

Abstract
One variable that may impact the ability of vitamin D to reduce colon cancer risk is the expression of its high affinity receptor, VDR. Here we show that vitamin D does not reduce tumor formation in ApcΔ14/+ mice and that VDR expression is lost in the majority of the colon tumor cells. The extent of VDR loss corresponded inversely to the level of β-catenin nuclear localization and could be observed in early lesions composed of just a few crypts. Analysis of reported VDR regulators showed that the repressing class I histone deacetylases (HDACs) were significantly elevated in the tumors (up to 4-fold), whereas the VDR-activating RXRs were down-regulated (∼50%). Expression of the Slug repressor was also increased, but was found primarily in stromal cells. Analysis of epigenetically active compounds on colon cell lines and intestinal organoids showed that HDAC inhibitors were particularly adept at stimulating VDR expression. Treatment of tumor-bearing ApcΔ14/+ mice with the HDAC inhibitor panobinostat increased VDR expression in the tumors and normal mucosa. The RXR agonist bexarotene failed to activate VDR expression, indicating that RXR ligands were not limiting. Analysis of human microarray data indicated that VDR mRNA is frequently down-regulated in colon adenomas, which correlated positively with RXRA expression and inversely with HDAC 2 and 8 expression. Human adenomas showed variable VDR protein expression levels, both between and within individual lesions. Determining the mechanisms of VDR regulation in colon neoplasms may significantly enhance our ability to utilize vitamin D as a cancer prevention agent.
Keywords: vitamin D, colon cancer, VDR, Slug, histone deacetylases, RXR
Introduction
Colon cancer is the third most common type of cancer in the United States and accounts annually for about 11% of all cancer deaths (CDC and American Cancer Society)[1, 2]. While early detection and polyp removal through screening colonoscopy has offered significant benefit [3], colon cancer continues to take a serious toll on the US population. Identifying dietary agents and supplements that reduce the risk of colon cancer development could be a powerful accompaniment to screening colonoscopy; high-risk individuals presenting multiple or advanced colon lesions could utilize chemopreventive agents to reduce the risk of “interval” cancers that develop in between examinations. The power of chemoprevention as a tool for colon cancer prevention is strongly supported by the finding that a selected chemopreventive agent combination, DFMO and NSAID, can reduce polyp occurrence by up to 90% [4]. The success of this trial was based in part on the identification of a complementary pair of agents. Ideally chemopreventive agents and agent combinations would be devised that treat molecular deficiencies and/or suppress aberrant growth regulatory pathways promoting cancer development in an individual [5]. It is therefore important to understand the activities and limitations of individual agents, and to identify complementary agent combinations that provide more complete protection.
Vitamin D has long been suspected to reduce the risk of colon cancer development. Initial positive results came from geographical correlation studies showing an inverse relationship between sunlight exposure and colorectal cancer death rates [6]. Subsequent studies identified an interaction between high dietary or plasma vitamin D levels with a reduced colon cancer risk [7] [8] [9]. Although some observational studies have not detected protection by vitamin D [10, 11], there is sufficient positive data to consider vitamin D as a potential chemopreventive agent. Preclinical experiments likewise generated some positive data. Vitamin D was first tested in carcinogen-induced rodent colon cancer models (MNU, MNNG and DMH rat models) where significant vitamin D protection was reported [12-17]. Vitamin D protection has also been observed in a diet-induced model of colon cancer; sporadic colon tumors induced by a Western-style diet high in fat and low in vitamin D, calcium and folate can be suppressed by increasing vitamin D and calcium [18-20]. However, preclinical models also revealed some limitations. Lamprecht and colleagues [16] reported a significant reduction in vitamin D receptor activity within the colonic mucosa ten weeks after DMH treatment, suggesting that the ability of vitamin D to elicit protection might become diminished under some circumstances. Evidence of tumor protection has been observed in the ApcMin/+, but the degree of protection has been found to vary depending upon the experimental conditions. In one study, tumor frequency was not significantly affected, but tumor burden was decreased when animals received 1,25-dihydroxyvitamin D3 injections while on a AIN-93G diet [21, 22]. More substantial protection was observed when animals on a vitamin D-deficient diet were given injections of 1,25-dihydroxyvitamin D3 [23]. The limitations of vitamin D protection are also evident in a number of human intervention trials. In the Women's Health Initiative polyp prevention trial total vitamin D intake was not found to significantly reduce the risk of recurrent adenomas [24]. In a large placebo-controlled trial in which postmenopausal women received a daily vitamin D and calcium supplement for seven years the incidence of invasive colorectal cancer did not differ significantly between the supplementation group and the placebo group [25]. These conflicting pre-clinical and clinical data prompted us to test the effect of vitamin D supplementation on the ApcΔ14/+ mouse model, since this model generates a truncated APC protein similar to that which occurs in human cancers [26]. In addition, we also incorporated vitamin D into the diet at levels that approximated the high and low range of typical human consumption. Finally, we assessed the expression of VDR and other important regulatory proteins that control VDR expression.
Much of the present data points to the high affinity vitamin D receptor (VDR) as being a critical mediator of vitamin D protection. Zheng et al. tested this directly and found that ApcMin/+ mice on a VDR null background developed larger tumors than wild type controls [27]. Cancer suppression by VDR may be mediated in part by binding and inhibiting β-catenin [27]. Numerous reports have investigated VDR expression at different stages of colon cancer development with a number of studies finding that VDR expression is frequently lost in advanced lesions. Initial studies of colon cancer cell lines showed that well-differentiated cell lines tend to maintain higher levels of VDR expression relative to poorly differentiated lines with a greater metastatic potential [28]. Studies of patient-derived colorectal carcinoma tissue extracts initially generated conflicting results [28-32], but later studies employing histological approaches have generally shown VDR expression to be lost in metastatic cancers [33, 34]. The loss of VDR expression in advanced cancers is linked to the EMT and appears to be mediated by expression of the SNAIL1 and SLUG transcription factors [35-37]. A connection between colonic inflammation and VDR repression has also been uncovered, suggesting a possible link between long standing ulcerative colitis (UC) and increased colorectal cancer risk. VDR expression was found to be significantly lower in inflamed colonic mucosa [38], with the greatest reduction observed in long-standing UC patients at the highest risk of colon cancer [38]. Complementary results have been obtained in a mouse model of inflammation-promoted colon cancer. Mice treated with AOM and DSS show reduced VDR expression in inflamed mucosa, and ultimately develop tumors with reduced VDR expression [39]. The mechanism underlying the VDR expression changes in this model are not entirely clear, but may involve SNAIL1 and SLUG expression during the acute inflammatory phase.
Here we show that VDR expression and vitamin D responsiveness is dramatically down-regulated in colon tumors in the ApcΔ14/+ mouse, and that reduced VDR expression is observed even in very small lesions. VDR down-regulation in this model appears to be driven in part by increased HDAC expression, as VDR expression can be stimulated by treatment with the HDAC inhibitor, panobinostat [40]. Our data suggest that cancer prevention by vitamin D might be limited to early stages of cancer development, and that vitamin D intervention alone might not be sufficient to slow the growth and progression of initiated lesions. However, our data also suggest that agents that stimulate VDR expression may effectively enhance and extend the protective effects of vitamin D.
Materials and Methods
Mice
Male and female ApcΔ14/+ mice were fed modified AIN-93G diet containing low (250 IU/kg) or high (2500 IU/kg) vitamin D3 (Harlan Laboratories, WI) from 5 to 16 weeks of age. Calcium levels were maintained at 0.5% for both diets. Body weight was measured once per week, with no significant weight changes between the groups observed. For drug treatments, mice on the high vitamin D3 diet were injected intraperitoneally (i.p.) with four doses of panobinostat (10 mg/kg) or 5% dextrose (vehicle control) over three consecutive days (once/day, twice/day, once/day) during the last week of the experiment [40]. Mice on the high vitamin D3 diet were also treated with three doses of Bexarotene (50 mg/kg) or sesame oil (vehicle control) by gavage over three consecutive days (once/day)[41]. Mice were sacrificed 2 to 3 hours after the last dose. Mice were maintained in a temperature-controlled, light-cycled room and allowed free access to drinking water and the diet. Animal experiments were conducted after approval of the Center for Comparative Medicine (CCM), University of Connecticut Health Center.
Tumor incidence and multiplicity
Mice were sacrificed at 16 weeks of age for tumor scoring and histological analyses as described previously [42]. The small intestine and colon were harvested and flushed with ice-cold PBS and excised longitudinally. Some of the colon tumors were snap frozen in liquid nitrogen. Remaining tissues were fixed flat in 10% neutral buffered formalin solution for overnight and stored in 70% ethanol. The tissues were stained with 0.2% methylene blue and the number and size of the tumors were scored under a dissecting microscope.
Immunohistochemistry (IHC)
Small intestine and colon were paraffin-embedded and sectioned at a 5-μm thickness. Tissue sections were deparaffinized, antigen retrieved in 10 mM sodium citrate and incubated with 3% hydrogen peroxide for 20 min at RT. Sections were blocked with 10% goat serum and incubated overnight at 4°C with anti-VDR (1:2000; Santa Cruz, CA), anti-β-catenin (1:2000; Sigma-Aldrich, MO) or anti-Slug (1:50; Cell Signaling, MA). Sections were then incubated with secondary antibody for 30 min at RT, followed by signal detection using DAB solution (Vector Laboratories Inc.). Tissues were counterstained with hematoxylin. No primary antibody controls were run for all antibodies. The specificty of the VDR D-6 mouse monoclonal antibody was previously reported [43]. The β-catenin antibody has been reported to show the expected patterns of stabilization and cellular translocation during cancer development in Apc mutant mouse models [42].
RNA analysis
Isolated colon tissue and tumors were places in 1 mL of TRIzol reagent (Invitrogen, Carlsbad, CA) and homogenized. Chloroform extraction and precipitation was performed as described by Invitrogen. RNA was was quantified with a Thermo Scientific NanoDrop 8000 Spectrophotometer (Waltham, MA). Two micrograms of RNA were reverse transcribed using the Applied Biosystems High Capacity cDNA Reverse Transcription reagents (Foster City, CA) and analyzed using TaqMan assays (Life Technologies, Carlsbad, CA) on a Applied Biosystems 7500 Fast Real-time PCR System. ACTB was used an an internal control for the RNA quantification. The Applied Biosystems 7500 Standard Real-time PCR System software was used to analyze the date. The comparative CT method was used to determine the relative gene expression levels. Statistical analysis was performed using Excel and GraphPad Prism software.
Cell culture
HT29 and HCT116 colon cancer cell lines were obtained from the American Type Culture Collection (Manassas, VA) and cultured in McCoy's 5A medium with 10% fetal bovine serum, non-essential amino acids and antibiotic/antimycotic (Life Technologies, Guilford, CT). Cell lines from ATCC are authenticated. YAMC and IMCE cells were a gift from Dr. Whitehead (who established them) and were maintained in RPMI medium as previously described [44, 45].
Establishment and analysis of intestinal organoids
ApcMin/+ ESCs were maintained and differentiated into intestinal organoids using 30% Wnt3A conditioned media as previously described [46, 47]. Organoids were transferred to a 96-well microwell non-treated flat-bottom dish (Thermo Scientific, Waltham MA) containing WntCM media with compounds obtained from the Cayman Epigenetics Screening Library (Cayman Chemical Company, Ann Arbor MI). The final concentration of each compound was 25 μM. Organoids were collected for gene expression analysis after 48 hours. Following cell lysis, mRNA was captured on poly-T RNA capture plates as previously described [47]. Pre-amplification was carried out with 5 μL cDNA, 5μL 0.2× TaqMan primer pool, and 10μL TaKaRa PCR mix (6.3μL nuclease-free H2O, 2μL 10X Buffer, 1.6μL dNTP mix, 0.1μL TaKaRa Taq)(TaKaRa-Bio, Shiga, Japan) per reaction, using an initial temperature cycle of 95°C/3min, 55°C/2min, 72°C/2min, followed by 15 cycles of 95°C/15sec, 60°C/2min, 72°C/2min. Amplified cDNA was diluted 1:20 in nuclease-free water for further qPCR analysis. Quantitative PCR was carried out in 384-well optical PCR plates using the 7900HT thermal cycler (all from Life Technologies, Carlsbad, CA). Resulting data was analyzed by Applied Biosystems RQ Manager 1.2, Microsoft Excel, and SAS JMP.
Microarray data analysis
GEO data set GDS2947 was downloaded from NCBI and analyzed using Excel and GraphPad Prism software [48]. The Pearson correlation coefficient and p values were determined to assess the potential relationships between VDR and: the class I HDACs (HDACs 1, 2, 3 and 8), RXRA and SNAI2/SLUG. Signifcance was set for p < 0.05.
Statistics
Bulk RNA data was analyzed using a Student's t test for comparing two treatment groups, or by using an analysis of variance test (ANOVA) when comparing more than two groups. A Tukey's post-hoc test was employed to determine the significance of differences between multiple groups, with p<0.05 considered significant.
Results
Dietary vitamin D and tumor protection
The ApcΔ14/+ mouse model harbors a frame-shift mutation within exon 14, which generates a truncated protein of a size similar to that frequently observed in both FAP patients and sporadic colon cancers [26]. We determined whether colon and small intestinal tumor formation in these animals could be suppressed by the level of dietary vitamin D. The vitamin D3 level in defined mouse diets is 1000 IU/kg diet (as set by the National Research Council). For our studies, 250 IU/kg was selected for the low dose, which is sub-optimal for VDR target gene activation, but sufficient to maintain animal bone growth and general health [49]. A ten-fold higher level of 2500 IU was used to model a “supplemented” diet that would be employed to achieve cancer prevention. However, as shown in Figure 1A and 1B, no difference in the number of small intestinal tumors was observed, nor did high levels of vitamin D have any impact on the size of small intestinal tumors formed. In the colon, where the vast majority of human APC-initiated tumors develop, there was a trend towards reduction in tumor frequency and load, but this effect did not reach significance (Figures 1C and 1D). Similar findings were obtained when these vitamin D levels were tested on a more promotional dietary background with higher fat and lower fiber levels (Supplementary, Figure S1). From these data we conclude that vitamin D supplementation is not a highly effective approach to cancer prevention in this pre-clinical mouse model.
Figure 1. No effect of dietary vitamin D levels on tumor formation in ApcΔ14/+ mice.
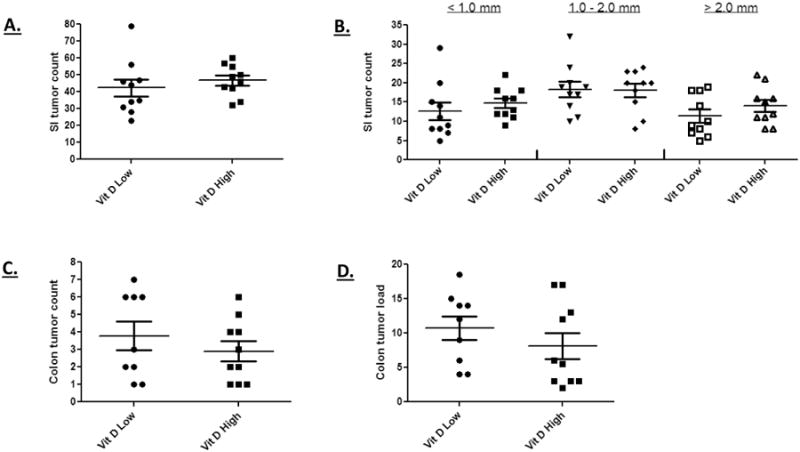
A high vitamin D3 diet did not reduce the overall incidence of small intestinal tumors in ApcΔ14/+ mice (A) nor did it effect the formation of larger tumors (B). The high vitamin D3 diet generated a small, non-significant decrease in colon tumor incidence (D) and load (E). These results were obtained from ten animals in both the high and low vitamin D supplementation group.
VDR loss in colon tumors
Given the lack of protection from tumor development by a high vitamin D diet observed in Figure 1, we determined the VDR expression status in colon tumors and adjacent normal tissue of mice on the low and high vitamin D diets. The data shown in Figure 2A indicates that VDR mRNA expression was reduced ∼5-fold in tumors relative to normal tissue. In addition, the high vitamin D diet stimulated VDR expression in normal tissue but not in the tumors. The VDR-target gene Cdh1 showed a similar pattern of expression, being vitamin D responsive in the normal tissue, but not in the tumors (Figure 2B)[50]. Analysis of VDR protein expression by IHC reflected the change in mRNA expression; normal regions of the tissue stained intensely for nuclear VDR, whereas large regions of the tumor were negative (Figure 3A). These findings are consistent with a model in which loss of VDR expression limits the ability of vitamin D to control tumor formation and growth.
Figure 2. Relative gene expression in normal and tumor tissue of mice on the low (250 IU) and high (2500 IU) vitamin D3 diet.

RNA was isolated from exophytic tumors and adjacent normal tissue at the 16 week time point. The relative expression of VDR (A) and Cdh1 (B) was determined using TaqMan assays. A significant reduction of VDR (p<0.0001) and Cdh1 (p<0.01) expression was observed in tumors. In addition, a vitamin D only significantly increased expression of these genes in normal tissue (p<0.01). A total of seven and five mice, respectively, were examined in the low and high vitamin D groups.
Figure 3. VDR protein expression is lost in colon tumors of ApcΔ14/+ mice.
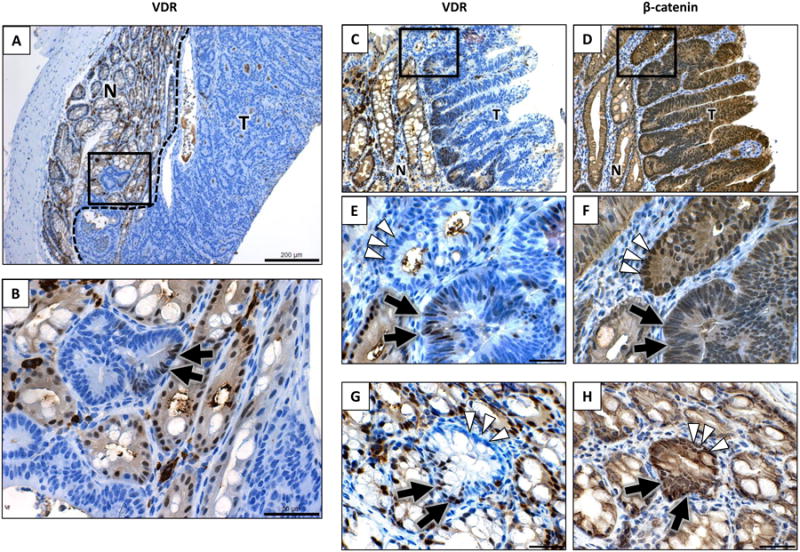
A and B) A representitive tumor from a 16-week old mouse maintained on the high vitamin D3 diet is shown. Panel A shows a low magnification view of positively staining normal tissue juxtaposed with a large, non-staining exophytic tumor (200 μm). The boxed region (at the tumor-normal interface) is shown at a higher magnification in panel B (50 μm). Most cancer cells in this lesion do not express VDR, except for limited pockets of expression (shown by the arrow). C through F) VDR expression and β-catenin nuclear localization in tumors of ApcΔ14/+ mice. The C and D panels show serial sections of tumor tissue stained for VDR or β-catenin, repectively (bar is 100 μm). Figures E and F show a higher magnification view of the top panels (bar is 30 μm). The arrowheads in panels E and F indicate an adenomatous crypt that is VDR negative with heavy β-catenin nuclear localization. The arrows indicate an adenomaotus crypt with lower β-catenin localization and sporadic VDR staining. G and H) An individual crypt showing elevated β-catenin nuclear localization (G) and reduced VDR expression (H). This crypt includes regions of low VDR expression (arrowheads), with other areas similar to staining as adjacent normal tissue (arrows).
Although large regions of the tumor were VDR negative, a closer analysis of the staining revealed that clusters of cells within the tumors retained VDR expression (Figure 3B). Another example of VDR staining is shown in Figure 3C, where a series of adenomatous colon crypts express VDR at their base. A higher magnification view shows that the VDR expression in these adenomatous crypts is variable (Figure 3E), with some crypts showing VDR-expressing cells (double arrow) and others being VDR-negative (white arrowheads). Since the extent of β-catenin nuclear localization can vary between adenomatous crypts, and antagonism between VDR and β-catenin has been reported, a serial section of tissue was analyzed for β-catenin nuclear localization (Figure 3D and 3F). An inverse association was observed between VDR expression and β-catenin nuclear localization in the majority of the adenomatous crypts examined, with co-expression apparent in relatively few lesions (less than 5%). Potential mechanisms underlying this inverse association are discussed below.
To determine whether VDR expression was reduced at an early stage of tumor development, the tissue was examined for very small lesions showing aberrant β-catenin stabilization and nuclear localization (arising from Apc loss of heterozygosity)[51]. Figure 3 shows a single crypt with increased levels of β-catenin stabilization and nuclear localization (Figure 3H; white arrowheads). This single crypt also showed reduced VDR levels (Figure 3G; white arrowheads), indicating that VDR expression can be lost at a very early stage of colon tumor development. Ten single crypt lesions were observed in the study group, and only one maintained VDR expression at normal levels (90% VDR-positive cells). The very early loss of VDR expression is consistent with the inability of vitamin D supplementation to reduce tumor incidence in this model.
Expression of VDR regulators in ApcΔ14/+ tumors
VDR expression is tightly regulated by a complex promoter and enhancer element. Several transcriptional regulatory proteins have been demonstrated to impact VDR expression under a number of different physiological contexts, so we investigated the potential role of these factors in the loss of VDR expression in tumors. Colon cancer cells have been reported to lose VDR expression as the result of increased Snail and Slug expression [35]. These transcription factors can be expressed in colon cancer cells following the epithelial to mesenchymal transition (EMT). The Snail and Slug transcription factors bind to an E-box element within the VDR promoter and inhibit its expression through the recruitment of transcriptional co-repressors. Although Snail1 expression was unchanged in the tumors, Slug was increased ∼2-fold (Figure 4A). Since Slug is normally expressed in stromal cells, an IHC was performed to determine if tumor epithelial cells within the lesion were expressing Slug. As shown in Figure 4B, Slug staining was limited to stromal cells positioned between the adenomatous crypts of the tumor. The decreased VDR expression in the tumor cells can therefore not be directly accounted for by an increased expression of Snail1 or Slug, although it is possible that the stromal cells may indirectly impact VDR expression in the tumors.
Figure 4. Expression of potential VDR regulators in normal and tumor tissue.
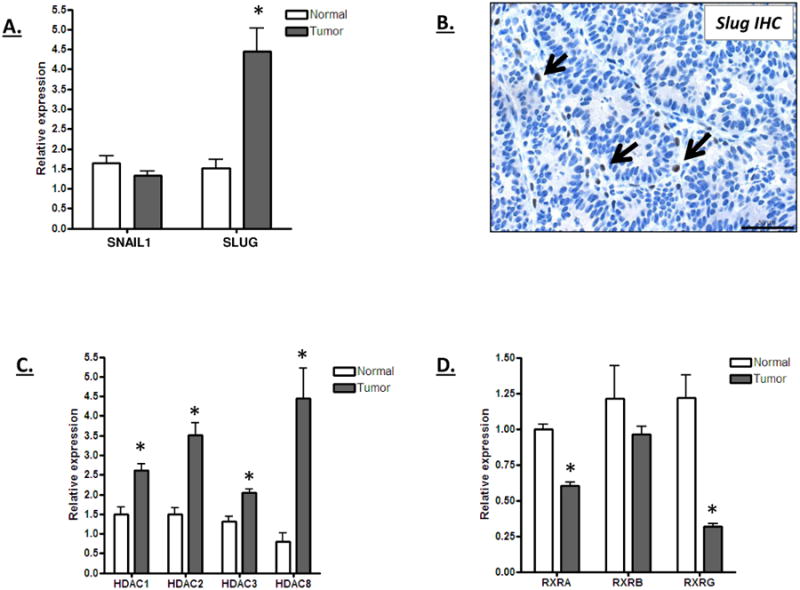
A) Relative Snail1 and Slug mRNA expression in normal and tumor tissue of mice on the high vitamin D3 diet. RNA was isolated from tumors and adjacent normal tissue at the 16 week time point and analyzed for Snail1 and Slug mRNA expression levels. The increased Slug expression in the tumors was significant (p<0.01). B) Representative IHC staining for Slug expression in an ApcΔ14/+ colon tumor. Sporadic Slug staining was observed in stromal cells (indicated by arrows), but not within the epithelial cells of the tumor. C and D) Relative HDAC (C) and RXR (D) mRNA expression in normal and tumor tissue of mice on the high vitamin D3 diet. All of the class I HDACs were expressed at a significantly higher level in tumors than in normal tissue (p<0.01) whereas RXRA and RXRG were expressed at a lower level in the tumor (p<0.001). Five animals were analyzed in each group.
Other negative regulators of VDR include the class I HDACs, which are commonly over-expressed in human and mouse colon tumors [52-56]. As shown in Figure 4C, all of the class I HDACs were increased in expression in the ApcΔ14/+ tumors. Conversely, all three retinoid X receptor genes, which are positive regulators of VDR expression that interact with the VDR promoter and activate its expression, were down-regulated (Figure 4D). These data suggest that an increase in HDAC expression and/or a decrease in RXR expression may contribute to the reduction in VDR expression observed in the ApcΔ14/+ tumor cells.
Reactivation of VDR expression by HDAC inhibitors
Given the association between increased HDAC expression and VDR repression, we assessed the ability of HDAC inhibitors to stimulate VDR expression in cancer cells. We first examined a number of cell culture models for their responsiveness to HDAC inhibition. In Figure 5A and 5B, HCT116 and HT29 cells were treated with SAHA and assayed for VDR expression. VDR was activated in HCT116 cells by SAHA, indicating that HDACs limit VDR expression in this human cell line. The level of VDR activation was found to be comparable to p21, a gene that has been well-documented for its regulation by HDACs in colon cancer cells [57, 58]. In contrast, VDR expression in HT29 cells was not activated by SAHA, even though p21 was responsive (Figure 5A). One possible explanation for the lack of VDR activation in HT29 cells is that the VDR promoter in these cells is subjected to higher level of epigenetic repression, such as DNA methylation. To determine whether DNA methylation was limiting VDR expression in HT29 cells, cells were treated with 5-aza-deoxycytidine for 48 hours. As shown in Figure 5B, 5-aza-deoxycytidine was able to activate VDR expression in HT29 cells, consistent with VDR silencing through DNA methylation in this cell line. (A longer 96-hour treatment with 5-aza-2′-deoxycytidine did not increase VDR expression in HCT116 cells.) Finally, to assess the role of HDACs in VDR regulation is mouse colon cells, we tested the ability of panobinostat to activate VDR expression in conditionally immortalized YAMC (Apc+/+) and IMCE (Apc-/+) cells [44, 45]. Panobinostat readily activated the expression of VDR in both cell lines, consistent with the role of HDACs in regulating VDR expression in colonic cells.
Figure 5. Epigenetic regulation of VDR in colon derived cell lines.

A and B) HCT116 and HT29 cells were treated with 10 μM SAHA or 5 μM 5-azacytidine for 48 hours. RNA was then prepared and analyzed for VDR and p21 expression. Significant changes are indicated by asterisks (p<0.01). C) YAMC and IMCE cells were treated with the indicated levels of panobinostat for 24 hours. The expression of VDR mRNA was determined. Increased mRNA expression was determined to be significant for both panobinostat concentrations tested (* p<0.001).
Given that HDACs are over-expressed in ApcΔ14/+ tumors (Figure 4C), and that VDR expression can be reactivated in some colon cancer cell lines with HDAC inhibitors, the effect of HDAC inhibition on VDR expression in vivo was tested. Panobinostat was chosen for these studies, given its broad spectrum of activity and favorable pharmacokinetic properties (relative to other hydroxyamic acid HDAC inhibitors)[40]. Four doses were used, each spaced approximately 12 hours apart (Figure 6A). Approximately 3 hours after the last dose, animals were euthanized and tumor and normal tissues were obtained. As shown in Figure 6B, VDR expression was increased in the tumors by panobinostat. We next compared the activation of VDR to that of p21, a gene that is readily activated in colon cancer cells by HDAC inhibitors (Fig. 5). The degree of VDR activation was comparable (albeit slightly lower) to p21. Finally, we tested the response of adjacent normal tissue to panobinostat treatment. VDR expression in normal tissue was also responsive to panobinostat, and was in fact more responsive than p21 within the normal tissue. These findings are consistent with HDACs contributing to the suppression of VDR in colon tumors and adjacent epithelium.
Figure 6. Pharmacological activation of VDR expression in colon tumors.
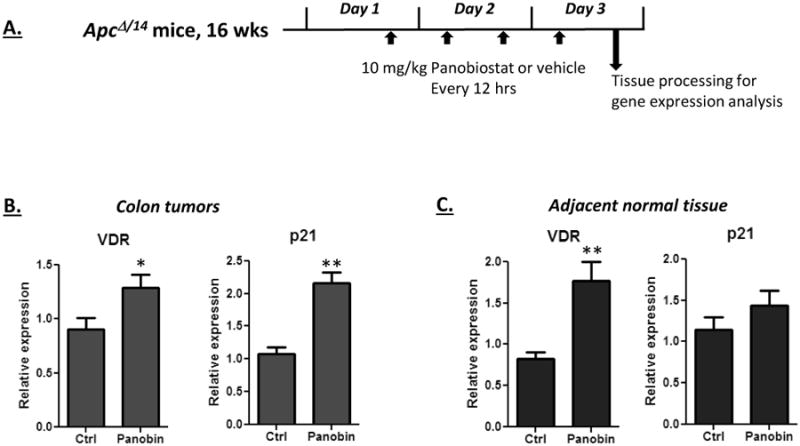
A) Schematic of dosing procedure. Tumor bearing ApcΔ14/+ mice on the high vitamin D3 diet received four i.p. injections of panobinostat over a 3 day period (10 mg/kg, spaced approximately twelve hours apart). Control animals received injections of 5% dextrose as the vehicle control. Three hours after the final injection, colon tumors and adjacent normal tissue was obtained and assayed for VDR and p21 expression. B and C) VDR and p21 expression was activated by panobinostat in the tumors (B) and the adjacent normal tissue (C). VDR and p21 increases were significant in the tumors, and VDR activation was significant in the normal tissue (* p<0.05, ** p<0.01). Five animals were in each group.
Histone acetylation frequently works in combination with other chromatin modifications to enforce epigenetic gene regulation. We therefore tested a panel of agents that target a variety of epigenetic modifiers, including histone deacetylases, methyltransferases and demethylases, on intestinal organoids generated from ApcMin/+ ESCs. (Intestinal organoids were employed here since they maintain a similar structural arrangement as normal tissue, and we have developed approaches that facilitate the medium throughput analysis of gene expression changes in this system [47].) Whereas p21 expression was responsive to many of the epigenetically active compounds, VDR was selectively and robustly activated only in response to HDAC inhibitors (Figure 7). As reported in other cell systems, the HDAC inhibitors also suppressed expression of the Wnt target gene Myc, whereas the effect of other epigenetic agents on Myc expression was more variable.
Figure 7. Analysis of epigenetically-active compounds on VDR expression in ApcMin/+ intestinal organoids.
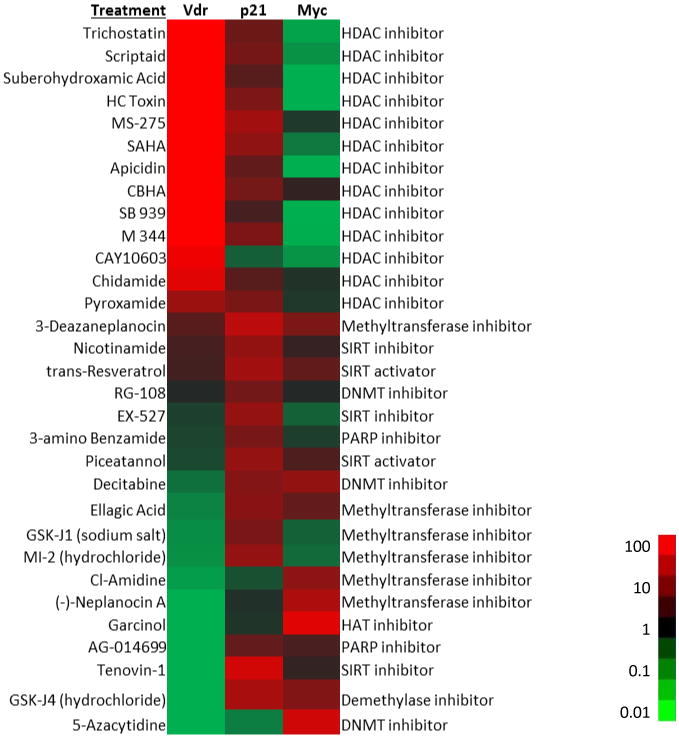
Intestinal organoids were generated from ApcMin/+ ESCs and treated with compounds from the Cayman Epigenetics Screening Library (48 hours) in a high throughput format. RNA was isolated and analyzed for the expression of VDR, p21 and Myc. The compound name is shown at the left of the heat map, with its activity shown on the right. The fold-activation of mRNA expression relative to control cells was determined and displayed on a green to red scale. Data were sorted by the fold activation of VDR.
Given that RXR expression is reduced in ApcΔ14/+ tumors, we also determined whether the pharmacological RXR agonist bexarotene may activate VDR expression in these tumors. Bexarotene was provided to the animals through gavage for three days, and three hours after the last dose, tissue was prepared and assessed for VDR expression. Bexarotene did not activate VDR expression, in tumors nor in normal tissue (Supplementary, Figure S2). Although this result does not exclude the potential contribution of RXR down-regulation to the reduced VDR expression in the colon tumors, it appears that the availability of RXR agonists does not limit VDR expression in this model.
Relationship between VDR expression and transcriptional regulators in human adenomas
Taking advantage of the NCBI GEO database, we utilized a published microarray data set comparing human adenomas and adjacent normal tissue to determine whether VDR mRNA expression was reduced in the adenomas [48]. As shown in Figure 8A, 29 out of 32 matched normal-adenoma pairs showed lower levels of VDR expression in the adenomas. The reduction in VDR expression was inversely associated with the expression of the class I HDACs, all of which were significantly increased in the adenomas. To further examine the relationship between VDR expression and potential regulatory proteins, a correlation analysis was performed. As shown in Figure 8B, a significant inverse correlation between VDR expression and HDACs 2 and 8 were observed. This association was less significant for HDAC1 (p<0.003) and non-significant for HDAC3 (p=0.074). These data suggest that the increased expression of a subset of HDACs may contribute to VDR repression in human adenomas. As found in the ApcΔ14/+ tumors, RXRA expression was significantly reduced in human adenomas, and this reduction was correlated with VDR expression, suggesting a potential co-regulation of these two nuclear receptors. Finally, the expression of SNAIL2/SLUG was lower in adenomas than normal tissue, and showed a positive correlation with VDR expression. This result argues against a role of SNAI2/SLUG in VDR repression in human adenomas.
Figure 8. VDR and class I HDAC expression levels in human adenomas and adjacent normal colonic tissue.
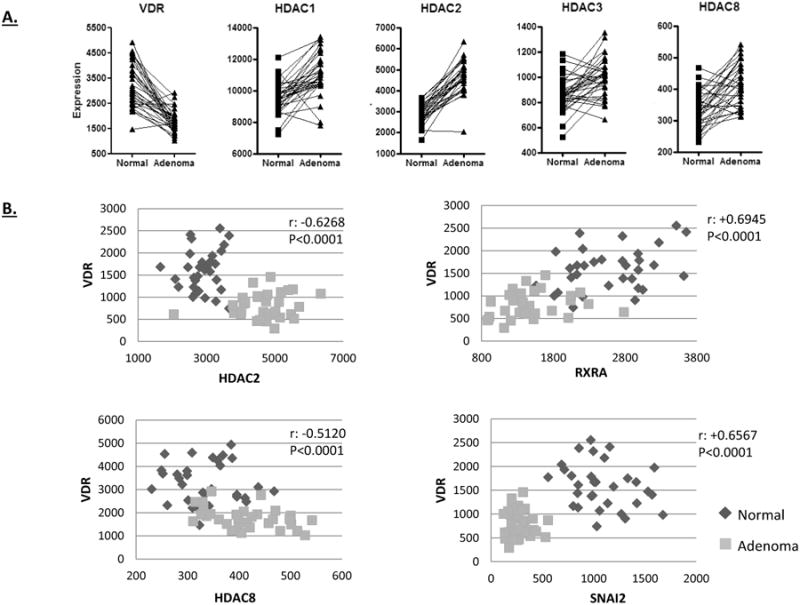
A) Data from the NCBI GEO database (GDS2947) was downloaded and plotted to show VDR and HDAC expression in the matched samples. A significant down-regulation in VDR expression was observed in the adenomas, which is in contrast to the up-regulation of the class I HDACs (p<0.0001). B) VDR expression negatively correlated with HDAC2 and HDAC8 expression, and positively correlated with RXRA and SNAI2/SLUG in human adenomas. Correlations were signficant as determined by Pearson, with the p values indicated on the graphs.
We also examined the expression of VDR in human adenomas by IHC. Representative examples of tubular adenomas are shown in Figures 9A and 9B. Close analysis of cells within individual lesions showed variable levels of VDR expression, and included cells with high to no detectable VDR expression. In this regard, the human adenomas are similar to the lesions formed in the ApcΔ14/+ mouse, although the VDR-positive cells in the mouse tumors were more likely to be found in restricted cell clusters. Implications of these findings with regard to cellular growth control and cancer prevention by vitamin D are discussed below.
Figure 9. Representative IHC images of human adenomas stained for VDR expression.
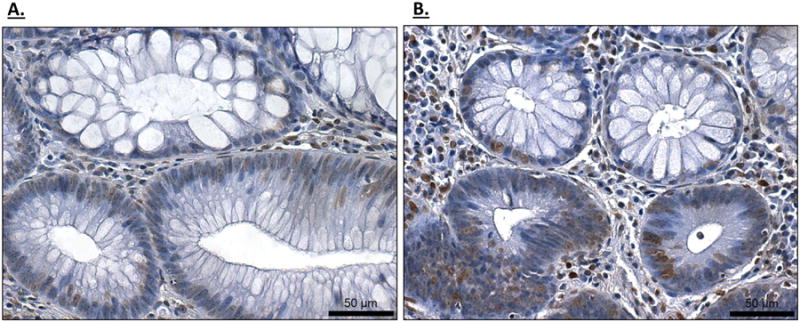
Two independent adenomas (A and B) showing VDR-expressing and non-expressing cells within a lesion.
Discussion
The effective utilization of chemopreventive agents to reduce the risk of colon cancer development could provide a powerful complement to colonoscopic screening efforts, particularly in high-risk patients. Epidemiological studies have suggested that vitamin D may prevent colon cancer [59-64]. However, intervention trials in patient cohorts supplemented with vitamin D have generated somewhat ambiguous results [25]. On the other hand, pre-clinical studies in mouse colon cancer models have shown moderate cancer protection. For example, in ApcMin/+ mice, three weekly injections of 1,25-dihroxyviatmin D3 supplementation did not reduce tumor incidence, but reduced tumor burden by almost half. In a study intended to optimize vitamin D as a dietary chemopreventive agent, we first attempted to demonstrate vitamin D protection in the ApcΔ14/+ mouse model [26]. Tumors formed in this model are genetically similar to those formed in FAP patients [26], and carry Apc truncation mutations like the majority of sporadic cancers. Interestingly we found no significant protection in ApcΔ14/+ mice, either in the small intestine or in the colon. Upon examination of normal mucosa and neoplastic tissue, we found an extensive loss of VDR expression in tumors formed in these animals. Previous work has shown that VDR expression can be reduced within inflamed colonic epithelium and in neoplastic lesions associated with ulcerated mucosa [39]. Our findings here show that the loss of VDR expression can occur even in the absence of inflammation. The lack of colon cancer protection afforded by vitamin D in this model may therefore be related to the lack of VDR expression in the lesions.
Examination of the VDR expression pattern in the ApcΔ14/+ tumors showed a number of interesting features. First, even though the majority of cancer cells within the tumor did not express VDR, a subpopulation of cells expressed VDR at levels similar to those found in normal crypts. Lineage tracing and transcription profiling experiments have revealed that colon cancers are composed of multiple cell types with varying proliferative capacities and differentiation profiles [65, 66]. Although it is not clear how VDR expression relates to the different cell types found in the tumors, it is likely that this cell heterogeneity is contributing to the variability in expression. Examination of human adenomas likewise showed variability in VDR expression within individual lesions. However, unlike the tumors formed in ApcΔ14/+ mice, where VDR-expressing cells were clustered within the lesion, VDR-expressing cells in human adenomas were intermingled amongst non-expressing cells. The relationship between the expressing and non-expressing cells within the mouse colon lesions and human adenomas is presently unclear, but identifying these cell types could provide insight into the chemopreventive potential of vitamin D. Another interesting feature of VDR expression in the ApcΔ14/+ tumors is that VDR-expression in adenomatous crypts is inversely associated with β-catenin nuclear localization. Although the basis of this inverse relationship is unclear, there are reports of VDR and β-catenin competition for nuclear binding sites, raising the possibility that this inverse expression may be mechanistically linked [67]. Specifically, VDR has been reported to compete with β-catenin for binding to the Lef1 transcription factor, suggesting that VDR might reduce the nuclear accumulation of β-catenin by blocking its binding to nuclear Lef1 [67]. Finally, a loss of VDR expression could be observed in very early lesions in the mouse, suggesting that a decrease in VDR expression occurs soon after Apc loss and β-catenin stabilization.
The loss of VDR expression in human colon cancers has previously been reported to occur after a lesion has undergone an EMT [35, 68-70]. For these advanced cancers, VDR expression is frequently repressed through the actions of the SNAIL transcription factors (SNAIL1 and SNAIL2/SLUG), which are transcription regulatory proteins central to EMT enforcement. However, mouse colon cancer models typically undergo a “truncated EMT” in which vimentin expression increases, E-cadherin decreases, but SNAIL expression is not activated [71]. Consistent with these reports, we did not observe increased Snail1 or Slug expression in the epithelial cells of the colon tumors of ApcΔ14/+ mice, although Slug expression was increased within the stroma. These findings indicate that some other mechanism is involved in repressing VDR expression. Our analysis of VDR and SNAIL expression in human colon adenomas is likewise inconsistent with SNAIL repressing VDR expression in these lesions. Whether the mechanism of VDR repression is similar in ApcΔ14/+ mice and human adenomas remains to be determined.
VDR expression is regulated by a number of different promoter response elements and transcription factors, so any one of a number of regulatory pathways may be responsible for the decreased VDR expression observed in the lesions. One pathway of interest is a positive feedback loop in which VDR-RXR complexes activate VDR transcription by binding vitamin D response elements (VDREs) in the VDR promoter [72]. Our data suggest that this positive feedback loop is shut down in ApcΔ14/+ tumors, since vitamin D supplementation did not activate VDR expression in the tumors. There are reports that RXR and RXR agonists can regulate VDR expression [72, 73]. Consistent with this possibility, we found that RXR expression was down-regulated in the tumors of ApcΔ14/+ mice (specifically RXRA and RXRG). RXR down-regulation has been reported in other mouse colon cancer models, and appears to be mediated by promoter methylation [74]. However, we were unable to activate VDR expression with the synthetic RXR agonist bexarotene, suggesting that the availability of RXR ligands is not limiting VDR expression in ApcΔ14/+ mice.
Another frequent molecular aberration in both mouse and human colon tumors that may be related to VDR repression is the increased expression of HDACs. HDACs catalyze the deacetylation of histone proteins, which can reduce the expression of target promoters. The class I nuclear HDACs (HDACs 1, 2, 3 and 8) are frequently increased in expression in colon cancer cells and we found a similar up-regulation in the ApcΔ14/+ mouse model and in human adenomas [55]. The increased expression of the HDACs may be related to hyper-activation of Wnt signaling in the tumors resulting from mutations to Apc. For example, HDAC2 is activated by the Myc transcription factor in response to mutations within the Apc gene and associated β-catenin stabilization [55]. Studies have shown that HDACs primarily regulate the expression level of active genes, or genes poised for activation [75]. VDR appears to fall into this category since its expression in both normal and tumor tissue of ApcΔ14/+ mice was stimulated by the HDAC inhibitor panobinostat. Increased HDAC expression in the mouse tumors may therefore contribute to the reduced level of VDR expression. HDACs are however unlikely to be entirely responsible for VDR repression in the tumors, since panobinostat did not completely restore VDR expression to normal levels. HDAC inhibitors were however generally capable of stimulating VDR expression in some human cancer cell lines, as well as conditionally immortalized mouse colon cell lines and a mouse intestinal organoid system. In one colon cancer cell line, VDR appeared to be repressed through DNA methylation, indicating that other mechanism for VDR silencing are also possible.
The finding that HDAC inhibitors activate VDR expression in tumors and normal tissue raises the possibility that HDAC inhibitory compounds may be capable of enhancing cancer chemoprevention by vitamin D. In one report, the selective HDAC2 inhibitor, OSU-HDAC42, reduced tumor formation in the large and small intestine of ApcMin/+ mice by 26 and 44%, respectively [76], and it would be of interest to know whether vitamin D supplementation might further reduce tumor formation. Although it is not feasible to use pharmacological HDAC inhibitors as chemopreventive agents, it has been reported that a number of natural compounds and dietary factors have intrinsic HDAC inhibitory activity, including fiber-derived short chain fatty acids and isothiocyanates derived from cruciferous vegetables [58, 77-79]. Although agents with HDAC inhibitory activity have been found to be protective in mouse colon cancer models, the contribution of VDR to this protection has thus far been untested. It is not clear which dietary compounds might be best suited for stimulating VDR expression in humans, or whether agents targeting other regulatory proteins might further promote expression. In this regard, recently developed intestinal organoid-based high throughput gene analysis approaches may be useful since a wide variety of compounds and compound combinations can be readily analyzed in this system [47].
Whether vitamin D is effective for reducing colon cancer risk in human populations is unclear. Observational and interventional studies have generated conflicting data on this topic [25, 59, 61, 64]. However, a recent analysis of serum vitamin D levels and colon cancer development from the Nurses' Health Study and the Health Professionals' Follow-up Study found that individuals with the highest serum vitamin D levels had approximately half the risk of developing colon cancer relative to the lowest quintile [80]. Interestingly, VDR expression in the resulting cancers was lost at the same rate, regardless of serum vitamin D levels. Although there are a number of potential explanations for this finding, it appears that there are a number of mechanisms by which cancer suppression by vitamin D can be overcome. ApcΔ14/+ mice appear to model VDR repression mediated through the increased expression of HDACs. These mice could therefore be useful for identifying compounds that alleviate HDAC-mediated repression. Understanding how vitamin D and VDR expression impacts colon cancer development could improve the way in which vitamin D is utilized in the clinic as a cancer chemopreventive agent. Given that the benefits and risks associated with vitamin D are well understood, it may turn out to be a highly effective cancer preventive agent when used in the proper dietary context (e.g., under conditions that suppress VDR silencing) and targeted to the appropriate patient population.
Supplementary Material
Acknowledgments
Financial support: These studies were supported by a University of Connecticut Diet and Health Initiative grant to D.W. Rosenberg and C. Giardina, grant R21CA158743 to C. Giardina and grant R01CA159976 to D.W. Rosenberg.
Footnotes
Conflicts of Interest: none
References
- 1.Vital signs: colorectal cancer screening, incidence, and mortality --- United States, 2002--2010. MMWR Morb Mortal Wkly Rep. 2011;60:884–9. [PubMed] [Google Scholar]
- 2.Edwards BK, Ward E, Kohler BA, Eheman C, Zauber AG, Anderson RN, et al. Annual report to the nation on the status of cancer, 1975-2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116:544–73. doi: 10.1002/cncr.24760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson RS, Thorson AG. Colorectal cancer screening. Curr Oncol Rep. 2009;11:482–9. doi: 10.1007/s11912-009-0065-8. [DOI] [PubMed] [Google Scholar]
- 4.Meyskens FL, Jr, McLaren CE, Pelot D, Fujikawa-Brooks S, Carpenter PM, Hawk E, et al. Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: a randomized placebo-controlled, double-blind trial. Cancer Prev Res (Phila) 2008;1:32–8. doi: 10.1158/1940-6207.CAPR-08-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meyskens FL, Jr, Szabo E. Diet and cancer: the disconnect between epidemiology and randomized clinical trials. Cancer Epidemiol Biomarkers Prev. 2005;14:1366–9. doi: 10.1158/1055-9965.EPI-04-0666. [DOI] [PubMed] [Google Scholar]
- 6.Garland CF, Garland FC. Do sunlight and vitamin D reduce the likelihood of colon cancer? Int J Epidemiol. 1980;9:227–31. doi: 10.1093/ije/9.3.227. [DOI] [PubMed] [Google Scholar]
- 7.McCullough ML, Robertson AS, Rodriguez C, Jacobs EJ, Chao A, Carolyn J, et al. Calcium, vitamin D, dairy products, and risk of colorectal cancer in the Cancer Prevention Study II Nutrition Cohort (United States) Cancer Causes Control. 2003;14:1–12. doi: 10.1023/a:1022591007673. [DOI] [PubMed] [Google Scholar]
- 8.Lieberman DA, Prindiville S, Weiss DG, Willett W, Group VACS. Risk factors for advanced colonic neoplasia and hyperplastic polyps in asymptomatic individuals. JAMA. 2003;290:2959–67. doi: 10.1001/jama.290.22.2959. [DOI] [PubMed] [Google Scholar]
- 9.Freedman DM, Looker AC, Chang SC, Graubard BI. Prospective study of serum vitamin D and cancer mortality in the United States. J Natl Cancer Inst. 2007;99:1594–602. doi: 10.1093/jnci/djm204. [DOI] [PubMed] [Google Scholar]
- 10.Lin J, Zhang SM, Cook NR, Manson JE, Lee IM, Buring JE. Intakes of calcium and vitamin D and risk of colorectal cancer in women. Am J Epidemiol. 2005;161:755–64. doi: 10.1093/aje/kwi101. [DOI] [PubMed] [Google Scholar]
- 11.Terry P, Baron JA, Bergkvist L, Holmberg L, Wolk A. Dietary calcium and vitamin D intake and risk of colorectal cancer: a prospective cohort study in women. Nutr Cancer. 2002;43:39–46. doi: 10.1207/S15327914NC431_4. [DOI] [PubMed] [Google Scholar]
- 12.Kawaura A, Takahashi A, Tanida N, Oda M, Sawada K, Sawada Y, et al. 1 Alpha-hydroxyvitamin D3 suppresses colonic tumorigenesis induced by repetitive intrarectal injection of N-methyl-N-nitrosourea in rats. Cancer Lett. 1990;55:149–52. doi: 10.1016/0304-3835(90)90025-s. [DOI] [PubMed] [Google Scholar]
- 13.Oda M, Kawaura A, Tanida N, Sawada K, Maekawa S, Kano M, et al. Effects of 1 alpha-hydroxyvitamin D3 on N-methyl-N-nitrosourea-induced colonic tumorigenesis, and on fecal bile acid profiles with respect to soluble and precipitated phases in rats. Tokushima J Exp Med. 1990;37:75–81. [PubMed] [Google Scholar]
- 14.Kawaura A, Tanida N, Sawada K, Oda M, Shimoyama T. Supplemental administration of 1 alpha-hydroxyvitamin D3 inhibits promotion by intrarectal instillation of lithocholic acid in N-methyl-N-nitrosourea-induced colonic tumorigenesis in rats. Carcinogenesis. 1989;10:647–9. doi: 10.1093/carcin/10.4.647. [DOI] [PubMed] [Google Scholar]
- 15.Kawaura A, Tanida N, Nishikawa M, Yamamoto I, Sawada K, Tsujiai T, et al. Inhibitory effect of 1alpha-hydroxyvitamin D3 on N-methyl-N′-nitro-N-nitrosoguanidine-induced gastrointestinal carcinogenesis in Wistar rats. Cancer Lett. 1998;122:227–30. doi: 10.1016/s0304-3835(97)00397-2. [DOI] [PubMed] [Google Scholar]
- 16.Belleli A, Shany S, Levy J, Guberman R, Lamprecht SA. A protective role of 1,25-dihydroxyvitamin D3 in chemically induced rat colon carcinogenesis. Carcinogenesis. 1992;13:2293–8. doi: 10.1093/carcin/13.12.2293. [DOI] [PubMed] [Google Scholar]
- 17.Sitrin MD, Halline AG, Abrahams C, Brasitus TA. Dietary calcium and vitamin D modulate 1,2-dimethylhydrazine-induced colonic carcinogenesis in the rat. Cancer Res. 1991;51:5608–13. [PubMed] [Google Scholar]
- 18.Newmark HL, Yang K, Kurihara N, Fan K, Augenlicht LH, Lipkin M. Western-style diet-induced colonic tumors and their modulation by calcium and vitamin D in C57Bl/6 mice: a preclinical model for human sporadic colon cancer. Carcinogenesis. 2009;30:88–92. doi: 10.1093/carcin/bgn229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang K, Kurihara N, Fan K, Newmark H, Rigas B, Bancroft L, et al. Dietary induction of colonic tumors in a mouse model of sporadic colon cancer. Cancer Res. 2008;68:7803–10. doi: 10.1158/0008-5472.CAN-08-1209. [DOI] [PubMed] [Google Scholar]
- 20.Newmark HL, Yang K, Lipkin M, Kopelovich L, Liu Y, Fan K, et al. A Western-style diet induces benign and malignant neoplasms in the colon of normal C57Bl/6 mice. Carcinogenesis. 2001;22:1871–5. doi: 10.1093/carcin/22.11.1871. [DOI] [PubMed] [Google Scholar]
- 21.Huerta S, Irwin RW, Heber D, Go VL, Moatamed F, Ou C, et al. Intestinal polyp formation in the Apcmin mouse: effects of levels of dietary calcium and altered vitamin D homeostasis. Dig Dis Sci. 2003;48:870–6. doi: 10.1023/a:1023083025595. [DOI] [PubMed] [Google Scholar]
- 22.Huerta S, Irwin RW, Heber D, Go VL, Koeffler HP, Uskokovic MR, et al. 1alpha,25-(OH)(2)-D(3) and its synthetic analogue decrease tumor load in the Apc(min) Mouse. Cancer Res. 2002;62:741–6. [PubMed] [Google Scholar]
- 23.Xu H, Posner GH, Stevenson M, Campbell FC. Apc(MIN) modulation of vitamin D secosteroid growth control. Carcinogenesis. 2010;31:1434–41. doi: 10.1093/carcin/bgq098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartman TJ, Albert PS, Snyder K, Slattery ML, Caan B, Paskett E, et al. The association of calcium and vitamin D with risk of colorectal adenomas. J Nutr. 2005;135:252–9. doi: 10.1093/jn/135.2.252. [DOI] [PubMed] [Google Scholar]
- 25.Wactawski-Wende J, Kotchen JM, Anderson GL, Assaf AR, Brunner RL, O'Sullivan MJ, et al. Calcium plus vitamin D supplementation and the risk of colorectal cancer. N Engl J Med. 2006;354:684–96. doi: 10.1056/NEJMoa055222. [DOI] [PubMed] [Google Scholar]
- 26.Colnot S, Niwa-Kawakita M, Hamard G, Godard C, Le Plenier S, Houbron C, et al. Colorectal cancers in a new mouse model of familial adenomatous polyposis: influence of genetic and environmental modifiers. Lab Invest. 2004;84:1619–30. doi: 10.1038/labinvest.3700180. [DOI] [PubMed] [Google Scholar]
- 27.Zheng W, Wong KE, Zhang Z, Dougherty U, Mustafi R, Kong J, et al. Inactivation of the vitamin D receptor in APC(min/+) mice reveals a critical role for the vitamin D receptor in intestinal tumor growth. Int J Cancer. 2012;130:10–9. doi: 10.1002/ijc.25992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evans SR, Nolla J, Hanfelt J, Shabahang M, Nauta RJ, Shchepotin IB. Vitamin D receptor expression as a predictive marker of biological behavior in human colorectal cancer. Clin Cancer Res. 1998;4:1591–5. [PubMed] [Google Scholar]
- 29.Kane KF, Langman MJ, Williams GR. Antiproliferative responses to two human colon cancer cell lines to vitamin D3 are differently modified by 9-cis-retinoic acid. Cancer Res. 1996;56:623–32. [PubMed] [Google Scholar]
- 30.Kane KF, Langman MJ, Williams GR. 1,25-Dihydroxyvitamin D3 and retinoid X receptor expression in human colorectal neoplasms. Gut. 1995;36:255–8. doi: 10.1136/gut.36.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cross HS, Bajna E, Bises G, Genser D, Kallay E, Potzi R, et al. Vitamin D receptor and cytokeratin expression may be progression indicators in human colon cancer. Anticancer Res. 1996;16:2333–7. [PubMed] [Google Scholar]
- 32.Vandewalle B, Adenis A, Hornez L, Revillion F, Lefebvre J. 1,25-dihydroxyvitamin D3 receptors in normal and malignant human colorectal tissues. Cancer Lett. 1994;86:67–73. doi: 10.1016/0304-3835(94)90181-3. [DOI] [PubMed] [Google Scholar]
- 33.Sheinin Y, Kaserer K, Wrba F, Wenzl E, Kriwanek S, Peterlik M, et al. In situ mRNA hybridization analysis and immunolocalization of the vitamin D receptor in normal and carcinomatous human colonic mucosa: relation to epidermal growth factor receptor expression. Virchows Arch. 2000;437:501–7. doi: 10.1007/s004280000275. [DOI] [PubMed] [Google Scholar]
- 34.Matusiak D, Murillo G, Carroll RE, Mehta RG, Benya RV. Expression of vitamin D receptor and 25-hydroxyvitamin D3-1[alpha]-hydroxylase in normal and malignant human colon. Cancer Epidemiol Biomarkers Prev. 2005;14:2370–6. doi: 10.1158/1055-9965.EPI-05-0257. [DOI] [PubMed] [Google Scholar]
- 35.Larriba MJ, Martin-Villar E, Garcia JM, Pereira F, Pena C, de Herreros AG, et al. Snail2 cooperates with Snail1 in the repression of vitamin D receptor in colon cancer. Carcinogenesis. 2009;30:1459–68. doi: 10.1093/carcin/bgp140. [DOI] [PubMed] [Google Scholar]
- 36.Larriba MJ, Munoz A. SNAIL vs vitamin D receptor expression in colon cancer: therapeutics implications. Br J Cancer. 2005;92:985–9. doi: 10.1038/sj.bjc.6602484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palmer HG, Larriba MJ, Garcia JM, Ordonez-Moran P, Pena C, Peiro S, et al. The transcription factor SNAIL represses vitamin D receptor expression and responsiveness in human colon cancer. Nat Med. 2004;10:917–9. doi: 10.1038/nm1095. [DOI] [PubMed] [Google Scholar]
- 38.Wada K, Tanaka H, Maeda K, Inoue T, Noda E, Amano R, et al. Vitamin D receptor expression is associated with colon cancer in ulcerative colitis. Oncol Rep. 2009;22:1021–5. doi: 10.3892/or_00000530. [DOI] [PubMed] [Google Scholar]
- 39.Knackstedt RW, Moseley VR, Sun S, Wargovich MJ. Vitamin D receptor and retinoid X receptor alpha status and vitamin D insufficiency in models of murine colitis. Cancer Prev Res (Phila) 2013;6:585–93. doi: 10.1158/1940-6207.CAPR-12-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crisanti MC, Wallace AF, Kapoor V, Vandermeers F, Dowling ML, Pereira LP, et al. The HDAC inhibitor panobinostat (LBH589) inhibits mesothelioma and lung cancer cells in vitro and in vivo with particular efficacy for small cell lung cancer. Mol Cancer Ther. 2009;8:2221–31. doi: 10.1158/1535-7163.MCT-09-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Janakiram NB, Mohammed A, Qian L, Choi CI, Steele VE, Rao CV. Chemopreventive effects of RXR-selective rexinoid bexarotene on intestinal neoplasia of Apc(Min/+) mice. Neoplasia. 2012;14:159–68. doi: 10.1593/neo.111440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakanishi M, Menoret A, Tanaka T, Miyamoto S, Montrose DC, Vella AT, et al. Selective PGE(2) suppression inhibits colon carcinogenesis and modifies local mucosal immunity. Cancer Prev Res (Phila) 2011;4:1198–208. doi: 10.1158/1940-6207.CAPR-11-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Becklund BR, DeLuca HF. Identification of a highly specific and versatile vitamin D receptor antibody. Arch Biochem Biophys. 2010;494:166–77. doi: 10.1016/j.abb.2009.11.029. [DOI] [PubMed] [Google Scholar]
- 44.Whitehead RH, VanEeden PE, Noble MD, Ataliotis P, Jat PS. Establishment of conditionally immortalized epithelial cell lines from both colon and small intestine of adult H-2Kb-tsA58 transgenic mice. Proc Natl Acad Sci U S A. 1993;90:587–91. doi: 10.1073/pnas.90.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.D'Abaco GM, Whitehead RH, Burgess AW. Synergy between Apc min and an activated ras mutation is sufficient to induce colon carcinomas. Mol Cell Biol. 1996;16:884–91. doi: 10.1128/mcb.16.3.884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cao L, Gibson JD, Miyamoto S, Sail V, Verma R, Rosenberg DW, et al. Intestinal lineage commitment of embryonic stem cells. Differentiation. 2011;81:1–10. doi: 10.1016/j.diff.2010.09.182. [DOI] [PubMed] [Google Scholar]
- 47.Cao L, Kuratnik A, Xu W, Gibson JD, Kolling Ft, Falcone ER, et al. Development of intestinal organoids as tissue surrogates: Cell composition and the Epigenetic control of differentiation. Mol Carcinog. 2013 doi: 10.1002/mc.22089. [DOI] [PubMed] [Google Scholar]
- 48.Sabates-Bellver J, Van der Flier LG, de Palo M, Cattaneo E, Maake C, Rehrauer H, et al. Transcriptome profile of human colorectal adenomas. Mol Cancer Res. 2007;5:1263–75. doi: 10.1158/1541-7786.MCR-07-0267. [DOI] [PubMed] [Google Scholar]
- 49.Fleet JC, Gliniak C, Zhang Z, Xue Y, Smith KB, McCreedy R, et al. Serum metabolite profiles and target tissue gene expression define the effect of cholecalciferol intake on calcium metabolism in rats and mice. J Nutr. 2008;138:1114–20. doi: 10.1093/jn/138.6.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palmer HG, Gonzalez-Sancho JM, Espada J, Berciano MT, Puig I, Baulida J, et al. Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J Cell Biol. 2001;154:369–87. doi: 10.1083/jcb.200102028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sheng H, Shao J, Williams CS, Pereira MA, Taketo MM, Oshima M, et al. Nuclear translocation of beta-catenin in hereditary and carcinogen-induced intestinal adenomas. Carcinogenesis. 1998;19:543–9. doi: 10.1093/carcin/19.4.543. [DOI] [PubMed] [Google Scholar]
- 52.Godman CA, Joshi R, Tierney BR, Greenspan E, Rasmussen TP, Wang HW, et al. HDAC3 impacts multiple oncogenic pathways in colon cancer cells with effects on Wnt and vitamin D signaling. Cancer Biol Ther. 2008;7:1570–80. doi: 10.4161/cbt.7.10.6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pilarsky C, Wenzig M, Specht T, Saeger HD, Grutzmann R. Identification and validation of commonly overexpressed genes in solid tumors by comparison of microarray data. Neoplasia. 2004;6:744–50. doi: 10.1593/neo.04277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spurling CC, Godman CA, Noonan EJ, Rasmussen TP, Rosenberg DW, Giardina C. HDAC3 overexpression and colon cancer cell proliferation and differentiation. Mol Carcinog. 2008;47:137–47. doi: 10.1002/mc.20373. [DOI] [PubMed] [Google Scholar]
- 55.Weichert W, Roske A, Niesporek S, Noske A, Buckendahl AC, Dietel M, et al. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo. Clin Cancer Res. 2008;14:1669–77. doi: 10.1158/1078-0432.CCR-07-0990. [DOI] [PubMed] [Google Scholar]
- 56.Wilson AJ, Byun DS, Popova N, Murray LB, L'Italien K, Sowa Y, et al. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem. 2006;281:13548–58. doi: 10.1074/jbc.M510023200. [DOI] [PubMed] [Google Scholar]
- 57.Archer SY, Johnson J, Kim HJ, Ma Q, Mou H, Daesety V, et al. The histone deacetylase inhibitor butyrate downregulates cyclin B1 gene expression via a p21/WAF-1-dependent mechanism in human colon cancer cells. Am J Physiol Gastrointest Liver Physiol. 2005;289:G696–703. doi: 10.1152/ajpgi.00575.2004. [DOI] [PubMed] [Google Scholar]
- 58.Hinnebusch BF, Meng S, Wu JT, Archer SY, Hodin RA. The effects of short-chain fatty acids on human colon cancer cell phenotype are associated with histone hyperacetylation. J Nutr. 2002;132:1012–7. doi: 10.1093/jn/132.5.1012. [DOI] [PubMed] [Google Scholar]
- 59.Davis CD, Milner JA. Vitamin D and colon cancer. Expert Rev Gastroenterol Hepatol. 2011;5:67–81. doi: 10.1586/egh.10.89. [DOI] [PubMed] [Google Scholar]
- 60.Giardina C, Madigan JP, Tierney CA, Brenner BM, Rosenberg DW. Vitamin D resistance and colon cancer prevention. Carcinogenesis. 2012;33:475–82. doi: 10.1093/carcin/bgr301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ma Y, Zhang P, Wang F, Yang J, Liu Z, Qin H. Association between vitamin D and risk of colorectal cancer: a systematic review of prospective studies. J Clin Oncol. 2011;29:3775–82. doi: 10.1200/JCO.2011.35.7566. [DOI] [PubMed] [Google Scholar]
- 62.Peterlik M, Grant WB, Cross HS. Calcium, vitamin D and cancer. Anticancer Res. 2009;29:3687–98. [PubMed] [Google Scholar]
- 63.Welsh J. Cellular and molecular effects of vitamin D on carcinogenesis. Arch Biochem Biophys. 2012;523:107–14. doi: 10.1016/j.abb.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yin L, Grandi N, Raum E, Haug U, Arndt V, Brenner H. Meta-analysis: longitudinal studies of serum vitamin D and colorectal cancer risk. Aliment Pharmacol Ther. 2009;30:113–25. doi: 10.1111/j.1365-2036.2009.04022.x. [DOI] [PubMed] [Google Scholar]
- 65.Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M, et al. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science. 2012;337:730–5. doi: 10.1126/science.1224676. [DOI] [PubMed] [Google Scholar]
- 66.Dalerba P, Kalisky T, Sahoo D, Rajendran PS, Rothenberg ME, Leyrat AA, et al. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat Biotechnol. 2011;29:1120–7. doi: 10.1038/nbt.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luderer HF, Gori F, Demay MB. Lymphoid enhancer-binding factor-1 (LEF1) interacts with the DNA-binding domain of the vitamin D receptor. J Biol Chem. 2011;286:18444–51. doi: 10.1074/jbc.M110.188219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Larriba MJ, Bonilla F, Munoz A. The transcription factors Snail1 and Snail2 repress vitamin D receptor during colon cancer progression. J Steroid Biochem Mol Biol. 2010;121:106–9. doi: 10.1016/j.jsbmb.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 69.Pena C, Garcia JM, Garcia V, Silva J, Dominguez G, Rodriguez R, et al. The expression levels of the transcriptional regulators p300 and CtBP modulate the correlations between SNAIL, ZEB1, E-cadherin and vitamin D receptor in human colon carcinomas. Int J Cancer. 2006;119:2098–104. doi: 10.1002/ijc.22083. [DOI] [PubMed] [Google Scholar]
- 70.Pena C, Garcia JM, Larriba MJ, Barderas R, Gomez I, Herrera M, et al. SNAI1 expression in colon cancer related with CDH1 and VDR downregulation in normal adjacent tissue. Oncogene. 2009;28:4375–85. doi: 10.1038/onc.2009.285. [DOI] [PubMed] [Google Scholar]
- 71.Chen X, Halberg RB, Burch RP, Dove WF. Intestinal adenomagenesis involves core molecular signatures of the epithelial-mesenchymal transition. J Mol Histol. 2008;39:283–94. doi: 10.1007/s10735-008-9164-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carlberg C, Dunlop TW, Saramaki A, Sinkkonen L, Matilainen M, Vaisanen S. Controlling the chromatin organization of vitamin D target genes by multiple vitamin D receptor binding sites. J Steroid Biochem Mol Biol. 2007;103:338–43. doi: 10.1016/j.jsbmb.2006.12.044. [DOI] [PubMed] [Google Scholar]
- 73.Carlberg C, Bendik I, Wyss A, Meier E, Sturzenbecker LJ, Grippo JF, et al. Two nuclear signalling pathways for vitamin D. Nature. 1993;361:657–60. doi: 10.1038/361657a0. [DOI] [PubMed] [Google Scholar]
- 74.Volate SR, Muga SJ, Issa AY, Nitcheva D, Smith T, Wargovich MJ. Epigenetic modulation of the retinoid X receptor alpha by green tea in the azoxymethane-Apc Min/+ mouse model of intestinal cancer. Mol Carcinog. 2009;48:920–33. doi: 10.1002/mc.20542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–31. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ravillah D, Mohammed A, Qian L, Brewer M, Zhang Y, Biddick L, et al. Chemopreventive effects of an HDAC2-selective inhibitor on rat colon carcinogenesis and APCmin/+ mouse intestinal tumorigenesis. J Pharmacol Exp Ther. 2014;348:59–68. doi: 10.1124/jpet.113.208645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Archer SY, Hodin RA. Histone acetylation and cancer. Curr Opin Genet Dev. 1999;9:171–4. doi: 10.1016/s0959-437x(99)80026-4. [DOI] [PubMed] [Google Scholar]
- 78.Myzak MC, Ho E, Dashwood RH. Dietary agents as histone deacetylase inhibitors. Mol Carcinog. 2006;45:443–6. doi: 10.1002/mc.20224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Myzak MC, Karplus PA, Chung FL, Dashwood RH. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 2004;64:5767–74. doi: 10.1158/0008-5472.CAN-04-1326. [DOI] [PubMed] [Google Scholar]
- 80.Jung S, Qian ZR, Yamauchi M, Bertrand KA, Fitzgerald KC, Inamura K, et al. Predicted 25(OH)D score and colorectal cancer risk according to vitamin D receptor expression. Cancer Epidemiol Biomarkers Prev. 2014;23:1628–37. doi: 10.1158/1055-9965.EPI-14-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.