype AML. In addition, we analyzed 170 dnAML patients published by TCGA to expand our findings.
Together, the genomic and clinical findings allow for the following principle conclusions: i) RUNX1 mutations, TP53 mutations, elevated SNP 6.0 array-based genomic complexity, SNP 6.0 array-defined deletions/cnLOH of 3p, 5q, 7p, 7q, 12q, 16q, 17p, and 17q, and complex and monosomal karyotypes all are strong predictors of partial or complete resistance to intensive induction therapy, resulting in dismal overall survival, ii) in all AML sub cohorts after exclusion of TP53 mutant cases, NPM1 mutations predicted for chemotherapy sensitivity (in addition, all AML cases carrying mutations of CEBPA, KRAS and t(8;21), inv(16) and 11q23 abnormalities achieved a CR), while mutations in RUNX1 and ASXL1, elevated SNP 6.0 array-based genomic complexity and SNP 6.0 array-defined deletions/cnLOH of 5q, 7q, 12p, and 16q were often associated with resistance to induction chemotherapy, and, iii) presence of any aCNA/cnLOH in patients carrying mutations in TET2, IDH1, NPM1, DNMT3A significantly worsened prognosis when compared with patients carrying such mutations but no aCNA/cnLOH, with similar trends observed in patients carrying either RUNX1 or FLT3 mutations.
Additional novel conclusions center on i) the finding that genomic losses or gains infrequently affect the loci of genes known to be recurrently mutated in AML, providing clear evidence that these lesion types serve largely independent biological functions in AML, ii) that genomic complexity markedly associates with TP53 mutation status, but that mutations in RUNX1 also associated with the presence of ≥1 aCNA/cnLOH in TP53 wildtype dnAML and that previous reports that mutations in TP53 and RUNX1 were mutually exclusive in dnAML (44) can be extended to all clinical subtypes, and iii) that NPM1, CEBPA, and FLT3-ITD mutations are significantly associated with stable genomes (absence of SNP-A lesions) and that marked differences exists in the aCNA/cnLOH load of AML subgroups stratified by specific gene mutations.
While this is the first study to integrate the majority of recurrently mutated genes with SNP-A data in a survival analysis, there are limitations to this analysis. First, our methodology did not evaluate all known recurrent mutations, and the use of Sanger sequencing, while capable of identifying clonally represented mutations, likely did not detect subclonal mutations that are of unclear clinical significance. In addition, no uniform therapy was used in this cohort, and while most patients were treated with intensive induction regimens, differences in specific induction chemotherapies may have influenced outcomes. Further, the smaller size of the cohort studied precluded the ability to comprehensively analyze all competing variables in multivariate analysis. The TCGA cohort provided additional patients for study, but the use of unpurified tumor DNA, a different sequencing methodology and lower rates of use of intensive therapies limit to some extent a direct comparison of genomic findings and may account for some of the differences observed.
Given that genomic complexity resulting from genomic instability as measured by SNP-A-based aCNA/cnLOH counts is strongly associated with poor survival and chemoresistance in AML and given that the effect of the presence of such genomic instability supersedes that of mutations in all commonly mutated genes in AML evaluated with the exception of TP53 and possibly TET2, multiple relevant questions arise. Is genomic complexity (and the underlying pathogenic mechanisms that lead to it) one of the defining characteristics upon which clinical and biological AML subgroups should be defined? Are recurrent AML associated gene mutations other than TP53 more associated with AML initiation or clonal outgrowth rather than with chemotherapy resistance? If so, are the observed relationships of these mutations with AML prognosis in part indirect and due to co-occurrence or conversely the lack of association with either TP53 mutations or genomic instability?
In summary, our data demonstrate that RUNX1 mutations, ASXL1 mutations, SNP 6.0 array-based deletions/cnLOH of 5q, 7q, 12p, and 16q and elevated SNP 6.0 array-based genomic complexity significantly predict partial or complete resistance to induction chemotherapy even in the absence of a co-occurring TP53 mutation. Furthermore, highly adverse prognostic information in AML can be derived from determinations of patient’s age, karyotype, TP53 mutation status and/or SNP 6.0 array-based genomic complexity, which should be measured routinely for all patients with AML at diagnosis.
Supplementary Material
TRANSLATIONAL RELEVANCE.
Several gene mutations, structural chromosomal aberrations, specific acquired copy number aberrations (aCNAs) and the total number of aCNAs as detected through SNP 6.0 array profiling have been associated with clinical outcome in acute myelogenous leukemia (AML). However, it remains incompletely understood how their interrelatedness affects their predictive and prognostic usefulness. In an AML cohort, failure to achieve a complete remission after one or two intensive induction regimens predicted for very poor long-term survival. The partial or complete resistance to induction chemotherapy significantly associated with mutations in RUNX1, ASXL1, and TP53, various recurrent aCNAs and elevated SNP-A-based genomic complexity. Prognostically, the presence of ≥1 SNP-A-based aCNA/cnLOH and all higher thresholds predicted for poor long-term survival irrespective of TP53 status. When combining this data with AML-TCGA results in de novo AML, the presence of ≥1 aCNA/cnLOH added negative prognostic information to knowledge of mutations in TET2, IDH1, NPM1, DNMT3A and RUNX1.
Acknowledgments
We are grateful for services provided by the microarray core and DNA sequencing core of the University of Michigan Comprehensive Cancer Center.
GRANT SUPPORT
Supported by the National Institutes of Health through CA171972 (SM). SM is a Scholar in Clinical Research of the Leukemia and Lymphoma Society of America (SM). This research is supported (in part) by the National Institutes of Health through the University of Michigan&s Cancer Center Support Grant (5 P30 CA46592) and the National Cancer Institute Oncology Research Training Grant (T32 CA009357-30) (BP).
Footnotes
INDIVIDUAL CONTRIBUTIONS
Brian Parkin, Peter Ouillette, Mehmet Yildiz, Kamlai Saiya-Cork and Sami Malek performed the laboratory research.
Brian Parkin and Sami Malek analyzed clinical data.
Kerby Shedden assisted with statistical analysis.
Sami Malek conceived the study and supervised the work.
Brian Parkin, Kerby Shedden and Sami Malek wrote the paper.
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts of interest to declare.
References
- 1.Slovak ML, Kopecky KJ, Cassileth PA, Harrington DH, Theil KS, Mohamed A, et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood. 2000;96:4075–83. [PubMed] [Google Scholar]
- 2.Breems DA, Van Putten WLJ, De Greef GE, Van Zelderen-Bhola SL, Gerssen-Schoorl KBJ, Mellink CHM, et al. Monosomal karyotype in acute myeloid leukemia: a better indicator of poor prognosis than a complex karyotype. J Clin Oncol. 2008;26:4791–7. doi: 10.1200/JCO.2008.16.0259. [DOI] [PubMed] [Google Scholar]
- 3.Haferlach C, Alpermann T, Schnittger S, Kern W, Chromik J, Schmid C, et al. Prognostic value of monosomal karyotype in comparison to complex aberrant karyotype in acute myeloid leukemia: a study on 824 cases with aberrant karyotype. Blood. 2012;119:2122–5. doi: 10.1182/blood-2011-10-385781. [DOI] [PubMed] [Google Scholar]
- 4.Döhner K, Schlenk RF, Habdank M, Scholl C, Rücker FG, Corbacioglu A, et al. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood American Society of Hematology. 2005;106:3740–6. doi: 10.1182/blood-2005-05-2164. [DOI] [PubMed] [Google Scholar]
- 5.Schnittger S, Schoch C, Kern W, Mecucci C, Tschulik C, Martelli MF, et al. Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood American Society of Hematology. 2005;106:3733–9. doi: 10.1182/blood-2005-06-2248. [DOI] [PubMed] [Google Scholar]
- 6.Thiede C, Koch S, Creutzig E, Steudel C, Illmer T, Schaich M, et al. Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML) Blood. 2006;107:4011–20. doi: 10.1182/blood-2005-08-3167. [DOI] [PubMed] [Google Scholar]
- 7.Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98:1752–9. doi: 10.1182/blood.v98.6.1752. [DOI] [PubMed] [Google Scholar]
- 8.Bacher U, Haferlach C, Kern W, Haferlach T, Schnittger S. Prognostic relevance of FLT3-TKD mutations in AML: the combination matters–an analysis of 3082 patients. Blood. 2008;111:2527–37. doi: 10.1182/blood-2007-05-091215. [DOI] [PubMed] [Google Scholar]
- 9.Whitman SP, Maharry K, Radmacher MD, Becker H, Mrózek K, Margeson D, et al. FLT3 internal tandem duplication associates with adverse outcome and gene- and microRNA-expression signatures in patients 60 years of age or older with primary cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Blood American Society of Hematology. 2010;116:3622–6. doi: 10.1182/blood-2010-05-283648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wouters BJ, Löwenberg B, Erpelinck-Verschueren CAJ, van Putten WLJ, Valk PJM, Delwel R. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood American Society of Hematology. 2009;113:3088–91. doi: 10.1182/blood-2008-09-179895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dufour A, Schneider F, Metzeler KH, Hoster E, Schneider S, Zellmeier E, et al. Acute myeloid leukemia with biallelic CEBPA gene mutations and normal karyotype represents a distinct genetic entity associated with a favorable clinical outcome. J Clin Oncol American Society of Clinical Oncology. 2010;28:570–7. doi: 10.1200/JCO.2008.21.6010. [DOI] [PubMed] [Google Scholar]
- 12.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–33. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thol F, Damm F, Lüdeking A, Winschel C, Wagner K, Morgan M, et al. Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol American Society of Clinical Oncology. 2011;29:2889–96. doi: 10.1200/JCO.2011.35.4894. [DOI] [PubMed] [Google Scholar]
- 14.Gaidzik VI, Schlenk RF, Paschka P, Stölzle A, Späth D, Kuendgen A, et al. Clinical impact of DNMT3A mutations in younger adult patients with acute myeloid leukemia: results of the AML Study Group (AMLSG) Blood American Society of Hematology. 2013;121:4769–77. doi: 10.1182/blood-2012-10-461624. [DOI] [PubMed] [Google Scholar]
- 15.Schnittger S, Dicker F, Kern W, Wendland N, Sundermann J, Alpermann T, et al. RUNX1 mutations are frequent in de novo AML with noncomplex karyotype and confer an unfavorable prognosis. Blood. 2011;117:2348–57. doi: 10.1182/blood-2009-11-255976. [DOI] [PubMed] [Google Scholar]
- 16.Gaidzik VI, Bullinger L, Schlenk RF, Zimmermann AS, Röck J, Paschka P, et al. RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML study group. J Clin Oncol. 2011;29:1364–72. doi: 10.1200/JCO.2010.30.7926. [DOI] [PubMed] [Google Scholar]
- 17.Mendler JH, Maharry K, Radmacher MD, Mrózek K, Becker H, Metzeler KH, et al. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and MicroRNA expression signatures. J Clin Oncol American Society of Clinical Oncology. 2012;30:3109–18. doi: 10.1200/JCO.2011.40.6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Metzeler KH, Maharry K, Radmacher MD, Mrózek K, Margeson D, Becker H, et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2011;29:1373–81. doi: 10.1200/JCO.2010.32.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu W-J, Tan X-H, Luo X-P, Guo B-P, Wei Z-J, Ke Q, et al. Prognostic significance of TET2 mutations in adult patients with acute myeloid leukemia: a meta-analysis. Leuk Lymphoma. 2014 doi: 10.3109/10428194.2014.893308. [DOI] [PubMed] [Google Scholar]
- 20.Chou W-C, Huang H-H, Hou H-A, Chen C-Y, Tang J-L, Yao M, et al. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood. 2010;116:4086–94. doi: 10.1182/blood-2010-05-283291. [DOI] [PubMed] [Google Scholar]
- 21.Metzeler KH, Becker H, Maharry K, Radmacher MD, Kohlschmidt J, Mrózek K, et al. ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN Favorable genetic category. Blood. 2011;118:6920–9. doi: 10.1182/blood-2011-08-368225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Krönke J, Bullinger L, et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. 2010;28:3636–43. doi: 10.1200/JCO.2010.28.3762. [DOI] [PubMed] [Google Scholar]
- 23.Schnittger S, Haferlach C, Ulke M, Alpermann T, Kern W, Haferlach T. IDH1 mutations are detected in 6.6% of 1414 AML patients and are associated with intermediate risk karyotype and unfavorable prognosis in adults younger than 60 years and unmutated NPM1 status. Blood American Society of Hematology. 2010;116:5486–96. doi: 10.1182/blood-2010-02-267955. [DOI] [PubMed] [Google Scholar]
- 24.Thol F, Damm F, Wagner K, Göhring G, Schlegelberger B, Hoelzer D, et al. Prognostic impact of IDH2 mutations in cytogenetically normal acute myeloid leukemia. Blood American Society of Hematology. 2010;116:614–6. doi: 10.1182/blood-2010-03-272146. [DOI] [PubMed] [Google Scholar]
- 25.Green CL, Evans CM, Zhao L, Hills RK, Burnett AK, Linch DC, et al. The prognostic significance of IDH2 mutations in AML depends on the location of the mutation. Blood American Society of Hematology. 2011;118:409–12. doi: 10.1182/blood-2010-12-322479. [DOI] [PubMed] [Google Scholar]
- 26.Bowen DT, Frew ME, Hills R, Gale RE, Wheatley K, Groves MJ, et al. RAS mutation in acute myeloid leukemia is associated with distinct cytogenetic subgroups but does not influence outcome in patients younger than 60 years. Blood American Society of Hematology. 2005;106:2113–9. doi: 10.1182/blood-2005-03-0867. [DOI] [PubMed] [Google Scholar]
- 27.Bacher U, Haferlach T, Schoch C, Kern W, Schnittger S. Implications of NRAS mutations in AML: a study of 2502 patients. Blood American Society of Hematology. 2006;107:3847–53. doi: 10.1182/blood-2005-08-3522. [DOI] [PubMed] [Google Scholar]
- 28.Wattel E, Preudhomme C, Hecquet B, Vanrumbeke M, Quesnel B, Dervite I, et al. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood. 1994;84:3148–57. [PubMed] [Google Scholar]
- 29.Parkin B, Erba H, Ouillette P, Roulston D, Purkayastha A, Karp J, et al. Acquired genomic copy number aberrations and survival in adult acute myelogenous leukemia. Blood. 2010;116:4958–67. doi: 10.1182/blood-2010-01-266999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rücker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2011 doi: 10.1182/blood-2011-08-375758. [DOI] [PubMed] [Google Scholar]
- 31.Wagner K, Damm F, Göhring G, Görlich K, Heuser M, Schäfer I, et al. Impact of IDH1 R132 mutations and an IDH1 single nucleotide polymorphism in cytogenetically normal acute myeloid leukemia: SNP rs11554137 is an adverse prognostic factor. J Clin Oncol American Society of Clinical Oncology. 2010;28:2356–64. doi: 10.1200/JCO.2009.27.6899. [DOI] [PubMed] [Google Scholar]
- 32.Hou H-A, Huang T-C, Lin L-I, Liu C-Y, Chen C-Y, Chou W-C, et al. WT1 mutation in 470 adult patients with acute myeloid leukemia: stability during disease evolution and implication of its incorporation into a survival scoring system. Blood. 2010;115:5222–31. doi: 10.1182/blood-2009-12-259390. [DOI] [PubMed] [Google Scholar]
- 33.Becker H, Marcucci G, Maharry K, Radmacher MD, Mrózek K, Margeson D, et al. Mutations of the Wilms tumor 1 gene (WT1) in older patients with primary cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Blood American Society of Hematology. 2010;116:788–92. doi: 10.1182/blood-2010-01-262543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thol F, Bollin R, Gehlhaar M, Walter C, Dugas M, Suchanek KJ, et al. Mutations in the cohesin complex in acute myeloid leukemia: clinical and prognostic implications. Blood American Society of Hematology. 2014;123:914–20. doi: 10.1182/blood-2013-07-518746. [DOI] [PubMed] [Google Scholar]
- 35.Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia. 2014 doi: 10.1038/leu.2014.55. [DOI] [PubMed] [Google Scholar]
- 36.Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–89. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen Y, Zhu Y-M, Fan X, Shi J-Y, Wang Q-R, Yan X-J, et al. Gene mutation patterns and their prognostic impact in a cohort of 1,185 patients with acute myeloid leukemia. Blood American Society of Hematology. 2011;118:5593–603. doi: 10.1182/blood-2011-03-343988. [DOI] [PubMed] [Google Scholar]
- 38.Schlenk RF, Döhner K, Krauter J, Fröhling S, Corbacioglu A, Bullinger L, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–18. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]
- 39.Tiu RV, Gondek LP, O&Keefe CL, Huh J, Sekeres MA, Elson P, et al. New lesions detected by single nucleotide polymorphism array-based chromosomal analysis have important clinical impact in acute myeloid leukemia. J Clin Oncol. 2009;27:5219–26. doi: 10.1200/JCO.2009.21.9840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bullinger L, Krönke J, Schön C, Radtke I, Urlbauer K, Botzenhardt U, et al. Identification of acquired copy number alterations and uniparental disomies in cytogenetically normal acute myeloid leukemia using high-resolution single-nucleotide polymorphism analysis. Leukemia. 2010;24:438–49. doi: 10.1038/leu.2009.263. [DOI] [PubMed] [Google Scholar]
- 41.Tiu RV, Gondek LP, O&Keefe CL, Elson P, Huh J, Mohamedali A, et al. Prognostic impact of SNP array karyotyping in myelodysplastic syndromes and related myeloid malignancies. Blood. 2011 doi: 10.1182/blood-2010-07-295857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jerez A, Gondek LP, Jankowska AM, Makishima H, Przychodzen B, Tiu RV, et al. Topography, Clinical, and Genomic Correlates of 5q Myeloid Malignancies Revisited. J Clin Oncol. 2012 doi: 10.1200/JCO.2011.36.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jerez A, Sugimoto Y, Makishima H, Verma A, Jankowska AM, Przychodzen B, et al. Loss of heterozygosity in 7q myeloid disorders: clinical associations and genomic pathogenesis. Blood American Society of Hematology. 2012;119:6109–17. doi: 10.1182/blood-2011-12-397620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Long J, Parkin B, Ouillette P, Bixby D, Shedden K, Erba H, et al. Multiple distinct molecular mechanisms influence sensitivity and resistance to MDM2 inhibitors in adult acute myelogenous leukemia. Blood. 2010;116:71–80. doi: 10.1182/blood-2010-01-261628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parkin B, Ouillette P, Wang Y, Liu Y, Wright W, Roulston D, et al. NF1 inactivation in adult acute myelogenous leukemia. Clin Cancer Res. 2010;16:4135–47. doi: 10.1158/1078-0432.CCR-09-2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parkin B, Ouillette P, Li Y, Keller J, Lam C, Roulston D, et al. Clonal evolution and devolution following chemotherapy in adult acute myelogenous leukemia. Blood American Society of Hematology. 2012;121:369–77. doi: 10.1182/blood-2012-04-427039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shih AH, Chung SS, Dolezal EK, Zhang S-J, Abdel-Wahab OI, Park CY, et al. Mutational analysis of therapy-related myelodysplastic syndromes and acute myelogenous leukemia. Haematologica. 2013;98:908–12. doi: 10.3324/haematol.2012.076729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pedersen-Bjergaard J, Andersen MK, Andersen MT, Christiansen DH. Genetics of therapy-related myelodysplasia and acute myeloid leukemia. Leukemia. 2008;22:240–8. doi: 10.1038/sj.leu.2405078. [DOI] [PubMed] [Google Scholar]
- 50.Hou H-A, Lin C-C, Chou W-C, Liu C-Y, Chen C-Y, Tang J-L, et al. Integration of cytogenetic and molecular alterations in risk stratification of 318 patients with de novo non-M3 acute myeloid leukemia. Leukemia. 2014;28:50–8. doi: 10.1038/leu.2013.236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.