

Abstract
The mechanisms leading to the development of remote lung injury after trauma remain unknown, although a central role for the gut in the induction of lung injury has been postulated. We hypothesize that the development of remote lung injury after trauma/hemorrhagic shock requires activation of Toll Like Receptor-4 (TLR4) in the intestinal epithelium and sought to determine the mechanisms involved. We now show that trauma/hemorrhagic shock caused lung injury in wild-type mice but not in mice lacking TLR4 in the intestinal epithelium, confirming the importance of intestinal TLR4 activation in the process. Activation of intestinal TLR4 after trauma led to increased endoplasmic reticulum (ER) stress, enterocyte apoptosis and the release of circulating HMGB1, while inhibition of ER stress attenuated apoptosis, reduced circulating HMGB1, and decreased lung injury severity. Neutralization of circulating HMGB1 led to reduced severity of lung injury after trauma, and mice lacking HMGB1 in the intestinal epithelium were protected from the development of lung injury, confirming the importance of the intestine as the source of HMGB1, whose release of HMGB1 induced a rapid pkc-zeta-mediated internalization of surface tight junctions in the pulmonary epithelium. Strikingly, the use of a novel small molecule TLR4 inhibitor reduced intestinal ER stress, decreased circulating HMGB1, and preserved lung architecture after trauma. Thus, intestinal epithelial TLR4 activation leads to HMGB1 release from the gut and the development of lung injury, while strategies that block upstream TLR4 signaling may offer pulmonary protective strategies after trauma.
Introduction
Trauma is the leading cause of death and disability in patients under 54 years of age(1). Of patients who survive their initial injury, a third develop remote organ injury(2), which may be reflective of immune dysregulation in response to the initial insult(3, 4). Of all the organs affected, injury to the lung is one of the leading causes of post-trauma morbidity(5), with an incidence of 79 per 100,000 persons/year, and carries a lethality of 40%(6),(7). Importantly, the mechanisms that mediate the induction of trauma-induced lung injury remain largely unexplained and represent a major gap in our knowledge in the field. In addressing how remote trauma can cause secondary lung injury, previous investigators have postulated that there may be a link between the intestine of the injured host and the development of injury in the lung. However, such a link between the gut the lung in the pathogenesis of lung injury remains unproven, and the identity of potential molecules involved remain purely speculative.
In seeking to understand the mechanisms contributing to trauma-induced acute lung injury, we now hypothesize that activation of the innate immune system of the intestine after trauma is directly responsible for the development of lung injury. Chief amongst the potential molecular sensors within the intestine of the injured host that are well positioned to respond to contribute to injury are the Toll-like receptors (8). In particular, toll like receptor 4 (TLR4), the receptor for lipopolysaccharide (9) and also for a variety of endogenous molecules that are released during trauma (10), is expressed on the intestinal epithelium (11), where it could readily serve as a sensor on the gut during trauma (12). While TLR4 on the intestinal epithelium has not been linked to the development of secondary lung injury after trauma, it is activated in response to stressors, which leads to the induction of ER stress within the gut epithelium and the loss of mucosal integrity through enterocyte apoptosis (13). Moreover, TLR4 activation leads to the release of damage associated molecules such as HMGB1, which can cause secondary organ injury through the recruitment of inflammatory cells into the lung parenchyma and/or the disruption of tight junctions between adjacent lung epithelial cells (14).
Based upon these findings, we now hypothesize that trauma leads to the development of secondary lung injury through a novel link between TLR4 activation in the intestinal epithelium and the release of HMGB1 which is required for the lung injury to occur. We further hypothesize that experimental strategies that pharmacologically inhibit TLR4 can attenuate lung injury after trauma. In support of these hypotheses, we now provide evidence for a novel link between TLR4-induced ER stress within the intestinal epithelium leading to enterocyte apoptosis, and HMGB1 release which is required for lung injury, while mice lacking TLR4 or HMGB1 in the gut are protected from these events, and the use of a novel TLR4 inhibitor reverses lung injury after trauma in mice.
Materials and Methods
Cell culture and reagents
Human lung alveolar cells were obtained from ATCC (HBE 135-E6e7). Where indicated, cells were treated with LPS (Escherichia coli 0111:B4 purified by gel filtration chromatography (>99% pure; Sigma-Aldrich) for 6 hours at a concentration of 25 μg/ml, or HMGB1 (generous gift of Dr. Kevin Tracey, Feinstein Institute, New York) for 6 hours at a concentration of 2.5 μg/ml, or with the protein kinase C (pkc)-zeta pseudo substrate and functional inhibitor from Promega at 150 nM for 6 hours as described by Warburton (15). C34 is a 2-acetamidopyranoside (MW 389) with the formula C17H27NO9 that we determined recently to be a novel TLR4 inhibitor (16), and was used at a concentration of 2.5 mg/kg in mice for 6 hours, and was dissolved in 1% DMSO, which was also used as vehicle control. C34 was purchased commercially from Enamine.
Antibodies used in this study are as follows: E-cadherin – R&D Systems Inc (Minneapolis, MN, mouse anti rat/mouse/human), DAPI (Life Sciences), BiP (Abcam, rabbit anti rat/mouse/human), cleaved caspase 3 (Biocare Medical, rabbit anti rat/mouse), myeloperoxidase (Thermo scientific, rabbit anti rat/mouse), HMGB1 (Millipore, rabbit anti rat/mouse/human), ZO-1 (Invitrogen, rabbit anti rat/mouse). Myeloperoxidase staining was performed using DAB peroxidase (HRP) substrate kit (Vector labs) as described by Krzyzaniak et al (17). IL-6 ELISA kits were obtained from R&D Laboratories.
To evaluate directly whether the apoptosis of intestinal epithelial cells is required for HMGB1 release, IEC-6 cells were treated with LPS (50 μg/ml for 6h) in the presence of either saline or the pan-caspase apoptosis inhibitor Z-VAD-FMK (10μM, Enzo Life Science, 30 min prior). In parallel, cells were also treated with the ER stress inducer thapsigargin (0.5 μM, Across Organics, 6h). After treatments, media was collected and assessed for the secretion of HMGB1 by ELISA (HMGB1 ELISA kit, IBL International), and the degree of apoptosis by TUNEL (Roche).
Immunohistochemistry
Immunohistochemistry was performed on 5μm paraffin sections as follows: paraffin sections were first warmed to 56°C in a vacuum incubator (Isotemp Vacuum Oven, Fisher Scientific) then washed immediately twice in xylene, gradually re-dehydrated in ethanol (100%, 95%, 70%, water), and then processed for antigen retrieval in citrate buffer (10mM pH6.0)/microwave (1000 watt, 6 minutes). Samples were then washed with PBS, blocked with 1% BSA/5% donkey-serum (1 hour, room temperature), then incubated overnight at 4°C with primary antibodies (1:200 dilutions in 0.5% BSA), washed 3 times with PBS, incubated with appropriate fluorescent labeled secondary antibodies (1:1000 dilution in 0.5% BSA, Life Technologies Inc) as well as the nuclear marker DAPI (Biolegend), and slides were then mounted using Gelvatol (Sigma-Aldrich) solution prior to imaging using a Zeiss LSM 710 Confocal microscope (Carl Zeiss, Jena, Germany) under appropriate filter sets.
Quantitative real-time PCR
Total RNA was isolated from whole lung or terminal ileum from mice that were either treated with saline, or which had been subjected to experimental trauma/hemorrhagic shock as described below using the RNeasy kit (Qiagen), and reverse transcribed (1 μg of RNA) using the QuantiTect Reverse Transcription Kit (Qiagen). Quantitative real-time PCR was performed as previously described using the Bio-Rad CFX96 Real-Time System (Biorad, Hercules, CA) using the primers described below, and are reported as fold change relative to RPLO as a housekeeping gene as in (18):
RPLO: Mouse: Forward 5′-GGCGACCTGGAAGTCCAACT-3′; Reverse 5′-CCATCAGCACCACAGCCTTC-3′,
IL-6: Mouse: Forward 5′-GGCTAAGGACCAAGACCATCCAA-3′; Reverse 5′-TCTGACCACAGTGAGGAATGTCCA-3′,
XBP1 (spliced) Mouse: Forward 5′-GAGTCCGCAGCAGGTGC; Reverse 5′-CAAAAGGATATCAGACTCAGAATCTGAA-3′, and
HMGB1 Mouse: Forward 5′-GGGAGGAGCACAAGAAGAAG-3′; Reverse 5′-TCATAACGAGCCTTGTCAGC-3′
MCP-1 mouse Forward 5′-ATGCAGTTAACGCCCCACTC-3′; Reverse 5′-CCCATTCCTTCTTGGGGTCA-3′
Transgenic mice generation and treatment
All mice experiments were approved by the Animal Care and Use Committee of the University of Pittsburgh. TLR4−/− mice and mice in which TLR4 was selectively deleted from the intestinal epithelium (TLR4ΔIEC) were generated in our laboratory as recently described (19). Mice in which HMGB1 was selectively deleted from the intestinal epithelium (HMGB1ΔIEC) were generated by breeding Hmgb1-loxP mice (obtained from Dr. Eugene Chang, University of Chicago) with villin-Cre transgenic mice. Progeny were fertile, healthy and were shown to be deficient in HMGB1 in the intestinal epithelium and not in the lamina propria, as revealed by immunohistochemistry (see Results).
Experimental models of trauma
All experiments were approved by the University of Pittsburgh Animal Care and Use Committee and the Institutional Review Board of the University of Pittsburgh and maintained under National Institutes of Health (NIH) animal care guidelines in specific pathogen-free conditions with a 12-h:12-h light/dark cycle and free access to standard laboratory feed and water. Male wild-type (i.e., C57/BL-6 mice) mice and mice deficient in TLR4 (TLR4ΔIEC) or HMGB1 (HMGB1ΔIEC) in the intestinal epithelium were studied as described below. All experiments were performed in 8–12-wk old males that weighted ~30 g.
In all cases, mice were anesthetized, subjected to tissue trauma consisting of bilateral femur fracture in association with hemorrhagic shock, and euthanized 6 h later. Animals were anesthetized with intraperitoneal pentobarbital sodium (50 mg/kg) and inhaled isoflurane (Abbott Laboratories, Chicago, IL). Under sterile conditions, a left-groin exploration was performed, and the left femoral artery was cannulated with tapered PE-10 tubing and connected to a blood pressure transducer (Micro-Med, Tustin, CA) for continuous mean arterial pressure monitoring for the duration of the experiment (6 h) as we have described (12, 20). A bilateral, closed, midshaft femur fracture was then performed using two Hemostats applied to the hind-limb region. Where indicated, mice were also subjected to hemorrhagic shock as described (21). To do so, after mice recovered from the inhalational anesthesia for 10 min, blood was withdrawn to allow the mean arterial pressure to drop to 25 mmHg over 5 min, and the blood pressure was maintained at this level for 150 min. The mice were then resuscitated over 10 min with their remaining shed blood plus two times the maximal shed blood in lactated Ringer’s solution (Baxter, Deerfield, IL). Sham-operated mice underwent anesthesia and femoral cannulation only. All mice were re-anesthetized with intraperitoneal pentobarbital sodium (20 mg/kg) as necessary throughout the experiment. At the end of 6 h (or after 4 h in cases in which mice underwent hemorrhage), mice were killed under inhalational anesthesia via cardiac puncture technique. Necropsy was performed to verify the presence of bilateral femur fractures and to ensure the absence of fracture-site hematomas. Immediately after euthanasia, the lungs were freshly harvested and either perfused with 4% paraformaldehyde to ensure complete preservation and to prevent airway collapse or freshly frozen for further biochemical analysis. The ileum was also harvested, and prepared immediately for processing in paraffin, or fresh frozen for subsequent biochemical analysis. Serum from postmortem blood samples was obtained for cytokine and blood chemistry analysis. Where indicated, C34 or anti-HMGB1 antibodies were administered 1 h before the initiation of the trauma/hemorrhagic shock model. Salubrinal (Tocris Biosciences) was administered intraperitoneally as a single dose, 30 minutes prior to the initiation of the model, at a dose of 1mg/kg. It was dissolved in DMSO stock diluted 20 times with 0.5% hydroxymethycellulose/0.1% Tween-80 solution.
Statistical Analysis
Statistical analysis was performed using SPSS 13.0 software. In experiments in which more than two experimental groups were present, all groups were first assessed by two-way ANOVA, and after determining significance, an unpaired t test was performed to determine the groups that were significantly different from control. Two-tailed student’s t-test was used for comparison for experiments consisting of two experimental groups. For mouse studies, at least 5 mice per group were included.
Results
Activation of TLR4 in the intestinal epithelium is required for the induction of secondary lung injury after trauma
We first evaluated directly whether the activation of TLR4 within the intestinal epithelium may be required for the development of secondary lung injury after trauma, and if so, sought to evaluate the potential mechanisms involved. To do so, we subjected wild-type mice and mice that we had generated to lack TLR4 within the intestinal epithelium (TLR4ΔIEC mice) (19) to a well validated model of trauma/hemorrhagic shock as described in Methods. Mice were then sacrificed at 6 hours from the induction of injury. As shown in Figure 1, wild-type mice subjected to experimental trauma/hemorrhagic shock displayed significant lung injury, which was manifest by thickening of the inter-alveolar septi and cellular infiltration (Figure 1A–B), by the recruitment of PMNs as revealed by staining for myeloperoxidase (MPO, Figure 1E–F), by apoptosis of pneumocytes as revealed by expression of cleaved-caspase 3 within the alveolar epithelium (Figure 1I–J, M), and by the increased expression of IL-6 and the chemokine (monocyte chemo-attractant protein 1) MCP1 in the lung (Figure 1N, O). Importantly, exposure of TLR4ΔIEC mice to the same trauma did not cause lung injury, as manifest by reduced histological evidence of inflammation (Figure 1C), reduced PMN infiltration (Figure 1G), reduced apoptosis (Figure 1K, M) and reduce IL-6 and MCP-1 expression (Figure 1N–O). These findings together indicate that TLR4 signaling in the gut is required for the induction of lung injury after remote trauma. We next examined the signaling mechanisms within the gut by which these events could occur.
Figure 1. Activation of TLR4 in the intestinal epithelium is required for the induction of secondary remote lung injury after trauma.

Representative micrographs showing sections of lung from either wild-type (A–B, E–F, I–J), TLR4ΔIEC mice (C, G, K), or wild-type mice pre-treated with the ER stress releasing agent salubrinal (1mg/kg, D, H, L) that were either untreated or subjected to trauma/hemorrhagic shock as indicated. Sections were stained for H&E (A–D), myeloperoxidase (E–H, arrows show MPO positive pneumocytes), or in panels I–L were stained for cleaved caspase 3 (CC3, red), DAPI (blue), or E-cadherin (green); arrows show apoptotic pneumocytes and examined via confocal microscopy. M: % positive CC3 pneumocytes per high power field N–O: qRT-PCR showing expression of IL-6 (N) or monocyte chemotactic protein 1 (MCP1) (O) in lung lysates in untreated mice (white bars) or mice subjected to trauma/hemorrhagic shock (red bars) under the indicated conditions.; for M–O: *p<0.05 wild-type control vs. wild-type trauma; +p<0.05 wild-type trauma vs. TLR4ΔIEC trauma; ** wild-type trauma vs. wild-type+salubrinal trauma. All data are mean±SEM; at least 6 animals per group. Representative of 3 independent experiments.
Trauma/hemorrhagic shock induces ER stress in the intestinal epithelium in a TLR4-dependent manner, causing enterocyte apoptosis and the release of HMGB1
We recently described that TLR4 activation in the intestinal epithelium induces ER stress within these same cells in the gut (13), which then plays a major role in the induction of epithelial apoptosis through activation of caspase 3 (13). In view of these findings, we next sought to evaluate whether TLR4 signaling in the intestinal epithelium could induce ER stress and intestinal epithelial apoptosis after trauma. As can be seen in Figure 2, compared with the normal intestinal epithelial architecture seen under control conditions (Figure 2A), the induction of trauma/hemorrhagic shock resulted in a marked derangement of the intestinal epithelium and a loss of the intact crypt villus (Figure 2B). The induction of trauma was also associated with a significant increase in ER stress - as manifest by an increase in the expression of BiP - within the intestinal epithelium (Figure 2E–F), as well as an increase in the expression of XBP1-spliced (XBP1s) by qRT-PCR (Figure 2Q). Importantly, the induction of trauma/hemorrhagic shock in TLR4ΔIEC mice did not lead to mucosal destruction (Figure 2C) and did not induce ER stress (Figure 2G, M). An apparent consequence of TLR4-induced ER stress within the intestinal epithelium after trauma was a dramatic increase in apoptosis of the intestinal epithelium in wild- type mice (Figure 2I–J) that was not seen in TLR4ΔIEC mice (Figure 2K, R). The destruction in the intestinal mucosa after trauma and induction of apoptosis could both be reversed through pre- treatment with the ER stress reducing agent salubrinal, which has been shown to protect cells from ER stress in other systems(22–24) (Figure 2D, H, L, Q–R). Shown in Figure 2 panels H and Q is evidence that ER stress is reduced in the intestinal epithelium after pre-treatment with salubrinal. The TLR4-dependent increase in ER stress and apoptosis were associated with an increase in the expression of the DAMP molecule HMGB1 in the intestinal epithelium, as manifest by immunostaining (Figure 2M–P) and expression by qRT-PCR (Figure 2S), as well as the release of HMGB1 into the serum (Figure 2T) in a pattern that mirrored the severity of gut inflammation as measured by IL-6 by qRT-PCR (Figure 2U). The release of HMGB1 into the serum was reduced after pre-treatment with salubrinal, suggesting that the increase in HMGB1 release may require the induction of ER stress (Figure 2P, T). Moreover, the pre-treatment of wild-type mice with salubrinal significantly attenuated the degree of lung injury in wild-type mice subjected to trauma/hemorrhagic shock, as manifest by reduced gross evidence of lung injury (Figure 1D), reduced staining for MPO (Figure 1H), reduced apoptosis (Figure 1L–M) and reduced expression of the pro-inflammatory cytokine IL-6 (Figure 1N) and the pro-inflammatory chemokine MCP-1 (Figure 1O). Taken together, these findings presented in aggregate in Figures 1 and 2 suggest that experimental trauma induces TLR4 activation within the intestinal epithelium leading ER stress, resulting in intestinal epithelial apoptosis, and the subsequent increase in HMGB1 expression in the gut and its release into the serum.
Figure 2. Trauma/hemorrhagic shock induces ER stress in the intestinal epithelium in a TLR4-dependent manner, causing enterocyte apoptosis and the release of HMGB1.

A–P: Representative micrographs showing sections of the ileum from either wild-type (A–B, E–F, I–J, M–N), TLR4ΔIEC (C, G, K, O), or wild-type mice pre-treated with salubrinal (1mg/kg D, H, L, P) that were either untreated or were subjected to trauma/hemorrhagic shock as indicated. Sections were stained for H&E (A–D), BiP (red stain in E–H), cleaved caspase 3 (CC3, red stain in I–L), or HMGB1 (green stain in M–P) and all immunofluorescent sections were co-stained with DAPI (blue). Arrows show apoptotic cells in the intestinal epithelium. Q–U: mean±SEM of data from mice that were either control (white bars) or subjected to trauma/hemorrhagic shock (red bars) under the indicated conditions. Shown is qRT-PCR for the ER stress gene XBP-1s in intestinal lysates (Q), the percent of cells per high power field that are CC3 positive out of 100 fields per group (R), qRT-PCR for HMGB1 in intestinal lysates (S), serum ELISA for HMGB1 (T), and qRT-PCR for IL-6 in intestinal lysates (U). In all groups, *p<0.05 wild-type control vs. wild-type trauma; +p<0.05 wild-type trauma vs. TLR4ΔIEC trauma; ** wild-type trauma vs. wild-type + salubrinal trauma. All data are mean±SEM; at least 6 animals per group repeated in three independent experiments.
Remote lung injury after trauma requires HMGB1 release from the intestinal epithelium
We next sought to determine in greater detail how TLR4 signaling in the intestinal epithelium after trauma could lead to acute lung injury, and investigated whether the TLR4-mediated release of HMGB1 from the intestinal epithelium may be required. To do so, we first generated mice lacking HMGB1 in the intestinal epithelium (HMGB1ΔIEC mice). As shown in Figure 3A–B, sections of the intestinal epithelium from untreated wild-type mice show the expression of HMGB1 in the lamina propria and the intestinal epithelium (Figure 3A), while HMGB1 expression was restricted to the lamina propria and not seen in the intestinal epithelium in HMGB1ΔIEC mice (Figure 3B), despite similar histological appearance of the small intestinal villi (Figure 3C–D) and lungs at baseline (Supplemental Figure 1A–B) in both strains. Importantly, HMGB1 release from the intestinal epithelium was found to be required for the induction of lung injury. In support of this conclusion, neutralization of HMGB1 using affinity purified anti-HMGB1 antibody prevented distal lung injury as manifest histologically (Figure 3E–F), by reduced expression of MPO (Figure 3I–J), by reduced expression of cleaved caspase 3 in the lung (Figure 3M–N, Q), and reduced lung IL-6 expression (Figure 3R). While HMGB1 may be released from many cell types including leukocytes and epithelial cells (25), HMGB1 release from the intestinal epithelium was critical for the development of remote lung injury, as mice lacking HMGB1 in the intestinal epithelium (HMGB1ΔIEC) were protected from the development of lung injury (Figure 3G, K, O, Q–S). As anticipated, the induction of trauma in HMGB1ΔIEC mice resulted in reduced circulating HMGB1 in the serum compared to wild-type mice after trauma (Figure 3T). It is noteworthy that the induction of intestinal epithelial apoptosis is at least partially required for the release of HMGB1, as treatment of intestinal epithelial cells in vitro with either LPS or the ER stress inducing agent thapsigargin induced apoptosis and the release of HMGB1 into the media which were both prevented by pre-treatment with the pan-caspase inhibitor Z-VAD (Supplemental Figure 2). To further support a role for HMGB1 in the development of lung injury, injection of recombinant HMGB1 (4μg/g body weight) into TLR4ΔIEC mice (which themselves had showed reduced HMGB1 levels in the plasma Figure 3T) reversed the protection from trauma induced lung injury that had been seen in the TLR4ΔIEC mice, which under these conditions displayed significant lung inflammation after trauma (Figure 3H, L, P–S; the corresponding serum levels of HMGB1 are shown in Figure 3T). Finally, HMGB1ΔIEC mice were found to be protected from gut injury after trauma/hemorrhagic shock as manifest by significantly reduced expression of the pro-inflammatory cytokine IL-6 in the ileum compared with wild-type mice (Supplemental Figure 1C). Taken together, these findings illustrate that TLR4 signaling in the intestinal epithelium leads to HMGB1 release which is required for the development of lung injury. We therefore next sought to define in greater detail the mechanisms by which HMGB1 release could lead to lung injury.
Figure 3. Remote lung injury after trauma requires HMGB1 release from the intestinal epithelium.

A–B: Representative confocal micrographs of the intestinal villi of untreated wild-type (A) and intestinal specific HMGB1 knockout mice (B, HMGB1ΔIEC), stained for HMGB1 (red), E-cadherin (green) and DAPI (blue); arrows show HMGB1 stained nuclei in intestinal epithelium and lamina propria in wild-type mice, but in lamina propria only in the HMGB1ΔIEC mice. C–D: Representative H&E stained histomicrographs showing sections obtained from the terminal ileum of wild type (C) and HMGB1ΔIEC mice (D). E–P: Representative micrographs showing sections of lung from either wild-type (E, I, M), wild-type + exogenous HMGB1 (4 mg/kg, F, J, N), HMGB1ΔIEC (G, K, O), or TLR4ΔIEC + exogenous HMGB1 (4 μg/g, H, L, P) mice that were either untreated or were subjected to trauma/hemorrhagic shock as indicated. Sections were imaged by bright field microscopy and stained for H&E (E–H) or myeloperoxidase (I–L, brown, arrows show MPO-positive cells), or were imaged by confocal microscopy and were stained for cleaved caspase 3 (red, arrows show apoptotic cells), DAPI (blue) or E-cadherin (green) in M–P). Q–R: mean±SEM of percent CC3-positive cells/high power field (Q) and expression of IL-6 (R) and monocyte chemotactic protein 1 (MCP1) (S) by qRT-PCR in wild-type, HMGB1ΔIEC or TLR4ΔIEC mice that were subjected to trauma/hemorrhagic in the presence or absence of exogenous anti-HMGB1 or recombinant HMGB1 (4μg/g) as indicated. In both panels, *p<0.05 wild-type vs. HMGB1ΔIEC, **p<0.05 wild-type vs. wild-type + anti-HGMB1, +p<0.005 TLR4ΔIEC vs. TLR4ΔIEC + recombinant HMGB1. All data are mean±SEM; at least 6 animals per group repeated in triplicate. T: Serum HMGB1 by ELISA in mice of the strain indicated subjected to trauma/hemorrhagic shock in the presence of salubrinal (10μM) or anti-HMGB1 as shown. All groups are significantly different from wild-type by ANOVA, p<0.05. Representative of 3 independent experiments.
HMGB1 signaling in the intestinal epithelium leads to internalization of the tight junction protein ZO-1 in the pulmonary epithelium
We next sought to define how HMGB1 release could induce lung injury in the setting of trauma/hemorrhagic shock findings. Previous authors have reported that acute lung injury requires the internalization of the tight junction protein ZO-1, resulting in a loss of the protective barrier (15, 26), which is thought to be an inciting event in the development of lung inflammation (15, 27, 28). These observations raise the distinct possibility that internalization of tight junctions in response to HMGB1 release from the gut after trauma/hemorrhagic shock could participate in the development of lung injury. Consistent with this possibility, we now show that exposure of primary human lung epithelial cells to HMGB1 resulted in an internalization of the tight junction protein ZO-1 as compared with untreated cells (Figure 4A–B). Importantly, the pre-treatment of cells with PKC-zeta pseudo-substrate (PKCi) (29, 30) as described in Methods reversed the internalization of ZO-1 from the cell surface (Figure 4C), while treatment with the inhibitor alone had no effect (Figure 4D), suggesting a role of PKC-zeta in ZO-1 trafficking in pneumocytes in response to HMGB1. In support of the in vivo significance for a role for HMGB1 in influencing the trafficking of ZO-1 in pneumocytes after trauma/hemorrhagic shock, the expression of ZO-1 on the pulmonary epithelium was found to be reduced on the cell surface compared to saline treated mice, while pre-treatment with HMGB1 inhibitory antibodies restored the ZO-1 on the cell surface, as revealed by confocal microscopy (Figure 4E–G; E′–G′ correspond to the magnification of areas defined by dotted lines). There was no effect on internalization of ZO-1 after pre-treatment with IgG (Figure 4F, F′). These findings suggest that HMGB1 release leads to the internalization of tight junctions in pneumocytes in the setting of trauma/hemorrhagic shock in which lung injury is seen.
Figure 4. HMGB1 signaling in the intestinal epithelium leads to internalization of the tight junction protein ZO-1 in the pulmonary epithelium.

A–D: Confocal micrographs of human lung alveolar epithelial cells treated with either saline (A), recombinant HMGB1 (2.5 μg/ml, B), recombinant HMGB1 in the presence of PKC-zeta pseudo substrate (PKCi, 150nM, C), or PKC-zeta pseudo substrate alone (150nM, D); cells were stained for zona occludens-1 (ZO-1, green) and DAPI (blue). Arrows point to ZO-1 positive tight junctions. E–G: Representative confocal micrographs of lung sections from wild-type mice treated with either saline under control conditions (E), or subjected to trauma/hemorrhagic shock and treated with IgG (F) or anti-HMGB1 (G) and stained for ZO-1 and DAPI. Arrows show regions of tight junctions in which ZO-1 staining is seen; the areas indicated by the dotted lines are shown at the indicated magnification. Representative of 3 independent experiments.
The novel TLR4 inhibitor C34 inhibits TLR4 and protects against lung injury after trauma in mice
The above findings - in which TLR4 signaling in the intestinal epithelium after trauma/hemorrhagic shock is required for the induction of lung injury - suggest that strategies that inhibit TLR4 could offer novel therapeutic approaches for the prevention of lung injury in trauma. To identify potential TLR4 inhibitors, we recently utilized a similarity search algorithm using the TLR4 inhibitor Eritoran (E5564)(31) in conjunction with a limited screening approach of small molecule libraries to identify compounds that could bind to the E5564 site (16). We successfully identified a lead compound, C34, which we recently published to be a 2-acetamidopyranoside (MW 389) with the formula C17H27NO9(16). As we recently reported, molecular docking of C34 to the hydrophobic internal pocket of MD-2 demonstrated a tight fit while C34 inhibited TLR4 in vivo and also inhibited TLR4 in the intestine in both mouse and human tissue (16). As shown in Figure 5, C34 markedly inhibited the degree of lung inflammation that developed in experimental trauma, as measured by a significant reduction in the degree of inflammation within the lung (Figure 5A–C) and by the expression of lung IL-6 by qRT-PCR (Figure 5J). In support of a role for TLR4-induced ER stress in the gut in the process leading to HMGB1 release and lung injury, C34 pre-treatment was found to reduce the degree of mucosal injury (Figure 5D–F) and ER stress within the gut (Figure 5G–I, K), and also to decrease HMGB1 release in the serum (Figure 5L), and to reduce the expression of TLR4 (Figure 5M) and HMGB1 (Figure 5N) within the intestinal mucosa after trauma/hemorrhagic shock. These findings raise the possibility that factors that regulate TLR4 signaling could suppress the degree of lung injury that develops after trauma/hemorrhagic shock, potentially through effects on ER stress and HMGB1 release.
Figure 5. The novel TLR4 inhibitor C34 inhibits TLR4 and protects against lung injury after trauma in mice.

A–I: Representative bright field H&E micrographs of lung (A–C) or terminal ileum (D–F) and confocal sections of terminal ileum (G–I) stained for BiP (red) and DAPI (blue) from wild-type mice that were either uninjured (A, D, G), subjected to trauma/hemorrhagic shock in the absence (B, E, H), or presence (C, F, I) of C34 (2.5μg/g IP). J–N: qRT-PCR showing the expression of IL-6 in the lung (J) or XBP1s in the ileum (K); L: serum ELISA for HMGB1 in mice that were uninjured or subjected to trauma/hemorrhagic shock in the absence or presence of C34; M–N: expression of TLR4 (M) and HMGB1 (N) in the ileum by qRT-PCR. *p<0.05 control vs. trauma; **p<0.005 trauma vs. trauma + C34. Representative of 3 separate experiments with over 5 mice per group.
Discussion
Three independent lines of evidence support the conclusion that TLR4 activation in the intestinal epithelium is required for the induction of secondary lung injury through the release of HMGB1 from the gut. First, mice lacking TLR4 in the intestinal epithelium released less HMGB1 and were protected from the development of lung injury after trauma/hemorrhagic shock, supporting the hypothesis that TLR4 in the intestinal epithelium is required for the development of lung injury. Second, mice lacking HMGB1 in the intestinal epithelium showed reduced concentrations of circulating HMGB1, and were protected from the development of lung injury. Finally, and perhaps most significantly from a therapeutic perspective, treatment of mice with a novel TLR4 inhibitor C34 C17H27NO9(16) reversed the development of lung injury after systemic trauma/hemorrhagic shock, and reduced the extent of HMGB1 release into the circulation (Figure 5). Taken together, these findings - summarized in the schematic diagram shown in Figure 6 - advance our understanding of how secondary lung injury may occur after remote injury, and provide a contextual framework by which novel therapeutic approaches for traumatized patients may be developed.
Figure 6. Trauma/hemorrhagic shock induces TLR4 signaling in the intestine leading to lung injury via the release of HMGB1 from the intestine.
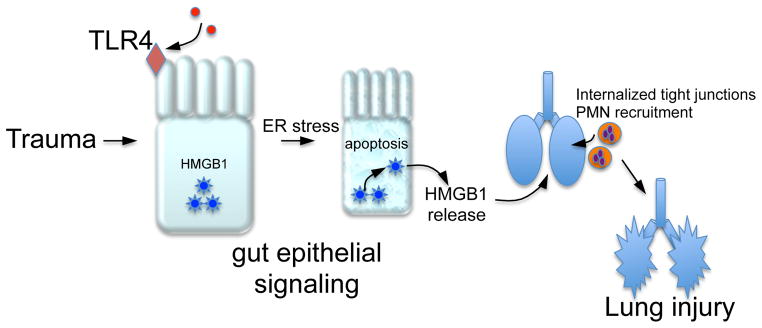
See text for details.
The current studies build upon a growing body of work that have shed light on the mechanisms leading to the development of acute lung injury after trauma, and on the role of HMGB1 in particular. For instance, Deitch and colleagues were among the earliest proponents of the importance of the gastrointestinal tract in driving lung injury after trauma (32, 33), and more recently have shown that components of the intestinal lymph play a major role in the development of lung injury in rats (34, 35), although the individual constituents within the lymph fluid that lead to the lung injury remain unknown (34). Further, Abraham and colleagues have shown that HMGB1 plays a critical role in the development of lung injury after trauma (14), as instillation of HMGB1 into the trachea causes lung inflammation (36), while antibody inhibition of HMGB1 attenuates the degree of lung injury and mortality in models of trauma (14, 37, 38). Reino et al recently showed that TLR4 mutant mice are protected from the development of trauma/hemorrhagic shock induced lung injury, although the specific TLR4 expressing cell that was responsible for this protection was not shown (39). The novelty and significance of the current work is therefore that our conclusions support these prior observations, but provide additional mechanistic details by showing that TLR4 signaling in the gut leading to intestinal HMGB1 release is critical to the lung injury that was observed, and that the consequences of exaggerated HMGB1 release can be therapeutically reversed by the use of a novel TLR4 inhibitor that we recently identified, namely C34 (16). Moreover, we have shown that the development of ER stress in the intestinal epithelium in response to TLR4 may be a requirement for lung inflammation to occur, which may explain in part why both lymphatic and circulatory routes can contribute to lung injury, as epithelial ER stress would be expected to influence the composition of both of these compartments. We readily acknowledge that although we now showed that trauma in mice leads to a significant increase of HMGB1 in the plasma that was not seen in HMGB1ΔIEC mice, we have not measured HMGB1 in the lymph, and cannot therefore exclude a potential role for HMGB1 acting on the lung via the lymphatic system. These studies therefore provide additional details onto the canvas of the “gut hypothesis” of lung injury, and shed light on potential therapeutic possibilities.
One of the more intriguing questions to arise from the current studies relates to what may be the source of TLR4 activation in the intestinal epithelium that leads to the downstream consequences that culminate in lung injury. On first inspection, it is tempting to postulate that microbial products present within the intestinal lumen - particularly those that are rich in lipopolysaccharide - may directly activate TLR4. However, it is not known whether TLR4 is on the apical or the basal surface of the pneumocyte, and the poor applicability of the currently available anti-TLR4 antibodies for immuno-localization do not readily allow for this determination. It is also plausible that factors within the blood stream may activate TLR4 within the lung epithelium, which could be either microbial or endogenous in origin. In this regard, a variety of host substrates have been shown in a variety of in vitro and in vivo studies to activate TLR4, including Hsp70, uric acid, S100, heparan sulfate, and HMGB1 itself (40). In support of a role endogenous molecules as central mediators in the development of trauma-induced lung injury, Deitch and colleagues recently showed that non-bacterial intraluminal molecules were responsible for the development of lung injury in traumatized rats (34). It is noteworthy that both bacterial and non-bacterial activation of TLR4 would be expected to induce ER stress equally within the intestinal epithelium(41–43), a process which we have shown to occur in the setting of intestinal epithelium in the newborn in the pathogenesis of necrotizing enterocolitis(13). The induction of ER stress within the gut is an attractive intermediate pathway to focus on in the release of HMGB1, as a variety of studies including from our group have shown that exaggerated ER stress in response to TLR4 signaling leads to enterocyte apoptosis(13), which may explain why TLR4 signaling so readily induced HMGB1 release from the gut. (13), which may explain why TLR4 signaling so readily induced HMGB1 release from the gut. Using gene array and Ingenuity Pathway Analysis, we have shown that ER stress pathways are rapidly upregulated in the liver in a murine model of hemorrhagic shock + trauma (44). We now posit that an assessment of the degree of ER stress induction in the gut or other tissues may provide novel biomarkers to determine who may be at risk for the development of further organ injury, and may thus benefit from additional therapeutic intervention. Further studies will be required in order to explore this in further detail.
In summary, we have now shown that development of lung injury after trauma requires the activation of TLR4 within the intestinal epithelium and a gut epithelial source of HMGB1, which is then required for the development of lung injury. We readily acknowledge that while the current findings are novel, there are several avenues for further investigation to address some of the questions raised from the current studies. These include the determination of the precise agonist that activates TLR4 in the first place as described above, as well as an assessment of whether animals that lack TLR4 in other cells may be similarly protected from lung injury after trauma, suggesting that hierarchical TLR4 signaling regulates the lung inflammatory response. It is our hope that by addressing these further questions within the context of the novel pathway that we summarize in Figure 6 that novel therapeutic approaches may be designed for these injured patients.
Supplementary Material
Acknowledgments
Funding sources: DJH is supported by R01GM078238 and R01DK083752 from the National Institutes of Health and the Hartwell Foundation for Biomedical Research. This work was also funded by National Institute of General Medical Sciences Grant P50-GM-53789 to DJH and TRB.
References
- 1.Krug EG, Sharma GK, Lozano R. The global burden of injuries. Am J Public Health. 2000;90:523–526. doi: 10.2105/ajph.90.4.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Minei JP, Cuschieri J, Sperry J, Moore EE, West MA, Harbrecht BG, O’Keefe GE, Cohen MJ, Moldawer LL, Tompkins RG, Maier RV. The changing pattern and implications of multiple organ failure after blunt injury with hemorrhagic shock. Crit Care Med. 2012;40:1129–1135. doi: 10.1097/CCM.0b013e3182376e9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vodovotz Y, Billiar TR. In silico modeling: methods and applications to trauma and sepsis. Crit Care Med. 2013;41:2008–2014. doi: 10.1097/CCM.0b013e31829a6eb4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mi Q, Constantine G, Ziraldo C, Solovyev A, Torres A, Namas R, Bentley T, Billiar TR, Zamora R, Puyana JC, Vodovotz Y. A dynamic view of trauma/hemorrhage-induced inflammation in mice: principal drivers and networks. PLoS One. 2011;6:e19424. doi: 10.1371/journal.pone.0019424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levy G, Fishman JE, Xu DZ, Dong W, Palange D, Vida G, Mohr A, Ulloa L, Deitch EA. Vagal nerve stimulation modulates gut injury and lung permeability in trauma-hemorrhagic shock. The journal of trauma and acute care surgery. 2012;73:338–342. doi: 10.1097/TA.0b013e31825debd3. discussion 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 7.Herridge MS, Cheung AM, Tansey CM, Matte-Martyn A, Diaz-Granados N, Al-Saidi F, Cooper AB, Guest CB, Mazer CD, Mehta S, Stewart TE, Barr A, Cook D, Slutsky AS. One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med. 2003;348:683–693. doi: 10.1056/NEJMoa022450. [DOI] [PubMed] [Google Scholar]
- 8.Sasai M, Yamamoto M. Pathogen recognition receptors: ligands and signaling pathways by Toll-like receptors. International reviews of immunology. 2013;32:116–133. doi: 10.3109/08830185.2013.774391. [DOI] [PubMed] [Google Scholar]
- 9.Park BS, Lee JO. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med. 2013;45:e66. doi: 10.1038/emm.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 11.Neal MD, Leaphart C, Levy R, Prince J, Billiar TR, Watkins S, Li J, Cetin S, Ford H, Schreiber A, Hackam DJ. Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J Immunol. 2006;176:3070–3079. doi: 10.4049/jimmunol.176.5.3070. [DOI] [PubMed] [Google Scholar]
- 12.Sodhi C, Levy R, Gill R, Neal MD, Richardson W, Branca M, Russo A, Prindle T, Billiar TR, Hackam DJ. DNA attenuates enterocyte Toll-like receptor 4-mediated intestinal mucosal injury after remote trauma. Am J Physiol Gastrointest Liver Physiol. 2011;300:G862–873. doi: 10.1152/ajpgi.00373.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Afrazi A, Branca MF, Sodhi CP, Good M, Yamaguchi Y, Egan CE, Lu P, Jia H, Shaffiey S, Lin J, Ma C, Vincent G, Prindle T, Jr, Weyandt S, Neal MD, Ozolek JA, Wiersch J, Tschurtschenthaler M, Shiota C, Gittes GK, Billiar TR, Mollen K, Kaser A, Blumberg R, Hackam DJ. Toll-like receptor 4-mediated endoplasmic reticulum stress in intestinal crypts induces necrotizing enterocolitis. J Biol Chem. 2014;289:9584–9599. doi: 10.1074/jbc.M113.526517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JY, Park JS, Strassheim D, Douglas I, Diaz del Valle F, Asehnoune K, Mitra S, Kwak SH, Yamada S, Maruyama I, Ishizaka A, Abraham E. HMGB1 contributes to the development of acute lung injury after hemorrhage. Am J Physiol Lung Cell Mol Physiol. 2005;288:L958–965. doi: 10.1152/ajplung.00359.2004. [DOI] [PubMed] [Google Scholar]
- 15.El-Hashash AH, Turcatel G, Varma S, Berika M, Al Alam D, Warburton D. Eya1 protein phosphatase regulates tight junction formation in lung distal epithelium. J Cell Sci. 2012;125:4036–4048. doi: 10.1242/jcs.102848. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Neal MD, Jia H, Eyer B, Good M, Guerriero CJ, Sodhi CP, Afrazi A, Prindle T, Jr, Ma C, Branca M, Ozolek J, Brodsky JL, Wipf P, Hackam DJ. Discovery and validation of a new class of small molecule Toll-like receptor 4 (TLR4) inhibitors. PLoS One. 2013;8:e65779. doi: 10.1371/journal.pone.0065779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krzyzaniak MJ, Peterson CY, Cheadle G, Loomis W, Wolf P, Kennedy V, Putnam JG, Bansal V, Eliceiri B, Baird A, Coimbra R. Efferent vagal nerve stimulation attenuates acute lung injury following burn: The importance of the gut-lung axis. Surgery. 2011;150:379–389. doi: 10.1016/j.surg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yazji I, Sodhi CP, Lee EK, Good M, Egan CE, Afrazi A, Neal MD, Jia H, Lin J, Ma C, Branca MF, Prindle T, Richardson WM, Ozolek J, Billiar TR, Binion DG, Gladwin MT, Hackam DJ. Endothelial TLR4 activation impairs intestinal microcirculatory perfusion in necrotizing enterocolitis via eNOS-NO-nitrite signaling. Proc Natl Acad Sci U S A. 2013;110:9451–9456. doi: 10.1073/pnas.1219997110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sodhi CP, Neal MD, Siggers R, Sho S, Ma C, Branca MF, Prindle T, Jr, Russo AM, Afrazi A, Good M, Brower-Sinning R, Firek B, Morowitz MJ, Ozolek JA, Gittes GK, Billiar TR, Hackam DJ. Intestinal epithelial Toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology. 2012;143:708–718. e701–705. doi: 10.1053/j.gastro.2012.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levy RM, Mollen KP, Prince JM, Kaczorowski DJ, Vallabhaneni R, Liu S, Tracey KJ, Lotze MT, Hackam DJ, Fink MP, Vodovotz Y, Billiar TR. Systemic inflammation and remote organ injury following trauma require HMGB1. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1538–1544. doi: 10.1152/ajpregu.00272.2007. [DOI] [PubMed] [Google Scholar]
- 21.Prince JM, Levy RM, Yang R, Mollen KP, Fink MP, Vodovotz Y, Billiar TR. Toll-like receptor-4 signaling mediates hepatic injury and systemic inflammation in hemorrhagic shock. Journal of the American College of Surgeons. 2006;202:407–417. doi: 10.1016/j.jamcollsurg.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 22.Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- 23.Sokka AL, Putkonen N, Mudo G, Pryazhnikov E, Reijonen S, Khiroug L, Belluardo N, Lindholm D, Korhonen L. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J Neurosci. 2007;27:901–908. doi: 10.1523/JNEUROSCI.4289-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang H, Antoine DJ, Andersson U, Tracey KJ. The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol. 2013;93:865–873. doi: 10.1189/jlb.1212662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caraballo JC, Borcherding J, Thorne PS, Comellas AP. Protein kinase C-zeta mediates lung injury induced by diesel exhaust particles. Am J Respir Cell Mol Biol. 2013;48:306–313. doi: 10.1165/rcmb.2012-0056OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Costantini TW, Loomis WH, Putnam JG, Drusinsky D, Deree J, Choi S, Wolf P, Baird A, Eliceiri B, Bansal V, Coimbra R. Burn-induced gut barrier injury is attenuated by phosphodiesterase inhibition: effects on tight junction structural proteins. Shock. 2009;31:416–422. doi: 10.1097/SHK.0b013e3181863080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mazzon E, Cuzzocrea S. Role of TNF-alpha in lung tight junction alteration in mouse model of acute lung inflammation. Respir Res. 2007;8:75. doi: 10.1186/1465-9921-8-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Willis CL, Meske DS, Davis TP. Protein kinase C activation modulates reversible increase in cortical blood-brain barrier permeability and tight junction protein expression during hypoxia and posthypoxic reoxygenation. J Cereb Blood Flow Metab. 2010;30:1847–1859. doi: 10.1038/jcbfm.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caraballo JC, Yshii C, Butti ML, Westphal W, Borcherding JA, Allamargot C, Comellas AP. Hypoxia increases transepithelial electrical conductance and reduces occludin at the plasma membrane in alveolar epithelial cells via PKC-zeta and PP2A pathway. Am J Physiol Lung Cell Mol Physiol. 2011;300:L569–578. doi: 10.1152/ajplung.00109.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rossignol DP, Lynn M. TLR4 antagonists for endotoxemia and beyond. Curr Opin Investig Drugs. 2005;6:496–502. [PubMed] [Google Scholar]
- 32.Magnotti LJ, Upperman JS, Xu DZ, Lu Q, Deitch EA. Gut-derived mesenteric lymph but not portal blood increases endothelial cell permeability and promotes lung injury after hemorrhagic shock. Ann Surg. 1998;228:518–527. doi: 10.1097/00000658-199810000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adams CA, Jr, Magnotti LJ, Xu DZ, Lu Q, Deitch EA. Acute lung injury after hemorrhagic shock is dependent on gut injury and sex. Am Surg. 2000;66:905–912. discussion 912–903. [PubMed] [Google Scholar]
- 34.Fishman JE, Sheth SU, Levy G, Alli V, Lu Q, Xu D, Qin Y, Qin X, Deitch EA. Intraluminal Nonbacterial Intestinal Components Control Gut and Lung Injury After Trauma Hemorrhagic Shock. Ann Surg. 2014 doi: 10.1097/SLA.0000000000000631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deitch EA. Gut-origin sepsis: evolution of a concept. Surgeon. 2012;10:350–356. doi: 10.1016/j.surge.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ. HMG-1 as a mediator of acute lung inflammation. J Immunol. 2000;165:2950–2954. doi: 10.4049/jimmunol.165.6.2950. [DOI] [PubMed] [Google Scholar]
- 37.Suda K, Kitagawa Y, Ozawa S, Saikawa Y, Ueda M, Ebina M, Yamada S, Hashimoto S, Fukata S, Abraham E, Maruyama I, Kitajima M, Ishizaka A. Anti-high-mobility group box chromosomal protein 1 antibodies improve survival of rats with sepsis. World J Surg. 2006;30:1755–1762. doi: 10.1007/s00268-005-0369-2. [DOI] [PubMed] [Google Scholar]
- 38.Yang R, Harada T, Mollen KP, Prince JM, Levy RM, Englert JA, Gallowitsch-Puerta M, Yang L, Yang H, Tracey KJ, Harbrecht BG, Billiar TR, Fink MP. Anti-HMGB1 neutralizing antibody ameliorates gut barrier dysfunction and improves survival after hemorrhagic shock. Mol Med. 2006;12:105–114. doi: 10.2119/2006-00010.Yang. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reino DC, Palange D, Feketeova E, Bonitz RP, Xu da Z, Lu Q, Sheth SU, Pena G, Ulloa L, De Maio A, Feinman R, Deitch EA. Activation of toll-like receptor 4 is necessary for trauma hemorrhagic shock-induced gut injury and polymorphonuclear neutrophil priming. Shock. 2012;38:107–114. doi: 10.1097/SHK.0b013e318257123a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McClure R, Massari P. TLR-Dependent Human Mucosal Epithelial Cell Responses to Microbial Pathogens. Frontiers in immunology. 2014;5:386. doi: 10.3389/fimmu.2014.00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang CC, Yao CA, Yang JC, Chien CT. Sialic Acid Rescues Repurified Lipopolysaccharide-Induced Acute Renal Failure via Inhibiting TLR4/PKC/gp91-mediated Endoplasmic Reticulum Stress, Apoptosis, Autophagy, and Pyroptosis Signaling. Toxicol Sci. 2014 doi: 10.1093/toxsci/kfu121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rao J, Yue S, Fu Y, Zhu J, Wang X, Busuttil RW, Kupiec-Weglinski JW, Lu L, Zhai Y. ATF6 mediates a pro-inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia-reperfusion injury. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2014;14:1552–1561. doi: 10.1111/ajt.12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anderson EK, Hill AA, Hasty AH. Stearic acid accumulation in macrophages induces toll-like receptor 4/2-independent inflammation leading to endoplasmic reticulum stress-mediated apoptosis. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:1687–1695. doi: 10.1161/ATVBAHA.112.250142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edmonds RD, Vodovotz Y, Lagoa C, Dutta-Moscato J, Yang Y, Fink MP, Levy RM, Prince JM, Kaczorowski DJ, Tseng GC, Billiar TR. Transcriptomic response of murine liver to severe injury and hemorrhagic shock: a dual-platform microarray analysis. Physiological genomics. 2011;43:1170–1183. doi: 10.1152/physiolgenomics.00020.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.