

Abstract
Because p38α plays a critical role in inflammation, it has been an attractive target for the development of anti-inflammation therapeutics. However, p38α inhibitors showed side effects including severe liver toxicity that often prevailed over the benefits in clinical studies, and the mechanism of toxicity is not clear. Here, we demonstrate that p38α regulates the inflammatory responses in the acute liver inflammation in a tissue-specific manner, and liver toxicity by p38α inhibitors may be resulted from the inhibition of protective activity of p38α in the liver. Genetic ablation of p38α in T and NKT cells protected mice from the liver injury in ConA-induced liver inflammation, while liver-specific deletion of p38α aggravated the liver pathology. We found that p38α deficiency in the liver increased the expression of chemokines to recruit more inflammatory cells, indicating that p38α in the liver plays a protective anti-inflammatory role during acute liver inflammation. Therefore, our results suggest that p38α regulates the inflammatory responses in a tissue-specific manner, and that the tissue-specific p38α targeting strategies can be used for the development of an effective anti-inflammation treatment with an improved side-effect profile.
Keywords: p38α MAPK, p38α inhibitor, liver, inflammation, hepatotoxicity
Introduction
The kinase p38α is a member of p38 MAPK family, and a key player of diverse biological processes including cell proliferation, differentiation, and survival as well as inflammatory responses (1-3). The significance of p38α-mediated signaling in the induction of inflammatory responses was originally identified as the target of the pyridinyl imidazole that inhibited the expression of inflammatory cytokines in LPS-treated monocytes (4). TLR-mediated p38α signaling leads to the activation of signaling cascades for the expression proinflammatory cytokines, enzymes and other inflammation-related molecules (5-10). Thus, p38α-mediated signaling is a critical process in inflammatory responses, and has been an attractive target for the development of anti-inflammatory therapeutic strategies.
Because p38α inhibitors efficiently blocked the expression of inflammatory cytokines and ameliorated the development of inflammatory diseases in animal studies (11), it has been postulated that treatment with inhibitor compounds would be beneficial in inflammatory diseases accompanied with elevated cytokine levels. Pharmacological inhibitors of p38α have been developed and tested for the treatment of inflammatory diseases including allergic, autoimmune, and other inflammatory diseases (12-16). Although p38α inhibitors efficiently block the activity of the kinase in inflammation, the chemicals showed severe toxicity in liver. For instance, pharmacological inhibitors VX-702 and VX-745 have been shown to be effective in an animal arthritis model and inhibited LPS-induced inflammation in peripheral blood mononuclear cells (17,18). In clinical studies, the p38α inhibitors reduced levels of inflammatory biomarkers, such as C-reactive protein (CRP), erythrocyte sediment rate, amyloid A, and soluble p55 TNFR. However, the inhibitors also showed severe hepatotoxicity, including significantly increased aminotransferase levels in blood, which resulted in delay or discontinuation of new drug development (19).
Therefore, assessment of the role p38α in liver may be of interest not only to better understand the tissue-specific function of p38α in inflammatory diseases, but also to explain potential hepatic side effects upon treatment with p38α inhibitors (20). In this study, we tested whether the unexpected and detrimental effects of p38α inhibitors resulted from the tissue-specific regulation of the kinase activity during inflammation. We demonstrated that ablation of p38α in T and NKT cells protected mice from ConA-induced acute liver injury while liver-specific p38α deficiency exacerbated the pathology of liver. In T and NKT cells, p38α regulated the expression of inflammatory cytokines that play an essential role for acute liver inflammation. However, p38α inhibited the expression of chemokines by negatively regulating the activation of JNK in hepatocytes, indicating that inhibition or deletion of p38α in liver facilitates the recruitment of inflammatory cells to the site of inflammation during acute liver injury and resulted in the severe liver damage. Therefore, it is suggested that tissue-specific p38α activity differentially controls the inflammation, and the tissue-specific therapeutic strategy of p38α inhibition may be effective for the treatment of inflammatory diseases with an improved side-effect profile.
Materials and Methods
Mice and acute liver injury model
C57Bl/6 background WT mice were obtained from the mouse breeding facility at The Scripps Research Institute. Macrophage-specific (LysMCre-p38αΔ/Δ), or T lymphocyte-specific p38α deficient mice (LckCre-p38αΔ/Δ) were generated previously (6,21). p38α floxed mice were crossed with liver-specific Cre-expressing mice (Alb-Cre, The Jackson Laboratory) to obtain a liver-specific p38α-deficient mouse strain. Protocols for the use of animals were approved by the Institutional Animal Care and Use Committee.
To induce acute liver injury in mice, ConA (20 mg/kg body weight in PBS) was intravenously administered via the lateral tail vein. Mice were euthanized, blood was collected, and livers were surgically removed.
Reagents
Con A was obtained from Sigma-Aldrich.; TNF was from Peprotech; α-Galactosyl ceramide (α-GalCer, KRN7000) was from Enzo Life Science; SP600125 was from Calbiochem. Anti-GAPDH antibody was from Chemicon; anti-TCR-FITC antibody was from BD Bioscience, and anti-CXCL10 antibody was purchased from Bioss Antibodies (Boston, MA). Other antibodies were purchased from Cell Signaling. APC-mCD1d/PBS57 ligand tetramer was generously provided by the National Institute of Allergy and Infectious Disease MHC Tetramer Core Facility (Atlanta, GA, USA).
ALT and AST Assays
Serum ALT and AST levels were analyzed by commercial assay kits (Pointe Scientific).
Histology
Liver tissues were fixed in 4% paraformaldehyde, and further processed for H&E staining. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed using In situ cell death detection kit according to the manufacturer's instructions (Roche).
Immunohistochemistry
Paraffin slides were stained with goat anti-TCR-FITC and rabbit anti-CXCL10 Abs followed by anti-rabbit PE incubation. For DAB staining of CXCL10 in the liver tissues, slides were incubated with rabbit anti-CXCL10 Ab, and followed by goat anti-rabbit-HRP Ab. The slides were developed with DAB substrate (Cell signaling) and incubated with Mayer's Hematoxylin (Sigma) for nuclei counterstaining.
Cell culture
HEK 293T and mouse hepatocyte Hepa 1-6 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS). Cell viability was measured by MTT assay according to the manufacturer's instruction (Roche).
Reverse transcription and quantitative PCR
RNAs from tissues or cells were prepared with Trizol reagent (Invitrogen), and qPCR was performed (21).
Hepatocyte preparation
Perfusion and digestion of liver was performed by sequential passing of pre-warmed perfusion buffer (Invitrogen) and pre-warmed digestion buffer via portal vein as previously described (22). Hepatocytes were isolated by centrifugation at 40g for 1 min at RT, and cell pellet was washed with PBS three times. Cell viability and purity were more than 95%.
Preparation of liver leukocytes
Blood was removed by injecting HBSS through portal vein. Liver was surgically removed and homogenate was incubated with 100 U/ml collagenase for 40 min at 37°C, and cell digest was passed through a 100-μm cell strainer. Cell pellets were obtained by centrifugation for at 700g for 10 min at RT, and further resuspended in 40 % isotonic Percoll. After centrifugation at 700g for 20 min, liver leukocytes were obtained. Approximately 55-60% of total lymphocyte population was CD3+ lymphocyte, 30% was CD3+CD4+ Th cells, and 10-15% was NKT CD3+/NK1.1+ NKT cells as previously reported (23).
Flow cytometry
To analyze the intracellular level of p38α in NKT cells, liver leukocytes were stained with CD3-FITC Abs and APC-CD1d ligand tetramer. Cells were further fixed and permeabilized by Cytofix/Cytoperm kit, and further stained with anti-p38α Abs.
Plasmids and shRNAs
The shRNA targeting mouse p38α constructs was designed, and lentiviral vectors expressing the shRNA were generated according to the manufacturer's instructions (Biosettia). The sequences of the oligonucleotides used to generate the shRNAs are as follows: mouse p38α: 5′-AAAAGCCTGTTGCTGACCCTTATTTGGATCCAAATAAGGGTCAGCAACAGGC-3′; A negative scramble sequence of 5′-AAAAGCTACACTATCGAGCAATTTTGGATCCAAAATTGCTCGATAGTGTAGC-3′ was used as a control.
Immunoblotting
Cells lysates were prepared by incubating cells in lysis buffer. Samples were resolved by SDS-PAGE and analyzed by immunoblotting using chemiluminescence substrate.
Measurement of cytokines
TNF or IFN-γ concentrations were measured by ELISA (eBioscience).
Statistical analysis
Student's t-test was performed to analyze the statistical significance. A P value of less than 0.05 was considered significant.
Results
Tissue-specific role of p38α in ConA-induced liver injury
First, we generated macrophage-, T cell-, or hepatocyte-specific p38α-deficient mouse strains, and confirmed deletion of p38α is specific in hepatocytes in albumin-cre-driven conditional knockout mice (6,21) (Supplemental Figure 1). Using ConA-induced acute liver injury model (24,25), we investigated the tissue-specific role of p38α in liver inflammation. Control (p38αfl/fl) and tissue-specific p38α-deficient mice were intravenously injected with ConA, and serum aminotransferase levels were measured. The ALT and AST levels in T cell-specific p38α-deficient (p38αΔT) mice were significantly reduced compared with control (Figure 1A). We also tested the liver injury by measuring ALT levels in the blood in a time-course manner. ALT levels were comparable between control, p38αΔT, and p38αΔliver mice until 4 hours after ConA injection. Then, ALT level in control mice significantly elevated, but not in p38αΔT mice. However, ablation of p38α in liver rather increased ALT levels. H&E staining showed that liver pathology characterized by infiltration of inflammatory cells, liver hemorrhage, and cell death was less severe in p38αΔT mice than control (Figure 1C). Additionally, TUNEL assay indicated that induction of apoptosis was significantly reduced in the liver tissue of p38αΔT mice (Figure 1D). Because T and NKT cells play an important role in ConA-induced liver damage (26-28), and Lck-cre-mediated deletion of p38α was observed in T cells as well as NKT cells of hepatic leukocytes (Supplemental Figure 1) (21,29), our results indicate that p38α promotes the activation of T and NKT cells in ConA-induced hepatitis.
Figure 1. Tissue-dependent role of p38α in ConA-induced acute liver inflammation.
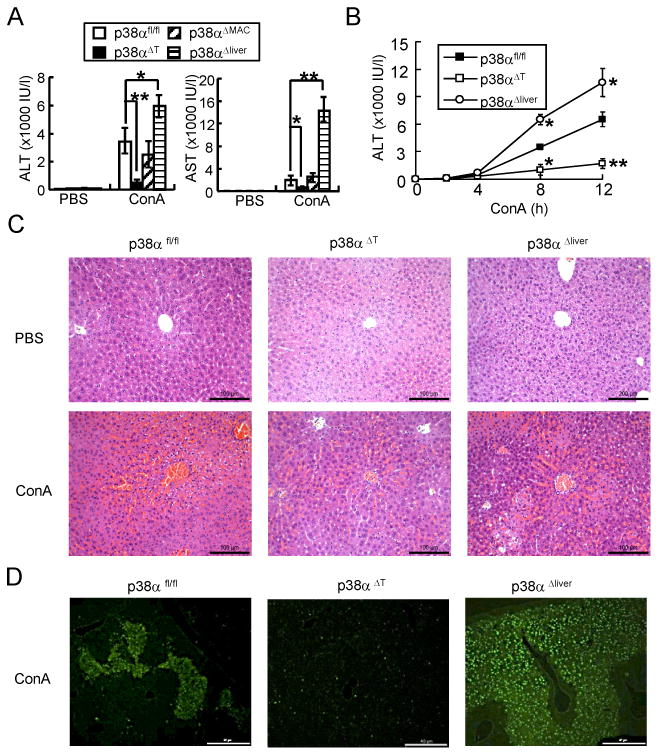
(A) p38αfl/fl, p38αΔT, p38αΔMAC, or p38αΔliver mice were injected intravenously with PBS or ConA. Serum ALT and AST levels were measured after 8 h (A, mean ± s.d., n=6). (B) ALT levels were measured after 2, 4, 8 or 12 h after ConA injection into p38αfl/fl, p38αΔT, or p38αΔliver mice (n=5-6) (B). Data represent means ± s.d.; *p < 0.05 and **p < 0.01 (Student's t-test). (C) H & E staining of the liver tissues from PBS- or ConA-injected mice. Scale bars, 100 μM. ×200. (D) TUNEL staining of liver from ConA-injected mice. Scale bars, 40 μM. ×100.
We next sought to identify the role of p38α in liver. Like the role for inflammation in T/NKT cells, we hypothesized that p38α deficiency in hepatocytes would ameliorate acute liver inflammation. Interestingly, the serum ALT/AST levels of ConA-injected p38αΔliver mice significantly increased, which indicated that ablation of p38α in liver aggravated the liver damage (Figure 1A). Histological analysis supported the result that the degree of inflammation and induction of apoptosis were higher in the liver tissues of p38αΔliver mice (Figure 1C & D). The ALT/AST levels in ConA-injected p38αΔMAC mice were comparable with p38αfl/fl mice, indicating that innate immune cells are not involved in this acute hepatitis model (Figure 1A) (26-28). Therefore, our results demonstrate that p38α in T and NKT cells promotes the induction of inflammation while hepatocyte p38α protects mice from ConA-induced liver damage, suggesting the tissue-specific role of p38α in inflammation.
p38α in T and NKT cells regulates the expression of inflammatory cytokines during ConA-induced liver inflammation
Next, we compared the cytokine levels in the sera of control, p38αΔT, or p38αΔliver mice to further examine the tissue-specific role of p38α. In ConA-induced acute liver injury, the inflammatory cytokines from T and NKT cells play a significant role in the induction of cell death of hepatocytes and liver damage. Intravenous injection of ConA promotes T and NKT cell activation to produce TNF and IFN-γ. TNF is quickly expressed and the serum concentration is highest at 1-2 hours, while IFN-γ expression is slower than TNF and reaches the maximal concentration at 6-8 hours (24,26,30,31). ConA-induced TNF and IFN-γ levels in the sera were significantly reduced in p38αΔT mice compared with control (Figure 2A). However, the cytokine levels were comparable between control and p38αΔliver mice (Figure 2B) as the activation of p38α in T/NKT cells was not affected by ablation of p38α in hepatocytes. This result is consistent with the finding that ablation of p38α in T/NKT cells protected mice from liver damage while the deficiency in liver aggravated acute liver injury.
Figure 2. Tissue-specific ablation of p38α differentially regulates the expression of cytokines during ConA-induced liver damage.
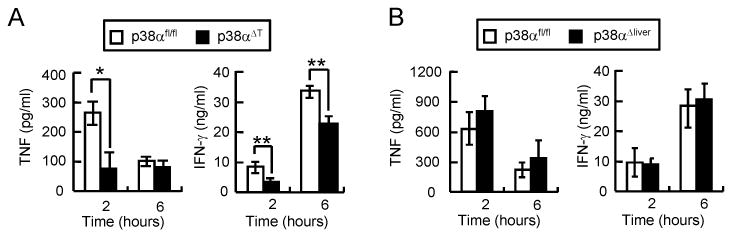
p38αfl/fl and p38αΔT mice (A), or p38αfl/fl and p38αΔliver mice (B) were injected with ConA. IFN-γ and TNF levels in the sera were determined by ELISA (n = 6). Data represent means ± s.d.; *p < 0.01 and **p < 0.05 (Student's t-test).
Tissue-specific p38α activation differentially regulates the inflammatory responses
To elucidate the tissue-specific regulation of inflammation by p38α. we compared the levels of inflammatory cytokines in the liver tissues and hepatic leukocytes in ConA-injected p38α-deficient mice. Expression of TNF was significantly increased in the liver tissue of ConA-injected control mice. However, TNF mRNA level was lower in the liver of p38αΔT mice at 2 hours of ConA administration while TNF levels was substantially increased in the liver tissues of p38αΔliver mice (Figure 3A). The mRNA levels of IFN-γ in the liver tissues were comparable between control and p38αΔliver mice after 2 hours of ConA injection; however, expression was significantly reduced in the liver of p38αΔT mice while increased in p38αΔliver mice after 6 hours (Figure 3B). We next tested the expression of inflammatory cytokines in hepatic leukocytes that include T and NKT cells. Expression of inflammatory cytokines was substantially reduced in hepatic leukocytes of p38αΔT mice while that in hepatic leukocytes of control and p38αΔliver mice was comparable (Figure 3C & D). Collectively, our results indicated that p38α in T and NKT cells promotes the inflammatory responses, whereas activation of p38α in hepatocytes negatively regulates the inflammation.
Figure 3. p38α regulates the inflammatory responses in liver and hepatic leukocytes during ConA-induced hepatitis.
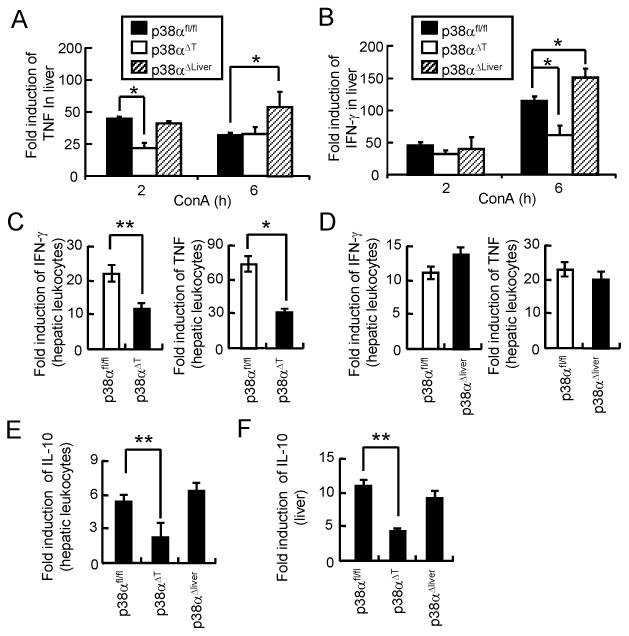
(A-D) p38αfl/fl, p38αΔT, or p38αΔliver mice received ConA. (A & B) Liver tissues were obtained after 2 or 6 h, or (C & D) hepatic leukocytes were after 4 h. IFN-γ and TNF mRNA levels were analyzed by qPCR (n = 4). Fold induction was calculated by comparing to the samples of PBS-injected mice. (E & F) Expression of IL-10 in hepatic leukocytes (E), and liver (F) of ConA-injected mice was analyzed by qPCR. Data shown are means ± s.d.; *p < 0.01 and **p < 0.05 (Student's t-test).
IL-10 is a potent anti-inflammatory cytokine that plays a hepatoprotective role in Con A-induced liver injury. During ConA-induced inflammation, IL-10 is also derived from T and NKT cells and produced within the liver (32,33). IL-10 expression was significantly reduced in hepatic leukocytes of p38αΔT mice compared with control cells while leukocytes of p38αΔliver mice did not show any significant changes (Figure 3E). Consistently, the mRNA level of IL-10 in the liver tissues was substantially reduced in the liver of p38αΔT mice but not in p38αΔliver mice (Figure 3F). These results indicate that IL-10 does not contribute to the protection of severe liver injury of ConA-injected p38αΔliver mice.
p38α promotes the inflammatory responses in T cells and NKT cells
Because T and NKT cells play an important role in the induction of ConA-induced liver damage (26-28), we next tested p38α-mediated regulation of inflammatory cytokine expression in T and NKT cells. Lck-cre-mediated deletion of p38α was observed in T cells as well as NKT cells of hepatic leukocytes (Supplemental Figure 1) (29). Hepatic leukocytes from control, p38αΔT or p38αΔliver mice were treated with ConA or α-galactosylceramide (α-GalCer) that activates T or NKT cells, respectively. We found that production of cytokines was significantly reduced in leukocytes of p38αΔT mice from the early time point after stimulation, but not in liver leukocytes of p38αΔliver mice (Figure 4A & B). Induction of cytokine mRNA by ConA was significantly reduced in p38α-deficient leukocytes (Figure 4C). Consistently, our study also indicated that TCR-induced expression of cytokine genes was also significantly reduced in p38α-deficient T cells compared with control cells (21) (data not shown). However, induction of cytokine mRNA by α-GalCer did not differ between control and p38α-deficient leukocytes (Figure 4D), implying that p38α does not directly regulate the induction of inflammatory cytokine expression in NKT cells, but is involved in the post-transcriptional regulation of gene expression as shown previously (29). Collectively, our results suggest that p38α promotes the inflammatory responses of T and NKT cells in a distinct mechanism (see discussion).
Figure 4. p38α regulates the expression of inflammatory cytokines in T and NKT cells.
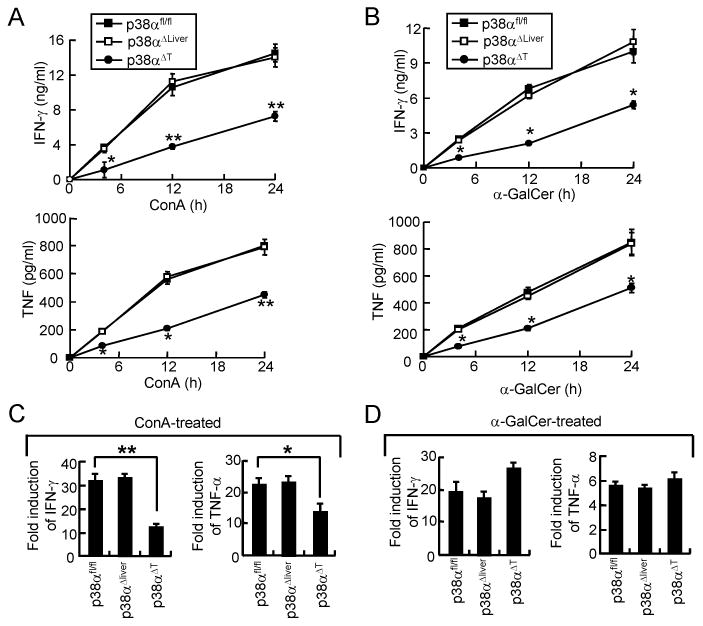
Hepatic leukocytes from p38αfl/fl, p38αΔliver, or p38αΔT mice were treated with medium or ConA (5 μg/ml, A & B), or α-GalCer (200 ng/ml, C & D). (A & C) Culture supernatants were obtained after 4, 12, or 24 h to determine IFN-γ and TNF concentrations by ELISA. (B & D) After 4 h, mRNA levels of IFN-γ and TNF were determined by qPCR. Fold induction was calculated by comparing the levels of unstimulated cells. Data shown are means ± s.d.; *p < 0.05 and **p < 0.01 compared with control cells (Student's t-test).
Deletion of p38α in hepatocytes does not affect the induction of cell death, but increases the activation of JNK
Because TNF plays a significant role in ConA-induced liver damage (25,34), we tested whether TNF-induced death of hepatocytes was affected by ablation of p38α. Previous studies have shown that actinomycin D is required to increase the sensitivity to TNF and to induce apoptosis of hepatocyte in vitro, which mimics the in vivo situation (35,36). Thus, incubation of hepatocytes with TNF plus actinomycin D is generally used to test TNF-induced apoptosis in vitro. TNF-induced cell death of WT and p38α-deficient primary hepatocytes, or control and p38α knockdown (KD) Hepa 1-6 cells was comparable (Figure 5A &B). Additionally, TNF-induced cleavage of caspases 3 and 9 was similar between control and p38α KD hepatocytes (Figure 5C), indicating that p38α does not regulate the induction of cell death in hepatocytes.
Figure 5. p38α signaling in hepatocytes is dispensable in TNF-induced cell death.
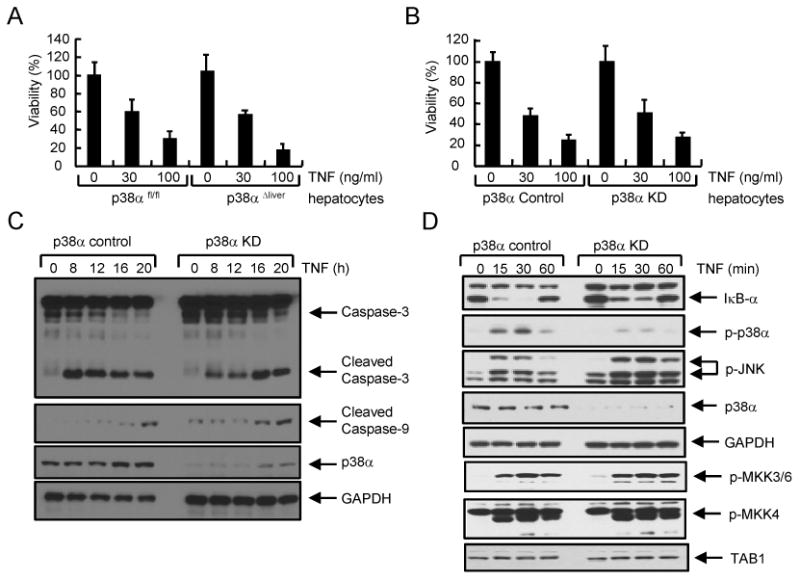
(A & B) Hepatocytes from p38αfl/fl or p38αΔliver mice (A), or control or p38α KD Hepa 1-6 hepatocyte cells (B) were incubated with actinomycin D (ActD, 1 μg/ml) and TNF. Cell viability was assessed after 24 h. (C & D) Control or p38α KD Hepa 1-6 cells were incubated with ActD and TNF (30 ng/ml). Cell lysates were prepared at the indicated times to examined the activation of caspases (C) or NF-κB/MAPK signaling (D). Knockdown of p38α was confirmed using anti-p38α antibody. GAPDH levels were determined as an internal control.
Next, we examined the role of p38α in TNFR signaling in hepatocytes. Degradation of IκB-α and phosphorylation of MAPK ERK by TNF treatment were comparable between control and p38α KD Hepa 1-6 cells, while phosphorylation of p38α was only detected in control cells (Figure 5D). However, knockdown of p38α rather increased and sustained the phosphorylation of JNK, implying an inhibitory function of p38α on JNK activation in hepatocytes (Figure 5D). Phosphorylation of JNK increased and sustained in p38α KD hepatocytes, but not in p38α-deficient macrophages or T cells (6,21). Additionally, it has been shown that activation of JNK increased in the liver of LPS-injected p38αΔliver mice (37). Since the activation of JNK can be directly regulated by the upstream kinase MKK4, we further tested whether p38α regulated the activation of MKK4. Phosphorylation of MKK4 was not affected in p38α KD hepatocytes, suggesting that p38α directly regulates the activation of JNK, but not by inhibiting MKK4 activity in hepatocytes (Figure 5D). Additionally, phosphorylation of MKK3/6 and the expression of TAB1 at the upstream of p38α were comparable between control and p38α KD Hepa 1-6 cells. Collectively, our results suggest that p38α is dispensable in TNF-induced cell death and negatively regulates the activation of JNK in hepatocytes.
p38α negatively regulates the expression of chemokines in hepatocytes
We demonstrated that expression of inflammatory cytokines was not affected in hepatic leukocytes of p38αΔliver mice, and p38α did not contribute to TNF-induced cell death of hepatocytes. However, histological analysis revealed that more inflammatory cells were observed in the liver tissues of ConA-injected p38αΔliver mice, which indicated that recruitment of inflammatory cells was enhanced in p38α-deficient liver during acute liver inflammation. Since chemokines play a crucial role in immune cell recruitment, we examined the expression patterns of chemokines in liver of ConA-injected control and p38αΔliver mice (Supplemental Figure 2). Expression of several chemokines was significantly induced in liver of control mouse. Moreover, expression of CXCL9, CXCL10, CXCL11, and CCL25 was further increased in the liver tissue of p38αΔliver mice compared with control (Figure 6A). Previous studies have shown that CXCL10 plays a role for the development of liver diseases and the recruitment of inflammatory immune cells to the site of inflammation (38-40). Thus, we tested whether increased expression of CXCL10 correlated with the recruitment of immune cells in the liver tissue of p38αΔliver mice. IHC result revealed that CXCL10 expression and T cell recruitment substantially increased in the liver tissues of ConA-injected p38αΔliver mice (Figure 6B & Supplemental Figure 3). Since expression of inflammatory cytokines by hepatic leukocytes were not affected in p38αΔliver mice, our result indicates that ablation of p38α in liver increased the expression of chemokines to recruit more inflammatory cells and resulted in severe inflammation and injury. Additionally, knockdown of p38α increased TNF-induced expression of chemokines in hepatocytes in vitro (Supplemental Figure 4). Collectively, our results suggest that p38α in liver negatively regulates the recruitment of inflammatory cells by suppressing the expression of chemokines during acute liver inflammation.
Figure 6. p38α negatively regulates the induction of chemokines by inhibiting JNK activity in hepatocytes.
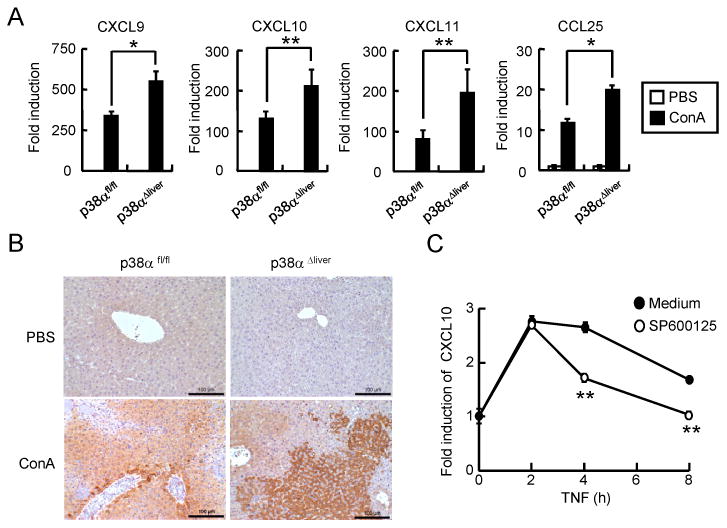
(A) Total RNAs were prepared after 4 h from the liver tissues of ConA-injected p38αfl/fl or p38αΔliver mice, and expression of chemokines was analyzed by qPCR. (B) Liver paraffin slides were stained with rabbit anti-CXCL10, and further incubated with goat anti-rabbit-HRP Abs, and incubated with DAB. (C) Hepa 1-6 cells were incubated with medium or SP600213 for 1 hour, and treated with TNF for 2, 4, or 8 h. Expression level of CXCL10 was measured by qPCR. Data shown are means ± s.d. *; p < 0.05, and **; p < 0.01 (Student's t-test).
JNK-mediated expression of chemokines is suppressed by p38α in hepatocytes
Next, we examined how chemokine expression was regulated by p38α in liver. Although activation of p38α induces the expression of chemokines in many types of cells, such as macrophages and intestinal epithelial cells (6,16,21,41), deletion of p38α increased the expression of chemokines in hepatocytes. Since activation of JNK increased in TNF-treated p38α KD hepatocytes (Figure 5D) and in the liver tissue of LPS-injected p38αΔliver mice (37), we tested the regulation of chemokine expression by JNK in hepatocytes. First, we examined whether CXL10 expression was affected by inhibition of JNK activity. TNF treatment induced the expression of CXCL10 in hepatocytes, which was significantly reduced by JNK inhibitor SP600125 (Figure 6C). Furthermore, increased expression of CXCL10 in p38α KD hepatocytes was significantly reduced by inhibition of JNK activity (Supplemental Figure 4), indicating that CXCL10 expression is down-regulated by inhibition of JNK activity by p38α. Therefore, it is suggested that p38α negatively regulates the activation of JNK during acute liver injury to inhibit the expression of chemokines that recruit the immune cells to the site of inflammation.
Discussion
p38α displays diverse functions in many biological processes. Since p38α is widely expressed in immune cells and involved in the induction of inflammatory responses such as expression of inflammatory cytokines and mediators, it has been considered as an important regulator of inflammatory diseases. Thus, p38α has been a potential target for the treatment of inflammatory diseases. However, clinical studies using p38α inhibitors have shown disappointing results and side effects of severe hepatotoxicity (11,17-19). In this study, we questioned whether the adverse liver damage by p38α inhibitor treatment is caused by inhibition of liver p38α activity that plays a protective role during inflammation.
Previous studies showed that p38α negatively regulates the proliferation of hepatocytes by antagonizing JNK pathway, and liver-specific p38α-deficient mice exhibited enhanced hepatocyte proliferation after partial hepatectomy and developed more liver tumors (42,43). In contrast, a study showed that liver-specific deletion of p38α reduced cell growth and caused cytokinesis failure during chronic biliary cirrhosis in mice (20). We demonstrated an opposing role of p38α in the induction of ConA-induced liver injury. Ablation of p38α in T and NKT cells protected mice from acute liver injury while p38α deficiency in liver rather aggravated the damage in the liver tissue. Therefore, the tissue-specific regulation of p38α activation is also dependent on the pathologic conditions.
Pro-inflammatory cytokines such as IFN-γ and TNF produced by T and NKT cells are the key molecules that cause liver damage in ConA-induced hepatitis (26,44,45). However, some studies also indicated that macrophage may be involved in this hepatitis model as depletion of Kupffer cells suppressed the liver injury induced by ConA (46,47). The mechanism of Kupffer cell involvement in this hepatitis model is not clear. A study suggested that Kupffer cells initiate intrasinusoidal sinusoidal thrombosis in collaboration with endothelial cells (47), indicating an indirect effect of Kuppfer cells on liver damage. Additionally, we observed that ConA treatment did not significantly induce the expression of TNF in control or p38α-deficient macrophages in vitro (data not shown), indicating that ConA administration does not directly activate macrophages in this acute liver damage model. Therefore, it is suggested that p38α in macrophages does not directly contribute to the induction of ConA-induced liver injury.
It has been well documented that p38α plays an essential role in the regulation of immune responses. TCR signaling-mediated expression of inflammatory cytokines is regulated by p38α (3,21). Inflammatory cytokine levels were significantly reduced in the sera of ConA-injected p38αΔT mice, suggesting that p38α regulates the production of cytokines in T and NKT cells. The mRNA levels of inflammatory cytokines were substantially reduced in hepatic leukocytes of p38αΔT mice, but not in leukocytes of p38αΔliver mice. Additionally, p38α deficiency resulted in the reduced expression of inflammatory cytokines in T and NKT cells in vitro. The mRNA levels and production of inflammatory cytokines were reduced in p38α-deficient T cells, whereas induction of cytokine mRNAs was comparable between control and p38α-deficient NKT cells. This result indicates that p38α may regulate the inflammatory responses of T and NKT cells by different mechanisms. In T cells, p38α regulates the expression of inflammatory cytokines by regulating the activation of downstream kinases and transcription factors that are essential for the transcription of cytokine genes (2,3). However, p38α seems to be important for the translational or post-translational regulation of cytokines in NKT cells. Similarly, a previous study indicated that p38α signaling regulates the cytokine mRNA translation by activation of MAPK-interacting kinases (MNKs) that activate the eukaryotic initiation factor eIF-4E for the translation of cytokine mRNAs (29). Taken together, we suggest that regulation of cytokine production by p38α in NKT cells is mediated by a mechanism that facilitates the translation of cytokine mRNAs. More studies are needed to clarify the mechanism.
Expression of chemokines increased and more inflammatory cells were recruited to the liver of p38αΔliver mice, which suggests a protective role of p38α in liver inflammation. Expression of chemokines increased in the liver tissue of ConA-injected mice. However, treatment of ConA did not increase the level of CXCL10 in hepatocytes while TNF or IFN-γ induced the expression (data not shown). Previous studies also indicate that ConA administration activates T cells to produce IFN-γ, which further increases the expression of CXCL10 in hepatocytes (38,48,49). Therefore, cytokines produced by T or NKT cells by ConA administration induce the expression of chemokines in the liver, and in turn, increased expression of chemokines recruits more inflammatory cells to the liver tissues.
Expression of CXCL10 was suppressed by p38α in liver, providing protection against acute liver injury. Previous studies have demonstrated that CXCL10 plays a role for the development of liver diseases such as tetrachloride-induced acute liver injury, ischemia, and reperfusion injury, and liver injury induced by hepatitis C virus infection (38-40). Furthermore, administration of neutralizing anti-CXCL10 antibody significantly ameliorated ConA-induced hepatitis, suggesting that CXCL10 plays an essential role in the induction of acute liver injury (40). Therefore, we suggest that p38α in liver inhibits the expression of chemokines to block the excessive recruitment of inflammatory cells during acute liver inflammation.
Our results demonstrated that phosphorylation of JNK by TNF significantly increased in p38α KD hepatocytes. Although the mitogen-activated kinase kinase 4 (MKK4) lies at the upstream of JNK, phosphorylation of MKK4 was comparable between control and p38α KD hepatocytes after TNF treatment, suggesting that p38α directly regulates JNK activity without regulating MKK4 activation. A study showed that activation of JNK increased in the liver tissue of LPS-injected p38αΔliver mice, indicating the negative regulation of JNK activity by p38α in hepatocytes (37). Many studies have confirmed the role of JNK in TNF-mediated liver injury. Inhibition of JNK activity by pharmacological inhibitor SP600125 blocked the TNF-induced cell death of hepatocytes (34), and JNK1- and JNK2-deficient mice were resistant to ConA-induced liver damage (50). However, it is not clear how p38α directly and selectively regulates the activation of JNK in hepatocytes.
Our findings suggest the tissue-specific function of p38α in inflammation. In ConA-induced acute liver inflammation model, genetic ablation of p38α in T and NKT cells protected mice from the liver injury while deletion of the kinase in liver augmented the pathology of liver inflammation, indicating the protective role of p38α in liver during the inflammatory diseases. Thus, inhibition of p38α activity in liver may result in the development of severe inflammation, which is often caused by the treatment of p38α inhibitors. Therefore, strategies that deliver the inhibitors specifically to the inflammatory cells, but not to the liver will help improve the treatment of inflammatory diseases.
Supplementary Material
Acknowledgments
We acknowledge the NIH Tetramer Core Facility (contract HHSN272201300006C) for provision of mCD1d/PBS57 ligand tetramer.
This work was supported by a grant from the National Institutes of Health (AI088229) to Y. J. K.
Abbreviations
- MAPK
mitogen-activated protein kinase
- LPS
lipopolysaccharide
- TLR
toll-like receptor
- ConA
concanavalin A
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- NKT
natural killer T cells
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
References
- 1.Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal. 2000;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 2.Rincon M, Davis RJ. Regulation of the immune response by stress-activated protein kinases. Immunol Rev. 2009;228:212–224. doi: 10.1111/j.1600-065X.2008.00744.x. [DOI] [PubMed] [Google Scholar]
- 3.Cook R, Wu CC, Kang YJ, Han J. The role of the p38 pathway in adaptive immunity. Cell Mol Immunol. 2007;4:253–259. [PubMed] [Google Scholar]
- 4.Lee JC, Kumar S, Griswold DE, Underwood DC, Votta BJ, Adams JL. Inhibition of p38 MAP kinase as a therapeutic strategy. Immunopharmacology. 2000;47:185–201. doi: 10.1016/s0162-3109(00)00206-x. [DOI] [PubMed] [Google Scholar]
- 5.Zhu W, Downey JS, Gu J, PF Di, Gram H, Han J. Regulation of TNF expression by multiple mitogen-activated protein kinase pathways. J Immunol. 2000;164:6349–6358. doi: 10.4049/jimmunol.164.12.6349. [DOI] [PubMed] [Google Scholar]
- 6.Kang YJ, Chen J, Otsuka M, Mols J, Ren S, Wang Y, Han J. Macrophage deletion of p38alpha partially impairs lipopolysaccharide-induced cellular activation. J Immunol. 2008;180:5075–5082. doi: 10.4049/jimmunol.180.7.5075. [DOI] [PubMed] [Google Scholar]
- 7.Guan Z, Buckman SY, Pentland AP, Templeton DJ, Morrison AR. Induction of cyclooxygenase-2 by the activated MEKK1 --> SEK1/MKK4 --> p38 mitogen-activated protein kinase pathway. J Biol Chem. 1998;273:12901–8. doi: 10.1074/jbc.273.21.12901. [DOI] [PubMed] [Google Scholar]
- 8.Da SJ, Pierrat B, Mary JL, Lesslauer W. Blockade of p38 mitogen-activated protein kinase pathway inhibits inducible nitric-oxide synthase expression in mouse astrocytes. J Biol Chem. 1997;272:28373–28380. doi: 10.1074/jbc.272.45.28373. [DOI] [PubMed] [Google Scholar]
- 9.Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal. 2000 Jan;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. 2000. 12: 1-13. [DOI] [PubMed] [Google Scholar]
- 10.Pietersma A, Tilly BC, Gaestel M, de JN, Lee JC, Koster JF, Sluiter W. p38 mitogen activated protein kinase regulates endothelial VCAM-1 expression at the post-transcriptional level. Biochem Biophys Res Commun. 1997;230:44–48. doi: 10.1006/bbrc.1996.5886. [DOI] [PubMed] [Google Scholar]
- 11.Gaestel M, Kotlyarov A, Kracht M. Targeting innate immunity protein kinase signalling in inflammation. Nat Rev Drug Discov. 2009;8:480–499. doi: 10.1038/nrd2829. [DOI] [PubMed] [Google Scholar]
- 12.Zhan Y, Kim S, Izumi Y, Izumiya Y, Nakao T, Miyazaki H, Iwao H. Role of JNK, p38, and ERK in platelet-derived growth factor-induced vascular proliferation, migration, and gene expression. Arterioscler Thromb Vasc Biol. 2003;23:795–801. doi: 10.1161/01.ATV.0000066132.32063.F2. [DOI] [PubMed] [Google Scholar]
- 13.Ma XL, Kumar S, Gao F, Louden CS, Lopez BL, Christopher TA, Wang C, Lee JC, Feuerstein GZ, Yue TL. Inhibition of p38 mitogen-activated protein kinase decreases cardiomyocyte apoptosis and improves cardiac function after myocardial ischemia and reperfusion. Circulation. 1999;99:1685–1691. doi: 10.1161/01.cir.99.13.1685. [DOI] [PubMed] [Google Scholar]
- 14.Bogoyevitch MA, Gillespie-Brown J, Ketterman AJ, Fuller SJ, Ben-Levy R, Ashworth A, Marshall CJ, Sugden PH. Stimulation of the stress-activated mitogen-activated protein kinase subfamilies in perfused heart. p38/RK mitogen-activated protein kinases and c-Jun N-terminal kinases are activated by ischemia/reperfusion. Circ Res. 1996;79:162–173. doi: 10.1161/01.res.79.2.162. [DOI] [PubMed] [Google Scholar]
- 15.Waetzig GH, Seegert D, Rosenstiel P, Nikolaus S, Schreiber S. p38 mitogen-activated protein kinase is activated and linked to TNF-alpha signaling in inflammatory bowel disease. J Immunol. 2002;168:5342–5351. doi: 10.4049/jimmunol.168.10.5342. [DOI] [PubMed] [Google Scholar]
- 16.Otsuka M, Kang YJ, Ren J, Jiang H, Wang Y, Omata M, Han J. Distinct effects of p38alpha deletion in myeloid lineage and gut epithelia in mouse models of inflammatory bowel disease. Gastroenterology. 2010;138:1255–65. 1265. doi: 10.1053/j.gastro.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sweeney SE. The as-yet unfulfilled promise of p38 MAPK inhibitors. Nat Rev Rheumatol. 2009;5:475–477. doi: 10.1038/nrrheum.2009.171. [DOI] [PubMed] [Google Scholar]
- 18.Gruenbaum LM, Schwartz R, Woska JR, Jr, DeLeon RP, Peet GW, Warren TC, Capolino A, Mara L, Morelock MM, Shrutkowski A, Jones JW, Pargellis AC. Inhibition of pro-inflammatory cytokine production by the dual p38/JNK2 inhibitor BIRB796 correlates with the inhibition of p38 signaling. Biochem Pharmacol. 2009;77:422–432. doi: 10.1016/j.bcp.2008.10.032. [DOI] [PubMed] [Google Scholar]
- 19.Goldstein DM, Kuglstatter A, Lou Y, Soth MJ. Selective p38alpha inhibitors clinically evaluated for the treatment of chronic inflammatory disorders. J Med Chem. 2010;53:2345–2353. doi: 10.1021/jm9012906. [DOI] [PubMed] [Google Scholar]
- 20.Tormos AM, Arduini A, Talens-Visconti R, I BB del, Nebreda AR, Sastre J. Liver-specific p38alpha deficiency causes reduced cell growth and cytokinesis failure during chronic biliary cirrhosis in mice. Hepatology. 2013;57:1950–1961. doi: 10.1002/hep.26174. [DOI] [PubMed] [Google Scholar]
- 21.Shim EJ, Bang BR, Kang SG, Ma J, Otsuka M, Kang J, Stahl M, Han J, Xiao C, Vallance BA, Kang YJ. Activation of p38alpha in T cells regulates the intestinal host defense against attaching and effacing bacterial infections. J Immunol. 2013;191:2764–2770. doi: 10.4049/jimmunol.1300908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li WC, Ralphs KL, Tosh D. Isolation and culture of adult mouse hepatocytes. Methods Mol Biol. 2010;633:185–196. doi: 10.1007/978-1-59745-019-5_13. [DOI] [PubMed] [Google Scholar]
- 23.Zhang J, Dong Z, Zhou R, Luo D, Wei H, Tian Z. Isolation of lymphocytes and their innate immune characterizations from liver, intestine, lung and uterus. Cell Mol Immunol. 2005;2:271–280. [PubMed] [Google Scholar]
- 24.Trautwein C, Rakemann T, Brenner DA, Streetz K, Licato L, Manns MP, Tiegs G. Concanavalin A-induced liver cell damage: activation of intracellular pathways triggered by tumor necrosis factor in mice. Gastroenterology. 1998;114:1035–1045. doi: 10.1016/s0016-5085(98)70324-5. [DOI] [PubMed] [Google Scholar]
- 25.Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. 2013;59:583–594. doi: 10.1016/j.jhep.2013.03.033. [DOI] [PubMed] [Google Scholar]
- 26.Tiegs G, Hentschel J, Wendel A. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J Clin Invest. 1992;90:196–203. doi: 10.1172/JCI115836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaneko Y, Harada M, Kawano T, Yamashita M, Shibata Y, Gejyo F, Nakayama T, Taniguchi M. Augmentation of Valpha14 NKT cell-mediated cytotoxicity by interleukin 4 in an autocrine mechanism resulting in the development of concanavalin A-induced hepatitis. J Exp Med. 2000;191:105–114. doi: 10.1084/jem.191.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takeda K, Hayakawa Y, Van KL, Matsuda H, Yagita H, Okumura K. Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc Natl Acad Sci U S A. 2000;97:5498–5503. doi: 10.1073/pnas.040566697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagaleekar VK, Sabio G, Aktan I, Chant A, Howe IW, Thornton TM, Benoit PJ, Davis RJ, Rincon M, Boyson JE. Translational control of NKT cell cytokine production by p38 MAPK. J Immunol. 2011;186:4140–4146. doi: 10.4049/jimmunol.1002614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kusters S, Gantner F, Kunstle G, Tiegs G. Interferon gamma plays a critical role in T cell-dependent liver injury in mice initiated by concanavalin A. Gastroenterology. 1996;111:462–471. doi: 10.1053/gast.1996.v111.pm8690213. [DOI] [PubMed] [Google Scholar]
- 31.Wahl C, Wegenka UM, Leithauser F, Schirmbeck R, Reimann J. IL-22-dependent attenuation of T cell-dependent (ConA) hepatitis in herpes virus entry mediator deficiency. J Immunol. 2009;182:4521–4528. doi: 10.4049/jimmunol.0802810. [DOI] [PubMed] [Google Scholar]
- 32.Louis H, Le MO, Peny MO, Quertinmont E, Fokan D, Goldman M, Deviere J. Production and role of interleukin-10 in concanavalin A-induced hepatitis in mice. Hepatology. 1997;25:1382–1389. doi: 10.1002/hep.510250614. [DOI] [PubMed] [Google Scholar]
- 33.Erhardt A, Biburger M, Papadopoulos T, Tiegs G. IL-10, regulatory T cells, and Kupffer cells mediate tolerance in concanavalin A-induced liver injury in mice. Hepatology. 2007;45:475–485. doi: 10.1002/hep.21498. [DOI] [PubMed] [Google Scholar]
- 34.Schwabe RF, Brenner DA. Mechanisms of Liver Injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol. 2006;290:G583–G589. doi: 10.1152/ajpgi.00422.2005. [DOI] [PubMed] [Google Scholar]
- 35.Xu Y, Bialik S, Jones BE, Iimuro Y, Kitsis RN, Srinivasan A, Brenner DA, Czaja MJ. NF-kappaB inactivation converts a hepatocyte cell line TNF-alpha response from proliferation to apoptosis. Am J Physiol. 1998;275:C1058–C1066. doi: 10.1152/ajpcell.1998.275.4.C1058. [DOI] [PubMed] [Google Scholar]
- 36.Leist M, Gantner F, Bohlinger I, Germann PG, Tiegs G, Wendel A. Murine hepatocyte apoptosis induced in vitro and in vivo by TNF-alpha requires transcriptional arrest. J Immunol. 1994;153:1778–1788. [PubMed] [Google Scholar]
- 37.Heinrichsdorff J, Luedde T, Perdiguero E, Nebreda AR, Pasparakis M. p38 alpha MAPK inhibits JNK activation and collaborates with IkappaB kinase 2 to prevent endotoxin-induced liver failure. EMBO Rep. 2008;9:1048–1054. doi: 10.1038/embor.2008.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoneyama H, Kai Y, Koyama J, Suzuki K, Kawachi H, Narumi S, Ichida T. Neutralization of CXCL10 accelerates liver regeneration in carbon tetrachloride-induced acute liver injury. Med Mol Morphol. 2007;40:191–197. doi: 10.1007/s00795-007-0371-x. [DOI] [PubMed] [Google Scholar]
- 39.Zhai Y, Shen XD, Gao F, Zhao A, Freitas MC, Lassman C, Luster AD, Busuttil RW, Kupiec-Weglinski JW. CXCL10 regulates liver innate immune response against ischemia and reperfusion injury. Hepatology. 2008;47:207–214. doi: 10.1002/hep.21986. [DOI] [PubMed] [Google Scholar]
- 40.Sahin H, Borkham-Kamphorst E, do ON, Berres ML, Kaldenbach M, Schmitz P, Weiskirchen R, Liedtke C, Streetz KL, Maedler K, Trautwein C, Wasmuth HE. Proapoptotic effects of the chemokine, CXCL 10 are mediated by the noncognate receptor TLR4 in hepatocytes. Hepatology. 2013;57:797–805. doi: 10.1002/hep.26069. [DOI] [PubMed] [Google Scholar]
- 41.Kang YJ, Otsuka M, van den Berg A, Hong L, Huang Z, Wu X, Zhang DW, Vallance BA, Tobias PS, Han J. Epithelial p38alpha controls immune cell recruitment in the colonic mucosa. PLoS Pathog. 2010;6:e1000934. doi: 10.1371/journal.ppat.1000934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hui L, Bakiri L, Stepniak E, Wagner EF. p38alpha: a suppressor of cell proliferation and tumorigenesis. Cell Cycle. 2007;6:2429–2433. doi: 10.4161/cc.6.20.4774. [DOI] [PubMed] [Google Scholar]
- 43.Hui L, Bakiri L, Mairhorfer A, Schweifer N, Haslinger C, Kenner L, Komnenovic V, Scheuch H, Beug H, Wagner EF. p38alpha suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat Genet. 2007;39:741–749. doi: 10.1038/ng2033. [DOI] [PubMed] [Google Scholar]
- 44.Toyabe S, Seki S, Iiai T, Takeda K, Shirai K, Watanabe H, Hiraide H, Uchiyama M, Abo T. Requirement of IL-4 and liver NK1+ T cells for concanavalin A-induced hepatic injury in mice. J Immunol. 1997;159:1537–1542. [PubMed] [Google Scholar]
- 45.Mizuhara H, O'Neill E, Seki N, Ogawa T, Kusunoki C, Otsuka K, Satoh S, Niwa M, Senoh H, Fujiwara H. T cell activation-associated hepatic injury: mediation by tumor necrosis factors and protection by interleukin 6. J Exp Med. 1994;179:1529–1537. doi: 10.1084/jem.179.5.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hatano M, Sasaki S, Ohata S, Shiratsuchi Y, Yamazaki T, Nagata K, Kobayashi Y. Effects of Kupffer cell-depletion on Concanavalin A-induced hepatitis. Cell Immunol. 2008;251:25–30. doi: 10.1016/j.cellimm.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 47.Tsutsui H, Nishiguchi S. Importance of Kupffer cells in the development of acute liver injuries in mice. Int J Mol Sci. 2014;15:7711–7730. doi: 10.3390/ijms15057711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Erhardt A, Wegscheid C, Claass B, Carambia A, Herkel J, Mittrucker HW, Panzer U, Tiegs G. CXCR3 deficiency exacerbates liver disease and abrogates tolerance in a mouse model of immune-mediated hepatitis. J Immunol. 2011;186:5284–5293. doi: 10.4049/jimmunol.1003750. [DOI] [PubMed] [Google Scholar]
- 49.Feng D, Mei Y, Wang Y, Zhang B, Wang C, Xu L. Tetrandrine protects mice from concanavalin A-induced hepatitis through inhibiting NF-kappaB activation. Immunol Lett. 2008;121:127–133. doi: 10.1016/j.imlet.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 50.Maeda S, Chang L, Li ZW, Luo JL, Leffert H, Karin M. IKKbeta is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFalpha. Immunity. 2003;19:725–737. doi: 10.1016/s1074-7613(03)00301-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.