

Abstract
Aberrant levels and function of the potent anti-inflammatory high-density lipoprotein (HDL) and accelerated atherosclerosis have been reported in patients with autoimmune inflammatory diseases. Whether HDL affects the development of an autoimmune response remains elusive. Herein, we utilized apolipoprotein A-I deficient (apoA-I-/-) mice, characterized by diminished circulating HDL levels, to delineate the role of HDL in autoimmunity. ApoA-I-/-mice exhibited increased severity of antigen-induced arthritis compared to wild-type mice and this was associated with elevated Th1 and Th17 cell reactivity in the draining lymph nodes (dLNs). Furthermore, reconstituted HDL (rHDL) attenuated IFN-γ and IL-17 secretion by antigen-specific T cells upon stimulation of dLNs in vitro. The suppressive effects of rHDL were mediated through modulation of dendritic cell (DC) function. Specifically, rHDL-treated DCs demonstrated an immature phenotype characterized by downregulated co-stimulatory molecules, the release of low amounts of pro-inflammatory cytokines, and failure to promote T cell proliferation in vitro. The mechanism of action involved the inhibition of NF-κB nuclear translocation and the decrease of Myd88 mRNA levels by rHDL. Finally, modulation of DC function by rHDL was critically dependent on the presence of SR-BIand ABCA1 but not of the ABCG1 transporter. These findings reveal a novel role of HDL in the regulation of adaptive inflammatory responses through suppression of DC function that could be exploited therapeutically in autoimmune inflammatory diseases.
Introduction
High-density lipoprotein (HDL) constitutes a heterogeneous population of particles that is characterized by great complexity in terms of structure and functionality (1). Although it has been best recognized for its inverse correlation with cardiovascular events (2, 3), accumulating evidence has revealed novel roles of HDL in cholesterol metabolism, endothelial integrity and inflammation (4, 5). HDL metabolism entails the successive interactions of apolipoprotein A-I (apoA-I), the major protein component of HDL, with the ATP-Binding Cassette (ABC) Transporters ABCA1, ABCG1 and the scavenger receptor class B type I (SR-BI)(6). Numerous studies have implicated each of these transporters in HDL's anti-atherogenic actions involving mechanisms such as cholesterol efflux and activation of HDL-induced signaling pathways (7-9). However, under certain conditions HDL can become functionally defective. In fact, HDL's protective functions are compromised in patients with coronary heart disease, diabetes and autoimmune diseases (10-12).
Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disease manifested by leukocyte infiltration into the synovial lining leading to cartilage destruction and bone erosion (13, 14). RA pathology is critically dependent on the presence of auto-reactive IFN-γ-producing Th1 and IL-17-releasing Th17 CD4+ cell subsets, and pro-inflammatory cytokines such as TNF-α, IL-1β and IL-6 (15). RA is associated with increased cardiovascular morbidity and mortality, and the majority of these patients are dyslipidemic (16). Interestingly, although HDL-cholesterol (HDL-c) levels are decreased, the levels of pro-inflammatory HDL are increased in RA patients (17). In support, anti-rheumatic drugs improve both HDL functionality and cholesterol levels (16). In vivo studies using animal models of arthritis have suggested a protective role of apoA-I and reconstituted HDL (rHDL). Particularly, treatment of rats with an apoA-I mimetic peptide inhibited collagen-induced arthritis and reduced inflammatory cytokine levels (18). Moreover, administration of apoA-I or rHDL attenuated peptidoglycan-polysaccharide (PG-PS)-induced arthritis in Lewis female rats in an ABCA1-dependent manner (19). Although HDL modulates the activity of various immune cell subsets (12), the mechanism through which HDL regulates autoreactive T cell responses in vivo remains elusive.
Dendritic cells (DCs) are professional APCs that carry antigens in the draining lymph nodes (dLNs) and promote the activation, differentiation and polarization of naïve T cells into effector Th cell subsets (20). Specifically, mature DCs present the antigen in the context of MHC and provide co-stimulatory signals that are required for efficient activation and priming of T cells. Furthermore, through secretion of pro-inflammatory cytokines, DCs direct the polarization of T cell towards the different T cell lineages. Antigen recognition in the presence of IL-12 favors the generation of Th1 cells, whereas IL-6 and IL-23 drive the generation of Th17 effector cells (21-23). Since Th1 and Th17 cells and pro-inflammatory cytokines orchestrate the autoimmune responses in RA, strategies aiming at modulation of DC function and subsequent suppression of autoreactive Th1/Th17 responses might provide novel targets in the design of therapeutic protocols for the treatment of this disease.
In this study, we show that rHDL exerts its anti-inflammatory properties through modulation of DC maturation and function, and that rHDL-exposed DCs suppress T cell proliferation in vitro. Of interest, apoA-I-/- mice that lack conventional HDL, develop severe arthritis and elevated Th1 and Th17 cell responses. Finally, our findings support a critical role of SR-BI and ABCA1 transporters in the rHDL-mediated anti-inflammatory function of DCs.
Materials and Methods
Animals
Wild-type (WT) C57BL/6 and apoA-I-/-(ApoA1tm1Unc) mice on a C57BL/6 background were obtained from the specific pathogen-free animal facility of the Institute of Molecular Biology and Biotechnology (Heraklion, Greece). Mice were maintained on a 12 h light/dark cycle and fed standard rodent chow. Femurs and tibias were obtained from ABCA1-/- (24), ABCG1-/-(DeltagenInc, San Carlos, California) and SR-BI-/- mice (25). All experimental protocols were in accordance with institutional guidelines and were approved by the Greek Federal Veterinary Office.
Reagents
Cell cultures were maintained in complete medium consisting of DMEM (Invitrogen), supplemented with 10% FBS (Biochrom) and antibiotic-antimycotic 100× (Invitrogen). For flow cytometric analysis, the following fluorescent-conjugated Abs were used: anti-CD11b (M1/70)and anti-MHC class II (AF6-120.1) were purchased from BD Biosciences. Antibodies against CD3 (145-2C11), CD4 (RM4-5), CD8a (53-6.7), CD44 (IM7), CD45R/B220 (RA3-6B2), Ly-6G/Ly-6C (Gr-1) (RB6-8C5), CD11c (N418), CD40 (3/23), CD80 (16-10A1), CD86 (GL-1) were all purchased from BioLegend.
Induction of arthritis and clinical assessment
Mice were immunized subcutaneously at the base of the tail with 100 μg methylated BSA (mBSA) (Sigma-Aldrich) in 50 μL PBS emulsified with equal volume of CFA (Sigma-Aldrich). On day 21 post immunization, mice were injected intra-articularly (i.a.) with 50 μg mBSA in 20 μL PBS into the leftknee joint. The swelling of the joint was evaluated by daily measurement (day 21-30) of the knee diameter (mm) using a digital vernier caliper (Cocraft). Nine days post i.a injection (day 30) mice were euthanized and blood and tissues were collected.
Histological grading
Tissues were fixed in 3.7% formaldehyde for 24 h and legs were decalcified in Schaefer solution for 2-3 days. Tissues were embedded in paraffin and 4 μm thicksections were stained with H&E. Specimens were blindly evaluated by two pathologists. Histological grade of arthritis was determined as follows: 0= normal knee joint, 1=normal synovium or mild synovial reaction with scarce inflammatory cells, 2= moderate synovial reaction and moderate density of inflammatory cells, 3= severe synovial hyperplasia and dense population of inflammatory cells, 4= pannus formation associated with articular cartilage erosion. The extent of skin inflammation was evaluated by scoring samples separately for the following parameters: severity of inflammation, extent of epidermal changes, and size of abscesses. A total histological score was assessed by summing the scores (0= normal, 1= mild, 2= moderate, 3=severe) for each parameter. The range of possible score was 0-9. Histopathology images were captured using a Nikon Eclipse E-400 microscope and Nikon Digital sight, DS-SM, photographic system. Images were auto-corrected in Windows Photo Manager.
Serum HDL preparation and cholesterol measurement
Mice were euthanized and blood was collected by cardiac puncture following a 4 h fast. Serum was isolated and used for HDL preparation. Briefly, equal volumes of dextran sulfate (MP biomedicals) and MgCl2 stock solutions were mixed to produce the precipitation solution consisting of 10 g/L dextran sulfate and 0.5 M MgCl2. Serum samples were mixed thoroughly with precipitation solution at a volume ratio of 10:1 and incubated for 10 min at room temperature. The mixture was centrifuged for 40 min at 14000 × g at 4 °C and the HDL-containing supernatant was collected for determination of cholesterol levels and functionality assays. Total and HDL-cholesterol was measured using a commercially available assay (Thermo Scientific) according to the manufacturer's recommendations.
Paraoxonase-1 (PON-1) activity assay
PON-1 activity was determined in HDL preparations from mouse serum using paraoxon as substrate. Briefly, 5 μL of HDL was added in 245 μL of buffer (100 mM Tris–HCl, pH 8.0 and 2 mM CaCl2) containing 1.1 mM paraoxon (Sigma-Aldrich) and the absorbance at 405 nm was measured every 40 sec for 20 min at room temperature in a microplate spectrophotometer. The rate of p-nitrophenol formed by the hydrolysis of paraoxon was determined by monitoring the increase in absorbance. PON-1 activity was expressed as units per liter (U/L) of HDL with 1 U defined as the activity that catalyzes the formation of 1 μmoL p-nitrophenol per min.
Preparation of rHDL containing apoA-I
Recombinant apoA-I was produced and purified from the culture medium of adenovirus-infected HTB-13 cells as previously described (26). rHDL particles were prepared by the sodium cholate dialysis method (27) using POPC/cholesterol/apoA-I/sodium cholate in a molar ratio of 100:10:1:100 as previously described (28). The rHDL preparation was extensively dialyzed against PBS and the formation of apolipoprotein-lipid complexes was verified by two-dimensional gel electrophoresis (29, 30). Concentration of rHDL was based on apoA-I content and was determined by DC Protein Assay (Bio-Rad).
In vitro proliferation of dLN cells (dLNCs)
Female mice (8-10 week old) were immunized subcutaneously at the base of the tail with 100 μg OVA (Sigma-Aldrich) emulsified in equal volume of CFA. dLNs were excised 9 days later and single cell suspensions were prepared. dLNCs were cultured in flat-bottom 96-well plates (5 × 105 cells/well) in the presence of OVA (15 μg/mL) and/or increasing concentrations of rHDL (1μM = 28 μg/mL) for 72 h. Cells were then pulsed with 1μCi [3H]thymidine (TRK120; Amersham Biosciences) for 18 h, and the incorporated radioactivity was measured using a Beckman beta counter. Cytokine levels in culture supernatants were determined by ELISA and cells were analyzed by flow cytometry after 48 h of stimulation.
Generation of mouse bone marrow-derived dendritic cells (BMDCs)
BMDCs were generated from bone marrow progenitors, as previously described (31). Briefly, bone marrow was isolated from femurs and tibias of female mice, treated with RBC lysis buffer (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM Na2EDTA) and plated at 2.5 × 106 cells per 100 mm cell culture dish in complete medium supplemented with 20% supernatant from a murine GM-CSF secreting cell line (X63Ag8; kindly provided by B. Stockinger, National Institute of Medical Research, London, U.K.) (32). The culture medium was half-renewed on days 3, 6 and 8. On day 9, non adherent cells were collected and cultured in 24-well tissue culture plates (1 × 106 cells/well) in the presence of either 0.5 μg/mL Lipopolysaccharide from Escherichia coli O111: B4 (LPS) (Calbiochem) or 20 μg/mL zymosan (InvivoGen) and/or 4 μM rHDL. BMDCs were harvested as indicated for RNA, Western blot and FACS analysis. Culture supernatants were collected at 12 h or 18 h for cytokine analysis by ELISA.
Co-cultures
BMDCs from WT mice were stimulated with 0.25 μg/mL LPS and pulsed with 20 μg/mL OVA in the presence or absence of 4 μM rHDL for 12 h. Cells were collected and washed to remove excess LPS, OVA and rHDL and were co-cultured with dLNCs were isolated from OVA-immunized mice (9d post immunization) as described above. Culture supernatants were harvested 48 h later.
ELISA
Cytokine levels in culture supernatants were measured by ELISA kits following the manufacturer's recommendations. Mouse IL-17, IFN-γ, IL-12 p70, IL-8, IL-6 and TNF-α were purchased from R&D Systems. Mouse IL-23, IL-2 were obtained from eBioscience, and IL-10 from BioLegend.
Flow cytometry
Cells were stained by incubation with fluorescent-conjugated antibodies for 20 min at 4 °C in PBS/5% FBS. Intracellular Foxp3 staining was performed using the Alexa Fluor® 488 anti-mouse Foxp3 antibody (BioLegend) and theFoxp3 / Transcription Factor Staining Buffer Set (eBioscience), according to the manufacturer's protocol. Cells were acquired on a FACSCalibur (BD Biosciences) and data were analyzed using the FlowJo software (Tree Star).
RNA isolation and quantitative PCR
Total RNA was prepared from mouse BMDCs using TRIzol reagent according to the manufacturer's protocol (Invitrogen). For each sample 1 μg of isolated RNA was used for cDNA synthesis with SuperScript II Reverse Transcriptase (Invitrogen). Quantitative PCR (qPCR) was performed on a StepOnePlus Real-Time PCR system (Applied Biosystems) using KAPA SYBR FAST qPCR Master Mix (KapaBiosystems). The primer sequences were as follows: mouse Hprt, forward 5′-CGAAGTGTTGGATACAGGCC-3′ and reverse 5′- GGCAACATCAACAGGACTCC-3′; mouse Myd88, forward 5′- CCACCCTTGATGACCCCCTA-3′ and reverse 5′-TGCGGCGACACCTTTTCTC-3′; mouse Trif, forward 5′-TCTACCAGCTCAAGACCCCT-3′ and reverse 5′- GTCAGCCTGTCCCTTTCCAA-3′; mouse Atf3, forward 5′- GAGCTGAGATTCGCCATCCA-3′ and reverse 5′-CCGCCTCCTTTTCCTCTCAT-3′;Melting curve analysis was performed to ensure primer specificity. The expression of the target genes was normalized to the expression of the housekeeping gene, Hprt, and the normalized Ct values were calibrated against the control condition (untreated BMDCs) for each sample. The relative gene expression levels were determined using the relative standard curve method and the comparative Ct method (ΔΔCt method) as described in Applied Biosystems Guide.
Western blot analysis
BMDCs were lysed on ice for 30 min in RIPA lysis buffer (50 mM Tris-HCl pH 8.0,150 mM NaCl,1% Triton X-100,0.1% SDS and0.5% sodium deoxycholate) supplemented with cOmplete protease inhibitor cocktail (Roche), 5 mM NaF, 1 mM Na3VO4 and centrifuged at 13,000 × g for 30 min at 4 °C. The supernatant was collected as total cell lysate and protein concentration was determined by DC Protein Assay (Bio-Rad). 30 μg of total cell protein per sample were denatured by boiling for 10 min, loaded onto a 10.5% SDS polyacrylamide gel and transferred to nitrocellulose membrane (GE Healthcare Life Sciences). Blots were incubated with primary antibodies to anti-pStat3 (Tyr705) (Cell Signaling), anti-STAT3 (BD Biosciences). Anti-actin antibody (Merck Millipore) was used as loading control. Following incubation with HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) proteins were visualized on a ChemiDoc XRS+ imaging system (BioRad) and band intensities were quantified using the Image Lab Software (BioRad).
Immunofluorescence
BMDCs were seeded onto poly-L-lysine (Sigma-Aldrich) coated coverslips in 24-well tissue culture plates (1 × 106 cells/well). 24 h later cells were stimulated with 0.5 μg/mL LPS and treated with 4 μM rHDL for 2 h. Cells were fixed with 4% paraformaldehyde (Sigma-Aldrich), permeabilized with PBS-Triton X100 0.5%, and incubated with anti-p65 (sc-109, Santa Cruz Biotechnology). Alexa Fluor 555 Goat Anti-Rabbit IgG (Molecular Probes) was used as secondary antibody. DNA was stained with 1 μg/mL DAPI and coverslips were mounted on Mowiol 4-88 (Sigma-Aldrich). Images were acquired using a confocal laser scanning microscope (TCS SP8, Leica Microsystems) and a HC PL APO CS2 40×/1.30 oil objective using identical settings within each experiment. Images were obtained using the LAS AF v3.3 software (Leica Microsystems) and processed with Adobe Photoshop 7.0 (Adobe Systems). A total of 100 cells from four different fields from each slide were analyzed for fluorescence intensity of nucleus and cytoplasm using the NIH ImageJ software.
Statistical analysis
Data are expressed as mean ± SEM unless stated otherwise. Statistical significance was determined using two-tailed Student's t test. The χ2 test was used for categorical variables. For all results, p<0.05 was considered statistically significant. Analysis was performed using GraphPad Prism software (version 5.00, GraphPad).
Results
Increased severity of antigen-induced arthritis (AIA) in apoA-I-/- mice
HDL-c levels are significantly reduced in various autoimmune conditions such asRA (16), however whether there is a role for HDL in induction and perpetuation of the autoimmune response remain selusive. We investigated the development of AIA in apoA-I-/-mice that are characterized by very low serum HDL-c levels (∼25% of normal) (33). AIA is induced upon intra-articular injection of mBSAin mBSA-sensitized animals (Fig.1A) and serves as a model for RA (34, 35). Interestingly, apoA-I-/- mice developed increased severity of AIA compared to WT animals (Fig. 1B). This result correlated well with the histological examination of injected knee joints of apoA-I-/- mice which developed extensive pannus formation (grade 4) contrary to WT mice which exhibited moderate to severe inflammation (grade 2-3) (*p=0.0393 for pannusvs non-pannus; Fig. 1C and 1D). A striking observation was the extensive inflammatory reaction in the skin of apoA-I-/- mice three weeks after immunization as opposed to WT mice (Fig. 1E), that was characterized by significant epidermis alterations at the site of the injection including ulceration, dense inflammation in the dermis with abundant neutrophil infiltration and formation of large abscesses (Fig. 1F and 1G; data not shown). In contrast, WT mice showed moderate inflammatory reaction in the dermis, and occasional small neutrophil aggregates (Fig. 1F and 1G). Collectively, these findings indicate that decreased levels of circulating HDL are associated with exacerbation of inflammatory arthritis and severe inflammatory reactions in the skin.
Figure 1.

ApoA-I-/-mice demonstrate an aggravated AIA phenotype. (A) WT and apoA-I-/-mice (n=13/group) were immunized subcutaneously (s.c.) with mBSA/CFA and arthritis was induced upon intra-articular (i.a.) injection of mBSA 21 days later. (B) Evaluation of arthritis by measurement of knee joint swelling daily after i.a. injection (n=8-9/group) (day 4: *p=0.0233, day 5: *p=0.0455, day 6: *p=0.0182). Results are expressed as mean ± SEM. (C) Representative H&E stained sections of inflamed joints from WT (moderate arthritis) and apoA-I-/- mice (pannus) as well as non-injected knee joint (normal) are shown (original magnification ×200). (D) Histological scores for AIA in WT and apoA-I-/- mice. The histopathologies of injected knee joints were scored as described in Methods;*p=0.0393 for pannusvs non-pannus.(E) Photos of skin lesions in apoA-I-/- mice 21 days after mBSA immunization. (F) Representative H&E stained sections of control and inflamed skin (original magnification: left side ×20, right side ×400). (G) Histological grading of skin inflammation developed in WT and apoA-I-/- mice (*p=0.0239). In all panels, the data are from two independent experiments.
HDL's anti-inflammatory functions are compromised in arthritic WT mice
We examined whether HDL-c levels were affected by the inflammatory response and if HDL functionality was altered during AIA development. According to our results total and HDL cholesterol levels of both mBSA-immunized (day 21) and arthritic (day 30 i.a.) mice showed no significant differences as compared to naïve WT mice (Fig. 2A). To assess the functional properties of HDL, we measured HDL-associated PON-1 activity at different time points during disease development. Our results indicated that there is a biphasic response of the immunized mice in terms of HDL activity: an early up-regulation of HDL functionality (day 10) which could be accounted for by an early attempt to attenuate the inflammation followed by a gradual decline in HDL functionality as the inflammation resolves (day 21 and day 30; Fig. 2B). Importantly, our data showed that upon the intra-articular antigenic challenge HDL functionality significantly decreased to even lower levels (day 21 vs day 30 i.a.; Fig. 2B). Although mice are still capable of responding to the intra-articular mBSA challenge by increasing PON-1 activity (day 30vs day 30i.a.), this potential is markedly compromised compared to day 10 (Fig. 2B). Collectively, our results signify that although the HDL-c levels remained unaffected, the anti-inflammatory/anti-oxidant properties of circulating HDL were attenuated in an inflammatory environment.
Figure 2.
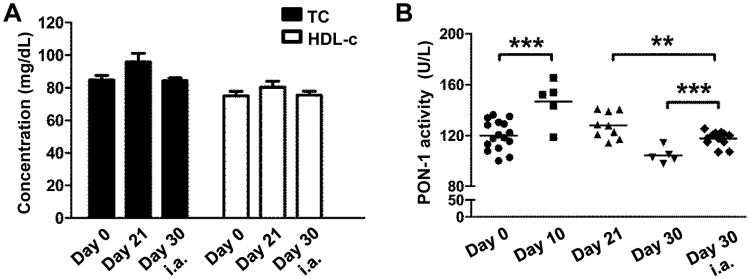
HDL functionality is altered during AIAin WTmice. (A) Total (TC) and HDL-cholesterol (HDL-c) levels in serum of naïve mice (day 0), mBSA-immunized (day 21) and arthritic mice (day 30 i.a.). (B) Characterization of HDL antioxidant/anti-inflammatory properties by assessment of HDL-associated PON-1 activity (**p=0.0084, ***p=0.0007). Results are expressed as mean ± SEM; data are combined of two independent experiments; n=5-16/group.
ApoA-I-/-mice exhibit exacerbated OVA-specific Th1 and Th17 immune responses
Development and progression of arthritis depends on the generation of autoreactive Th1 and Th17 cells (36, 37). To monitor the induction of T cell responses in apoA-I-/- mice we first examined whether T cell lymphopoiesis was affected. Analysis of CD4+ and CD8+ T cell subsets in thymus and peripheral lymphoid organs revealed no differences between naïve apoA-I-/- and WT mice indicating that loss of apoA-I did not cause alterations in T cell development and homeostasis (Fig. 3A). In addition, the frequency of peripheral CD4+Foxp3+ T regulatory cells (Tregs) was similar in apoA-I-/- and WT mice (Fig. 3B). We next assessed whether decreased HDL levels could affect the induction of antigen-specific Th1 and Th17 responses. Interestingly, dLNs of apoA-I-/- mice showed increased cellularity 9 days after the antigenic challenge compared to WT mice (53.54 ± 2.556 versus 42.13 ± 2.020 × 106 cells; Fig.4A). Detailed analysis of the dLNs for the different cells subsets revealed a significant increase of CD11c+ DCs in apoA-I-/-, 3.5 and 9 days after the antigenic challenge, compared to WT mice, and to a lesser extent of CD4+ T cells (Fig. 4B). Of note, no significant differences were observed in the frequencies of B220+ B cells and CD11b+ macrophages whereas CD8+ T cells were slightly decreased in dLNs of apoA-I-/- mice (Fig. 4B). Moreover, the absolute numbers of CD4+Foxp3+ Tregs were significantly increased in immunized apoA-I-/-mice (Fig. 4B). Although both groups of mice exhibited similar frequencies of activated CD3+CD4+CD44+ T cells after in vitro re-stimulation with OVA (Fig.4C), dLNCs from apoA-I-/- mice exhibited increased proliferation as indicated by IL-2 levels (181.3 ± 14.1 versus 74.7 ± 7.9; Fig.4D). This finding was accompanied by markedly increased production of both IL-17 (773.1 ± 105.9 versus 292.5 ± 85.8; Fig.4E) and IFN-γ (708.9 ± 114.8 versus 172.4 ± 40.0; Fig.4F) in apoA-I-/-OVA-stimulated dLNC supernatants whereas IL-10 was undetectable (data not shown). Taken together, these data demonstrate that apoA-I deficiency promotes T cell proliferation and enhances antigen-specific Th1 and Th17 immune responses.
Figure 3.

T cell development and homeostasis is not affected in apoA-I-/- mice.(A and B) Flow cytometric analysis of thymocytes, slpenocytes and lymph node cells from naïve WT and apoA-I-/- mice (n=3/group) are shown. Gates were set as indicated. Data are representative of two independent experiments.
Figure 4.

OVA-specific Th1 and Th17 immune responses are modulated by HDL. (A) LN cellularity in WT and apoA-I mice (n=12-13/group) 9 days after OVA immunization (**p=0.0018). Results are expressed as mean ± SEM; data are combined of five independent experiments.(B) dLNCs from WT and apoA-I-/- mice (n=4-6/group) were collected 3.5 and 9 days post OVA immunization (p.i.). Gating strategy and frequencies or absolute numbers of CD11c+ DCs, B220+ B cells, CD11b+ macrophages, CD4+ and CD8+ T cells and Foxp3+ Tregs are shown. Numbers on the gates denote frequencies. Results are expressed as mean ± SEM. (C) Flow cytometric analysis of OVA-primed dLNCs from WT and apoA-I-/-- mice (n=6/group) after in vitro re-stimulation with 15 μg/mL OVA for 48 h. Gates were set as indicated. Data are representative of three independent experiments. (D-F) Levels of IL-2 (***p<0.0001), IL-17 (**p=0.0055) and IFN-γ (***p=0.0008) in culture supernatants of OVA-primed dLNCs are shown (n=6-7/group). Results are expressed as mean ± SEM; data are combined of at least four independent experiments. (G) OVA-primed dLNCs from WT mice (n=3/group) were re-stimulated with OVA (15 μg/mL) in vitro in the presence of rHDL (1 μM) for 72 h and then pulsed with 1 μCi [3H]thymidine for 18 h. The incorporated radioactivity was measured. Results are expressed as mean ± SEM. (H-J), dLNCs from OVA-immunized WT mice (n=6-8/group) were cultured with increasing concentrations of rHDL in the presence of OVA (15 μg/mL). Levels of IL-2 (***p≤0.0004), IL-17 (**p=0.001, ***p=0.0006) and IFN-γ (**p≤0.0081) were determined in culture supernatants 48 h later. Results are expressed as mean ± SEM; data are combined of at least three independent experiments.
rHDL inhibits antigen-specific Th1 and Th17 immune responses in vitro
Next, we examined whether HDL could directly affect the induction of Th1 and Th17 immune responses. To address this, we performed in vitro re-stimulation assays of dLNCs from OVA-immunized WT animals in the presence of titrating amounts of rHDL. Treatment with rHDL led to impaired proliferation of OVA-specific T cells as a response to the antigenic stimulus (Fig. 4G). In support, IL-2 secretion was decreased in culture supernatants of rHDL-treated dLNCs (Fig. 4H). Furthermore, decreased proliferation of dLNCs in the presence of rHDL was accompanied by a dose-dependent decrease in secretion of IL-17 (Fig.4I) and IFN-γ (Fig. 4J). Overall, these results provide direct evidence for a potent role of rHDL in the suppression of Th1 and Th17 immune responses in vitro.
rHDL downregulates co-stimulatory molecule expression and suppresses pro-inflammatory cytokine production in stimulated BMDCs
T cell activation, polarization and proliferation in vivo require antigen presentation by professional APCs. DCs serve as the best candidate that upon maturation provide the necessary co-stimulatory molecules and secrete pro-inflammatory cytokines to instruct T cell responses (20, 38). We asked whether the suppressive effect of rHDL on T cell responses is mediated through modulation of DC function. To this end, we first examined the maturation status of DCs in dLNs and spleen of immunized apoA-I-/- and WT mice. Our findings demonstratedan increase of MHC class II and CD40 expression in CD11c+ DCs from apoA-I-/-compared to WT mice and a more prominent increase in the expression of the co-stimulatory molecule CD86 in both dLNs and spleen (Fig. 5). On the other hand, no differences were observed in the expression levels of CD80 (Fig. 5). To examine a direct role of HDL on DC maturation, we generated BMDCs from WT mice and treated them with rHDL during LPS stimulation. As shown in Fig.6A, treatment with rHDL led to downregulation of MHC class II (MHC-II) and CD40, CD86 and CD80. To determine whether this effect was specific to the TLR4-mediated response in DCs, we stimulated BMDCs from WT mice with zymosan, a TLR2 ligand, and treated them with rHDL. Although expression of MHC-II was not altered, a significant down-regulation of CD40, CD86 and CD80 expression was observed in rHDL-treated zymosan-activated BMDCs compared to zymosan alone (Fig. 6B). These results indicate that rHDL's suppressive effect on DC maturation is not limited to TLR4-induced immune responses. Furthermore, secretion of pro-inflammatory cytokines IL-6, IL-12, IL-23, chemokine IL-8 and the anti-inflammatory cytokine IL-10 were reduced in rHDL-treated LPS-stimulated BMDCs, whereas TNF-α production was unaffected by rHDL treatment (Fig.6C). Of note, rHDL suppressed the secretion of IL-6, IL-8 and TNF-α in immature DCs indicating that the anti-inflammatory properties of rHDL could be exerted independent of LPS. Collectively, these data demonstrate an inhibitory effect of rHDL on activation and maturation of and cytokine secretion by DCs in vitro.
Figure 5.

The expression of DC maturation markers is increased in immunized apoA-I-/-mice. dLNCs and slpenocytes from WT and apoA-I-/- mice (n=4-8/group) were collected 3.5 and 9 days post OVA immunization. Expression levels of MHC-II, CD40, CD86 and CD80 were evaluated by flow cytometry. Representative FACS histograms are shown. Gates were set on CD11c+ cells.
Figure 6.

rHDL prevents activation and maturation of BMDCs. BMDCs from WTmice (n=2-6) were stimulated with LPS (0.5 μg/mL) or zymosan (20 μg/mL) and treated with 4 μM rHDL for 18 h. (A) Expression levels of MHC-II (*p=0.026), CD40 (**p=0.0076), CD86 (*p=0.0226) and CD80 (**p=0.0047) were evaluated by flow cytometry. Representative FACS histograms are shown. Gates were set on CD11c+ cells. Dot plots represent the averages of the geometric mean fluorescence intensity (MFI) of at least two independent experiments.. (B) Expression levels of MHC-II, CD40, CD86 and CD80 were evaluated by flow cytometry. Representative F ACS histograms are shown. Gates were set on CD1 1c+ cells. (C) Levels of IL-6, IL-23, IL-12 p70, IL-8, IL-10 and TNF-α in culture supernatants were measured by ELISA. Results are expressed as mean ± SEM; data are combined of three independent experiments; *p=0.0229, **p≤0.0038, ***p≤0.0008; ns; not significantly different. ND; not detectable.
rHDL treatment of mature BMDCs suppresses antigen-specific T cell proliferation
To assess the functional importance of our findings, we performed co-culture experiments with rHDL-treated OVA-pulsed BMDCs and OVA-primed dLNCs as shown in Fig. 7A. To this end, WT BMDCs were activated with LPS and pulsed with OVA in the presence or absence of rHDL for 12 h. BMDCs were then co-cultured with dLNCs isolated from OVA-immunized WT syngeneic mice. 48 h later culture supernatants were collected and IL-2 production was assessed. Interestingly, rHDL-treated OVA-pulsed BMDCs significantly reduced IL-2 production by OVA-primed LNCs as compared to OVA-pulsed BMDCs (Fig.7B). These findings provide evidence for the impaired capacity of rHDL-treated DCs to promote T cell proliferation.
Figure 7.
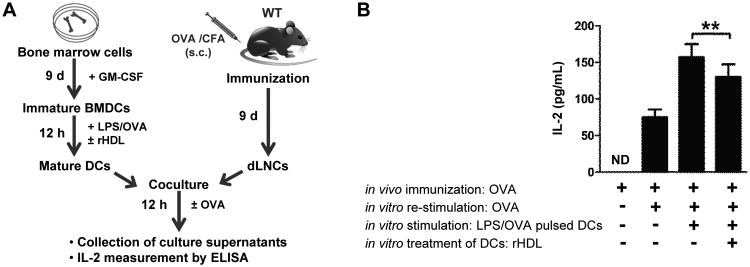
rHDL-treated DCs suppress antigen-specific T cell proliferation in vitro.(A) Outline of the co-culture experimental setup. LPS-stimulated OVA-pulsed BMDCs from WT mice (n=5/experiment) were treated with rHDL for 12 h and co-cultured with OVA-primed dLNCs. Cells were re-stimulated with OVA and 48 h later (B) IL-2 secretion in culture supernatants was assessed by ELISA. Results are expressed as mean ± SEM; data are combined of two independent experiments (**p=0.005).
rHDL prevents activation of the MyD88-dependent pathway in stimulated BMDCs
LPS binding to TLR4leads to NF-κB nuclear translocation and induction of inflammatory cytokine gene transcription (39). To gain insights into the molecular events leading to HDL-mediated suppression of DC function, we examined whether rHDL suppressed LPS-induced pro-inflammatory cytokine secretion by interfering with the NF-κB pathway. To this end, we performed immunofluorescence studies to assess nuclear translocation of the p65 subunit of NF-κB in LPS-stimulated BMDCs. Our findings showed a marked increase in nuclear p65 compared to unstimulated cells, whereas LPS-induced translocation of p65 to the nucleus was impaired after rHDL treatment (Fig.8A and 8B). In TLR4 signaling, activation of NF-κB is initiated by interactions with either MyD88 or TIR-domain-containing adapter-inducing interferon-β (TRIF) adaptor protein (39). In order to determine whether these two pathways were disrupted by rHDL, we assessed mRNA levels in LPS-stimulated BMDCs. We found that, rHDL decreased Myd88 mRNA levels in LPS-stimulated BMDCs after 18h of treatment (Fig. 8C). In contrast, relative to LPS-stimulated BMDCs' levels, rHDL treatment did not affect Trif expression neither at an early nor at a later time point (Fig. 8D). These data suggest a role for rHDL in modulating inflammatory responses through interference with the MyD88-dependent TLR4 signaling pathway.
Figure 8.
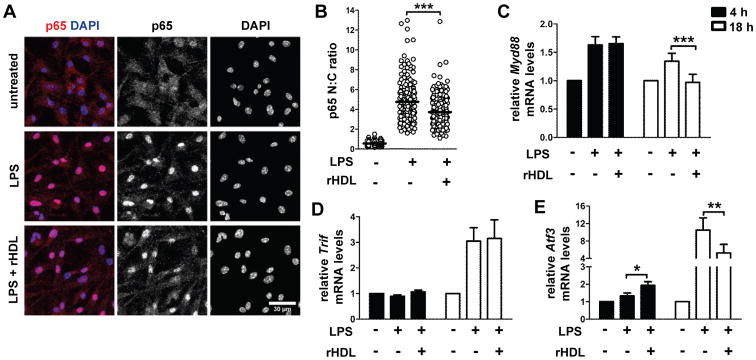
rHDL inhibits activation of BMDCs by interfering with the TLR4 pathway. WTBMDCs were stimulated with LPS (0.5 μg/mL) and treated with 4 μM rHDL for the indicated time periods. (A) Confocal immunofluorescence microscopy for p65 (red) translocation into the nucleus (DAPI stained, blue) in BMDCs stimulated with LPS for 2 h. Maximum projections of image stacks are shown; representative of two independent experiments. (B) Quantification of p65 cell distribution by determining nuclear: cytoplasmic (N: C) ratios of signal intensity at single cell level. Data are combined of two independent experiments; ***p<0.0001. (C-E) Relative mRNA levels of Myd88 (***p=0.0004), Trif and Atf3 (*p=0.0177, **p=0.0016) after treatment of WT BMDCs (n=6-8) with rHDL for 4h and 18h. Results are expressed as mean ± SEM; data are combined of at least three independent experiments.
Recently, activating transcription factor 3 (ATF3), a transcriptional regulator that provide negative feedback on TLR-induced inflammation, was identified as a critical mediator of HDL's anti-inflammatory effects in macrophages (40). Thus, we assessed Atf3 mRNA levels in rHDL-treated BMDC's. Indeed, after 4 h of treatment with rHDL, LPS-stimulated BMDCs demonstrated increased expression of Atf3 compared to non-treated cells (Fig.8E), whereas expression was decreased at 18h (Fig.8E). This finding suggests that an HDL-inducible ATF3-dependent mechanism may operate in DCs, acting as a restrainer of the TLR-stimulated response by preventing excessive pro-inflammatory cytokine production.
The anti-inflammatory effects of rHDL on stimulated BMDCs are mediated through ABCA1 and SR-BI
It is well established that HDL exerts its pleiotropic functions through interactions with SR-BI, ABCA1 and ABCG1 transporters (7, 9). To investigate the contribution of each transporter in rHDL's anti-inflammatory effects on DCs, we assessed cytokine secretion in LPS-stimulated ABCG1-/-, ABCA1-/- and SR-BI-/- BMDCs in the presence or absence of rHDL. Deficiency in any of these transporters in BMDCs did not affect their ability to efficiently secrete pro-inflammatory cytokines upon LPS stimulation (data not shown). Interestingly, rHDL suppressed the levels of IL-12, IL-8 or IL-23 in ABCG1-/- BMDCs as efficiently as in BMDCs from WT mice and to an even greater extent the secretion of IL-6, indicating that this transporter is not involved in rHDL's inhibitory effect on DC activation (Fig. 9A). In contrast, rHDL was unable to efficiently impair pro-inflammatory cytokine secretion in ABCA1-/- and SR-BI-/- BMDCs suggesting that rHDL-mediated modulation of DC function is ABCA1- and SR-BI-dependent (Fig. 9A). Notably, the decrease in IL-10 levels that was observed upon rHDL treatment of LPS-stimulated WT BMDCs (34.5 ± 1.6%) was retained independent of ABCG1, ABCA1 or SR-BI deficiency (ABCG1-/-: 30.5 ± 1.2%; ABCA1-/-: 38.0 ± 1.4%; SR-BI-/-: 35.9 ± 2.6%; Fig. 9A). This result suggests either that the ABCG1 transporter does not contribute to rHDL's inhibitory actions or that in the case of IL-10 there is a redundancy in the functions of the three transporters.
Figure 9.

The suppressive effect of rHDL on BMDCs activation is dependent on ABCA1 and SR-BI. BMDCs from WT, ABCG1-/-, ABCA1-/- and SR-BI-/- mice (n=4-6/group) were stimulated with LPS (0.5 μg/mL) and treated with 4 μM rHDL for 12 h. (A) Levels of IL-6, IL-12 p70, TNF-α, IL-23, IL-8 and IL-10 in culture supernatants of stimulated BMDCs. Percentages of cytokine secretion are shown for LPS-stimulated BMDCs after treatment with rHDL, normalized against untreated LPS-stimulated BMDCs of the same genotype. (B) Representative western blot images of pSTAT3, total STAT3 and actin are shown for each genotype. (C) Levels of pSTAT3 and actin were quantified by densitometry and the pSTAT3/actin ratio was calculated. In panels A and C results are expressed as mean ± SEM; data are combined of at least two independent experiments. *p<0.05, **p<0.01, ***p<0.001; ns; not significantly different.
Further support of these findings was obtained upon assessment of rHDL's effects on signaling events downstream of the NF-κB pathway in BMDCs from WT mice or mice deficient for any of the above transporters. Upon LPS stimulation STAT3 signaling, as indicated by increased STAT3 phosphorylation, was efficiently induced in BMDCs from all groups as compared with non stimulated cells (WT: 6.0 ± 0.8 fold; ABCG1-/-: 4.3 ± 0.5 fold; ABCA1-/-: 4.2 ± 0.5 fold; SR-BI-/-: 3.9 ± 0.5 fold; Fig. 9B and 9C) and treatment with rHDL was able to diminish STAT3 activation in WT (4.5 ± 0.7fold; *p=0.0283) and ABCG1-/-BMDCs (2.5 ± 0.2 fold; *p=0.0127). However, rHDL-treated ABCA1-/- and SR-BI-/- BMDCs demonstrated similar levels of LPS-induced STAT3 phosphorylation (ABCA1-/-: 3.9 ± 0.7 fold;SR-BI-/-: 4.2 ± 0.7 fold; Fig. 9B and 9C) as compared with cells treated with LPS alone. Overall, our data provide evidence for an important role of ABCA1 and SR-BI but not ABCG1 in the HDL-mediated anti-inflammatory function on DCs.
Discussion
Although numerous studies have reported the anti-inflammatory properties of HDL, the mechanisms involved in HDL-mediated suppression of autoimmune inflammatory responses are not well understood. Our results demonstrate that HDL suppresses the activation, maturation of and cytokine secretion by DCs resulting in the establishment of a “semi-mature phenotype” of these cells. To this end, treatment of BMDCs with rHDL resulted in reduced expression of MHC-II and co-stimulatory molecules that are required for efficient antigen-presentation and activation of T lymphocytes. This is in line with previous reports revealing a role of HDL and apoA-I on human DC differentiation and function (41, 42). Antigen loading to MHC-II is mediated either through the classical or via the autophagy pathway (43, 44). Recent data suggest that HDL inhibits autophagy induced by oxidized low-density lipoproteins (oxLDL) in endothelial cells (45). Whether HDL could suppress the induction of autophagy in DCs and delivery of antigens to MHC loading compartments remains to be shown.
The functional importance of the anti-inflammatory effects of HDL on DCs is demonstrated by the decreased ability of rHDL-treated BMDCs to promote T cell proliferation in vitro. DCs with a “semi-mature phenotype” have the ability to down-regulate immune responses and to ameliorate autoimmunity upon adoptive transfer in vivo (46, 47). In particular, it was shown that TNF-α-treated DCs are tolerogenic and suppress experimental autoimmune thyroiditis through induction of CD4+CD25+Tregs in vivo and in vitro (47). Since Tregs, play a prominent role in the re-establishment but also maintenance of self-tolerance it remains to be examined whether rHDL-treated BMDCs possess the ability to induce and/or expand Tregs in vivo. Interestingly, absolute numbers of CD4+Foxp3+ Tregs in the dLNs of immunized apoA-I-/-were elevated compared to WT mice. However, the suppressive potential of Foxp3+ Tregs in an inflammatory environment is currently under debate (48, 49).
According to our data, absence of apoA-I in mice resulted in exacerbated inflammation in the knee joints during AIA and caused extensive inflammatory reactions in the skin. These findings are in line with previous studies demonstrating that lack of apoA-Iin low-density lipoprotein receptor (LDLR) deficient mice fed with high-fat diet was associated with skin lesion development due to enlarged skin dLNs that contained expanded populations of cholesterol-enriched lymphocytes including T cells, B cells and DCs (50). Moreover, when fed an atherogenic diet these double knockout mice exhibited increased T cell activation, proliferation and production of autoantibodies in the plasma. Importantly, this autoimmune phenotype was restored after treatment of mice with either lipid-free apoA-I oradenovirus-mediated gene transfer of apoA-I (50, 51).
The capacity of HDL to bind LPS and neutralize its inflammatory activity has been demonstrated in multiple in vitro and in vivo studies (52, 53). Although this possibility cannot be excluded, the data presented in this study using BMDCs from ABCA1, ABCG1 or SR-BI deficient mice do not support such a mechanism of action for rHDL. In our experiments, rHDL did not efficiently suppress LPS-induced secretion of IL-6, IL-12 or IL-23 in mice lacking ABCA1 or SR-BI, indicating that a specific interaction between rHDL and these transporters, rather than sequestration of LPS by rHDL, is responsible for its anti-inflammatory functions on BMDCs.
LPS stimulation of DCs activates NF-κB, via either the MyD88- or the TRIF-dependent pathway, which is then recruited to the nucleus to induce inflammatory cytokine gene transcription (39). Our data demonstrate that rHDL's anti-inflammatory effects on LPS-stimulated BMDCs are mediated through interference with the MyD88/NF-κB pathway. Although the inhibitory actions of apoA-I and HDL on TLR signaling have been previously reported (19, 54), a mechanism by which HDL selectively impedes the MyD88-dependent signaling in DCs has not yet been established. Based on our findings we speculate that interaction of apoA-I on HDL with cell surface lipid transporters such as ABCA1 or SR-BI, leads to changes in downstream signaling events that in turn compromise NF-κB activation and transcriptional upregulation of Myd88 by LPS. This could be mediated via two alternative, but not necessarily mutually exclusive pathways. One mechanism could involve depletion of lipid rafts from plasma membranes as a result of cholesterol efflux initiated by apoA-I/ABCA1 or apoA-I/SR-BI interactions. Lipid rafts are dynamic structures that have been critically implicated in a variety of cellular processes including signal transduction, endocytosis, vesicular transport, and immunoregulation (55). Treatment of APCs with either HDL or apoA-I has been associated with cholesterol depletion and lipid raft disruption leading to suppressed T cell activation and down-regulation of pro-inflammatory cytokine secretion (56, 57). The second mechanism could involve activation of ABCA1- or SR-BI-induced signaling that results in inhibition of the TLR-induced response. According to previous studies, interaction of HDL or apoA-I with SR-BI present on endothelial cells activates downstream signaling cascades stimulating a variety of protective cellular responses (58). In addition, apoA-I binding to ABCA1 activates the JAK2/STAT3 pathway in macrophages leading to suppression of LPS-induced inflammatory cytokine production through the action of the mRNA-destabilizing protein, tristetraprolin (59). To date no signaling events have been reported as a result of interactions between apoA-I or HDL and ABCG1, a transporter known to greatly contribute to the cholesterol efflux process in macrophages. Notably, in our study the inhibitory properties of rHDL on BMDCs were found to be independent of ABCG1. These data argue against the hypothesis that the anti-inflammatory effects of rHDL on BMDCs are due to cholesterol depletion leading to lipid raft remodeling and favor a model which implicates transporter-specific downstream signaling events.
In a recent studyATF3, a transcriptional repressor of TLR-stimulated inflammation, was identified as an HDL-inducible target gene that mediated HDL's anti-inflammatory actions in macrophages (40). It was shown that the protective effects of HDL on TLR responses were fully dependent on ATF3 in vitro and in vivo. We show here that treatment of LPS-stimulated BMDCs with rHDL caused an upregulation in Atf3 expression. These data support the notion that an ATF3-dependent mechanism may also be activated by HDLin DCs to constrain the inflammatory response.
In summary, our findings provide evidence for a novel role of HDL in shaping the autoimmune responses both in vivo and in vitro. HDL suppresses adaptive T cell responses by modulating the pro-inflammatory function of DCs, in an ABCA1- and SR-BI-dependent fashion. The results presented here shed light on the mechanism underlying the anti-inflammatory properties of HDL that could aid the development of new therapeutic approaches for autoimmune and other inflammatory diseases.
Acknowledgments
We thank members of the Kardassis and Verginis labs for discussions and technical assistance; Prof. Joanna Moschandreas for assistance with biostatistical analyses (University of Crete Medical School); Dr Joanna Charlton for FACS antibodies (Institute of Molecular Biology and Biotechnology); Dr Panagiotis Fotakis (University of Crete Medical School), Christina Gkolfinopoulou and Dr Angeliki Chroni (both at Demokritos National Center for Scientific Research) for production and purification of human apolipoprotein A-I and for protocols; and Ronald van der Sluis and Mara Kröner (Leiden Academic Center for Drug Research) for preparing bone samples from ABCA1-/-, ABCG1-/- and SR-BI-/- mice.
This work was supported by grants from the Greek General Secretariat of Research and Technology (SYNERGASIA 09SYN-12-897 and ARISTEIA II grant #4220) to D.K. and by the European Union project Innovative Medicine Initiative 6 (‘BeTheCure’, contract number 115142-2) to P.V. and D.B. VIZ is supported by an NIH grant (HL48739). M. van Eck is an Established Investigator of the Netherlands Heart Foundation (grant 2007T056) and supported by The Netherlands Organization for Scientific Research (VICI Grant 91813603).
Abbreviations used in this article
- ABCA1
ATP Binding Cassette Transporter A1
- ABCG1
ATP Binding Cassette Transporter G1
- AIA
antigen-induced arthritis
- apoA-I
apolipoprotein A-I
- ATF3
activating transcription factor 3
- BMDC
bone marrow-derived dendritic cell
- DC
dendritic cell
- dLN
draining lymph node
- dLNC
draining lymph node cell
- Foxp3
forkhead box P3
- HDL
high-density lipoprotein
- LPS
lipopolysaccharide
- mBSA
methylated BSA
- PON-1
paraoxonase 1
- RA
rheumatoid arthritis
- rHDL
reconstituted HDL
- SR-BI
scavenger receptor class B type I
- Treg
regulatory T cell
- TRIF
TIR-domain-containing adapter-inducing interferon-β
- WT
wild-type
References
- 1.Shah AS, Tan L, Long JL, Davidson WS. Proteomic diversity of high density lipoproteins: our emerging understanding of its importance in lipid transport and beyond. Journal of lipid research. 2013;54:2575–2585. doi: 10.1194/jlr.R035725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gordon DJ, Rifkind BM. High-density lipoprotein--the clinical implications of recent studies. The New England journal of medicine. 1989;321:1311–1316. doi: 10.1056/NEJM198911093211907. [DOI] [PubMed] [Google Scholar]
- 3.Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. The American journal of medicine. 1977;62:707–714. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 4.Riwanto M, Landmesser U. High density lipoproteins and endothelial functions: mechanistic insights and alterations in cardiovascular disease. Journal of lipid research. 2013;54:3227–3243. doi: 10.1194/jlr.R037762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sorci-Thomas MG, Thomas MJ. High density lipoprotein biogenesis, cholesterol efflux, and immune cell function. Arteriosclerosis, thrombosis, and vascular biology. 32:2561–2565. doi: 10.1161/ATVBAHA.112.300135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zannis VI, Chroni A, Krieger M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. Journal of molecular medicine. 2006;84:276–294. doi: 10.1007/s00109-005-0030-4. [DOI] [PubMed] [Google Scholar]
- 7.Yvan-Charvet L, Wang N, Tall AR. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:139–143. doi: 10.1161/ATVBAHA.108.179283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoekstra M, Van Eck M, Korporaal SJ. Genetic studies in mice and humans reveal new physiological roles for the high-density lipoprotein receptor scavenger receptor class B type I. Current opinion in lipidology. 2012;23:127–132. doi: 10.1097/MOL.0b013e3283508c09. [DOI] [PubMed] [Google Scholar]
- 9.Mineo C, Shaul PW. Regulation of signal transduction by HDL. Journal of lipid research. 2013;54:2315–2324. doi: 10.1194/jlr.R039479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luscher TF, Landmesser U, von Eckardstein A, Fogelman AM. High-density lipoprotein: vascular protective effects, dysfunction, and potential as therapeutic target. Circulation research. 114:171–182. doi: 10.1161/CIRCRESAHA.114.300935. [DOI] [PubMed] [Google Scholar]
- 11.Hahn BH, Grossman J, Ansell BJ, Skaggs BJ, McMahon M. Altered lipoprotein metabolism in chronic inflammatory states: proinflammatory high-density lipoprotein and accelerated atherosclerosis in systemic lupus erythematosus and rheumatoid arthritis. Arthritis research & therapy. 2008;10:213. doi: 10.1186/ar2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Catapano AL, Pirillo A, Bonacina F, Norata GD. HDL in innate and adaptive immunity. Cardiovascular research. 2014;103:372–383. doi: 10.1093/cvr/cvu150. [DOI] [PubMed] [Google Scholar]
- 13.Feldmann M, Brennan FM, Maini RN. Rheumatoid arthritis. Cell. 1996;85:307–310. doi: 10.1016/s0092-8674(00)81109-5. [DOI] [PubMed] [Google Scholar]
- 14.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 15.McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- 16.Robertson J, Peters MJ, McInnes IB, Sattar N. Changes in lipid levels with inflammation and therapy in RA: a maturing paradigm. Nature reviews Rheumatology. 2013;9:513–523. doi: 10.1038/nrrheum.2013.91. [DOI] [PubMed] [Google Scholar]
- 17.Charles-Schoeman C, Watanabe J, Lee YY, Furst DE, Amjadi S, Elashoff D, Park G, McMahon M, Paulus HE, Fogelman AM, Reddy ST. Abnormal function of high-density lipoprotein is associated with poor disease control and an altered protein cargo in rheumatoid arthritis. Arthritis and rheumatism. 2009;60:2870–2879. doi: 10.1002/art.24802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Charles-Schoeman C, Banquerigo ML, Hama S, Navab M, Park GS, Van Lenten BJ, Wagner AC, Fogelman AM, Brahn E. Treatment with an apolipoprotein A-1 mimetic peptide in combination with pravastatin inhibits collagen-induced arthritis. Clinical immunology. 2008;127:234–244. doi: 10.1016/j.clim.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 19.Wu BJ, Ong KL, Shrestha S, Chen K, Tabet F, Barter PJ, Rye KA. Inhibition of arthritis in the Lewis rat by apolipoprotein A-I and reconstituted high-density lipoproteins. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:543–551. doi: 10.1161/ATVBAHA.113.302832. [DOI] [PubMed] [Google Scholar]
- 20.Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. 2002;20:621–667. doi: 10.1146/annurev.immunol.20.100301.064828. [DOI] [PubMed] [Google Scholar]
- 21.Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, Murphy TL, Murphy KM. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nature immunology. 2002;3:549–557. doi: 10.1038/ni794. [DOI] [PubMed] [Google Scholar]
- 22.Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 23.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 24.McNeish J, Aiello RJ, Guyot D, Turi T, Gabel C, Aldinger C, Hoppe KL, Roach ML, Royer LJ, de Wet J, Broccardo C, Chimini G, Francone OL. High density lipoprotein deficiency and foam cell accumulation in mice with targeted disruption of ATP-binding cassette transporter-1. Proc Natl Acad Sci U S A. 2000;97:4245–4250. doi: 10.1073/pnas.97.8.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rigotti A, Trigatti BL, Penman M, Rayburn H, Herz J, Krieger M. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc Natl Acad Sci U S A. 1997;94:12610–12615. doi: 10.1073/pnas.94.23.12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chroni A, Kan HY, Kypreos KE, Gorshkova IN, Shkodrani A, Zannis VI. Substitutions of glutamate 110 and 111 in the middle helix 4 of human apolipoprotein A-I (apoA-I) by alanine affect the structure and in vitro functions of apoA-I and induce severe hypertriglyceridemia in apoA-I-deficient mice. Biochemistry. 2004;43:10442–10457. doi: 10.1021/bi049782p. [DOI] [PubMed] [Google Scholar]
- 27.Matz CE, Jonas A. Micellar complexes of human apolipoprotein A-I with phosphatidylcholines and cholesterol prepared from cholate-lipid dispersions. The Journal of biological chemistry. 1982;257:4535–4540. [PubMed] [Google Scholar]
- 28.Laccotripe M, Makrides SC, Jonas A, Zannis VI. The carboxyl-terminal hydrophobic residues of apolipoprotein A-I affect its rate of phospholipid binding and its association with high density lipoprotein. The Journal of biological chemistry. 1997;272:17511–17522. doi: 10.1074/jbc.272.28.17511. [DOI] [PubMed] [Google Scholar]
- 29.Asztalos BF, Sloop CH, Wong L, Roheim PS. Two-dimensional electrophoresis of plasma lipoproteins: recognition of new apo A-I-containing subpopulations. Biochimica et biophysica acta. 1993;1169:291–300. doi: 10.1016/0005-2760(93)90253-6. [DOI] [PubMed] [Google Scholar]
- 30.Freeman LA. Native-native 2D gel electrophoresis for HDL subpopulation analysis. Methods Mol Biol. 2013;1027:353–367. doi: 10.1007/978-1-60327-369-5_17. [DOI] [PubMed] [Google Scholar]
- 31.Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 32.Zal T, Volkmann A, Stockinger B. Mechanisms of tolerance induction in major histocompatibility complex class II-restricted T cells specific for a blood-borne self-antigen. J Exp Med. 1994;180:2089–2099. doi: 10.1084/jem.180.6.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williamson R, Lee D, Hagaman J, Maeda N. Marked reduction of high density lipoprotein cholesterol in mice genetically modified to lack apolipoprotein A-I. Proc Natl Acad Sci U S A. 1992;89:7134–7138. doi: 10.1073/pnas.89.15.7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iliopoulos D, Kavousanaki M, Ioannou M, Boumpas D, Verginis P. The negative costimulatory molecule PD-1 modulates the balance between immunity and tolerance via miR-21. Eur J Immunol. 41:1754–1763. doi: 10.1002/eji.201040646. [DOI] [PubMed] [Google Scholar]
- 35.van den Berg WB, Joosten LA, van Lent PL. Murine antigen-induced arthritis. Methods Mol Med. 2007;136:243–253. doi: 10.1007/978-1-59745-402-5_18. [DOI] [PubMed] [Google Scholar]
- 36.Lubberts E, Koenders MI, van den Berg WB. The role of T-cell interleukin-17 in conducting destructive arthritis: lessons from animal models. Arthritis research & therapy. 2005;7:29–37. doi: 10.1186/ar1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schulze-Koops H, Kalden JR. The balance of Th1/Th2 cytokines in rheumatoid arthritis. Best Pract Res Clin Rheumatol. 2001;15:677–691. doi: 10.1053/berh.2001.0187. [DOI] [PubMed] [Google Scholar]
- 38.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 39.Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 40.De Nardo D, Labzin LI, Kono H, Seki R, Schmidt SV, Beyer M, Xu D, Zimmer S, Lahrmann C, Schildberg FA, Vogelhuber J, Kraut M, Ulas T, Kerksiek A, Krebs W, Bode N, Grebe A, Fitzgerald ML, Hernandez NJ, Williams BR, Knolle P, Kneilling M, Rocken M, Lutjohann D, Wright SD, Schultze JL, Latz E. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nature immunology. 2014;15:152–160. doi: 10.1038/ni.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim KD, Lim HY, Lee HG, Yoon DY, Choe YK, Choi I, Paik SG, Kim YS, Yang Y, Lim JS. Apolipoprotein A-I induces IL-10 and PGE2 production in human monocytes and inhibits dendritic cell differentiation and maturation. Biochemical and biophysical research communications. 2005;338:1126–1136. doi: 10.1016/j.bbrc.2005.10.065. [DOI] [PubMed] [Google Scholar]
- 42.Perrin-Cocon L, Diaz O, Carreras M, Dollet S, Guironnet-Paquet A, Andre P, Lotteau V. High-density lipoprotein phospholipids interfere with dendritic cell Th1 functional maturation. Immunobiology. 2012;217:91–99. doi: 10.1016/j.imbio.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 43.Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A, Mizushima N, Grinstein S, Iwasaki A. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity. 32:227–239. doi: 10.1016/j.immuni.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol. 2005;23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538. [DOI] [PubMed] [Google Scholar]
- 45.Muller C, Salvayre R, Negre-Salvayre A, Vindis C. HDLs inhibit endoplasmic reticulum stress and autophagic response induced by oxidized LDLs. Cell Death Differ. 18:817–828. doi: 10.1038/cdd.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Menges M, Rossner S, Voigtlander C, Schindler H, Kukutsch NA, Bogdan C, Erb K, Schuler G, Lutz MB. Repetitive injections of dendritic cells matured with tumor necrosis factor alpha induce antigen-specific protection of mice from autoimmunity. The Journal of experimental medicine. 2002;195:15–21. doi: 10.1084/jem.20011341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Verginis P, Li HS, Carayanniotis G. Tolerogenic semimature dendritic cells suppress experimental autoimmune thyroiditis by activation of thyroglobulin-specific CD4+CD25+ T cells. J Immunol. 2005;174:7433–7439. doi: 10.4049/jimmunol.174.11.7433. [DOI] [PubMed] [Google Scholar]
- 48.Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, Mauri C. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. The Journal of experimental medicine. 2004;200:277–285. doi: 10.1084/jem.20040165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, Backstrom BT, Sobel RA, Wucherpfennig KW, Strom TB, Oukka M, Kuchroo VK. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilhelm AJ, Zabalawi M, Grayson JM, Weant AE, Major AS, Owen J, Bharadwaj M, Walzem R, Chan L, Oka K, Thomas MJ, Sorci-Thomas MG. Apolipoprotein A-I and its role in lymphocyte cholesterol homeostasis and autoimmunity. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:843–849. doi: 10.1161/ATVBAHA.108.183442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilhelm AJ, Zabalawi M, Owen JS, Shah D, Grayson JM, Major AS, Bhat S, Gibbs DP, Jr, Thomas MJ, Sorci-Thomas MG. Apolipoprotein A-I modulates regulatory T cells in autoimmune LDLr-/-, ApoA-I-/- mice. The Journal of biological chemistry. 2010;285:36158–36169. doi: 10.1074/jbc.M110.134130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Levine DM, Parker TS, Donnelly TM, Walsh A, Rubin AL. In vivo protection against endotoxin by plasma high density lipoprotein. Proc Natl Acad Sci U S A. 1993;90:12040–12044. doi: 10.1073/pnas.90.24.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y, Zhu X, Wu G, Shen L, Chen B. Effect of lipid-bound apoA-I cysteine mutants on lipopolysaccharide-induced endotoxemia in mice. Journal of lipid research. 2008;49:1640–1645. doi: 10.1194/jlr.M700446-JLR200. [DOI] [PubMed] [Google Scholar]
- 54.Suzuki M, Pritchard DK, Becker L, Hoofnagle AN, Tanimura N, Bammler TK, Beyer RP, Bumgarner R, Vaisar T, de Beer MC, de Beer FC, Miyake K, Oram JF, Heinecke JW. High-density lipoprotein suppresses the type I interferon response, a family of potent antiviral immunoregulators, in macrophages challenged with lipopolysaccharide. Circulation. 122:1919–1927. doi: 10.1161/CIRCULATIONAHA.110.961193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Horejsi V, Hrdinka M. Membrane microdomains in immunoreceptor signaling. FEBS Lett. 588:2392–2397. doi: 10.1016/j.febslet.2014.05.047. [DOI] [PubMed] [Google Scholar]
- 56.Smythies LE, White CR, Maheshwari A, Palgunachari MN, Anantharamaiah GM, Chaddha M, Kurundkar AR, Datta G. Apolipoprotein A-I mimetic 4F alters the function of human monocyte-derived macrophages. Am J Physiol Cell Physiol. 298:C1538–1548. doi: 10.1152/ajpcell.00467.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang SH, Yuan SG, Peng DQ, Zhao SP. HDL and ApoA-I inhibit antigen presentation-mediated T cell activation by disrupting lipid rafts in antigen presenting cells. Atherosclerosis. 2012;225:105–114. doi: 10.1016/j.atherosclerosis.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 58.Al-Jarallah A, Trigatti BL. A role for the scavenger receptor, class B type I in high density lipoprotein dependent activation of cellular signaling pathways. Biochimica et biophysica acta. 1801:1239–1248. doi: 10.1016/j.bbalip.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 59.Tang C, Liu Y, Kessler PS, Vaughan AM, Oram JF. The macrophage cholesterol exporter ABCA1 functions as an anti-inflammatory receptor. The Journal of biological chemistry. 2009;284:32336–32343. doi: 10.1074/jbc.M109.047472. [DOI] [PMC free article] [PubMed] [Google Scholar]