

Abstract
Background
The molecular mechanisms underlying the early development of atrial fibrillation (AF) remain poorly understood. Emerging evidence suggests that abnormal epigenetic modulation via microRNAs (miRNAs) might be involved in the pathogenesis of paroxysmal AF (pAF).
Objective
To identify key molecular changes associated with pAF, we conducted state-of-the-art transcriptomic studies to identify the abnormal miRNA-mRNA interactions potentially driving AF development.
Methods
High-quality total RNA including miRNA was isolated from atrial biopsies of age-matched and sex-matched pAF patients and control patients in sinus rhythm (SR; n=4 per group) and used for RNA-sequencing and miRNA microarray. Results were analyzed bioinformatically and validated using quantitative real-time (qRT)-PCR and 3’UTR luciferase reporter assays.
Results
113 genes and 49 miRNAs were differentially expressed (DE) in pAF versus SR patients. Gene ontology analysis revealed that most of the DE genes were involved in the “gonadotropin releasing hormone receptor pathway” and “p53 pathway”. Of these DE genes, bioinformatic analyses identified 23 pairs of putative miRNA-mRNA interactions that were altered in pAF (involving 15 miRNAs and 17 mRNAs). Using qRT-PCR and 3’UTR luciferase reporter assays, the interaction between upregulation of miR-199a-5p and downregulation of FKBP5 was confirmed in samples from pAF patients.
Conclusion
Our combined transcriptomic analysis and miRNA microarray study of atrial samples from pAF patients revealed novel pathways and miRNA-mRNA regulations that may be relevant in the development of pAF. Future studies are required to investigate the potential involvement of the gonadotropin releasing hormone receptor and p53 pathways in AF pathogenesis.
Keywords: microRNA, RNA sequencing, transcriptomic study, paroxysmal atrial fibrillation
1. Introduction
Atrial fibrillation (AF) is the most common cardiac arrhythmia seen in clinics [1]. The molecular mechanisms underlying AF pathogenesis are complex and not well understood [2]. Because of this, a number of studies in the last decade have utilized microarray technologies to look for transcriptomic changes in both AF patients and animal models of AF [3-9]. Through these studies, a number of key pathways were found to be altered in AF patients such as Ca2+-dependent signaling pathways [3], inflammatory and immune pathways [5], and apoptotic and cell cycle pathways [7]. These prior studies all focused on patients with persistent or permanent AF, otherwise known as chronic AF (cAF), where extensive AF-induced atrial remodeling may have confounded the findings and thereby precluded a clear distinction between AF causes and consequences. By contrast, much less is known about the early changes that may occur in the atria of patients with paroxysmal AF (pAF), which is an earlier stage of AF development where atrial remodeling is still mostly limited [10].
MicroRNAs (miRNAs) are a class of small non-coding RNAs (ncRNAs) (containing 21-24 nucleotides) that function in RNA silencing and post-transcriptional regulation of gene expression [11]. Since miRNAs may potentially drive mRNA changes observed in AF patients, some studies performed microarray experiments using samples from AF patients[12, 13] while others have validated key roles for miRNAs such as miR-1, miR-328, and miR-26 in AF pathogenesis [14-16]. However, since these studies were all performed in patients with cAF, they were limited in their contribution to our understanding of the factors involved in early AF development.
With this in mind, and in light of the recent advance in RNA-sequencing (RNA-Seq) technology, we were motivated to conduct an unbiased transcriptomic study using atrial samples from pAF patients. The objective of this study is to identify pathways and miRNA-mRNA interactions that may play a role in early AF pathogenesis. Using both RNA-Seq and miRNA microarray, we uncovered 113 genes and 49 miRNAs that are differentially expressed in patients with pAF compared to those in sinus rhythm (SR). Additional bioinformatic analyses identified 23 pairs of putative miRNA-mRNA interactions (involving 15 miRNAs and 17 mRNAs) that are dysregulated in pAF patients. Several candidates were validated using quantitative real-time (qRT)-PCR, and one miRNA-mRNA pair (miR-199a-5p/FKBP5) was further validated using 3’UTR luciferase assay. Together, our study revealed novel signaling pathways and miRNA-mRNA interactions that are associated with the early development of AF.
2. Materials and methods
Detailed information is provided in the Online Supplement.
2.1. Human atrial samples
After written informed consent was obtained, the tip of the right atrial appendage was removed as part of the general surgical procedure in patients undergoing open-heart surgery for bypass grafting, valve replacement or a combination of both. All biopsies were taken at the same site, which is at the tip of the right atrial appendage. After collection, the samples were flash frozen in liquid nitrogen, under protocols approved by the ethics committee of the Faculty of Medicine, University Duisburg-Essen (12-5268-BO). Characteristics of the patients are listed in Tables 1 and 2.
Table 1.
Characteristics of patients for the transcriptomic studies.
| SR (RNA-Seq)* | SR (miR microarray) | pAF | |
|---|---|---|---|
| Patients, n | 4 | 4 | 4 |
| Gender, m/f | 2/2 | 3/1 | 3/1 |
| Age, y | 65.0±4.1 | 70.5±1.9 | 71.3±2.9 |
| Body mass index, kg/m2 | 27.4±1.5 | 26.9±1.5 | 25.2±1.9 |
| CAD, n | 3 | 3 | 3 |
| MVD/AVD, n | 0 | 0 | 0 |
| CAD+MVD/AVD, n | 1 | 1 | 1 |
| Hypertension, n | 4 | 4 | 4 |
| Diabetes, n | 2 | 1 | 1 |
| Hyperlipidemia, n | 4 | 4 | 3 |
| LVEF, % | 49.7±12.9 | 51.5±9.8 | 51.3±8.1 |
| Digitalis, n | 1 | 1 | 1 |
| ACE inhibitors, n | 3 | 2 | 4 |
| AT1 blockers, n | 0 | 0 | 0 |
| β-Blockers, n | 4 | 3 | 3 |
| Dihydropyridines, n | 1 | 1 | 3 |
| Diuretics, n | 2 | 2 | 1 |
| Nitrates, n | 0 | 0 | 1 |
| Lipid-lowering drugs, n | 4 | 3 | 3 |
With the exception of one SR sample, all the other SR and pAF samples were used for both the RNA-Seq and miR microarray experiments. Values are presented as mean±SEM or number of patients. SR, patients in sinus rhythm; pAF, paroxysmal atrial fibrillation patients; CAD, coronary artery disease, MVD/AVD, mitral/aortic valve disease; LVEF, left ventricular ejection fraction; ACE, angiotensin-converting enzyme; AT, angiotensin receptor. No statistical differences were found between SR (RNA-Seq) vs. pAF or SR (miR microarray) vs. pAF using two-tailed Student’s t-test for continuous variables and Fisher’s exact test for categorical variables (all P>0.1).
Table 2.
Characteristics of patients for the qRT-PCR experiments.
| SR* | pAF | |
|---|---|---|
| Patients, n | 8 | 10 |
| Gender, m/f | 6/2 | 5/5 |
| Age, y | 64.5±3.0 | 67.5±3.8 |
| Body mass index, kg/m2 | 27.2±1.0 | 30.5±2.9 |
| CAD, n | 6 | 6 |
| MVD/AVD, n | 1 | 2 |
| CAD+MVD/AVD, n | 1 | 1 |
| Hypertension, n | 7 | 10 |
| Diabetes, n | 3 | 3 |
| Hyperlipidemia, n | 6 | 5 |
| LVEF, % | 55.0±6.0 | 44.6±6.2 |
| Digitalis, n | 1 | 2 |
| ACE inhibitors, n | 6 | 6 |
| AT1 blockers, n | 0 | 1 |
| β-Blockers, n | 6 | 6 |
| Dihydropyridines, n | 1 | 5 |
| Diuretics, n | 3 | 4 |
| Nitrates, n | 0 | 2 |
| Lipid-lowering drugs, n | 5 | 6 |
The qRT-PCR experiments included additional samples not used for the RNA-Seq and miR microarray experiments. Values are presented as mean±SEM or number of patients. SR, patients in sinus rhythm; pAF, paroxysmal atrial fibrillation patients; CAD, coronary artery disease, MVD/AVD, mitral/aortic valve disease; LVEF, left ventricular ejection fraction; ACE, angiotensin-converting enzyme; AT, angiotensin receptor. No statistical differences were found between SR and pAF groups using two-tailed Student’s t-test for continuous variables and Fisher’s exact test for categorical variables (all P>0.1).
2.2. RNA sequencing and miRNA microarray
High quality total RNA including miRNAs was isolated from frozen human atrial samples using the miRNeasy Mini Kit from Qiagen (Venlo, The Netherlands). A portion of the RNA was converted to barcoded cDNA libraries for RNA-Seq using Ion Total RNA-Seq Kit v2.0 and sequenced on the Ion Proton™ System (Life Technologies, CA). Data analysis and visualization were accomplished using SNP & Variation Suite (Version 8; Golden Helix, Bozeman, MT) while gene ontology (GO) analysis was performed using PANTHER 9.0.[17] Another portion of the RNA was used for miRNA microarray, performed by LC Sciences (Houston, TX) on their in-house μParaflo® technology platform that included all human mature miRNAs available in the latest version of the miRBase database (Release 20).
2.3. Quantitative real-time PCR
To measure levels of mRNAs, reverse transcription was performed using iScript™ cDNA Synthesis Kit (Bio-Rad, Hercules, CA). To measure levels of miRNAs, reverse transcription and quantitative real-time (qRT)-PCR were carried out using a modified protocol as described.[18-20] qRT-PCR reactions were performed in triplicate in 96-well plates using PerfeCTa® SYBR® Green FastMix® (Quanta Biosciences, Gaithersburg, MD) in Mastercycler ep realplex (Eppendorf, Hamburg, Germany). Relative expression levels were calculated using the CT (cycle number) method after normalization to RPL7. All primers used for mRNA and miRNA detection are listed in Supplemental Tables S1 and S2, respectively.[21]
2.4. Mutagenesis and luciferase assay
The Homo sapiens FKBP5 3’UTR luciferase construct was purchased from SwitchGear Genomics (S811120, Active Motif). The putative interaction site between hsa-miR-199a-5p and the FKBP5 3′UTR was searched using both TargetScanHuman 6.2[22] and microRNA.org.[23] Mutagenesis was carried out using QuikChangeII (Agilent Technologies, Santa Clara, CA) to disrupt this putative interaction using forward primer 5′-AATAAAAACTAATTTAAAATATAACTGCGAATTCTTTTTAGGATTTAAAAAGGTGAGATGTTC CAGGTTTAAGCAAATGG -3′ and reverse primer 5′-CCATTTGCTTAAACCTGGAACATCTCACCTTTTTAAATCCTAAAAAGAATTCGCAGTTATATT TTAAATTAGTTTTTATT-3′, according to the manufacturer’s instructions.
3’UTR luciferase reporter assays were performed as described previously, with modifications.[20] Hsa-miR-199a-5p and a non-targeting scramble miRNA mimic were purchased from Life Technologies (Carlsbad, CA) and used at a final concentration of 10 nM. The luciferase constructs and miRNA mimics were co-transfected in pairs into HEK293 cells according to the protocol provided by SwitchGear Genomics (Active Motif) using LipoD293 (SignaGen Laboratories, Rockville, MD). Cells were harvested after 24 hours and assayed using the LightSwitch Luciferase Assay Reagents (Active Motif) according to the manufacturer’s instructions. Luciferase signals were normalized to the protein concentration of the respective lysates and to the mimics’ effect on the empty 3’UTR vector (S890005, Active Motif).
2.5. Statistical analysis
Two-tailed student’s t-test was used where appropriate. Data are presented as mean ± SEM and a P-value less than 0.05 was considered statistically significant, except for the miRNA microarray data analyzed by LC Sciences (Houston, TX) using a validated method.
3. Results
3.1. RNA-Seq of atrial samples from pAF patients
In order to perform an unbiased survey of changes in the transcriptome of pAF patients, we performed RNA-Seq using polyA+ mRNA isolated from right atrial appendages of both pAF patients and control patients in SR (Table 1). Over 20 million single-end reads were obtained per sample with an average length of 102 bp using the Ion Torrent Proton sequencer. 79% of RNA sequencing reads was mapped to the human genome. Gene expression was evaluated based on the number of reads located within all its exons. Total read counts were normalized and compared between SR and pAF samples (n=4 per group; Supplemental Table S3). The boxplots in Figure 1 show comparable read count distribution across all 8 samples. The results were then visualized in a volcano plot where the x-axis is the log2 ratio of the fold-change (pAF/SR) and the y-axis is the –log10 ratio of the P-value (Figure 2A). As shown by the red circles in Figure 2A and in Supplemental Table S3, there are 113 transcripts (out of 13,992 transcripts) that are differentially expressed (DE) between SR and pAF samples (P<0.05, fold change > 1.5). These 113 transcripts were further visualized in a heat-map as shown in Figure 2B and listed in Supplemental Table S4. Eighty-one of the 113 transcripts were downregulated in pAF samples whereas 32 were upregulated (Supplemental Table S4).
Figure 1. Columnwise side-by-side boxplots of normalized samples showing comparable count distribution across samples in the RNA-Seq experiment.
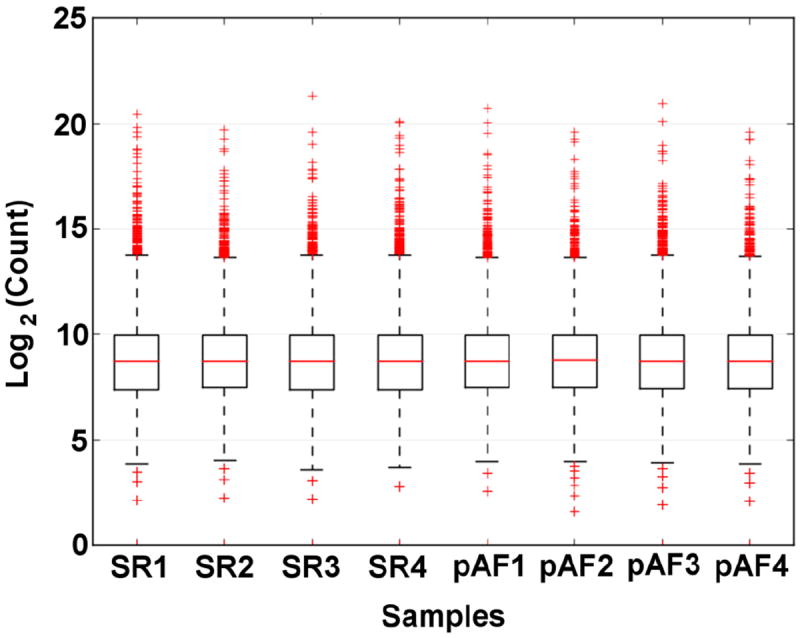
SR=control patients in sinus rhythm; pAF=paroxysmal atrial fibrillation.
Figure 2. RNA-Seq of atrial samples from sinus rhythm (SR) and paroxysmal AF (pAF) patients.
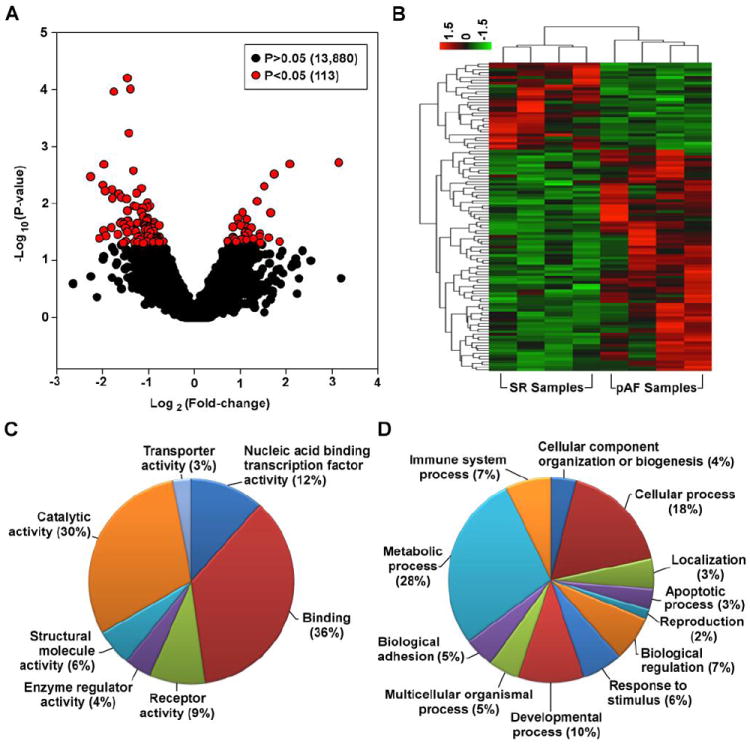
A. Volcano plot of all the transcripts detected by RNA-Seq highlighting the 113 transcripts that are differentially expressed between SR and pAF samples (red circles). B. Heat-map of the 113 differentially expressed transcripts. Gene ontology pie charts of the differentially expressed transcripts based on the “Molecular Function” (C) and “Biological Process” (D) categories.
To gain a deeper understanding of these DE transcripts, gene ontology (GO) analysis was performed using PANTHER 9.0.[17] The transcripts were categorized based on their “Molecular Function”, “Biological Process”, and “Cellular Component” as listed in Supplemental Table S4. In addition, the categories of “Molecular Function” and “Biological Process” were also plotted as pie charts in Figure 2C and 2D, respectively. The top 3 categories of “Molecular Function” were “Binding” (36%), “Catalytic activity” (30%), and “Nucleic acid binding transcription factor activity” (12%; Figure 2C) while the top 3 categories of “Biological Process” were “Metabolic process” (28%), “Cellular process” (18%), and “Developmental process” (10%; Figure 2D). In addition, the transcripts were also categorized based on known pathways in PANTHER 9.0 and the most notable pathways were “Gonadotropin releasing hormone receptor pathway”, “p53 pathway”, and some signaling pathways such as TGF-β and PDGF (Supplemental Table S4).
3.2. miRNA microarray of atrial samples from pAF patients
Using the same samples as for the RNA-Seq (except for one SR sample which had to be replaced for technical reasons; Table 1), we performed miRNA microarray and analysis (LC Sciences, Houston, TX) in order to examine all human mature miRNAs available in the miRBase database (Release 20). A total of 667 miRNAs were detected with 181 considered as significant (P<0.1) based on LC Sciences’ tested algorithm (Supplemental Table S5). These 181 were further filtered for those with high signals (>500), resulting in 49 miRNAs that are both significantly altered in pAF samples and with high signals on the microarray (Figure 3A). Of these 49 miRNAs, 23 were upregulated in pAF samples while 26 were downregulated, as shown in the heat-map in Figure 3B and Supplemental Table S6.
Figure 3. miRNA microarray of atrial samples from sinus rhythm (SR) and paroxysmal AF (pAF) patients.
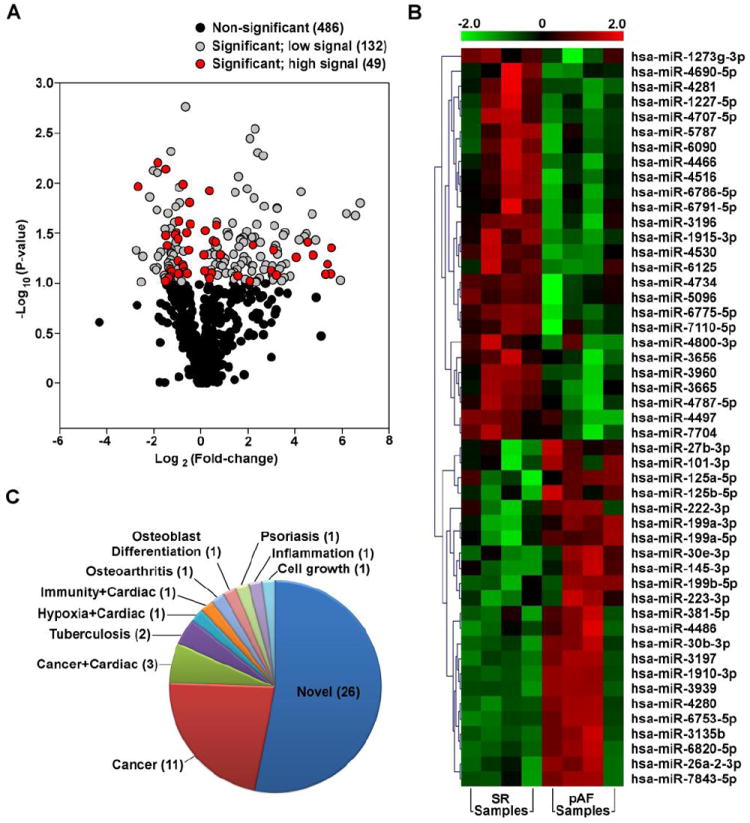
A. Volcano plot of all the miRNAs detected by microarray highlighting the 49 miRNAs that are both differentially expressed between SR and pAF samples and have a high signal on the microarray (red circles). B. Heat-map of the 49 differentially expressed miRNAs. C. Pie chart showing known roles of the differentially expressed miRNAs. “Novel” means no known function has been established for the miRNAs.
We searched the literature for known or established role(s) for the 49 DE miRNAs from our microarray (Figure 3B). Of these, only 23 miRNAs have a known or established role in physiology or human disease while the majority (26 miRNAs) do not and are therefore considered “Novel” (Figure 3C; Supplemental Table S6). Of the 23 known miRNAs, 14 were found to play a role in cancer while only 5 (hsa-miR-145-3p, hsa-miR-199a-3p, hsa-miR-199a-5p, hsa-miR-223-3p, hsa-miR-26a-2-3p) were known to play a role in cardiac diseases (Figure 3C; Supplemental Table S6).[24]
3.3. Putative miRNA-mRNA interactions in pAF patients
Post-transcriptional regulation plays an important role in controlling gene expression. To gain a deeper understanding of the potential interactions between key miRNAs and mRNAs which may play a role in pAF pathogenesis, we analyzed the RNA-Seq and miRNA microarray data in conjunction using a bioinformatics approach. As outlined in Figure 4A, RNA-Seq yielded 91 DE mRNAs (out of 113 DE transcripts after excluding the ncRNAs) while miRNA microarray found 49 DE miRNAs. We scanned the 3’UTR of all the DE mRNAs for high-confident miRNA seed sites using TargetScanHuman6.2.[22] Then of all the predicted miRNA-mRNA pairs, we filtered for pairs where the miRNA and mRNA were changed in the opposite direction, since an upregulated miRNA should lead to the downregulation of its target mRNA and vice versa. This analysis yielded 23 miRNA-mRNA pairs with putative interaction, consisting of 15 miRNAs and 17 mRNAs (Figure 4A; Table 3).
Figure 4. miRNA-mRNA interactions.
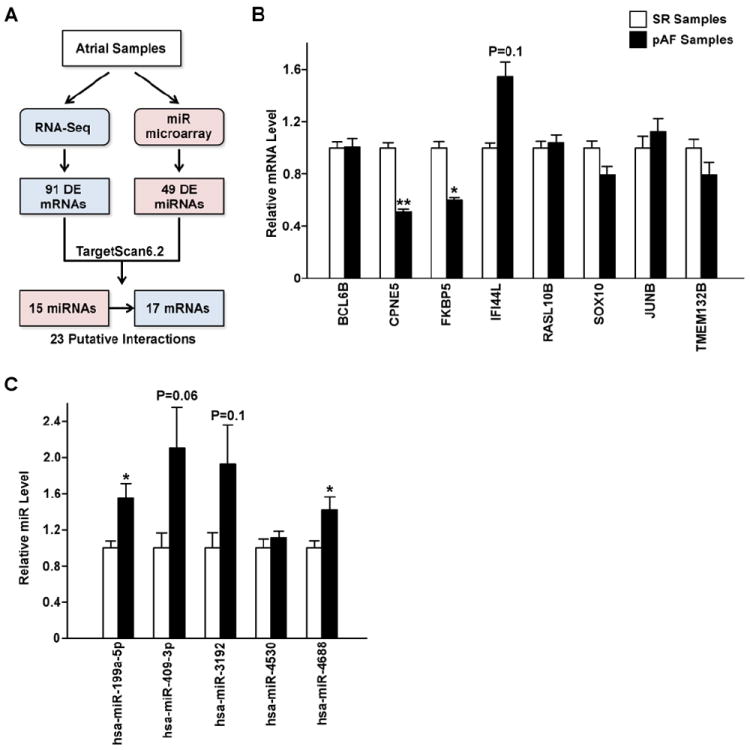
A. Schematic of the workflow to identify putative miRNA-mRNA interactions relevant for AF pathogenesis. DE=differentially expressed. B. qRT-PCR validation of 8 mRNAs selected from the 23 putative miRNA-mRNA interaction pairs. N=6-8 per group. C. qRT-PCR validation of 5 miRNAs selected from the 23 putative miRNA-mRNA interaction pairs in pAF vs. SR samples. N=4 per group. *P<0.05, **P<0.01, vs. SR.
Table 3.
Putative miRNA-mRNA interaction pairs in pAF patients.
| miRNA* | mRNA* | miRNA Log2(pAF/SR) | mRNA Log2(pAF/SR) |
|---|---|---|---|
| hsa-miR-93 | DPP3 | 0.67 | -0.81 |
| hsa-miR-199a-5p | FKBP5 | 0.63 | -1.02 |
| hsa-miR-199a-5p | JUNB | 0.63 | -1.15 |
| hsa-miR-199a-5p | ZFP2 | 0.63 | -1.79 |
| hsa-miR-409-3p | FKBP5 | 1.12 | -1.02 |
| hsa-miR-762 | OAS3 | -0.73 | 1.17 |
| hsa-miR-922 | JUNB | 6.19 | -1.15 |
| hsa-miR-3192 | FKBP5 | 2.69 | -1.02 |
| hsa-miR-3192 | SOX10 | 2.69 | -1.07 |
| hsa-miR-3652 | VSTM2L | 2.09 | -0.93 |
| hsa-miR-4296 | GATC | 2.65 | -1.02 |
| hsa-miR-4296 | SNX22 | 2.65 | -1.46 |
| hsa-miR-4311 | SPON1 | 2.29 | -0.87 |
| hsa-miR-4311 | TMEM132B | 2.29 | -1.99 |
| hsa-miR-4481 | TMEM132B | 2.43 | -1.99 |
| hsa-miR-4516 | IFI44L | -0.94 | 1.66 |
| hsa-miR-4516 | RASL10B | -0.94 | 0.72 |
| hsa-miR-4519 | GATC | 4.73 | -1.02 |
| hsa-miR-4530 | BCL6B | -1.49 | 1.14 |
| hsa-miR-4688 | CIDEC | 1.19 | -1.94 |
| hsa-miR-4688 | CPNE5 | 1.19 | -0.77 |
| hsa-miR-4688 | SOX10 | 1.19 | -1.07 |
| hsa-miR-4691-3p | BAIAP3 | 4.40 | -1.40 |
A total of 23 pairs of miRNA-mRNA interactions were predicted bioinformatically and based on RNA-Seq and miRNA microarray data using samples from control patients in sinus rhythm (SR) vs. patients with paroxysmal atrial fibrillation (pAF).
Next, we selected 8 mRNAs (BCL6B, CPNE5, FKBP5, IFI44L, RASL10B, SOX10, JUNB, and TMEM132B) for further validation based on at least one of the following considerations: 1) they were targeted by multiple miRNAs which could suggest a greater role in molecular regulation; 2) the miRNAs that targeted them also targeted other mRNAs; 3) the large magnitude of the changes of either these mRNAs or the miRNAs that targeted them in pAF based on the RNA-Seq and miR microarray experiments; and 4) they play key roles in other systems but were not previously studied in the heart. Using quantitative real time (qRT)-PCR, we found that only CPNE5 and FKBP5 were significantly downregulated (P<0.05) while IFI44L was non-significantly upregulated (P=0.1) in pAF patients, in agreement with the RNA-Seq data (Figure 4B and Supplemental Table S4). Based on these results, we selected a few of the predicted miRNAs that target CPNE5 and FKBP5 for further validation, including hsa-miR-199a-5p, hsa-miR-409-3p, hsa-miR-3192, hsa-miR-4530, and hsa-miR-4688 (Table 3). Figure 4C shows that only hsa-miR-199a-5p and hsa-miR-4688 reached statistical significance (P<0.05) while hsa-miR-409-3p (P=0.06) and hsa-miR-3192 (P=0.1) were non-significantly upregulated, all four recapitulating microarray findings (Supplemental Table S6). Since nothing is known about the role of FKBP5 in the heart other than that it is expressed there [25], we selected the miR-199a-5p/FKBP5 putative interaction for further study.
3.4. Validation of the miR-199a-5p/FKBP5 interaction
Based on both TargetScanHuman 6.2[22] and microRNA.org[23], hsa-miR-199a-5p is predicted to target the 3’UTR of human FKBP5 at position 1483 (Figure 5A). Furthermore, based on the conservation score calculated by TargetScanHuman 6.2,[26] this seed site is also conserved across species (Figure 5B). To validate this interaction, we performed luciferase assay using both the wild-type (WT) 3’UTR of FKBP5 and a mutant (Mut) 3’UTR of FKBP5 where the seed site for hsa-miR-199a-5p was mutated (Figure 5A). What we found was that the normalized luciferase signal from the WT 3’UTR of FKBP5 was significantly reduced by 20% in the presence of the hsa-miR-199a-5p mimic versus scramble (P<0.01, n=4; Figure 5C). However, this effect was rescued with the FKBP5 3’UTR mutant (P=N.S.; Figure 5C). This supported the bioinformatic prediction that hsa-miR-199a-5p targets the 3’UTR of human FKBP5 at position 1483.
Figure 5. Interaction between hsa-miR-199a-5p and FKBP5 3’fUTR.
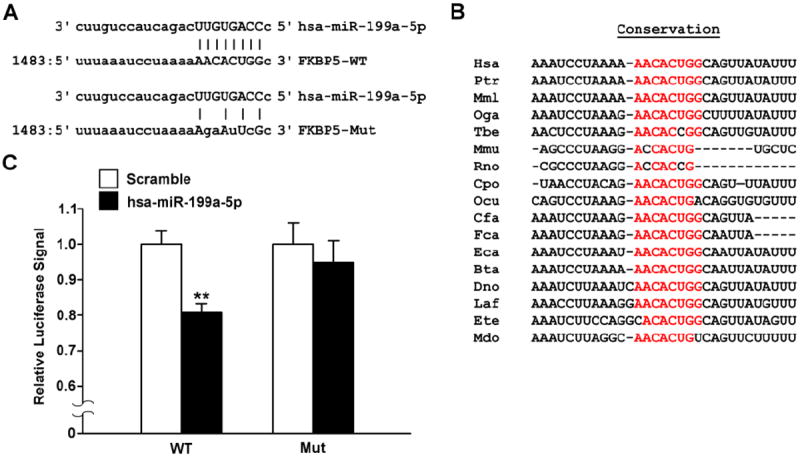
A. Schematic of bioinformatically predicted interaction site between hsa-miR-199a-5p and Homo sapiens FKBP5 3’UTR based on microRNA.org.[23] Top panel: wild-type (WT) 3’UTR of FKBP5. Bottom panel: mutant (Mut) 3’UTR of FKBP5 where the seed site for hsa-miR-199a-5p is partially ablated. B. Conservation of the hsa-miR-199a-5p seed site based on TargetScanHuman 6.2.[26] C. 3’UTR luciferase assay showing a relative decrease in the normalized luciferase signal from the WT FKBP5 3’UTR luciferase construct in the presence of hsa-miR-199a-5p but not from the mutant (Mut) construct. N=4 replicates per group. **P<0.01 vs. Scramble.
4. Discussion
To the best of our knowledge, this is both the first transcriptomic study in patients with pAF using RNA-sequencing technology and the first study to simultaneously analyze both mRNA and miRNA expression levels in AF. Using state-of-the-art RNA-sequencing and miRNA microarray, we discovered 113 genes and 49 miRNAs that were differentially expressed in patients with pAF. Of these, bioinformatic analysis further revealed 23 putative miRNA-mRNA regulations (consisting of 15 miRNAs and 17 mRNAs) that may play a role in AF pathogenesis. We selected one of these pairs of interaction and showed that the upregulation of miR-199a-5p may downregulate FKBP5 in pAF patients.
4.1. Comparison to previous transcriptomics studies in AF
Previously, studies have employed mRNA microarray technology in studying samples from AF patients [3-9]. In the present study, we chose instead to use the newer RNA-Seq technology. Compared to microarray technologies, RNA-Seq is superior in quantitatively evaluating gene expression, detecting low abundance transcripts, differentiating biologically critical isoforms, and allowing for the identification of genetic variants [27]. RNA-Seq also has a much greater dynamic range of detection and is inherently quantitative, unlike microarray technologies which rely on qRT-PCR validation [27].
To date, a few transcriptomic studies in AF have provided useful information in gene profiling of AF [3-9]. However, these studies were focused on cAF patients, in whom extensive, complex, and chronic atrial remodelings have already taken place. As a result, many molecular changes in cAF patients may be secondary to these remodeling processes and thereby complicating and confounding the interpretation of their findings. In contrast, our study is the first transcriptomic analysis focused on pAF instead of cAF patients, which we believe holds the key to identification of pathways involved in the initial disease development without confounding structural remodeling. The genes and pathways identified in our study could potentially be the key signaling pathways that initiate the pathological changes underlying AF development. In addition, by coupling the findings between RNA-Seq and miRNA microarray, we were able to discover potentially important miRNA-mRNA regulations that may drive AF pathogenesis.
Since miRNA microarrays were performed in cAF patients in 2 recent studies [12, 13], we were interested in comparing these findings with our results from pAF patients. Using right atrial appendage from patients with valvular heart disease with or without cAF, Cooley et al.[12] found 47 miRNAs that are DE in the cAF group. Of these 47 miRNAs, only 1 miRNA (hsa-miR-223-3p) was also changed in our DE list of miRNAs, which was upregulated in both Cooley’s[12] and our studies (Supplemental Table S6) [12]. A similar study but using left atrial appendage from patients with aortic stenosis with or without cAF found 21 miRNAs that were DE in the cAF group [13]. Of these 21 miRNAs, 3 miRNAs were also altered in our dataset (hsa-miR-3196, hsa-miR-4497, and hsa-miR-4707-5p) yet they were all upregulated in their study but downregulated in our study (Supplemental Table S6) [13]. These discrepant observations may be the result of the different stages of AF under study (pAF vs. cAF), which further supports the notion that atrial remodeling associated with AF development may affect gene regulations.
4.2. Novel findings and potential clinical implications
Gene ontology analysis of the DE mRNAs in pAF revealed that the top 3 categories of changes in 1) “Molecular Function” were “Binding”, “Catalytic activity”, and “Nucleic acid binding transcription factor activity” and in 2) “Biological Process” were “Metabolic process”, “Cellular process”, and “Developmental process” (Figure 1C-D). This suggests that even during the earlier stage of AF development, in the absence of gross atrial remodeling, the molecular pathways involved with gene transcription and growth and development are already in play. This is further confirmed with Pathway analysis of the same set of genes which showed that the most dysregulated pathways were the “Gonadotropin releasing hormone (GnRH) receptor pathway”, “p53 pathway”, and some signaling pathways such as TGF-β and PDGF (Supplemental Table S4), all of which are involved with transcriptional activation and growth.
The role of TGF-β and PDGF signaling cascades in cardiac diseases including AF has been well established previously [1]. In particular, these two pathways are critical in regulating fibrosis and inflammation, and may also affect ion channel function [28, 29]. On the other hand, little is known to date about the role of the GnRH receptor pathway and the p53 pathway in cardiac molecular physiology. Studies have suggested that GnRH receptor activation is associated with increased coronary heart disease and myocardial infarction [30]. Also, Dong et al. have shown that GnRH is capable of regulating cardiac contractile function via a GnRH receptor/PKA-dependent mechanism [31]. However, unlike in coronary heart disease where the GnRH signaling pathway is hyperactive, this pathway was found to be downregulated in the pAF patients in our study. Specifically, we have found that several proteins involved in the GnRH receptor pathway such as JUNB, JUND, and ADIPOQ were all downregulated in pAF patients (Supplemental Table S4). This suggests that these proteins (JUNB, JUND, and ADIPOQ) may contribute to AF development via mechanisms distinct from that in contractile dysfunction in the setting of coronary heart disease, but these mechanisms would require further investigation.
In terms of FKBP5, less is known about its function in the heart [32]. FKBP5, or FK506-binding protein 5, is a 51 kDa protein with peptidyl-prolyl cis-trans isomerase (PPIase) and co-chaperone activities. Through interaction with heat-shock protein 90 (HSP90), FKBP5 plays a role in intracellular trafficking and microtubule stabilization [32]. The known functions of FKPB5 in other organs include the regulation of steroid hormone receptor function [33], inhibition of apoptosis in cancer cells [34], promotion of Akt dephosphorylation and downregulation of Akt pathways [35], etc. Moreover, FKBP5 contains c-terminal tetratricopeptide repeat (TRP) domain and may inhibit store-operated calcium entry through the ISOC channel [32]. Nevertheless, to understand the role of FKBP5 in the heart and how its downregulation is involved in AF pathogenesis would require much future investigation.
In terms of the key ion channels and transporters known to affect cardiac excitability in AF, our study did not detect significant changes in their transcript levels (even though RNA-Seq can detect low abundance transcripts). Instead, we found changes in the aforementioned pathways controlling transcription, apoptosis, and extracellular matrix generation and turnover, all of which could eventually affect the functions of key ion channels and transporters. Together, these changes may contribute to both electrical and structural remodeling and predispose the cardiomyocytes to atrial fibrillation. Future studies are needed to elucidate these molecular mechanisms and develop interventions targeting components of these pathways.
4.3. Limitations of our study
Firstly, as with most human AF studies, this study was limited by the sample size of the atrial biopsies obtained from the patients and by the number of samples available, which may have led to increased statistical variability among the patients. Nevertheless, we did our best to match the patients in terms of gender, age, and underlying pathologies to minimize confounding factors (Tables 1 and 2). Secondly, this study was limited to patients with paroxysmal AF, which means our findings may not be generalized to include patients with chronic AF. Lastly, all of the pAF patients in this study suffered from hypertension pointing to hypertensive heart disease as a potential predisposing factor to their AF episodes (Tables 1 and 2). Thus our findings may not hold true for individuals with lone AF. Nevertheless, we believe that our findings may have captured unique changes in pAF which may be drivers of AF pathogenesis, although future studies are needed to validate and understand the significance of these findings.
5. Conclusions
We discovered 113 genes including 22 ncRNAs and 49 miRNAs that are differentially expressed in patients with paroxysmal AF compared to control patients in sinus rhythm. Of these, we identified 23 putative miRNA-mRNA interactions that may be involved in AF pathogenesis. We validated one of these pairs to demonstrate in principle the validity of our experimental and bioinformatic approach. Taken together, this study may open up new avenues of research to improve our understanding of early AF pathogenesis for the development of novel pharmacologic interventions that are both specific and effective.
Supplementary Material
Highlights.
We analyzed the transcriptomic and microRNA changes associated with the early stage of atrial fibrillation in patients.
We discovered 113 mRNAs and 49 miRNAs that were dysregulated in paroxysmal AF patients.
We discovered 23 pairs of putative microRNA-mRNA dysregulations in paroxysmal AF patients.
We identified new pathways implicated in the early stage of AF pathogenesis.
Acknowledgments
The authors would like to thank Paul G. Swinton and Jordan Showell (Baylor College of Medicine, Houston, TX) for technical assistance on the RNA-sequencing and mutagenesis, respectively.
Sources of funding: This study is supported by grants 12PRE11700012 (to D.Y.C.), 13EIA14560061 (to X.H.T.W.), and 14SDG20080008 (to N.L.) from the American Heart Association. This work was supported in part by NIH-grants HL089598, HL091947, and HL117641 (to X.H.T.W.), HL18761 (to J.F.M.), the Fondation Leducq networks ‘European North-American Atrial Fibrillation Research Alliance’ and the European Network for Translational Research in Atrial Fibrillation (EUTRAF: 261057) (to D.D.), Vivian L. Smith Foundation (to J.F.M.), and the Juanita P. Quigley endowed chair in cardiology to (X.H.T.W.). D.Y.C. was also supported by the Baylor College of Medicine Medical Scientist Training Program Caskey Scholarship.
ABBREVIATIONS
- AF
Atrial fibrillation
- cAF
Chronic atrial fibrillation
- DE
Differentially expressed
- miRNA
microRNA
- pAF
Paroxysmal atrial fibrillation
- qRT-PCR
Quantitative real-time polymerase chain reaction
- RNA-Seq
RNA sequencing
- SR
Sinus rhythm
Footnotes
Conflicts of Interest: Dr. Wehrens is a founding partner of Elex Biotech.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jalife J. Mechanisms of persistent atrial fibrillation. Current opinion in cardiology. 2014;29:20–7. doi: 10.1097/HCO.0000000000000027. [DOI] [PubMed] [Google Scholar]
- 2.Nattel S. Experimental evidence for proarrhythmic mechanisms of antiarrhythmic drugs. Cardiovasc Res. 1998;37:567–77. doi: 10.1016/s0008-6363(97)00293-9. [DOI] [PubMed] [Google Scholar]
- 3.Barth AS, Merk S, Arnoldi E, Zwermann L, Kloos P, Gebauer M, et al. Reprogramming of the human atrial transcriptome in permanent atrial fibrillation: expression of a ventricular-like genomic signature. Circ Res. 2005;96:1022–9. doi: 10.1161/01.RES.0000165480.82737.33. [DOI] [PubMed] [Google Scholar]
- 4.Kharlap MS, Timofeeva AV, Goryunova LE, Khaspekov GL, Dzemeshkevich SL, Ruskin VV, et al. Atrial appendage transcriptional profile in patients with atrial fibrillation with structural heart diseases. Ann N Y Acad Sci. 2006;1091:205–17. doi: 10.1196/annals.1378.067. [DOI] [PubMed] [Google Scholar]
- 5.Ohki R, Yamamoto K, Ueno S, Mano H, Misawa Y, Fuse K, et al. Gene expression profiling of human atrial myocardium with atrial fibrillation by DNA microarray analysis. Int J Cardiol. 2005;102:233–8. doi: 10.1016/j.ijcard.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 6.Lamirault G, Gaborit N, Le Meur N, Chevalier C, Lande G, Demolombe S, et al. Gene expression profile associated with chronic atrial fibrillation and underlying valvular heart disease in man. J Mol Cell Cardiol. 2006;40:173–84. doi: 10.1016/j.yjmcc.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 7.Kim NH, Ahn Y, Oh SK, Cho JK, Park HW, Kim YS, et al. Altered patterns of gene expression in response to chronic atrial fibrillation. Int Heart J. 2005;46:383–95. doi: 10.1536/ihj.46.383. [DOI] [PubMed] [Google Scholar]
- 8.Lai LP, Lin JL, Lin CS, Yeh HM, Tsay YG, Lee CF, et al. Functional genomic study on atrial fibrillation using cDNA microarray and two-dimensional protein electrophoresis techniques and identification of the myosin regulatory light chain isoform reprogramming in atrial fibrillation. J Cardiovasc Electrophysiol. 2004;15:214–23. doi: 10.1046/j.1540-8167.2004.03423.x. [DOI] [PubMed] [Google Scholar]
- 9.Cardin S, Libby E, Pelletier P, Le Bouter S, Shiroshita-Takeshita A, Le Meur N, et al. Contrasting gene expression profiles in two canine models of atrial fibrillation. Circ Res. 2007;100:425–33. doi: 10.1161/01.RES.0000258428.09589.1a. [DOI] [PubMed] [Google Scholar]
- 10.Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, et al. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation. 2014;129:145–56. doi: 10.1161/CIRCULATIONAHA.113.006641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–31. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 12.Cooley N, Cowley MJ, Lin RC, Marasco S, Wong C, Kaye DM, et al. Influence of atrial fibrillation on microRNA expression profiles in left and right atria from patients with valvular heart disease. Physiol Genomics. 2012;44:211–9. doi: 10.1152/physiolgenomics.00111.2011. [DOI] [PubMed] [Google Scholar]
- 13.Liu H, Chen GX, Liang MY, Qin H, Rong J, Yao JP, et al. Atrial fibrillation alters the microRNA expression profiles of the left atria of patients with mitral stenosis. BMC Cardiovasc Disord. 2014;14:10. doi: 10.1186/1471-2261-14-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Girmatsion Z, Biliczki P, Bonauer A, Wimmer-Greinecker G, Scherer M, Moritz A, et al. Changes in microRNA-1 expression and IK1 up-regulation in human atrial fibrillation. Heart Rhythm. 2009;6:1802–9. doi: 10.1016/j.hrthm.2009.08.035. [DOI] [PubMed] [Google Scholar]
- 15.Lu Y, Zhang Y, Wang N, Pan Z, Gao X, Zhang F, et al. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation. 2010;122:2378–87. doi: 10.1161/CIRCULATIONAHA.110.958967. [DOI] [PubMed] [Google Scholar]
- 16.Luo X, Pan Z, Shan H, Xiao J, Sun X, Wang N, et al. MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J Clin Invest. 2013;123:1939–51. doi: 10.1172/JCI62185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beavers DL, Wang W, Ather S, Voigt N, Garbino A, Dixit SS, et al. Mutation E169K in junctophilin-2 causes atrial fibrillation due to impaired RyR2 stabilization. J Am Coll Cardiol. 2013;62:2010–9. doi: 10.1016/j.jacc.2013.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reynolds JO, Chiang DY, Wang W, Beavers DL, Dixit SS, Skapura DG, et al. Junctophilin- 2 is necessary for T-tubule maturation during mouse heart development. Cardiovasc Res. 2013;100:44–53. doi: 10.1093/cvr/cvt133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Varkonyi-Gasic E, Wu R, Wood M, Walton EF, Hellens RP. Protocol: a highly sensitive RTPCR method for detection and quantification of microRNAs. Plant Methods. 2007;3:12. doi: 10.1186/1746-4811-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiang DY, Cuthbertson DW, Ruiz FR, Li N, Pereira FA. A Coregulatory Network of NR2F1 and microRNA-140. PLoS One. 2013;8:e83358. doi: 10.1371/journal.pone.0083358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang F, Hajkova P, Barton SC, Lao K, Surani MA. MicroRNA expression profiling of single whole embryonic stem cells. Nucleic Acids Res. 2006;34:e9. doi: 10.1093/nar/gnj009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 23.Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149–53. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Condorelli G, Latronico MV, Cavarretta E. microRNAs in Cardiovascular Diseases: Current Knowledge and the Road Ahead. J Am Coll Cardiol. 2014;63:2177–87. doi: 10.1016/j.jacc.2014.01.050. [DOI] [PubMed] [Google Scholar]
- 25.Aye TT, Scholten A, Taouatas N, Varro A, Van Veen TA, Vos MA, et al. Proteome-wide protein concentrations in the human heart. Mol Biosyst. 2010;6:1917–27. doi: 10.1039/c004495d. [DOI] [PubMed] [Google Scholar]
- 26.Garcia DM, Baek D, Shin C, Bell GW, Grimson A, Bartel DP. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat Struct Mol Biol. 2011;18:1139–46. doi: 10.1038/nsmb.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao S, Fung-Leung WP, Bittner A, Ngo K, Liu X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS One. 2014;9:e78644. doi: 10.1371/journal.pone.0078644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kiryu M, Niwano S, Niwano H, Kishihara J, Aoyama Y, Fukaya H, et al. Angiotensin IImediated up-regulation of connective tissue growth factor promotes atrial tissue fibrosis in the canine atrial fibrillation model. Europace : European pacing, arrhythmias, and cardiac electrophysiology : journal of the working groups on cardiac pacing, arrhythmias, and cardiac cellular electrophysiology of the European Society of Cardiology. 2012;14:1206–14. doi: 10.1093/europace/eus052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yeh YH, Kuo CT, Chan TH, Chang GJ, Qi XY, Tsai F, et al. Transforming growth factorbeta and oxidative stress mediate tachycardia-induced cellular remodelling in cultured atrialderived myocytes. Cardiovasc Res. 2011;91:62–70. doi: 10.1093/cvr/cvr041. [DOI] [PubMed] [Google Scholar]
- 30.Saigal CS, Gore JL, Krupski TL, Hanley J, Schonlau M, Litwin MS, et al. Androgen deprivation therapy increases cardiovascular morbidity in men with prostate cancer. Cancer. 2007;110:1493–500. doi: 10.1002/cncr.22933. [DOI] [PubMed] [Google Scholar]
- 31.Dong F, Skinner DC, Wu TJ, Ren J. The heart: a novel gonadotrophin-releasing hormone target. Journal of neuroendocrinology. 2011;23:456–63. doi: 10.1111/j.1365-2826.2011.02119.x. [DOI] [PubMed] [Google Scholar]
- 32.Cioffi DL, Hubler TR, Scammell JG. Organization and function of the FKBP52 and FKBP51 genes. Current opinion in pharmacology. 2011;11:308–13. doi: 10.1016/j.coph.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schulke JP, Wochnik GM, Lang-Rollin I, Gassen NC, Knapp RT, Berning B, et al. Differential impact of tetratricopeptide repeat proteins on the steroid hormone receptors. PLoS One. 2010;5:e11717. doi: 10.1371/journal.pone.0011717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Romano S, D’Angelillo A, Pacelli R, Staibano S, De Luna E, Bisogni R, et al. Role of FK506-binding protein 51 in the control of apoptosis of irradiated melanoma cells. Cell death and differentiation. 2010;17:145–57. doi: 10.1038/cdd.2009.115. [DOI] [PubMed] [Google Scholar]
- 35.Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, et al. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer cell. 2009;16:259–66. doi: 10.1016/j.ccr.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.