

Abstract
Many theories of autoimmune disease have been proposed since the discovery that the immune system can attack the body. These theories include the hidden or cryptic antigen theory, modified antigen theory, T cell bypass, T cell-B cell mismatch, epitope spread or drift, the bystander effect, molecular mimicry, anti-idiotype theory, antigenic complementarity, and dual-affinity T cell receptors. We critically review these theories and relevant mathematical models as they apply to autoimmune myocarditis. All theories share the common assumption that autoimmune diseases are triggered by environmental factors such as infections or chemical exposure. Most, but not all, theories and mathematical models are unifactorial assuming single-agent causation of disease. Experimental and clinical evidence and mathematical models exist to support some aspects of most theories, but evidence/models that support one theory almost invariably supports other theories as well. More importantly, every theory (and every model) lacks the ability to account for some key autoimmune disease phenomena such as the fundamental roles of innate immunity, sex differences in disease susceptibility, the necessity for adjuvants in experimental animal models, and the often paradoxical effect of exposure timing and dose on disease induction. We argue that a more comprehensive and integrated theory of autoimmunity associated with new mathematical models is needed and suggest specific experimental and clinical tests for each major theory that might help to clarify how they relate to clinical disease and reveal how theories are related.
Keywords: autoimmune disease theories and modeling, myocarditis, infection, sex differences, antigenic complementarity
1.1 Introduction
In this review we have four primary goals. One is to test the adequacy of autoimmune theories that were largely derived in animal models to describe clinical disease (Table 1). Secondly, we examine the mathematical models that have been developed for each major theory of autoimmunity. Third, we argue that there is a need for more integration across theories, across mathematical models, and between theories and mathematical models, particularly in light of our more recent understanding of the importance of innate immunity in the development of autoimmune disease. And our final goal is to highlight problems with individual theories and mathematical models that may lead to the development of novel or hybrid theories of greater explanatory and predictive power. In an ideal world, a good theory of autoimmunity combined with insightful modeling should lead to new and better approaches to effective translational research.
Table 1.
Theories on causes of autoimmune disease
| Theory | Description | References |
|---|---|---|
| Hidden/cryptic antigen | Tissue damage releases hidden antigens | 40, 41 |
| Epitope spread | Multiple epitopes against self needed before autoimmune disease develops | 57, 58, 59 |
| Anti-idiotype | Cellular receptor targets induce crossreactive autoAbs | 74, 89 |
| Molecular mimicry | Accidental crossreactivity | 28, 97–102, 135 |
| Bystander or adjuvant effect | Microbial or cytokine activation of pre-existing autoreactive immune cells | 135, 162–166 |
| Dual TCR | Non-specific activation of 2nd TCR | 28, 178 |
| Antigenic complementarity | Multiple infections by microbes that share antigenic complementarity/cross-reactivity | 185–191, 202 |
| Co-infection (or co-exposure) | Releases self tissue and activates immune response, may involve crossreactivity or antigenic complementarity |
In order to achieve our goals in a relatively short review article such as this one we have imposed three constraints on our content. The first is to limit our discussion of autoimmune theories to myocarditis. Myocarditis is clinically and experimentally well-characterized, and most of the major theories of autoimmunity have been tested using it making it well suited to our purpose. The second constraint is to limit our discussion to data that represent critical tests of assumptions that underpin specific theories or that can differentiate between theories. We will not, therefore, make any attempt at completeness, nor does this seem necessary in light of the many previous reviews on the topic. The third constraint is to focus these critical tests of theories to points that have potential clinical relevance or future treatment implications for myocarditis patients. Because all of the theories that are used to direct research on myocarditis and to explain the resulting data are also used to understand other autoimmune diseases, we are reasonably confident that the general conclusions that we reach in this review will be applicable to a much wider range of experimental and clinical autoimmune diseases.
It is important to note from the onset that for most theories described here there is a significant body of literature substantiating its case and in some cases mathematical models to explore its mechanisms. However, data “proving” each theory are open to different interpretations according to at least two, and often more, theories of autoimmunity and their mathematical models. Although it would be convenient to have a “crucial experiment” that clearly “proves” one theory or model correct and all the rest wrong, such is not the case. Theories are, in reality, built on systems of experimental studies and models assume the validity of the theories they mathematize. The value of a theory is based on three fundamental functions: one is to connect the most data in the most meaningful way; the second, to do so with the fewest assumptions; and the third, to predict connections (and therefore testable phenomena) that have yet to be observed. Good mathematical models facilitate these three functions. Thus, the value of a theory (and its mathematical and animal models) is not found in whether there are data that support it, but rather how much data have accumulated for which it cannot account and how many predictions it makes that cannot be validated. Because these are the most important aspects of theory evaluation, we have focused our review on what each theory has not accomplished and the data and predictions each makes that differentiate it from other theories. In this sense, our review is not about what we know regarding autoimmunity, but rather about the problematic aspects that reveal what we do not know.
1.2 Myocarditis
Before discussing theories, a brief summary of clinical and experimental models of myocarditis is needed. Clinically, myocarditis is defined as inflammation of the myocardium and is a relatively rare autoimmune disease. Myocarditis is also frequently associated with inflammation of the pericardium, a single cell layer on the outside of the heart, and termed perimyocarditis or myopericarditis [1]. No formal epidemiology studies exist on the incidence of myocarditis, but based on autopsy records myocarditis occurs in approximately 10% of cases of sudden death [2]. However, it is thought that myocarditis is likely to occur asymptomatically in a larger percentage of individuals [1]. This is at least partly because so many different environmental agents, and particularly infections, are known to be able to cause myocarditis like viruses, bacteria, parasites, and drugs [3, 4]. Myocarditis is a leading cause of sudden death in individuals under age 40 [5] and may lead to dilated cardiomyopathy (DCM) and chronic heart failure predominantly in men (women with myocarditis are far more likely to recover without progressing to DCM) [3, 6].
Myocarditis can be induced experimentally in mice using infections such as coxsackievirus B3 (CVB3), murine cytomegalovirus (MCMV), encephalomyocarditis virus (EMCV), reovirus, influenza virus, parvovirus, and the parasite Trypanosuma cruzi (modeling Chagas disease) or adjuvants (i.e., complete Freund’s adjuvant/CFA supplemented with inactivated Mycobacterium tuberculosis and/or pertussis toxin) with self-peptide (usually cardiac myosin) [7–9, reviewed in 10]. Myocarditis induced by adjuvant and self peptide is termed experimental autoimmune myocarditis (EAM). Interestingly, the time-course of disease progression from myocarditis to DCM is similar between animal models and human disease. Regardless of the agent used to induce myocarditis, the primary infiltrate during the acute stage of disease in patients and mice are macrophages (about 80% of infiltrate) followed by T and B cells (around 10–15% of the infiltrate) [11–13]. Autoimmune diseases have historically been considered as T and B cell-mediated diseases, but more recently the importance of innate cells like macrophages is being understood. For example, T cells have been considered to be the primary cells mediating damage in the classic autoimmune disease model experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis, but a new understanding of the importance of resident brain macrophages, termed microglia, is beginning to be appreciated [14].
The severity of inflammation during acute and chronic myocarditis and the progression to DCM is more severe in male mice with myocarditis, similar to myocarditis patients [6, 12, 15, 16]. Macrophages and mast cells, which are elevated in males, play a central role in driving the cardiac fibrosis that leads to DCM and chronic heart failure [12, 15]. Autoantibodies (autoAbs) against cardiac myosin are present in myocarditis and DCM patients and experimental models where they can contribute to cardiomyopathy [17–19]. In animal models, AutoAbs to cardiac myosin arise during acute myocarditis and are thought to contribute to chronic pathology by deposition of immune complexes (ICs) on/in the heart [17, 18].
1.3 Innate vs. Adaptive Immunity
Before discussing theories it is also important to emphasize the role of innate immunity in the pathogenesis of autoimmune disease. Many of the theories that we will be discussing focus on the role of an antigen-specific adaptive immune response in the development of autoimmune disease. But it is important to realize that many of these theories were devised before the realization of the essential role the innate immune response plays in the development of adaptive immunity; an understanding that began quite recently around 2000 [17, 20]. Now we know that the innate response “specifically” directs the adaptive immune response, not with antigen/epitope specificity but it directs the type of immune response (i.e., T helper (Th)1 vs. Th2) based on innate receptor activation such as Toll-like receptors (TLRs) and the inflammasome. Recent examination of the initiation of immune responses in autoimmune disease animal models reveals that innate mechanisms like danger-associated molecular patterns (DAMPs) and TLRs strongly drive reactivity to self and determine the type of adaptive immune response (i.e., Th1, Th2) [21–26]. However, most review articles and manuscripts discussing possible theories of autoimmunity do not reinterpret theories in light of the new understanding for the role of innate immunity in the process (Table 2). This is critically needed. The focus of many investigators (and review articles) continues to be on either innate or adaptive immunity, rather than integrating both.
Table 2.
Issues that need to be addressed in revised theories and mathematical models of autoimmune disease
|
Another underlying assumption of many theories is that autoimmune disease results because of a “defect” in the adaptive (or innate) immune response (Table 2) [22, 27, 28]. Autoimmune disease was originally defined in this way because in the 1950s, when theories were first being proposed, it was believed that the immune response would not attack “self” [29]. However, we now realize that self-reactivity is part of the normal regeneration and healing process [30]. Cellular debris (i.e., damaged self) must be removed when cells are damaged or die of natural causes, and the innate immune system (especially macrophages) plays a central maintenance and healing role in this process. AutoAbs and ICs are also important in clearing damaged cells/self from the body by binding to receptors on macrophages. Thus, we need to realize that autoreactive T and B cells can mediate homeostasis and healing as well as disease, and are not necessarily “defective”.
We now also have a new understanding of the role “adjuvant” plays in experimental models of autoimmune disease. Historically, the oil component of adjuvant was thought to provide a “depot” of antigen for antigen presentation, and the Mycobacterium and/or pertussis component thought to nonspecifically stimulate the immune response and allow vascular access to the injection site. Recent evidence indicates that many adjuvants, such as Mycobacterium, aluminum hydroxide (Alum) and Pertussis toxin, used to induce autoimmune disease in animal models stimulate specific TLRs and the inflammasome [31–33]. This means that the adjuvant does not just provide a “non-specific” stimulus to the immune system as previously thought (Table 2). This also explains why incomplete Freund’s adjuvant (IFA), without Mycobacterium, is not able to induce autoimmune disease in animal models [31]. Additionally, adjuvant and self peptide must be administered at the same time (i.e., day 0) or autoimmune disease does not develop [8], further indicating the important role the adjuvant plays in driving the innate immune response to self peptide. Many discussions on theories of autoimmunity continue to “ignore” the role the adjuvant plays in driving disease and focus only on the self peptide. We need to revise our theories based on this new understanding of the essential role of the innate immune system in driving disease.
We desperately need mathematical models that integrate the innate immune system into considerations of autoimmune disease as well. We have found only one paper that investigates mathematically the possible role of innate immunity in conjunction with adaptive immunity in the induction of autoimmune disease. Most mathematical models of autoimmunity assume that two key factors of the adaptive immune response control the outcome of responses to autoantigens, effector T cells and regulatory T cells (Treg). Segal and Bar-Or developed a model of autoimmunity that dispenses with Treg and incorporates instead the influence of cytokines [34]. The model suggests that cytokine levels are critical determinants of whether any particular immunological trigger results in a Th1 or Th2 response and, in turn, whether autoimmunity ensues. The model further proposes that cytokines are components of immunological memory and that their manipulation can prevent or treat certain autoimmune diseases like acute myocarditis. In addition, the model makes the unique prediction that presenting an auto-immunogenic trigger under cytokine conditions that favor a Th2 response can prevent autoimmune disease. This modeling approach begins to incorporate the concept of innate immune influence (e.g., innate cytokines) over adaptive immunity and ideas like this merit significant development in the future.
1.4 Theories
If evidence of our ignorance concerning the causes of autoimmunity is needed, it should suffice to observe that at least a dozen different theories currently vie to explain the phenomena. Of these, we will address the following theories: 1) hidden or cryptic antigen theory; 2) epitope spread or drift; 3) anti-idiotype theory; 4) molecular mimicry; 5) the bystander or adjuvant effect; 6) dual-affinity T cell receptors (TCR); 7) antigenic complementarity theory; and 8) co-infections or co-exposures (Table 1). All of these theories have the common assumption that while there is undoubtedly a genetic predisposition to autoimmune disease [35–37], predisposition requires environmental triggers [38, 39]. We will focus on the role different autoimmune theories propose for environmental agents, and infections in particular, in triggering autoimmunity. We will not address autoimmune theories that attribute autoimmunity to altered or modified antigens, processes such as T cell bypass, or T cell-B cell discordance. These theories have very little evidence to support them and few clear clinical implications.
We have chosen to address some experimental and clinical variables separately from the individual theories of autoimmunity, especially epidemiology, sex differences in susceptibility to disease, the role of innate immunity, and how the timing of exposure affects the immune response. Experimental investigation of these four factors appear to us to be capable of providing clear tests to distinguish between theories, to expose fundamental gaps that may require elaboration of new theories, and to provide novel clinical and therapeutic opportunities.
Finally, we distinguish between autoimmunity, a natural production of antibody and T/B cell responses to self antigens as part of immune surveillance, and autoimmune disease, which is the production of self-reactive antibodies and T/B cells that result in abnormal cell and tissue destruction leading to chronic inflammation.
1.4.1 Hidden Antigen Theory (HAT)
The hidden or cryptic antigen theory (HAT) is the oldest theory regarding the origins of autoimmune disease, and dates to the discovery of autoimmune disease itself at the end of the nineteenth century [29]. According to HAT, some self antigens are “cryptic” or “sequestered” and therefore not “seen” by the innate immune system. Because such self antigens are “hidden”, potentially autoreactive T and B cell clones against them are not deleted or tolerized. In most cases, such “hidden” antigens are expressed within immunologically privileged tissues or organs such as the testes, eyes and brain. Tissue or organ damage that results in the release of hidden antigens is posited to result in the activation of autoreactive clones and the induction of autoimmune disease (Figure 1) [40, 41].
Figure 1. Hidden Antigen Theory (HAT).

Left: Some antigens (black pentagons) are sequestered within cells or tissues that are inaccessible to the developing immune system so that their corresponding T cells are not deleted or tolerized. Tissue damage or infection (black dots) activates an immune response (stellated cells). Center: Cellular or tissue damage releases hidden antigens, which provoke a second immune response (antibody shapes). Right: Autoimmune attack directed at the cells harboring the hidden antigens. The tissue damage or infection that provoked the release of hidden antigen is likely to be resolved long before the autoimmune effects are observed and the initiating cause therefore remaining obscure [40, 41].
There are a number of considerations regarding HAT and the development of myocarditis. The heart is not an immunologically privileged site, but the specific targets of autoimmune myocarditis are generally thought to be intracellular proteins, including cardiac myosin, actin and troponin, which are not usually “seen” by the immune system. However, some strains of mice express cardiac myosin in the matrix and experimental myocarditis can be induced with transfer of autoAbs that form ICs in the heart [41]. This data suggest that if autoAbs against hidden antigens are present and the antigens are made available, possibly through physical, infectious or chemical damage to the heart [42–45], then autoimmune disease could develop. Evidence that cardiac myosin and troponins are primary targets of the immune response comes from the ability of these proteins to induce myocarditis if administered with an adjuvant like CFA in mice [8, 46]. AutoAbs against all of these major cardiac antigens are found during myocarditis [44]. Additionally, troponin I is used as a clinical biomarker for cardiovascular damage during myocardial infarction, myocarditis, and DCM [47, 48], indicating that troponin I is released systemically during acute myocarditis. However, there is a problem of “timing”. AutoAbs appear in viral models of myocarditis at the same time as damaged cardiac tissue is being released due to peak viral replication. [17, 44].
Another unresolved question when evaluating evidence for or against HAT is that most “healthy” people have low levels of cardiac autoAbs. Cardiac infarction, heart surgery, and cardiac transplantation result in the production of actin and cardiac myosin autoAbs that correlate with the risk of acute cardiac transplant rejection [49]. Data from cardiac trauma patients suggest that factors other than the mere presence of autoAbs against cardiac antigens are needed for the development of myocarditis. If release of self antigen was sufficient to induce autoimmune disease on its own, as the HAT theory suggests, then administration of damaged self tissue alone should be able to cause autoimmune disease in animal models. However, this is not the case. Administration of damaged self tissue, antigens and/or peptides always requires adjuvants that contain inactivated microbes and/or toxins to initiate disease (Table 2).
In recent years it has been discovered that DAMPs like interleukin (IL)-33, heat shock proteins (HSPs), and high-mobility group box family (HMGB) proteins are released from tissues when they are damaged and stimulate the innate immune response [50]. IL-33 is present at high levels in cardiac tissue and has been shown to be able to induce pericarditis and heart failure when administered to mice on its own, without viral infection [51]. When IL-33 was administered with CVB3, mice developed severe myocarditis and rapidly progressed to DCM and heart failure [51]. In fact the model used in these experiments pairs a mild CVB3 infection with injection of damaged heart protein, rather than purified cardiac myosin [8]. This damaged heart includes proteins like IL-33, cardiac myosin and actin. We have found that this strain of CVB3 (Nancy strain) induces little or no inflammation in mice without the concurrent administration of damaged heart protein. Thus, experiments need to be conducted to determine the role of cardiac derived-DAMPs in the development of myocarditis in the presence of cardiac proteins and/or infection.
Cardiac damage may be caused by viral or bacterial infection. Notably, the degree of cardiac damage experienced in autoimmune rheumatic fever is not different than that experienced in scarlet fever, which does not result in chronic autoimmune heart disease [52]. Interestingly, in animal models it is possible to induce autoimmune heart disease without the use of active infection. Valvulitis (inflammation of the cardiac valves) can be induced with recombinant group A streptococci (GAS) M protein (the immunodominant protein of GAS) combined with CFA [8, 53]. Although valvulitis is considered to be a clinically distinct disease from myocarditis, these experiments suggest that damage to cardiac tissue resulting in release of self peptide may not need to be present for induction of autoimmune heart disease. To the contrary, these experiments suggest that the tissue destruction that occurred in this case was caused by the immune system itself.
Importantly, inactivated CVB3, M protein from GAS, or cardiac myosin in the absence of adjuvants are not capable of inducing autoimmune heart disease. Non-infectious antigens require “adjuvants” for the development of autoimmune disease. The requirement for adjuvants to induce autoimmune disease in animal models is badly in need of explanation by HAT and many other theories, which often overlook the requirement for adjuvants when describing their theories (Table 2). Thus, HAT is unlikely to explain the development of autoimmune disease on its own, but may work in combination with other theories.
One final drawback to the requirement for cryptic epitopes in clinical autoimmunity is how such epitopes can be generated in sufficient amounts to be immunogenic and how they can be loaded onto MHC molecules for antigen presentation. The fact that exogenously delivered cryptic epitopes of cardiac myosin in the form of syngeneic peptides can cause myocarditis in the presence of appropriate adjuvants [54] does not address how syngeneic peptides would be generated naturally. The classical explanation for cryptic epitope generation given by immunologists is that certain activation states might result in novel proteases being expressed that cleave self-proteins into unique peptide sets compared to standardly expressed proteases. It is possible that infectious agents that trigger autoimmune diseases may encode their own proteases that also cleave self-proteins into novel autoantigenic peptide fragments. Such novel cleavage of dystrophin has been demonstrated by Badorff and Knowlton in enterovirus-induced myocarditis [55]. The major problem left by the novel-cleavage-by-pathogen-proteases mechanism is why cryptic autoantigen-release leads to autoimmune disease in only a tiny fraction of infected individuals.
In summary, the development of anti-cardiac protein autoAbs does correlate with the degree of preceding cardiac tissue damage but not with induction of autoimmune disease. The presence of autoantibodies following many types of heart damage suggests that acute autoimmunity usually plays a role in tissue healing rather than producing further damage (Table 2). Thus, the presence of autoantibodies, even at high titers, and to presumably hidden antigens, is neither necessary nor sufficient to induce autoimmune myocarditis. Although in some cases autoAbs directly cause disease (i.e., Graves’ disease, myasthenia gravis), for most autoimmune diseases the role of autoAbs and/or ICs as the “initiator” of disease remains unclear. The role of hidden antigens in the induction of autoimmune disease therefore appears questionable.
Mathematical Models of HAT
As far as we know, only one mathematical model assumes that hidden antigens are a necessity for inducing autoimmune disease. Borghans and DeBoer propose that “autoreactive T lymphocytes [to hidden antigens] are neither activated nor negatively selected” so that tolerance to hidden host antigens is effectively a passive state [56]. Exposure to hidden host antigens can activate autoreactive T cell clones, resulting in autoimmune disease. Alternatively, sub-pathogenic exposures to antigens or passively transferred autoreactive T cells, will stimulate Treg cells that protect against autoimmune disease. The obvious limitation of this model is that, as discussed above, not all targets of autoimmune disease are hidden antigens. The model shares with HAT the less obvious problem that release of hidden antigens following tissue or organ damage should induce autoimmune disease, but does not. A model that explicitly addresses how the immune response to self-antigen is well-regulated while exposure to pathogens or other environmental agents trigger autoimmune disease would be very welcome.
Another area where new mathematical models might be of value would be in exploring the theory that novel autoantigens are generated by pathogen-produced proteases. Obviously, this possibility would be difficult to model mathematically as it depends on factors like being able to detect microbial levels (or at least their protease levels), the type of cells infected, and the number of host cells infected (i.e., myocytes). Importantly, persistent viral infection may still generate virus proteases and cryptic epitopes even if the virus is not actively replicating.
Novel Experiments and Clinical Studies
It is important to determine whether cardiac surgery, artificially-induced heart attack, or exposure to cardiac-damaging chemicals preceding infection with CVB3 or other infections result in a higher likelihood of myocarditis in mice, and if disease increases whether this is due to the release of hidden antigens. These experiments must take into consideration the effect on the immune response of DAMPs released by cardiac injury. Similarly, is there any evidence that individuals who develop myocarditis were exposed to cardiac injury or cardiac-damaging drugs (e.g., chemotherapies, antibiotics) preceding the onset of myocarditis? The protease-produced novel autoantigen mechanism is also testable: epitopes obtained from the MHC of antigen presenting cells after infection could be compared to cardiac myosin epitopes known to be capable of inducing EAM.
1.4.2 Epitope Spread Theory (EST)
An epitope is a single antigenic site targeted by one specific antibody or T cell. The antigenic drift or epitope spread theory of autoimmune disease was put forward in 1992 by Lehmann et al. to explain a common observation that the dominant self-epitope/antigen targeted in an autoimmune disease is often different (and non-crossreactive) with those epitopes that were targeted during the initial stage of the autoimmune process [57–59]. Epitope spread occurs as part of the normal immune response to control infections. Initially the immune response recognizes a dominant antigen of the infectious agent and produces a T and B cell-specific response against it. When it later reencounters the same pathogen it produces an immune response against a second dominant antigen of the pathogen so that the adaptive immune response becomes better able to prevent infection with each future event, recognizing increasing numbers of epitopes for each microbial agent. This is the main reason why influenza vaccines must be changed each year because the virus evades the immune response by changing the “dominant” antigens on its surface membrane. It is well known, and often part of an autoimmune disease diagnosis, that autoimmune diseases usually only present clinically after several autoAbs directed against the target organ are present [60]. These observations suggest that infections or other agents that can cause release of and/or induce the immune system to target self antigens must be re-occurring so that the immune response spreads sufficiently to counteract them. The effect of recurrent infections on myocarditis is virtually unstudied (Table 2) [61].
One question is why does the immune system increasingly recognize different self-antigens over time? This could occur for a number of reasons. Aging itself has been found to increase B cell autoreactivity [62]. Dysregulation of the immune response could lead to epitope spread. Or, the immune system could be attempting to more effectively respond to tissue damage using epitope spread similar to the epitope spread that occurs during infections such as influenza, when the immune system responds to increasing numbers of viral antigens with each outbreak. While epitope spread of autoAbs has been studied in detail in animal models of EAE [63] and Theiler’s virus infection of the central nervous system [64], very little is known about epitope spread in myocarditis either in animal models or patients. Using a cardiac C-protein/CFA model of autoimmune myocarditis, Matsumoto et al. demonstrated that B cell epitope spreading was important for the development of DCM in Lewis rats [65]. Moreover, B cell epitope spreading was dependent on pathogenic T cells, the activation of which required additional inoculations of the initiating C-protein epitope in combination with antisera. Thus, epitope spread may require multiple pathogenic triggers for disease induction or maintenance.
The possibility that multiple pathogenic effectors may be required for epitope spread leads us to place particular emphasis on the fact that EST requires that the initiating antigen be different from and non-crossreactive with the target antigen in autoimmune disease. If this requirement is not met, then EST is no different than molecular mimicry where one antigen resembles another, which we will discuss below. Consider the case of myocarditis induced by CVB3 infection. Most investigators agree that the main autoimmune epitope targeted in the disease process is cardiac myosin, yet the initial antibodies produced in direct response to CVB3 do not crossreact with cardiac myosin nor do myosin antibodies recognize CVB3 [66, 67]. Other autoAbs have been found that target cardiac proteins and receptors during the chronic stage of disease (i.e., DCM) like the β2 adrenergic receptor (β2AR) and muscarinic acetylcholine receptor (AChR) [68–73], but it is not known whether any of these autoAbs appear prior to cardiac myosin autoAbs or vice-versa. Thus, it is not known whether EST is generally applicable let alone necessary for the induction of human autoimmune disease, and the conditions under which epitope spreading occur are still essentially unknown.
Mathematical Models of EST
We have found no mathematical models of EST. Given the extensive experimental and clinical evidence for EST, mathematical models of this phenomenon would be highly desirable but will be difficult to develop given current uncertainties about the necessity for repeated antigen exposure, T cell activation, and so on.
Novel Experiments and Clinical Studies
Does epitope drift occur in viral myocarditis or EAM animal models? Does autoAb epitope specificity differ in molecular targets or over time in EAM vs. myocarditis induced by infections? Do multiple cardiac autoAbs increase the risk for acute myocarditis or chronic DCM and heart failure in patients? Is T cell activation required in models of EAM other than the cardiac C-protein-induced model?
1.4.3 Anti-Idiotype Theory (AIT)
We noted in our discussion of EST that autoAbs in DCM patients are often induced to cell surface proteins including receptors like the β2AR and muscarinic AChR. The anti-idiotype theory (AIT) of autoimmune disease proposes that receptors used by infectious agents, particularly viruses, are the primary targets of the immune response resulting in autoimmune disease [74, 75]. Antibodies directed against the viral ligand that binds the cell receptor could then bind to the cell receptor on host cells becoming a so-called anti-idiotype autoAb. Anti-idiotype antibodies would thereby target the same tissue or cell type as the infectious agent. In this way, an anti-idiotype response to an infectious agent could lead to autoimmune disease (Figure 2). This theory suggests that the viral or microbial ligand would be a dominant antigen targeted by the immune response.
Figure 2. Anti-Idiotype Theory (AIT).
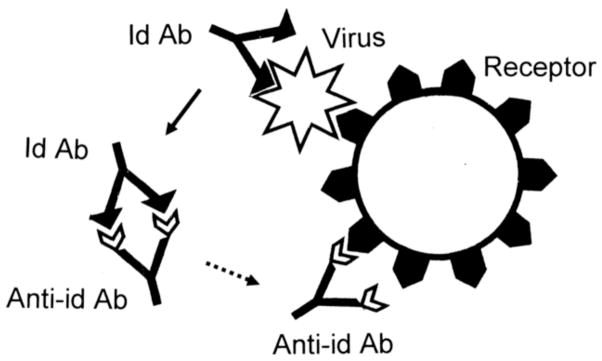
Viruses and other microbes (white stellate forms) utilize molecularly complementary cell surface receptors (black pentagons) in order to target specific cell types. Some idiotypic antibodies (Id Ab) against such microbes will be complementary to the microbial ligands used to target infection. Such idiotypic antibodies will therefore mimic the cell surface receptors. Autoimmune disease may arise if the idiotypic antibodies induce an anti-idiotypic response (Anti-Id Ab) because the anti-idiotypic response will mimic the microbial ligand, therefore attacking the cell surface receptors [74, 89].
It is important to pause for a moment to consider the differences between EST and AIT. In epitope spread, immunodominance shifts from one antigen to an unrelated one as the immune system adapts to the range of antigens presented to it. In contrast, AIT proposes that the shift from one antigenic target to another is mediated not by the presence of multiple antigens (cryptic or not), but by the induction of the idiotypic antibody response itself. If this idiotypic response is to an epitope complementary to a “self” epitope, then the induction of an anti-idiotype has the potential to induce autoimmunity. Thus, while both theories have the potential to shift the antibody response over time, they do so through very different mechanisms.
The application of AIT to myocarditis is straightforward. Clinically, what appear to be anti-idiotype antibodies are found in both patients and in animal models of myocarditis. Paque and Miller characterized the development of anti-idiotype antibodies in CVB3-induced myocarditis, demonstrating that while idiotypic antibody against CVB3 is present by 7 days after infection, anti-idiotype antibodies arise only at two weeks and peak at three [67, 76–79]. Similar to Neu et al. [66], Paque and Miller observed that idiotypic anti-CVB3 antibodies do not crossreact with cardiac myosin [67]. Rather, Paque and Miller maintain that anti-cardiac myosin reactivity arises only with the induction of the anti-idiotypic antibodies [67]. If anti-cardiac myosin antibodies are actually anti-idiotypic, it would follow that the original receptor for CVB3 would mimic myosin. In fact, both coxsackie-adenovirus receptor (CAR) and decay accelerating factor (DAF) also called CD55, which are used by some strains of CVB3 as receptors, have multiple and statistically significant similarities to cardiac myosin [80]. Several examples are illustrated in Figure 3. Alternatively, CVB3 may itself induce anti-idiotypic antibodies that recognize cardiac myosin: CVB3 mimics actin; actin is complementary to cardiac myosin so that anti-CVB3 (actin) antibodies mimic myosin [81]. Thus, anti-anti-CVB3 (actin) antibodies would behave like anti-cardiac myosin antibodies.
Figure 3. Similarity between Coxsackievirus and Adenovirus Receptor (CAR) and Human Cardiac Myosin (MYH6).

A similarity search using LALIGN reveals that CAR has multiple regions mimicking cardiac myosin (two of which are shown) so that putative anti-cardiac myosin antibodies in CVB3-induced myocarditis may originate as anti-idiotypic antibodies directed at CAR, as predicted by AIT. According to the anti-idiotype theory (AIT), a coxsackievirus infection might lead to the production of anti-idiotype antibodies that mimic the receptor it uses to infect heart tissue (see Figure 2). One of these receptors is CAR. No evidence of antibodies against CAR have been reported in myocarditis, but we suggest that perhaps antibodies have been misidentified as anti-cardiac myosin antibodies since CAR shares significant similarities to cardiac myosin and anti-cardiac myosin antibodies are common in myocarditis [80, 81].
While the presence of anti-idiotype autoAbs in myocarditis would seem to support the utility of AIT for understanding the initiation of disease, a number of problems complicate the matter. The first difficulty concerns lack of evidence for antibodies against cardiac cellular receptors used by initiating viral or bacterial pathogens. Two of the best-characterized infectious agents associated with autoimmune heart disease are CVB and GAS, both of which can infect cardiac tissue. Some, but not all, of the cellular receptors used by these infectious agents are known. GAS uses a number of extracellular proteins to adhere to cardiac myocytes including laminins [82], while CVB3 and adenoviruses often use CAR and DAF to enter target cells [83]. While knocking out the CAR gene does prevent CVB3 infection of cardiac muscle and subsequent myocarditis in mice by preventing active infection [84], we can find no experimental or clinical evidence that antibodies, idiotypic or anti-idiotypic, are generated against CAR or DAF in myocarditis. Moreover, myocarditis caused by strains of CVB3 that use CAR as a receptor for viral entry lead to high viral replication in the heart, massive necrosis, and rapid death of mice but only low myocardial inflammation [85, 86], a pathogenesis that does not translate well to most myocarditis patients. In contrast, other strains of CVB3 like the Nancy strain do not appear to use CAR or DAF when infecting Vero cells, which do not express these receptors. The receptor(s) used by CVB3 in this circumstance is not known, but produces a mild viral infection in mice that closely resembles human disease when injected with damaged heart proteins [8, 13, 86]. Thus, if AIT applies to myocarditis, it probably involves CVB receptors that have not yet been identified. Possibilities include the β2AR and AChR, which have been found to target the chronic phase of myocarditis (i.e., DCM) and might fit the profile of anti-idiotype autoantibodies [68–73]. However, several investigators have isolated at least five additional, as yet unidentified, proteins to which CVB3 binds that may also represent receptors [87, 88].
The second problem with applying AIT to myocarditis is that while Plotz proposed that anti-idiotype antibodies are pathogenic [74], Paque and Miller [76–79] and Weremeichik et al. [89] have shown that anti-idiotypic antibodies produced during CVB3-induced myocarditis down regulate disease. Weller et al. further demonstrated that induction of an anti-idiotype response by immunoglobulins is part of the mechanism by which immunoglobulin therapy can prevent CVB3-induced myocarditis in mice [90]. These results suggest that anti-idiotype antibodies in myocarditis may be palliative rather than causative, similar to rheumatoid factor [91].
In sum, anti-idiotype autoantibodies do appear in myocarditis, but have not been demonstrated to target the receptor proteins that CVB3 is known to use to infect cells. Thus, the receptors that lead to anti-idiotypic autoAbs remain unclear and the possibility that they play a protective, rather than pathogenic, role in myocarditis complicates matters.
Mathematical Models of AIT
No mathematical model of AIT, per se, appears to have been attempted. On the other hand, disturbances in idiotype-anti-idiotype networks have been mathematically modeled by several groups under the assumption that anti-idiotype immune responses are regulatory, either preventing or moderating disease. Thus, the current mathematical modeling literature suggests, if only by implication, that anti-idiotypes are not causes of autoimmune disease, but an attempt to control autoimmunity. For example, in Cohen and Atlan’s automata-based theory antigen-specific T cells induce anti-idiotypes that cause autoimmune disease, but these anti-idiotypes “vaccinate” against disease [92]. Several differential-equation-based models of anti-idiotype function model similar behavior and have been developed specifically to address the mechanism(s) by which T-cell vaccination can be accomplished in various animal models of disease [93, 94]. The implications of these models is that anti-idiotypes are regulatory, which is the opposite of Plotz’s theory that anti-idiotypic responses initiate autoimmune disease.
Mathematical models have also cast doubt on whether idiotype-anti-idiotype networks play a role in immune regulation. DeBoer and Hogeweg concluded that idiotype-anti-idiotype network models based on proliferation of antibody production due to antigen stimulation were unable to explain basic immunological phenomena such as regulation, immunity (memory), and self-non-self discrimination making them poor candidates to explain autoimmunity [95]. Whether the inability to successfully model the immune system using idiotype-anti-idiotype network approaches is a mathematical problem or an intrinsic failure of Jerne’s network theory to describe immune system behavior remains to be determined. In contrast, Sulzer and Weisbuch [96] developed a differential equation-based model of an immune system regulated by idiotype-anti-idiotype interactions that resolves many of the intrinsic problems that DeBoer and Hogeweg [95] had attributed to network models. Sulzer and Weisbuch found that in the instance where one or both of the idiotype anti-idiotype clones are self-reactive, the system could take on any of three states [96]. If clonal activation is low, then it is suppressed by the anti-idiotype; if clonal activation is moderate, then the system is tolerant; and if clonal activation is high, then autoimmunity results. Sulzer and Weisbuch’s model therefore suggests that AIT is plausible, the key factor being the degree to which the pathogen stimulates the idiotype-anti-idiotype network and the extent to which host antigens continue to drive the autoimmune process. However, the issue of whether Plotz’s version of anti-idiotype stimulation can produce autoimmune disease remains an open one for those mathematically modeling the immune system.
Novel Experiments and Clinical Studies
AIT needs to be evaluated with a range of new experiments and clinical tests. First, it would seem possible to produce anti-idiotype antibodies in inbred mice against key antibodies associated with myocarditis (e.g., anti-CVB, anti-cardiac myosin, etc.) and to inoculate naïve mice of the same strain with these anti-idiotype antibodies. If AIT is correct, then these anti-idiotype antibodies should be sufficient to induce myocardial inflammation. Alternatively, the effects of these anti-idiotype antibodies on the course of myocarditis could be studied to determine whether they exacerbate or ameliorate disease symptoms. Finally, a dedicated search for anti-idiotype antibodies directed against CAR, DAF, the β2AR, AChR, and other novel CVB receptor candidates [87, 88] should be undertaken. The possibility that these receptor anti-idiotypes might correspond to anti-cardiac myosin autoAbs should also be considered [80, 81].
1.4.4 Molecular Mimicry (MM) Theory
The theory of molecular mimicry (MM) provides one way out of many problems posed by HAT, EST and AIT. The concept of “molecular mimicry” was first posited by Damian in the early 1960s to explain how parasites evolved proteins that mimic host proteins in order to camouflage themselves from the immune system [97, 98]. The concept was broadened substantially by Lane and Koprowski who described increasingly frequent reports that antibodies induced by various pathogens could crossreact with host cellular proteins [99]. Pathogens in general, they suggested, have evolved to display proteins that have antigenic similarity to the proteins of their hosts. The possibility that molecular mimicry might lead to autoimmune disease was proposed a year later by Kaprowski in collaboration with Fujinami and Oldstone [100–102].
The essence of MM theory as a cause of autoimmune disease is that antigens of infectious agents mimic host proteins sufficiently to result in antibodies, B cells or T cells that crossreact with host cells (Figure 4). Srinivasappa et al. reported that 3% of antibodies against human pathogens result in crossreactivity [103]. It is hypothesized that if the crossreactivity is sufficiently robust, there may be loss of tolerance and autoimmune disease may result. An issue that remains unresolved is whether antigen-host similarity is determined by linear or conformational epitopes, or both [99]. Most studies of molecular mimicry have relied on linear epitope similarities since simple tools for comparing protein conformations are generally lacking.
Figure 4. Molecular Mimicry (MM) Theory of Autoimmune Disease.
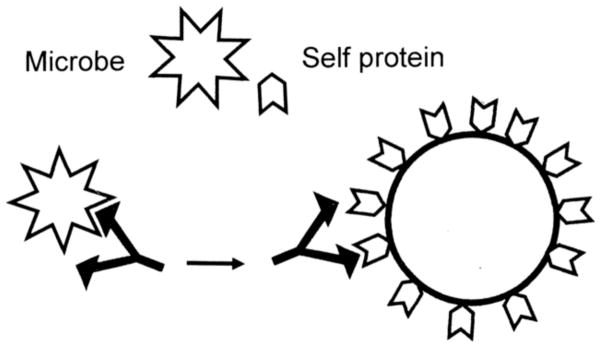
Many microbial proteins mimic host proteins resulting in epitope mimicry. Antibodies or T cells activated against a microbial epitope may therefore share weak affinity for the corresponding host epitope so that infection may induce autoimmune disease directed at cells displaying the epitope mimic [28, 97–102, 135].
Crossreactivity between GAS and cardiac proteins was established by Kaplan [104–107] about the same time that Damian first described the concept of molecular mimicry. Wood et al. subsequently identified similar crossreactivity between T. cruzi (the cause of Chagas disease, a type of myocarditis) and cardiac proteins [108]. Williams [109] integrated these studies within the newly proposed concept of molecular mimicry and Froude et al. [110] reviewed accumulating data that Streptococcal antigens mimicked not only host cardiac proteins but also host antigens expressed in many organs. The fact that most pathogens mimic several proteins from multiple organs and tissues is a point that is often lost in discussions of MM and one to which we will return below.
Cunningham et al. provided a major breakthrough in understanding MM as a cause of rheumatic heart disease by demonstrating that a critical locus of mimicry existed between specific peptides of GAS M protein and cardiac myosin [53, 111–113] (Figure 5). Recombinant M protein, M protein fragements, and cardiac myosin have each been used as antigens to induce valvulitis [8, 53, 114, 115].
Figure 5. Molecular (or Epitope) Mimicry of the Streptococcal M Protein for Human Cardiac Myosin.

Cunningham et al. demonstrated that the M protein of group A streptococci has many significant regions of homology with human cardiac myosin (of which only one is displayed here), that this sequence mimicry translates into immunologic cross-reactivity between the epitopes, and that such epitope mimics can be used (with CFA) to induce an animal model of EAM [53, 111–113].
Molecular mimicry is also relevant to CVB-induced myocarditis. Srinivasappa’s team demonstrated that antibodies raised against CVB4 crossreact with cardiac actin and myosin [116]. Several groups extended this work by showing that CVB3 induced antibody and T cell responses against actin, cardiac myosin, tropomyosin, and vimentin during myocarditis [117–121]. Subsequent studies have shown that CVB proteins mimic cardiac actin (Figure 6), that antibodies against CVB3 recognize cardiac actin as an antigen and, conversely, that anti-actin antibodies recognize CVB3 proteins [80, 81].
Figure 6. Molecular (or Epitope) Mimicry of Coxsackievirus B3 for Human Cardiac Actin.

One of many similar protein sequences shared by coxsackieviruses with human cardiac actin that may also act as epitope mimics in autoimmune myocarditis [80, 81].
As noted above, there has been a realization from the outset that not all mimicry may involve linear epitopes. Indeed, Cunningham’s group isolated a monoclonal antibody from a rheumatic heart disease patient that recognized the N-acetyl-βD-glucosamine (GlcNAc) epitope of Streptococci and, with increasing affinity, laminin and cardiac myosin as well [122]. This cross-reactivity displayed by GlcNAc, laminin and cardiac myosin may explain how an antibody response to Streptococci can target not only bacteria but also cardiac valves. The initial destruction of valve endothelial cells may be mediated by antibodies induced by GAS GlcNAc producing, as a side effect, an attack on laminin and cardiac myosin. Notably, the same scenario just described for GAS induction of vavlulitis could theoretically apply to CVB-induced myocarditis as well. Shikhman et al. demonstrated that antibodies against GAS GlcNAc crossreact with the viral protein (VP)1 of CVB3 [123]. Thus, a CVB infection could trigger anti-laminin antibodies causing cardiac myosite destruction, release of cardiac myosin, and subsequent epitope spread toward cardiac myosin as the main target of autoimmune disease. This form of MM therefore integrates some of the most compelling features of HAT and EST while clarifying how a particular tissue is targeted and specific antigens become the focus of subsequent autoimmunity (Figure 7).
Figure 7. Modified Version of Molecular Mimicry (MM) Theory.

One of the difficulties faced by the theory of molecular mimicry as it is applied to autoimmune myocarditis is that cardiac myosin is effectively a hidden antigen (see HAT, Figure 1), represented here by “Self Protein 2”, which should not be “visible” to the immune system. In order for the immune system to attack a hidden antigen such as cardiac myosin, the cells harboring it must be damaged. One way to create such cellular damage is by a viral infection. Another way is if there is a second cell surface protein (“Self Protein 1”) that mimics both the microbial trigger of the disease as well as the hidden antigen. In this modified version of MM, the immune response (antibody shapes) initiated by the microbe will cross-react with the cell surface host protein (“Self Protein 1”) damaging the cell and releasing the more antigenic hidden host protein (“Self Protein 2”). Thus, molecular mimicry between GAS and the coxsackievirus and adenovirus receptor (CAR) (Figure 3) could initiate an attack on cardiomyocytes resulting in damage that releases cardiac myosin; the similarities between GAS, CAR and cardiac myosin would then drive the subsequent autoimmune disease process.
Whether in its original or modified form, MM theory faces a number of problems. One is the question of why only a small proportion of the population who develop immunity to a molecular mimic such as GAS or CVB3 go on to develop myocarditis. While it is estimated that 14% of the population carry genes making them susceptible to rheumatic heart disease [124, 125], only three in a thousand actually develop valvulitis following GAS infection [126, 127]. If GAS were sufficient to induce valvulitis among genetically susceptible people, then this figure should be fourteen in one hundred. Similarly, only about one in 500 people who develop a Coxsackie viral infection develop autoimmune myocarditis [128]. Part of the discrepancy may be due to the fact that the true prevalence of myocarditis is unkown, and likely to be underdiagnosed [129, 130]. But underdiagnosis is unlikely to explain a discrepancy of more than two orders of magnitude. Thus, while molecular mimicry may be necessary to induce autoimmunity, the incidence of autoimmune disease following exposure to mimics suggests that it is not sufficient.
Because MM is so common between microbes and human antigens, one way out of this conundrum of rarity is the possibility that autoimmune disease requires multiple exposures to molecular mimics. Shared mimicking epitopes between multiple pathogens or environmental factors (for example, streptococcal M protein and enterovirus proteins that both mimic cardiac myosin) could result in serial reactivation of autoimmune T memory cells cross-reactive to cardiac myosin with ever increasing pathology [113]. This re-exposure could potentially mitigate the need for adjuvant as multiple reactivation of memory T cells ultimately requires less cytokine and accessory molecule stimulation.
Several other problems with the theory of MM also suggest its insufficiency. One is that despite thirty years of clinical studies, evidence of MM in patients with autoimmune diseases other than myocarditis has been very sparse [131–134]. Even for myocarditis, those studying this disease have been unable to induce disease experimentally using a purified molecular mimic antigen without adjuvant. M protein of GAS is not adequate, by itself, to produce valvulitis without an adjuvant. As with other theories of autoimmunity, there is nothing in MM theory that specifically explains why an adjuvant should be necessary or what role it plays in disease initiation. Thus, even Fujinami, one of the inventors of MM theory has expressed the view that perhaps additional factors, such as a bystander effect or dual affinity TCR, may be required in addition to molecular mimics to induce autoimmune disease [135].
A final problem with MM theory is that mimicry with human cardiac antigens is not limited to pathogens such as CVB or GAS, but occurs indescriminately throughout living organisms. Antigens capable of inducing EAM are found in common foods such as Zea mays (corn) and non-pathogenic pond bacteria [115, 136]. The fact that corn and pond bacterial antigens can induce low level myocardial inflammation when inoculated with an appropriate adjuvant suggests that the way in which a molecular mimic is presented to the immune system, and how the immune system is co-stimulated, determines the response. These additional response-determining factors need to be understood if the causes of autommune disease are to be elucidated. In particular, it would seem logical that non-infectious agents such as corn might not be able to activate cross-reactive T cells in the absence of an appropriate infectious trigger or adjuvant. It seems more likely that exposure to non-infectious agents would induce tolerance since oral presentation of cardiac proteins have themselves been shown to protect mice from myocarditis [137]. How the presence or absence of cofactors such as infections and adjuvants alters antigenicity is an important gap in our knowledge that needs to be rectified.
Indeed, the issue of non-pathogenic inducers of experimental forms of myocarditis raises one final issue regarding the theory of MM, the resolution of which would greatly benefit the field of autoimmune disease research, and that is a rigorous definition, either theoretical, experimental or clinical, of what constitutes disease-relevant mimicry. It is well established that MHC, TCR and antibodies each recognize a more-or-less limited range of epitopes [138, 139], but how closely this recognition is related to the ability of any particular microbial antigen to elicit an immune response capable of inducing autoimmune disease is not understood. Lacking a practical immunological definition of MM, how can the utility of the theory be properly evaluated?
Mathematical Models of MM
A range of mathematical models have addressed various aspects of the theory of MM. One set explores the evolutionary advantages and disadvantages of MM as a strategy for pathogens to evade the host immune system. This literature demonstrates that there is an interesting balance that must be struck between immunological evasion, “costly autoimmunity”, and “functional trade-offs” in proteins that evolve to mimic host proteins. Immunological evasion clearly benefits the pathogen by increasing replication and transmissibility. If mimicry results in autoimmunity in the host, however, pathogen fitness may be lowered by decreasing transmissibility. In addition, modeling suggests that MM may decrease optimal pathogen protein functionality, decreasing infectivity, replication, and transmission. Thus, host autoimmunity may be seen in an evolutionary context as a brake on pathogen exploitation of MM as a means to avoid immune surveillance [140].
A second set of mathematical models has explored the mechanism by which MM results in autoimmune disease. Blyuss and Nicholson, for example, developed a mathematical model for the dynamics of an immune response to a viral infection and the development of autoimmunity, which takes into account T cells with different activation thresholds and cross-reactivity between pathogen and host antigens [141]. The model provides conditions under which infection can be cleared by the immune system, as well as how it can lead to a chronic infection or recurrent infection with relapses and remissions. One assumption underlying the model is that chronic, relapsing forms of autoimmunity may require chronic or recurrent infection. Pinto et al. have provided an alternative model where the major determinant of whether autoimmune disease results from MM is how Treg respond to infection [142]. A greater description and discussion of these and related models is provided by Blyuss and Nicholson [141].
One very notable mathematical model suggests, contrary to MM theory that MM may actually be a means of promoting tolerance to potential antigens. Using an experimentally validated mathematical algorithm for determining MM, Kristóf et al. have modeled the conditions under which gut bacteria mimicking host antigens induce tolerance [143]. Two of their most striking findings were that there is “a strong negative correlation between the similarity of autoantigens to intestinal bacteria and the production of specific autoantibodies” and that “autoantigen length inversely correlated with the production of autoantibodies.” From these findings, Kristóf et al. conclude that “as a longer chain with more epitopes associates with an increased possibility of mimicry to any proteome, MM in general - regarding at least major tissue-specific autoantigens - seems to be rather protective.” Whether their results can be generalized beyond the gut microbiome remains to be seen, but this type of research is certainly worth pursuing because it has the potential to provide insight into our preconceptions about the role of MM as a cause of autoimmune disease.
Novel Experiments and Clinical Studies
The critical issue for testing MM theory in the context of myocarditis is testing its limitations. One outstanding question is whether cardiac myosin mimicry is necessary to induce myocarditis or whether CVB and GAS antigens that mimic other cardiac proteins such as the β2AR, CAR, DAF, actin, troponin, laminin, etc., can also induce experimental myocarditis [80]. It is quite possible that the failure to observe clinical correlates of MM in many diseases is due to using too narrow a focus on a single dominant antigen.
Potentially falsifying experiments also need to be run. GAS and CVB antigens that do not have any observable similarity to cardiac proteins should also be isolated or synthesized. The most important of these would be proteins that GAS or CVB use to bind to cardiac myocytes, which are therefore complementary to, rather than mimics of, cardiac proteins. For example, Dinkla et al. have identified an octapeptide from the GAS M protein that does not mimic cardiac myosin or induce antibodies crossreactive with cardiac myosin, but still produces rheumatic heart disease in animals [144–146]. Notably, this octapeptide binds to collagen IV on cardiomyocytes resulting in collagen IV autoantibodies. The ability of CVB or GAS antigens that do not mimic cardiac proteins to induce disease may force the field to rethink the role of mimicry by suggesting that the presence of crossreactive autoantibodies is a result of cellular damage rather than a cause of it [82].
A second set of potentially falsifying experiments would involve determining whether there are infectious agents (rather than just specific antigens in such agents) associated with either clinical or experimental forms of autoimmune myocarditis that do not exhibit MM with cardiac antigens. Many pathogens besides CVB and GAS are associated with autoimmune myocarditis. These include: T. cruzi (the cause of Chagas disease) [147], smallpox virus [148], varicella zoster virus (VZV) [149], CMV [150], hepatitis C virus [151], ECHO virus, adenovirus, Epstein-Barr virus (EBV), and parvovirus B19 [150, 152–155]. Of these, both T. cruzi and MCMV are often cited as classic cases of molecular mimicry. Some T. cruzi antigens mimic a variety of cardiac proteins including cardiac myosin [156, 157]. Rose, however, notes that purified and recombinant antigens derived from T. cruzi do not cause myocarditis nor are the crossreactive antibodies associated with autoimmune damage [158]. Similarly, while MCMV infection can lead to autoimmune myocarditis in rodents and a MCMV polypeptide appears to mimic cardiac myosin [7, 159, 160], this polypeptide has not been identified or sequenced, nor have attempts been made to induce EAM using it. Thus, the case for molecular mimicry of T. cruzi or MCMV as a cause of autoimmune heart disease is incomplete. More importantly, there is at this time no evidence for or against MM for the other pathogens associated with autoimmune myocarditis and thus the case remains pinned on a handful of examples.
1.4.5 Bystander or Adjuvant Effect Theory
Perhaps the most important question raised by the theory of MM is why it has not been possible to induce autoimmune disease with crossreactive inactivated pathogens or purified antigens without the use of an adjuvant. Rose et al. notes that neither inactivated CVB3 nor T. cruzi can induce EAM, despite the presence of molecular mimics [132, 158]. The addition of lipopolysaccharides (LPS) to the inactivated agents or the use of an appropriate adjuvant can, however, enable them to induce autoimmune disease, which has been termed the “adjuvant effect”. Similarly, inactivated GAS or GAS M protein cannot induce valvulitis, but the addition of CFA allows autoimmunogenicity [8]. These findings have led some researchers to suggest that autoimmune disease induction may require a second signal at the time a molecular mimic is encountered, like those provided by cytokines or innate immune activation caused by the adjuvant [132, 135]. As mentioned earlier, it is now realized that adjuvants activate antigen presenting cells via TLRs and other innate receptors eliciting “specific” innate immune responses. For example, activation of TLR2 by the M. tuberculosis component of CFA drives a Th17-type immune response [161].
The bystander effect theory (BET) or adjuvant theory of autoimmune disease developed following the observation by Tough and Sprent that various viruses, virus mimetics, bacteria, and bacterial products such as LPS can induce cytokine production resulting in the activation of heterologous polyclonal T cells [162, 163]. Beginning in 2001, Fujinami began hypothesizing that autoimmune diseases are induced by a combination of MM and bystander activation [135, 164–166]. The essential idea is that under normal conditions autoreactive immunity is kept under tight regulation that prevents disease initiation. In the presence of a bystander or adjuvant effect, cytokines are produced that non-specifically activate autoreactive T cells inducing pathogenesis. In some ways, the bystander effect is similar to the hidden antigen theory in relying on co-stimulation to initiate autoimmune disease. Whether the bystander effect works in conjunction with a molecular mimic is an issue of contention in the field, with groups such as Fujinami’s [135] and Rose’s [132] arguing for the compatibility of the two theories and Tandon’s group [82] arguing against it. As predicted by BET and noted above, neither cardiac myosin nor GAS M protein is sufficient to induce EAM in the absence of CFA. The use of IFA in combination with M protein or cardiac myosin also fails to produce EAM. These facts would suggest that a second signal is indeed necessary to induce autoimmunity using molecular mimicry antigens. On the other hand, the ability to induce rheumatic heart disease in animals using adjuvant in combination with an octapeptide that is complementary to, rather than a mimic of, cardiac proteins [144–146] suggests that specific innate activation rather than MM is the main trigger of autoimmunity [82].
One critical test of BET concerns the question of whether the adjuvant effect is a specific or non-specific phenomenon. If the adjuvant effect merely involves non-specific cytokine stimulation and therefore not specific to the particular inflammatory agent employed (i.e., M. tuberculosis and TLR2 for example), then it should be possible to replace CFA with other adjuvants in the induction of EAM. Thus far, experiments substituting CFA with other adjuvants have failed. As noted above, IFA which is missing M. tuberculosis is insufficient. More telling are experiments by Ketheesan’s group [167] who replaced CFA with Emulsigen® (MVP Laboratories Inc, USA) or Montanide ISA50V (SEPPIC, Paris, France) in a valvulitis model using GAS M5 protein as the antigen. Emulsigen and Montanide produced inflammatory responses equivalent to CFA in terms of stimulating T cell proliferation to antigen, edema, erythema, and necrosis at the injection site, but neither Emulsigen nor Montanide in combination with M5 protein produced lesions characteristic of valvulitis. These results suggest that the M. tuberculosis in CFA may be producing a specific, rather than a non-specific form of innate immune response. Evidence in support of this idea includes the data indicating that M. tuberculosis activates TLR2 on innate antigen presenting cells resulting in a Th17 and, to a lesser degree, a Th1-type immune response [161]- pathways that are known to increase myocarditis.
The question of adjuvant specificity is also raised by another unusual observation regarding the use of CFA. Fohlman et al. produced inactivated, attenuated, and subunit forms of CVB3 for use as vaccines against CVB-induced myocarditis [168]. The attenuated and inactivated virus vaccines were each tested alone and combined with each of three adjuvants: Quil A matrix, Alum, or CFA. Both the Quil A matrix and Alum were useful in promoting significantly enhanced antibody responses to the killed virus vaccine, and Alum also promoted antibody to the attenuated virus vaccine. Surprisingly, and in the words of the authors, “most remarkable is that Freund’s adjuvant did neither induce high neutralizing antibody titers nor protection” [168]. Since CFA clearly enhances the immune response to GAS M protein and to cardiac myosin, why did it not have a similar effect with CVB3? Well, what was not known at the time of Fohlman’s manuscript is that CFA drives a dominant Th17 response, while a Th1 response is needed for protection against CVB3 infection. Another important aspect of vaccine development is that adjuvants are tested for their efficacy at producing antibody responses, while the efficacy of CFA in autoimmune animal models is based on its ability to produce a cell-mediated immune response (i.e., inflammation). It should also be noted that Alum, similar to LPS, is known to activate TLR4 and the inflammasome, and to drive a mixed Th1/Th2 response that elevates antibody production [169]. Activation of TLR4 and the inflammasome is critically important for the development of myocarditis and DCM [12, 15, 16].
Another EAM model demonstrates again that different adjuvants can have very different effects. EAM can be produced in rodents by actin or cardiac C-protein in combination with Klebsiella pneumoniae O-3 LPS [170, 171]. This combination produced significant heart lesions typical of an autoimmune process, but substitution of LPS derived from other Klebsiella species, Escherichia coli, or Salmonella produced no autoimmune response in combination with heart protein extract. Further research is certainly merited to determine why differences in LPS significantly modify disease outcome. One possibility is that other types of LPS stimulate different TLRs beside TLR4, which is important for the development of myocarditis [12, 15].
Additional information about the adjuvant effect may be gleaned from two different models of CVB3-induced myocarditis. The traditional model uses strains of CVB3 that use CAR and/or DAF for viral entry and replicate at very high levels in the heart causing major necrosis but produce very low levels of cardiac inflammation (often less than 10%) while around 70% of the mice die during acute myocarditis [86]. Although this “virus only” model is thought to resemble fulminant myocarditis cases, biopsies from fulminant myocarditis patients typically have high inflammation but little evidence of necrosis. To develop a mouse model that more closely resembled most lymphocytic myocarditis patients we used a mild strain of CVB3 that does not use CAR or DAF for viral entry that had been passaged through the heart and so contained damaged heart protein [8, 17]. This combination results in a high level of acute inflammation in male mice (around 80% of heart inflamed), no apparent necrosis, and no deaths while 100% of BALB/c mice progress to develop DCM [12, 15]. We termed this model a “hybrid” CVB3 model because virus and damaged heart are used to induce disease [86]. So, what can the hybrid CVB3 model tell us about the adjuvant effect?
Well, for one thing the viral strain used in the hybrid model does not induce myocarditis if injected without damaged heart proteins. This suggests that two signals are needed-activation of the innate response to 1) live virus and to 2) damaged cardiac tissue. Damaged self is known to activate TLR4 and the inflammasome [172], and this is likely to be important for disease induction. Damaged cardiac tissue also contains cytokines like IL-33, which is a DAMP and part of the IL-1R/TLR4 pathway [173]. In the hybrid CVB3 model, the virus most likely provides a strong “adjuvant” signal while the immune response also targets cardiac self proteins.
There are a number of similarities between the hybrid CVB3 model and myocarditis induced using MCMV. MCMV is cultivated in the salivary glands of mice and damaged salivary gland tissue containing infectious MCMV is injected into mice to induce myocarditis [7, 17, 159, 160]. The disease course is very similar between the hybrid CVB3 model and MCMV-induced myocarditis [17]. Interestingly, one of the major components of salivary glands is contractile tissue composed of actomyosin [174]. So in this case also, activation of the innate immune response with a mild viral infection combined with damaged self (proteins found in cardiac tissue) produces a disease that closely resembles clinical myocarditis. In a similar manner Fujinami et al. used MCMV as a bystander infection to trigger central nervous system autoimmunity in conjunction with a proteolipid protein (PLP) expressing vaccinia virus (VV) [175]. In these experiments, MCMV by itself did not cause autoimmunity, nor did wild type VV or PLP-expressing VV. Lymphochoriomeningitis virus (LCMV) also failed to induce autoimmunity by itself or in combination with VV or PLP-VV, and wild-type VV was unable to initiate autoimmune disease following a PLP-VV infection. While the fact that PLP-VV in combination with MCMV did induce CNS autoimmunity could be interpreted as evidence for the bystander effect, the failure of LCMV and wild-type VV to do so could equally be interpreted as evidence against it.
In summary, there are a number of areas that need clarification in the bystander or adjuvant effect as a cause of autoimmune disease. If cytokines produce non-specific activation of autoreactive T cells, then one would expect any bystander cytokine or adjuvant to have a similar effect in inducing autoimmune disease. In other words, adjuvants should be able to substitute for each other if the effect is non-specific. It is increasingly clear however that adjuvants and even damaged self drive TLR-specific innate immune responses and cannot be equally substituted for each other.
Mathematical Models of BET
We have found only one mathematical model that incorporates the bystander effect into its explicit considerations [176]. As with most of the clinical and experimental literature on the bystander effect, Burroughs et al.’s model assumes that autoimmunity is made possible by the pre-existence of MM between the stimulating antigen and one or more host proteins [142, 176]. The effect of bystander stimulation in the model, as in BET itself, is to lower the threshold of antigen stimulation required to induce autoimmunity or to increase the rate at which autoimmune processes develop at any given level of antigen stimulation. Burroughs et al.’s model therefore confirms the plausibility of BET specifically as an adjunct to the theory of MM. The limitation of the mathematical model, like BET itself, is that both the model and the theory explicitly assume that any immunological stimulus (e.g., infection, adjuvant or environmental insult) can provide the bystander effect for any given antigen, whereas experimental and clinical evidence suggests that agents that produce bystander effects tend to be limited and specific to the antigenic trigger. Mathematical models are needed that investigate the extent to which the bystander effect varies as a function of how general or specific the interaction between bystander and trigger must be.
Novel Experiments and Clinical Studies
The main challenge that BET faces is to explain the apparent specificity of bystander cytokines and adjuvants for induction of autoimmune disease. Further characterization of the specificity of the “adjuvant” effect in EAM is needed. How specific are the adjuvant requirements for EAM induction using any specific antigen preparation? Along similar lines, and in light of Folhman et al.’s vaccine experiments [168], it would be worth inoculating animals with an attenuated or killed CVB preparation with CFA to determine whether the combination is capable of inducing myocarditis.
Another testable implication of BET is that it should be possible to produce autoimmune disease using a pure antigen (without adjuvant) such as M protein or inactivated CVB3 supplemented with the cytokines that would be produced by the adjuvant (or damage) like IL-1β or IL-33. Fohlman et al. noted, for example, that the successful adjuvants for CV vaccines stimulated antigen presenting cells to produce more IL-1 but not more IL-6 or tumor necrosis factor (TNF)-α than vaccines without adjuvants [168]. Can EAM be induced using inactivated CVB3 with IL-1β and/or some other combination of cytokines?
Finally, BET has testable clinical implications as well. BET predicts that autoimmune myocarditis will not be produced with co-infections like CVB and GAS, but requires infection plus self antigen. Thus, clinicians could test myocarditis patients for multiple infections.
1.4.6 Dual T Cell Receptor (DTCR) T Cells
Fujinami et al. proposed that some of the limitations of the theory of MM and BET could be accounted for by the recent discovery that many T cells have TCR with affinities for two different antigens called dual TCR (DTCR) [28]. This theory provides a mechanism for how self tolerance could be broken.
In 1993, Padovan et al. demonstrated that up to a third of all human T cells express two different V alpha TCR [177, 178]. Subsequent research demonstrated that about 1% of human T cells also express two different V beta TCR [179, 180]. In some cases, one of these TCR is self-reactive. The lower-than-normal expression of each TCR due to competition for space on the cell membrane is hypothesized to increase the probability that self-reactive clones will avoid clonal deletion resulting in increased susceptibility to autoimmunity. In particular, it has been hypothesized that activation of the non-autoreactive TCR by a pathogen or other antigen could stimulate clonal expansion inadvertently activating the autoreactive TCR on the same clone to cause autoimmune disease. In other words, a truly non-specific bystander effect could operate whereby the environmental trigger of an autoimmune disease could theoretically have no structural or sequence relationship to the autoimmune target.
Unfortunately for the purposes of this review, no investigator has yet applied DTCR to myocarditis so that our discussion of the theory’s possibilities and limitations must use other autoimmune disease models. We were also unable to find any evidence that DTCR has been linked to susceptibility or pathogenesis of human autoimmune disease. The following discussion is therefore speculative.
To begin with, the high prevalence of V alpha and V beta dual TCR T cells may be misleading. A recent study by Kekäläinen et al. found that 95% of dual TCR T cells are non-functional and may actually play a role in preventing autoimmunity [181]. So far increased susceptibility to autoimmune disease has been demonstrated only in transgenic mice engineered to express dual TCR T cells [28]. Even in transgenic models, dual TCR expression is not required for development of autoimmune disease, leading Auger et al. to write of their own experiments that “it is thus not necessary in this model to invoke a scenario in which a dual TCR T cell is activated by one TCR and then provokes autoimmunity through its second TCR” [182]. Based on these findings what expression of dual TCR does in the best case scenario is to increase susceptibility to the induction of autoimmunity.
Importantly, the presence of dual TCR T cells does not appear to increase susceptibility in all animal models of autoimmunity. While there is clearly a significant increase in susceptibility to diabetes in NOD mice with dual TCR T cells, Elliott and Altmann reported that “the incidence of EAE and of SLE is not affected by the absence of dual TCR-α cells [183].” Corthay et al. similarly found no increased susceptibility to collagen-induced arthritis by dual TCR [184]. Thus, in several experimental models of autoimmune disease DTCR do not appear to increase disease.
Mathematical Models of DTCR
We have found no mathematical models that address DTCR. This theory is therefore ripe for mathematical modeling.
Novel Experiments and Clinical Studies
A number of experiments and clinical studies should be conducted to define the role of DTCR. Do dual TCR T cells proliferate in response to cardiac antigens in myocarditis? If they do proliferate, are the pairs of TCR expressed on them randomly paired or do they occur specifically? Do the dual TCR expressed on these T cells reflect the triggering agent (i.e., CVB, GAS, T. cruzi, etc.), the host target (e.g., cardiac myosin), or both? How do dual TCR T cells respond to the M protein of GAS or to cardiac myosin in the absence or presence of an appropriate adjuvant?
1.4.7 Antigenic Complementarity Theory (ACT)
Antigenic complementarity theory (ACT) was first proposed by Westall and Root-Bernstein [185, 186]. ACT proposes that autoimmune diseases are caused by specific combinations of antigens, at least one of which mimics a self antigen. These antigens must be molecularly complementary to each other. As a result of this antigenic complementarity, the pair of T and/or B cells that are activated by the pair of antigens are also complementary to each other, which is to say that they have an idiotype-antiidiotype relationship although each is induced as an idiotypic response to its antigen. The result of antigenic and immunological complementarities is a complex network of misdirections. Each immunological response targets not only its appropriate antigen, but also the antigenic “self” mimic and the complementary immunological response as well. Each immune response must simultaneously address a non-self target, a self-antigen, and a complementary response by the immune system itself. Thus, autoimmune disease, according to ACT, is triggered when the immune system is tricked into attacking itself. The confusion as to what is antigenic within the immune system itself results in the inability to distinguish self from nonself (Figure 9). The ACT mechanism works equally well at the T cell and antibody levels (Figure 10).
Figure 9. Antigenic Complementarity Theory of Autoimmunity (ACT).

The antigenic complementarity theory postulates that autoimmunity results from co-infection with pairs of pathogens, at least one of which mimics a host protein [185–191, 202]. On the left, Microbe 1 has antigens that are molecularly complementary to antigens on Microbe 2. The antigens on Microbe 1 induce Antibody 1 (Ab1). The antigens on Microbe 2 induce Antibody 2 (Ab2). Because of the antigenic complementarity, Ab1 will be complementary to Ab2, which is to say that these two idiotypic antibodies will act as if they are an idiotype-anti-idiotype pair (see Figure 2). Thus, at the center, Ab1 will bind to both Microbe 1 and Ab2, while Ab2 will bind to both Microbe 2 and Ab1, thus producing circulating immune ICs. If, in addition, either Microbe 1 or Microbe 2 mimics a host protein (center top and bottom), then the antibodies induced by the microbes will also target these host proteins (right), just as is the case in Molecular Mimicry theory (see Figure 4). Thus, ACT combines basic elements of AIT with MM. ACT, however, also suggests a mechanism by which tolerance is broken in autoimmunity, which is that each idiotypic immune response mimics both a host and a microbial antigen so that the immune system itself becomes “confused” as to what is “self” and “nonself” and engages in an internal immunological civil war (center). Another unique prediction of ACT is that the molecular targets of autoimmune disease will themselves be complementary (center right).
Figure 10. Antigenic Complementarity Theory Applied to T Cell-Mediated Autoimmunity.
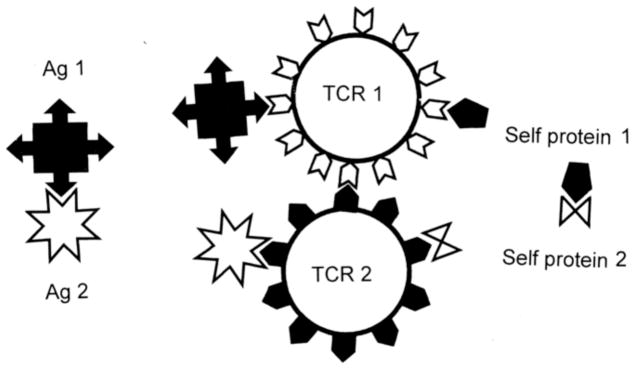
The same logic just outlined in Figure 9 with regard to antibody-mediated autoimmunity can also be applied to explaining T cell-mediated autoimmunity provoked by pairs of microbes bearing complementary antigens. The result will be to induce pairs of T cells bearing complementary T cell receptors (TCR-1 and TCR-2) that will act as if they are idiotype-anti-idiotype pairs. Such complementary idiotypic T cells will attack each other, forming perivascular cuffs or other cellular aggregates (the cellular equivalent of circulating ICs). If the antigens mimic host proteins, then these complementary T cells will also attack the host tissue. As in Figure 9, ACT predicts that autoimmune disease begins with an immunologic civil war.
Recently, Pendergraft and Preston have suggested a modified version of ACT in which autoimmunity is induced by pathogenic antigens that mimic antisense proteins (Figure 11) [187–191]. Antisense proteins are proteins that would be encoded by naturally occurring antisense RNAs or, more basically, by the non-coding strand of DNA that is complementary to any given exon. According to some investigators, antisense proteins are molecularly complementary to their sense proteins and are capable of binding directly to each other [192, 193]. A pathogen that expresses a protein that is an antisense mimic to a host protein could therefore produce antigenic complementarity that could, either directly, or by means of antiidiotype antibodies, result in the same consequences as ACT.
Figure 11. Antigenic Complementarity Theory 2 (ACT2).
Preston and Pendergraft have proposed an alternative version of ACT in which antigenic complementarity is mediated through antisense proteins [187–191]. Every genetically-encoded protein has, according to antisense protein theory, a complementary protein encoded in the complementary (non-coding) strand of DNA. If a microbe displays a protein that is an antisense protein to a genetically encoded host protein, and this microbial protein also mimics a host protein, then all of the effects outlined above in Figure 9 will follow and autoimmunity disease will be initiated against the tissue that encodes the antisense protein mimic.
One feature distinguishing ACT from other theories of autoimmunity is that it interprets adjuvant effects in the induction of autoimmune disease as resulting from specific antigenic complementarity rather than as non-specific inflammatory agents or innate TLR events as in BET. In ACT, adjuvants are not non-specific immune potentiators of an antigen in the induction of autoimmune disease; rather the so-called “antigen” and so-called “adjuvant”, because they induce molecularly complementary immune responses, are co-activators of each other’s immune responses. Each acts as a specific “adjuvant” for the other, but for no other antigen. Immunological complementarity drives a synergistic, positive feedback system of immune activation. In other words, each antigen acts as a specific adjuvant for the other. There are no nonspecific bystander effects according to ACT. ACT maintains that only specific infections or agents that display complementarity can act as triggers for any particular complementary antigen.
The ACT mechanism explains why the “adjuvant” in specific antigen-adjuvant pairs is rarely substitutable. Contrary to BET, ACT argues that the factor initiating autoimmunity is not simply cytokines or lymphokines acting in a non-specific manner, but the molecular complementarity of antigen and adjuvant. Adjuvants that are not complementary to their antigen will produce an immune response but not autoimmune disease. Complementarity between antigens (or between antigen and “adjuvant”) will result in autoimmunity, producing costimulation as a concomitant of the dual activation of the mutually-stimulating immune responses.
ACT has been applied theoretically to understanding a number of autoimmune diseases including EAE [185, 186, 194–200], idiopathic thrombocytopenia purpura [201, 202], and autoimmune myocarditis [81].
ACT applies to the induction of autoimmune myocarditis in the following manner. Both cardiac myosin [8, 53] and cardiac actin [170, 171] are capable of inducing autoimmune heart disease when inoculated with appropriate adjuvants. GAS is associated with the development of valvulitis and GAS mimics cardiac myosin. CVB is able to induce autoimmune myocarditis when inoculated with damaged cardiac tissue, and CV mimics cardiac actin. Cardiac myosin and actin are molecularly complementary, their combination forming active actomyosin. It follows that GAS, which mimics myosin, and CV, which mimics actin, must have complementary antigens. In fact polyclonal antibodies against cardiac myosin precipitate polyclonal antibodies against actin; polyclonal antibodies against CVB recognize cardiac actin and vice versa; and polyclonal antibodies against GAS recognize cardiac myosin and vice versa. Most importantly, polyclonal antibodies against GAS precipitate polyclonal antibodies against CVB [81]. In other words, idiotypic CV antibodies act like antiidiotypic antibodies against GAS antibodies; and idiotypic GAS antibodies act like antiidiotypic antibodies against CV antibodies. In short, CV and GAS have all of the characteristics of being complementary antigens as predicted by ACT (Figure 12). Pearce [203] and Kogut et al. [204] demonstrated experimentally that combinations of CVB with GAS produced much more severe viral infection and myocarditis in far greater numbers of mice and rabbits than did either infection alone. CV and GAS, in short act both as antigens and as “adjuvants” for each other.
Figure 12. Antigenic Complementarity in Autoimmune Myocarditis.
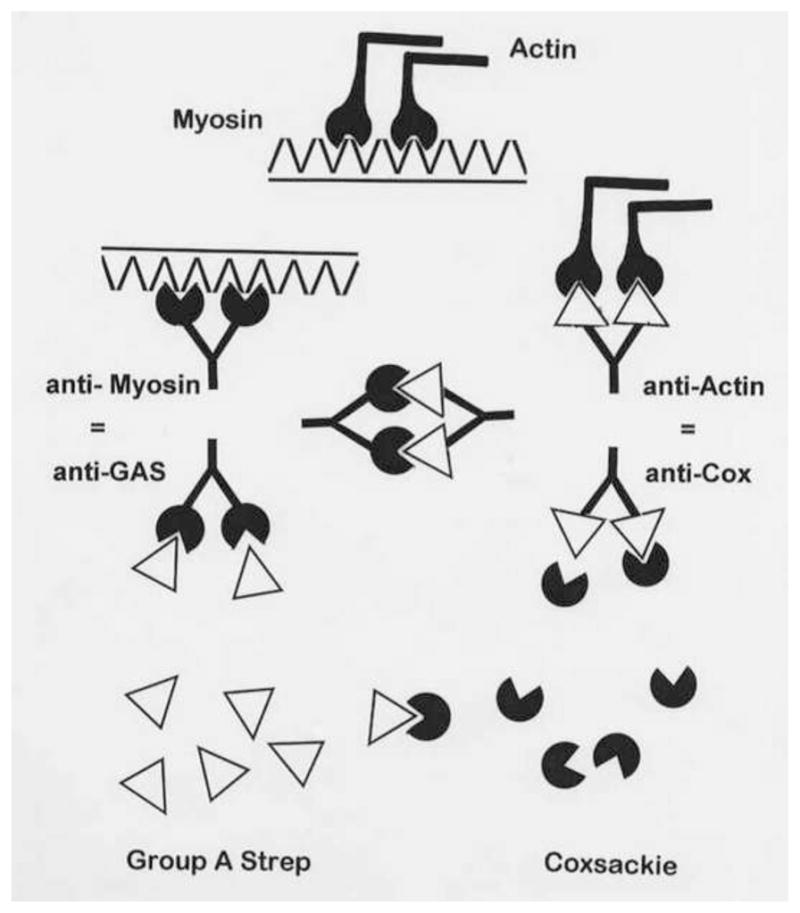
Root-Bernstein, et al. [81] have provided experimental evidence for most of the key assumptions implicit in ACT. This figure summarizes the experimental findings. As noted in Figure 5, the M protein of group A streptococci (GAS) mimics cardiac myosin. As noted in Figure 6, proteins of coxsackieviruses (CV) mimic cardiac actin. GAS antibodies bind to CV antibodies forming immune complexes. GAS antibodies recognize cardiac myosin. CV antibodies recognize actin. Actin and cardiac myosin are molecularly complementary to each other, forming actomyosin.
Another experiment also suggests that antigenic complementarity could be involved in the induction of autoimmunity. CVB is molecularly complementary to its receptor CAR. Dörner et al. demonstrated that antibodies against CAR can reduce CV infection [205], and Goodfellow et al. that soluble CAR also blocks CV infection in culture [206]. What surprised Dörner et al. was that inoculating mice with a combination of soluble CAR with CV resulted in greatly enhanced myocarditis despite the significantly lower infectivity of the virus [205]. As Dörner et al. conclude, the presence of soluble CAR (a potential antigen) clearly enhanced CV antigenicity increasing myocardial inflammation. These results are difficult to reconcile with theories of autoimmunity other than ACT. But there could be an alternative explanation for these findings. When CAR is not available for CV to use it could use other receptors [87, 88]. In our experience, when CAR is not used as a receptor viral replication in the heart is lower but inflammation is far more severe (80% vs. 10%) [12, 15].
Finally, evidence for antigenic complementarity in the induction of autoimmune disease also comes from Matsumoto et al.’s experiments on epitope spreading [65]. Lewis rats inoculated with peptide fragments of cardiac C-protein in CFA developed acute cardiac inflammation but no chronic autoimmunity. In contrast, animals inoculated with a combination of cardiac C-protein/CFA with antisera went on to develop chronic autoimmunity and DCM. The limitation of these experiments, like those involving CVB3-plus-CAR is that they are quite artificial and unlikely to mimic natural mechanisms by which human autoimmune disease is induced. If ACT is correct, then it will need to be implemented via naturally occurring complementary antigen mixtures such as the CVB3-GAS combination mentioned above.
If ACT is causing autoimmune disease many experiments previously described in this review would need to be reinterpreted, especially the evidence supporting AIT, MM theory and BET. According to ACT all of the evidence for the theory of MM is correct, but incomplete. Single molecular mimics should, according to ACT, be incapable of inducing autoimmunity on their own (without adjuvant) as is the case. ACT reinterprets “adjuvants” as being complementary antigens, which would explain why one adjuvant can rarely be substituted for another in EAM (or any other experimental autoimmune disease). ACT also reinterprets evidence supporting AIT to suggest that what are often described as anti-idiotype antibodies are actually complementary idiotypic antibodies.
ACT also suffers from some of the same inadequacies of the theories that it incorporates. For example, what is the functional definition of an “anti-idiotype” or a “molecular complement”? Since ACT depends on MM, what functionally defines a molecular mimic? In the absence of such functional definitions, ACT, like the AIT and theory of MM, remains ambiguous and difficult to evaluate.
Each of these differences between ACT and other theories of the causes of autoimmune disease provides specific predictions that can be tested to differentiate between the theories. None of these tests have yet been carried out, leaving the theory without substantial experimental support. Moreover, the original application of ACT to myocarditis is probably incorrect in identifying cardiac myosin and actin as the primary pathogen mimics that are targeted to initiate disease. As noted in the discussion of HAT, actin and cardiac myosin are unlikely to be available to the immune system until cellular damage has already commenced, making mimicry to extracellular proteins such as CAR, DAF, laminin, collagen IV, etc. more likely candidates [80].
Additionally, ACT does not explain why CV infections are associated with myocarditis but not valvulitis, while GAS infections are associated with valvulitits but not myocarditis. And similar to most other theories, ACT does not explain the requirement for activation of specific elements of innate immunity like TLR during disease induction nor does it address differences in susceptibility to disease associated with sex.
Finally, there is a general issue that neither ACT nor any other theory of autoimmunity adequately address and that is whether myocarditis (or other autoimmune diseases) is actually one disease or a spectrum of diseases. Myocarditis, for example, is defined and primarily diagnosed histologically. Therefore, it is possible that various inflammatory cardiovascular diseases are not single diseases but multiple subtypes of the same disease (i.e., myocarditis, valvulitis, atherosclerosis), which are quite distinct at the individual patient level. It is possible that different disease phenotypes are caused by distinct agents or sets of agents that trigger each disease. It is therefore conceivable that all the possible mechanisms for autoimmune disease induction described here occur, but some are more common than others. The major clinical question is whether any of the mechanisms/theories really reflect clinical disease and what proportion of total myocarditis patients fall into each category.
Mathematical Models of ACT
ACT has not formally been modeled mathematically. But it is interesting to note that independent of the specific mathematics used in idiotype-anti-idiotype models of the immune system, when idiotype and anti-idiotype are symmetrically activated (that is to say, when both the idiotype and the anti-idiotype are stimulated simultaneously and equally), the system loses the ability to regulate itself [95, 96, 207]. Since these mathematical models have assumed that idiotype-anti-idiotype interactions in a naïve immune system would naturally start from a state of symmetrical activation, the failure of the models to produce self-regulated behavior has generally been characterized as a failure of the mathematical model [95, 207]. On the other hand, one of the unique predictions made by ACT that sets it apart from other theories of autoimmunity is precisely that pairs of complementary idiotypes (i.e., the equivalent of idiotype-anti-idiotype pairs) will be symmetrically activated, resulting in loss of immunological regulation. Thus, the conditions under which previous mathematical models of autoimmunity have apparently failed may actually be clues to how a dysregulated immune system leads to autoimmune disease. In this case, further mathematical investigation of symmetrical activation of idiotype-anti-idiotype (or complementary idiotype) pairs, or of complementary pairs of molecular mimics, may be warranted.
Novel Experiments and Clinical Studies
The most incisive test of ACT would be to combine two of the main animal models of autoimmune heart disease, experimental valvulitis induced by GAS M protein/CFA and the hybrid CVB3 model of myocarditis induced by CVB3 and damaged heart tissue. If ACT is correct, then it should be possible to produce autoimmune myocarditis with a combination of GAS M protein and CVB3 without adjuvant or damaged heart tissue. Neither GAS M protein nor the Nancy strain of CVB3 induce myocarditis in BALB/c mice on their own (i.e., Nancy strain CVB3 requires damaged heart with the inoculation to induce disease in the “hybrid” model). The clinical implication of these studies is that people developing rheumatic heart disease or myocarditis should be characterized by having specific combinations of infections such as GAS and CVB. Similarly, in myocarditis cases associated with smallpox vaccination the individuals that develop myocarditis are predicted to have a co-infection at the time of exposure.
ACT can be differentiated experimentally from BET, DTCR, etc. by the specificity of the combination of antigens required to induce EAM. An “adjuvant” (e.g., CFA) that can induce autoimmune disease with one antigen (e.g., cardiac myosin) should not be replaceable by another “adjuvant” (e.g., Klebsiella LPS).
Another prediction of ACT is that some antigens expressed by pairs of inducing pathogens should be molecularly complementary to each other. The active agent within an “adjuvant” should bind directly to the “antigen”. This prediction is testable using a number of methods, including physicochemical techniques and has been demonstrated with nuclear magnetic resonance spectroscopy for the complex of muramyl dipeptide adjuvant with the encephalitogenic peptide that causes EAE in guinea pigs [195]. Another way to test for antigenic complementarity is to perform enzyme-linked immunoadsorption assays (ELISA) or Ouchterlony immunodiffiusion experiments to determine whether antibodies against one antigen or pathogen bind specifically to antibodies against another [81]. Alternatively, TCR sequences specific to the potential pathogen pairs can be synthesized and the ability to recognize each other determined [200]. None of these experiments should be successful according to other theories.
1.4.8 Co-infections, co-exposures, damaged self and timing
Experimental autoimmune models like EAM reveal that damage to self or availability of self-peptide to stimulate the innate immune response must occur at the same time (day 0) as the infection or adjuvant. If adjuvant is provided earlier or later than self-peptide autoimmune disease does not develop. Most discussions on the role of infections, and particularly viruses, in triggering autoimmune disease posit that infections cause damage to the tissue as well as stimulating the immune response and that this release of self-peptide could lead to autoimmune disease. However, the timing of the events does not coincide. Day 0 of infection does not coincide with release of damaged self, which for most infections occur about 5–7 days later. In the case of CVB3, Nancy strain alone is unable to induce myocarditis but requires damaged heart tissue to be inoculated at the same time as the infection to induce disease [8]. This “timing” issue indicates that two coinciding stimuli are necessary at time zero. One explanation could be that a co-infection is needed, with one infection providing the damaged self at the same time as the other infection activates the innate immune response. Or a chemical or drug co-exposure could release damaged self-tissue just as the individual gets an infection. The requirement for exact timing of presentation of damaged self and infectious or adjuvant stimulation of the innate immune response could explain at least in part why common infections do not often lead to autoimmune disease. An additional constraint could be that particular innate activation is important for certain autoimmune diseases to occur, like TLR4 and the inflammasome for myocarditis, and so precise timing and a precise stimulus would reduce the incidence of disease. A requirement for MM or antigenic complementarity to induce disease could provide an additional constraint.
Mathematical Models of Co-Infections, Timing, Etc
We know of no models that incorporate the need for co-exposures of antigen-adjuvant, why timing of inoculations with antigen and adjuvant should matter, etc. These are phenomena that are badly in need of theoretical modeling in autoimmunity.
Novel Experiments and Clinical Studies
This idea requires further study in animal models as there are virtually no publications examining re-infections, co-infections, co-exposures or the importance of “timing” in myocarditis or other autoimmune diseases. However, there is some indication that CVB3 and GAS co-infections occur at the same time in some patients [128, 204, 208–211]. Additionally, patients with viral myocarditis have been found to be infected with multiple cardiotropic viruses [154, 155, 212]. One of the challenges in determining whether co-infections or co-exposures occur in patients is that they may occur weeks or months (possibly years) before the clinical symptoms occur, and so it is difficult to examine this issue clinically. Perhaps advances in identifying infectious “signatures” from molecular screening of the microbiome could be applied to the study of infectious or other environmental exposures and autoimmune diseases. One important consideration about co-exposures or co-infections as a cause of autoimmune disease is that they do not necessarily require molecular crossreactivity or antigenic complementarity to induce disease.
1.5 Sex differences in autoimmune diseases and theories
One major gap in all theories and mathematical models for the causes of autoimmune disease is the lack of analysis of the reasons for sex differences in disease prevalence and severity (Table 2). It is well recognized and substantiated by epidemiological data that all major autoimmune diseases occur more frequently in one sex or the other [213–216]. Autoimmune diseases with a recognized sex difference in women compared to men include multiple sclerosis (2:1), dermatomyositis (2:1), rheumatoid arthritis (3:1), systemic sclerosis (4:1), autoimmune hepatitis (6:1), Graves’ disease (7:1), SLE (9:1), Sjogren’s syndrome (9:1), and Hashimoto’s thyroiditis (20:1) [213, 214, 216]. Autoimmune diseases that occur more frequently in men compared to women include myocarditis (2:1) and idiopathic pulmonary fibrosis (11:7) [16, 216]. Understanding the role of sex hormones on immune function and inflammatory diseases is an emerging area of investigation. Critically important is the realization that every organ, cell, and perhaps antigen has a sex [217]. At the least, antigens are recognized in the context of the sex of the antigen presenting cell (Table 2).
An unresolved question is why most autoimmune diseases occur more frequently in women than men. It is well known that immune responses to antigens differ between men and women. For example, women respond to infection, vaccination, and trauma with increased antibody production [218–221]. Although increased antibody levels protect women from infections, they also increase the risk of developing autoimmune diseases. Estrogen activates B cells resulting in increased levels of antibodies, autoantibodies and ICs, while androgens decrease B cell maturation, reduce B cell synthesis of antibody and suppress autoantibody production in humans [222–224]. Sex steroid hormone receptors such as estrogen receptor (ER)-α, ER-β, and the androgen receptor are expressed on the cell surface as well as intracellularly in immune cells. Likewise, cytokine receptors like IL-1R are found on classic hormone-producing tissues, indicating bi-directional regulation of the immune response [225]. Collectively, data support the idea that estrogen elevates autoantibodies, Th2 responses, and promotes fibrosis by stimulating profibrotic IL-4, TGFβ, and fibroblast growth factor, all of which contribute to the increased incidence of autoimmune diseases in women (Table 2) [12, 16, 216, 222, 226, 227]. In contrast, androgens increase the cell-mediated arm of the immune response associated with elevated Th1- and Th17-type immune responses (Table 2) [12, 16, 216]. Large differences in the direction of the immune response to the same antigens/infections in the context of sex are due to 100’s or 1000’s of genes that are differentially regulated by estrogen vs. androgen response elements. Although sex hormone receptors on/in cells of the adaptive immune response also influence immunity, the greatest impact of sex hormones is in initiating immune responses to antigens during the innate immune response [16, 216]. The role of sex hormones in influencing the pathogenesis of myocarditis and DCM has recently been reviewed [see 16, 228, 229].
All major theories and mathematical models concerning the mechanisms by which infections or other environmental agents could cause autoimmune diseases need to address the issue of sex differences. Why would exposure of a hidden antigen, cross-reactive epitope, or molecular complementarity promote autoimmune disease in predominantly one sex? EST and BET have less difficulty addressing how sex differences could fit their theories, although most investigators have not tried to explain sex differences in light of their theory. It is fundamentally important that new and revised theories of autoimmune disease incorporate an explanation for sex differences into their theories if researchers are going to be able to make a meaningful impact on clinical disease.
1.6. Summary of differences between and comparisons of theories
The eight theories of autoimmunity summarized in this review share some features and differ in others. All agree that infectious triggers are critical components of disease induction but differ on the antigenic relationship of infection to the tissue targeted. HAT, BET and EST posit no direct antigenic relationship (i.e., crossreactivity or antigenic complementarity) between infection and host target other than co-localization of a damaging immune response within the target tissue. Presumably, the focus of autoimmunity is determined by the tissue specificity of the inducing microbial agent.
AIT, MM theory, DTCR, and ACT all assume, on the other hand, some sort of specific relationship between the disease agent triggering autoimmunity and the target of autoimmune disease. AIT and ACT share the assumption that a disease agent complementary to the targeted host tissue must be present. MM theory, DTCR and ACT all share the assumption that a disease agent mimicking a host tissue antigen must be present.
BET, DTCR and ACT differ from the other theories of autoimmunity in proposing that autoimmunity requires two agents or co-exposures. BET hypothesizes that the second agent can be anything that causes significant cytokine production. DTCR hypothesizes that the second agent is whatever will trigger a self-reactive T cell bearing dual TCRs, one of which is autoreactive. ACT hypothesizes that the second agent must be antigenically complementary to the “primary” trigger and either a complement or mimic of a host antigen.
While all non-infectious animal models of myocarditis and rheumatic heart disease (and most other autoimmune diseases) that have so far been developed require some sort of “adjuvant”, only two of the theories explicitly address the role of “adjuvants” in disease induction, BET and ACT. BET attributes non-specific tissue damage to bystander infection. ACT requires a pair of molecularly complementary co-infections that act as “co-adjuvants”. ACT directly addresses the issue of why the “adjuvant” effect in autoimmune disease models appears to be antigen specific; BET does not.
The question of how “self” tolerance is abrogated in autoimmune disease is also one of the key features differentiating theories of autoimmunity and their mathematical models. HAT and EST share the general assumption that autoimmunity is mainly directed against inaccessible antigens to which the immune system is not tolerized and that autoimmunity results from tissue damage or infection that exposes these antigens. The difficulty with HAT and EST is that they do not explain why neither physical damage (e.g., heart attacks and surgery) nor the vast majority of CVB and GAS infections result in autoimmune disease.
Neither AIT nor MM directly address how tolerance is abrogated. Given that GAS and CVB each use multiple cardiomyocyte receptors and have proteins that mimic multiple cardiac proteins, it would seem logical from AIT and MM that anyone with a cardiac infection involving either pathogen would develop autoimmune disease unless other factors determine susceptibility. Genetics is one determinant of susceptibility, of course; the other factors that are sometimes called upon to explain why autoimmunity is so rare include T cell bypass, B cell-T cell mismatch, and incomplete clonal selection/deletion. While such stochastic mechanisms may indeed underlie susceptibility to autoimmune disease, they are not amenable to experimental investigation or clinical study which makes them less than satisfactory explanations. In addition, the fact that essentially every animal in an experimental protocol can be made to develop autoimmune disease with the right mixture of antigen and “adjuvant” argues strongly against random failures in deletion mechanisms as key events governing whether an animal (or person) develops autoimmune disease.
BET, DTCR, and ACT provide a different approach to understanding abrogation of tolerance. Each assumes that autoimmunity (as opposed to autoimmune disease) is a normal immunological function that is held in check by various mechanisms such as regulatory T cells, idiotypic networks, etc. DTCR postulates, in addition, that because multiple TCR or BCR are expressed on dual-affinity lymphocytes, there are too few of the highly active receptors to activate the deletion mechanism. The result for each theory is a pool of well-regulated, low-activity, but potentially highly reactive T and B cells. BET proposes that tolerance is broken when a bystander infection induces activation of autoreactive clones. DTCR proposes that infection with the antigen activating the co-expressed receptor on dual-affinity TCR and BCR also activates expression of autoreactivity. And ACT proposes that autoimmune disease follows from the co-adjuvant effect of being infected by pairs of complementary infections, whose mimicry confuses the immune system’s ability to differentiate “self” from “nonself”. The shared feature of these three theories is that autoimmune disease requires multiple concurrent stimuli or co-exposures.
Thus far, mathematical models of the eight theories discussed in this paper have failed to enlighten the issue of how tolerance is abrogated in autoimmunity. As noted above, there are mathematical models for five of the eight theories, each based on a different set of assumptions. Unfortunately, the mathematical modeling community has not yet compared these assumptions nor worked through their implications in a systematic manner. The models of each theory therefore provide little insight as to what needs to be done to select among the theories, or develop a more integrative, synthetic theory.
We must emphasize that although we have analyzed each of the theories of autoimmunity and their mathematical models separately here, it is also possible to mix and match them. Thus, a number of investigators have combined the theory of MM with BET and/or DTCR to provide a mechanism by which MM only rarely produces autoimmune disease. Similarly, ACT is, in some senses, a combination of AIT, MM theory and BET in which idiotypic, anti-idiotypic and “adjuvant” effects all play out simultaneously.
Specific testable differences between these theories for induction of autoimmune disease can be found in their implications for etiology, epidemiology, timing of antigen presentation, the role of circulating ICs in disease, and the role of innate immunity. One failure of all of the theories of autoimmunity analyzed here is that none addresses sex differences in disease susceptibility, which poses a general challenge to the entire field.
1.7 Etiological and epidemiological predictions
The theories and their mathematical models can be divided into two classes of etiologies. One set (HAT, EST, AIT, MM theory) are mono-causal, which is to say that they are based on the assumption that a single antigen is both necessary and sufficient to induce autoimmune disease in genetically susceptible individuals or animals. The rest of the theories (BET, ACT and DTCR) are multi-causal, which is to say that they are based on the assumption that two or more immunological agents are required to induce autoimmune disease in genetically susceptible people and animals. The multi-causal theories can, in turn, be divided into those that require non-specific immunological enhancement of the response to the primary antigen (BET and DTCR) and those that require a specific pair of complementary antigens (ACT). The requirement for adjuvants in the induction of animal models of myocarditis and valvulitis and many other models of autoimmune disease would seem to argue for a multi-causal etiology.
These etiological differences lead to important differences in the predictions the two classes of theories make about the epidemiology of myocarditis. The mono-causal theories predict that the incidence of myocarditis would be a direct function of the number of genetically susceptible individuals in the population and their probability of acquiring an infection capable of triggering autoimmune disease. In contrast, the multi-causal theories predict that the incidence of myocarditis would be a function of the number of susceptible individuals in the population and their probability of acquiring two or more simultaneous infections capable of inducing autoimmune disease. In general, then, multi-causal theories predict that myocarditis should be far less prevalent (probably by several orders of magnitude) than mono-causal theories. Since only very small fractions of people infected with GAS or CVB develop rheumatic heart disease or myocarditis, respectively, epidemiological data seem to favor a multi-factorial etiology.
Notably, the issue of monocausality versus multifactorial causality of myocarditis has been debated for almost fifty years. Burch raised the question of why most cases of GAS and CVB co-infections do not subsequently develop autoimmune disease [see also 81, 230], first postulating the possibility that rheumatic heart disease or myocarditis may be due to multifactorial processes [231–233]. Additionally, Pongpanitch et al. reported that 14 of 15 rheumatic heart disease cases were co-infected with CVB identified by viral deposition on the heart valve, while control hearts did not have deposition [234]. Other studies found similarly significant increases in CVB3 or CVB4 antibody titers among rheumatic heart disease patients compared with controls or other direct evidence of co-infections [208, 235–237]. Some studies have also found a strong, but not statistically significant, correlation between rheumatic heart disease and the presence of CVB infections [209, 210, 238]. Conversely, in studies that have looked for more than one infection in patients diagnosed with CVB-associated myocarditis, 65–80% presented with a concurrent GAS infection [128, 204, 211].
In addition to the observation of combined GAS-CVB infections, between 12% and 25% of patients with viral myocarditis have been found to be infected with multiple cardiotropic viruses including varicella zoster, adenovirus and parvovirus B19 [154, 155, 212]. EBV DNA was also isolated from the heart of patients with rheumatic heart disease, but EBV antibody titers were not significantly associated with disease, nor were titers of antibodies against hepatitis B virus, hepatitis C virus, rubella virus, or herpes simplex virus group 1 [230]. In other words, the GAS-CVB combination may be a common one associated with cardiac autoimmunity, but is unlikely to be the only important co-infection/co-exposure combination.
To the extent that autoimmune disease is caused by combined infections, then theories such as BET, DTCR and ACT currently have a better chance of providing explanatory frameworks than do the mono-infectious theories. On the other hand, the existing data also seem to indicate that not just any infection can promote autoimmune heart disease, so that, just as has been observed in animal experiments using adjuvants, some specificity of co-infection or exposure seems to be necessary.
Experimental models of autoimmune heart disease also provide tentative support for a multi-infectious etiology. Kogut et al. demonstrated in mice that combined infections of GAS with CVB were far more likely to produce greater viral replication and damage in the heart [204]. Pearce showed that while either Streptococcus pyogenes toxins or CVB infections could independently cause small percentages of rabbits to develop minor myocarditic lesions, a combination of the two produced severe lesions in nearly 100 percent of animals [203, 239]. Taken in conjunction with other animal studies demonstrating that streptococcal antigens and CVB in the absence of heart tissue or adjuvants fails to induce valvulitis or myocarditis, these combined GAS-CVB studies provide a strong rationale for further studies of combined infections. One caveat is that there may, of course, be combinations of infections other than GAS with CVB that produce myocarditis, since CMV, EBV, VZV, smallpox and other pathogens are also associated with myocarditis. One limiting factor may be that the combinations of infections will likely involve pairs of cardiotropic pathogens regardless of which theory turns out to be the best explanation for autoimmune myocarditis. Another area that needs to be investigated is the effect of co-exposure of chemicals and infections on the induction of myocarditis and other autoimmune diseases [240].
Multifactorial theories of autoimmune disease have several clinical implications. One is that new studies focused on the possibility of combined infections in people at risk for autoimmune diseases are needed. One outcome is that these studies will bolster current mono-factorial approaches by demonstrating absence of concurrent infections. If not, such studies will identify particular combinations of infections associated with particular autoimmune diseases. Such identification may, in turn, lead to the development of animal models that better mirror the human etiology of autoimmune diseases and so provide new clues for prevention and treatment. The evidence that rheumatic heart disease and myocarditis can involve combinations of GAS with CVB yields one final clinical possibility, which is that myocarditis may be preventable or treatable in at least some CVB-infected individuals with antibiotics.
The possibility that the etiologies of rheumatic heart disease and myocarditis are multifactorial has one final implication of great importance. Except for the handful of experiments just described, all attempts to model autoimmune diseases in animals have proceeded in accordance with Koch’s postulates, which explicitly assume monocausality. If autoimmune diseases turn out to be multi-causal, then a different set of postulates are required to prove disease causation. It will be necessary to demonstrate that individual infections or antigens are not sufficient to cause autoimmune disease and that combinations of them or co-exposures are. In addition, if autoimmune diseases do, in fact, require co-infections or exposures, then using animals from genetically identical strains it should be possible to inoculate one set of animals with one infection or exposure (which will not develop autoimmune disease), another with the other infection or exposure (which will not develop autoimmune disease), and then produce the autoimmune disease in a third set of animals by passive transfer of lymphocytes and/or antibodies obtained from both exposures.
1.8 Conclusions
We conclude that there is some evidence supporting all of the theories of autoimmunity that we have reviewed here; that most experimental and clinical data are open to more than one interpretation according to different theories; and that no single theory accounts for all of the clinical and experimental evidence. New, more comprehensive theories and mathematical models are needed. Table 2 summarizes the main challenges that a new theory will need to resolve. A major challenge will be to break the current pattern of each investigator attempting to gather data that supports their particular theory, and to begin the much more difficult quest to develop critical models, experiments and clinical observations that can differentiate between theories. Even more important will be the search for theories that integrate the very extensive data that now support one or more existing theories with the equally extensive set of anomalies and phenomena that do not support current theories. Perhaps the most important of these phenomena involve the very low rates of incidence of autoimmune diseases following exposure to common environmental triggers; the role that innate immunity plays in determining the effect of environmental triggers; how biologic sex skews disease risk; and how host tolerance is abrogated as a result of the concatenation of all of these factors. This will not be a simple challenge to address, but it is one that is much more likely to be achieved successfully if investigators with opposing views work together. This will also require new mathematical modeling approaches for exploring multifactorial etiologies as a cause of autoimmune disease.
Figure 8. Dual TCR (DTCR) Theory of Autoimmunity.
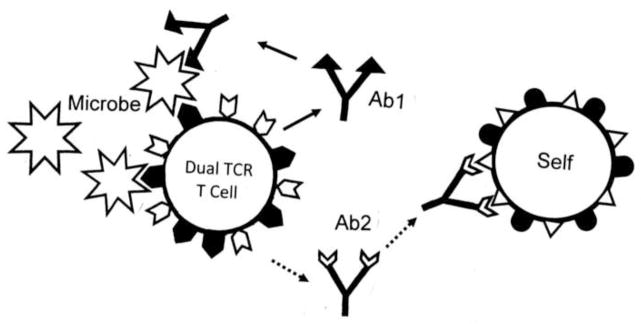
Some T cells display more than one T cell receptor (TCR) so that they can be activated by more than one antigen. Activation of a T cell by a microbe and subsequent production of antibodies could therefore inadvertently initiate activation of an unrelated immune response that crossreacts with host tissues [28, 135, 178].
Highlights.
Clinical and experimental evidence support some aspects of all theories
Critical comparative studies differentiating between theories and models needed
Theories monocausal but animal models suggest multi-factorial cause of disease
Theories do not adequately explain adjuvant, innate immunity or sex differences
New synthetic theory needed integrating anomalies, innate and adaptive immunity
Acknowledgments
Funding Sources
This work was supported by National Institutes of Health awards from the National Heart, Lung and Blood Institute [R01 HL111938] and National Institute of Environmental Health Sciences [R21 ES024414] to D.F., and an American Heart Association Grant-in-Aid [12GRNT12050000] to D.F. These funding agencies were not involved in writing the review article nor the decision to submit the article for publication.
Footnotes
Author Disclosure
D. Fairweather and R. Root-Bernstein both contributed to the planning, writing and editing of the manuscript. Both authors approved the final version of the article. Neither author has any financial or other conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Imazio M, Cooper LT. Management of myopericarditis. Expert Rev Cardiovasc Ther. 2013;11:193–201. doi: 10.1586/erc.12.184. [DOI] [PubMed] [Google Scholar]
- 2.Fabre A, Sheppard MN. Sudden adult death syndrome and other non-ischaemic causes of sudden cardiac death. Heart. 2006;92:316–320. doi: 10.1136/hrt.2004.045518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elamm C, Fairweather D, Cooper LT., Jr Pathogenesis and diagnosis of myocarditis. Heart. 2012;98:835–840. doi: 10.1136/heartjnl-2012-301686. [DOI] [PubMed] [Google Scholar]
- 4.Kindermann I, Barth C, Mahfoud F, et al. Update on myocarditis. J Am Coll Cardiol. 2012;59:779–792. doi: 10.1016/j.jacc.2011.09.074. [DOI] [PubMed] [Google Scholar]
- 5.Gupta S, Markham DW, Drazner MH, et al. Fulminant myocarditis. Nat Clin Pract Cardiovasc Med. 2008;5:693e706. doi: 10.1038/ncpcardio1331. [DOI] [PubMed] [Google Scholar]
- 6.McNamara DM, Starling RC, Cooper LT, et al. Clinical and demographic predictors of outcomes in recent onset dilated cardiomyopathy: results of the IMAC (Intervention in Myocarditis and Acute Cardiomyopathy)-2 study. J Am Coll Cardiol. 2011;58:1112–1118. doi: 10.1016/j.jacc.2011.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fairweather D, Lawson CM, Chapman AJ, Brown CMS, Booth TWM, Papadimitriou JM, Shellam GR. Wild isolates of murine cytomegalovirus induce myocarditis and antibodies that cross-react with virus and cardiac myosin. Immunology. 1998;94:263–270. doi: 10.1046/j.1365-2567.1998.00500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Myers JM, Fairweather D, Huber SA, Cunningham MW. Autoimmune myocarditis, valvulitis, and cardiomyopathy. Curr Protoc Immunol Chapter. 2013;15(Unit 15.14):1–51. doi: 10.1002/0471142735.im1514s101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esper L, Utsch L, Soriani FM, et al. Regulatory effects of IL-18 on cytokine profiles and development of myocarditis during Trypanosoma cruzi infection. Microbes Infect. 2014 doi: 10.1016/j.micinf.2014.03.007. pii. S1286-4579(14)00036-7. [DOI] [PubMed] [Google Scholar]
- 10.Pankuweit S, Klingel K. Viral myocarditis: from experimental models to molecular diagnosis in patients. Heart Fail Rev. 2013;18:683–702. doi: 10.1007/s10741-012-9357-4. [DOI] [PubMed] [Google Scholar]
- 11.Afanasyeva M, Georgakopoulos D, Belardi DF, et al. Quantitative analysis of myocardial inflammation by flow cytometry in murine autoimmune myocarditis: correlation with cardiac function. Am J Pathol. 2004;164:807–815. doi: 10.1016/S0002-9440(10)63169-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frisancho-Kiss S, Davis SE, Nyland JF, Frisancho JA, Cihakova D, Rose NR, Fairweather D. Cutting edge: cross-regulation by TLR4 and T cell Ig mucin-3 determines sex differences in inflammatory heart disease. J Immunol. 2007;178:6710–6714. doi: 10.4049/jimmunol.178.11.6710. [DOI] [PubMed] [Google Scholar]
- 13.Fairweather D, Coronado MJ, Garton AE, Dziedzic JL, Bucek A, Cooper LT, Jr, Brandt JE, Alikhan FS, Wang H, Endres CJ, Choi J, Pomper MG, Guilarte TR. Sex differences in translocator protein 18 kDa (TSPO) in the heart: implications for imaging myocardial inflammation. J Cardiovasc Trans Res. 2014;7:192–202. doi: 10.1007/s12265-013-9538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rawji KS, Yong W. The benefits and detriments of macrophages/microglia in models of multiple sclerosis. Clin Dev Immunol. 2013;2013:948976. doi: 10.1155/2013/948976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coronado MJ, Brandt JE, Kim E, Bucek A, Bedja D, Abston ED, Shin J, Gabrielson KL, Mitzner W, Fairweather D. Testosterone and interleukin-1β increase cardiac remodeling during acute coxsackievirus B3 myocarditis via serpin A 3n. Am J Physiol Heart Circ Physiol. 2012;302:H1726–H1736. doi: 10.1152/ajpheart.00783.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fairweather D, Cooper LT, Jr, Blauwet LA. Sex and gender differences in myocarditis and dilated cardiomyopathy. Curr Probl Cardiol. 2013;38:7–46. doi: 10.1016/j.cpcardiol.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fairweather D, Kaya Z, Shellam GR, Lawson CM, Rose NR. From infection to autoimmunity. J Autoimm. 2001;16:175–186. doi: 10.1006/jaut.2000.0492. [DOI] [PubMed] [Google Scholar]
- 18.Fairweather D, Frisancho-Kiss S, Njoku DB, Nyland JF, Kaya Z, Yusung SA, Davis SE, Frisancho JA, Barrett MA, Rose NR. Complement receptor 1 and 2 deficiency increases coxsackievirus B3-induced myocarditis and heart failure by increasing macrophages, IL-1β and immune complex deposition in the heart. J Immunol. 2006;176:3516–3524. doi: 10.4049/jimmunol.176.6.3516. [DOI] [PubMed] [Google Scholar]
- 19.Mascaro-Blanco A, Alvarez K, Yu X, et al. Consequences of unlocking the cardiac myosin molecule in human myocarditis and cardiomyopathies. Autoimmunity. 2008;41:442–453. doi: 10.1080/08916930802031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 21.Zhang P, Cox CJ, Alvarez KM, Cunningham MW. Cutting edge: cardiac myosin activates innate immune responses through TLRs. J Immunol. 2009;183:27–31. doi: 10.4049/jimmunol.0800861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mills KHG. TLR-dependent T cell activation in autoimmunity. Nat Rev Immunol. 2011;11:807–822. doi: 10.1038/nri3095. [DOI] [PubMed] [Google Scholar]
- 23.Kawasaki T, Kawai T, Akira S. Recognition of nucleic acids by pattern-recognition receptors and its relevance in autoimmunity. Immunol Rev. 2011;243:61–73. doi: 10.1111/j.1600-065X.2011.01048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanamsagar R, Hanke ML, Kielian T. Toll-like receptor (TLR) and inflammasome actions in the central nervous system. Trends Immunol. 2012;33:333–342. doi: 10.1016/j.it.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masters SL. Specific inflammasomes in complex diseases. Clin Immunol. 2013;143:223–228. doi: 10.1016/j.clim.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 26.Rosenbaum JT, Kim HW. Innate immune signals in autoimmune and autoinflammatory uveitis. Int Rev Immunol. 2013;32:68–75. doi: 10.3109/08830185.2012.750132. [DOI] [PubMed] [Google Scholar]
- 27.Blander JM, Torchinsky MB, Campisi L. Revisiting the old link between infection and autoimmune disease with commensals and T helper 17 cells. Immunol Res. 2012;54:50–68. doi: 10.1007/s12026-012-8311-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cusick MF, Libbey JE, Fujinami RS. Molecular mimicry as a mechanism of autoimmune disease. Clinic Rev Allerg Immunol. 2012;42:102–111. doi: 10.1007/s12016-011-8293-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rose NR. The discovery of thyroid autoimmunity. Immunol Today. 1991;12:167–168. doi: 10.1016/S0167-5699(05)80047-7. [DOI] [PubMed] [Google Scholar]
- 30.Nahrendorf M, Swirski FK, Aikawa E, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shakya AK, Nandakumar KS. Applications of polymeric adjuvants in studying autoimmune responses and vaccination against infectious diseases. J R Soc Interface. 2012;10:20120536. doi: 10.1098/rsif.2012.0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stubgen J-P. Immune-mediated myelitis following hepatitis B vaccination. Autoimmun Rev. 2012;12:144–149. doi: 10.1016/j.autrev.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 33.Yeter D, Deth R. ITPKC susceptibility in Kawasaki syndrome as a sensitizing factor for autoimmunity and coronary arterial wall relaxation induced by thimerosal’s effects on calcium signaling via IP3. Autoimmun Rev. 2012;11:903–908. doi: 10.1016/j.autrev.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 34.Segal LA, Bar-Or RL. On the role of a possible dialogue between cytokine and TCR presentation mechanisms in the regulation of autoimmune disease. J Theor Biol. 1998;190:161–178. doi: 10.1006/jtbi.1997.0545. [DOI] [PubMed] [Google Scholar]
- 35.Caforio AL, Iliceto S. Genetically determined myocarditis: clinical presentation and immunological characteristics. Curr Opin Cardiol. 2008;23:219–226. doi: 10.1097/HCO.0b013e3282fbf572. [DOI] [PubMed] [Google Scholar]
- 36.Li HS, Ligons DL, Rose NR. Genetic complexity of autoimmune myocarditis. Autoimmun Rev. 2008;7:168–173. doi: 10.1016/j.autrev.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guilherme L, Kohler KF, Postol E, Kalil J. Genes, autoimmunity and pathogenesis of rheumatic heart disease. Ann Pediatr Cardiol. 2011;4:13–21. doi: 10.4103/0974-2069.79617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Germolic D, Kono DH, Pfau JC, Pollard KM. Animal models used to examine the role of environment in the development of autoimmune disease: findings from an NIEHS Expert Panel Workshop. J Autoimm. 2012;39:285–293. doi: 10.1016/j.jaut.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mallampalli MP, Davies E, Wood D, et al. Role of environment and sex differences in the development of autoimmune diseases: a roundtable meeting report. J Women’s Health. 2013;22:578–586. doi: 10.1089/jwh.2013.4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rigante D, Mazzoni MB, Esposito S. The cryptic interplay between systemic lupus erythematosus and infections. Autoimmun Rev. 2014;13:96–102. doi: 10.1016/j.autrev.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 41.Liao L, Sindhwani R, Rojkind M, et al. Antibody-mediated autoimmune myocarditis depends on genetically determined target organ sensitivity. J Exp Med. 1995;181:1123–1131. doi: 10.1084/jem.181.3.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pearce JM. Cardiac lesions in rabbits produced by a filterable virus (virus Ill) Arch Pathol (Chicago) 1939;28:827–845. [Google Scholar]
- 43.Moraska A, Huber SA. Synergism between adriamycin and coxsackievirus group B type 3 (CVB3) in induction of myocardial injury: potential role for gamma/delta TCR+ T lymphocytes in pathogenesis. Clin Immunol Immunopathol. 1993;68:124–128. doi: 10.1006/clin.1993.1107. [DOI] [PubMed] [Google Scholar]
- 44.Latif N, Zhang H, Archard LC, et al. Characterization of anti-heart antibodies in mice after infection with coxsackie B3 virus. Clin Immunol. 1999;91:90–98. doi: 10.1006/clim.1998.4679. [DOI] [PubMed] [Google Scholar]
- 45.Melo TG, Almeida DS, Meirelles MN, Pereira MC. Disarray of sarcomeric alpha-actinin in cardiomyocytes infected by Trypanosoma cruzi. Parasitology. 2006;133(Pt 2):171–178. doi: 10.1017/S0031182006000011. [DOI] [PubMed] [Google Scholar]
- 46.Kaya Z, Katus HA, Rose NR. Cardiac troponins and autoimmunity: their role in the pathogenesis of myocarditis and of heart failure. Clin Immunol. 2010;134:80–88. doi: 10.1016/j.clim.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Agewall S, Giannitsis E, Jernbert T, Katus H. Troponin elevation in coronary vs. non-coronary disease. Eur Heart J. 2011;32:404–411. doi: 10.1093/eurheartj/ehq456. [DOI] [PubMed] [Google Scholar]
- 48.Fairweather D, Abston ED, Coronado MJ. Biomarkers of heart failure in myocarditis and dilated cardiomyopathy. In: Cihakova D, editor. Myocarditis. InTech Open Access Publisher; Rijeka, Croatia: 2011. pp. 323–348. [Google Scholar]
- 49.De Scheerder IK, de Buyzere ML, Delanghe JR, Clement DL, Wieme RJ. Anti-myosin humoral immune response following cardiac injury. Autoimmunity. 1989;4:51–58. doi: 10.3109/08916938909034359. [DOI] [PubMed] [Google Scholar]
- 50.Tam JC, Jacques DA. Intracellular immunity: finding the enemy within-how cells recognize and respond to intracellular pathogens. J Leukoc Biol. 2014 doi: 10.1189/jlb.4RI0214-090R. pii. jib.4RI0214–090R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abston ED, Barin JG, Cihakova D, Bucek A, Coronado MJ, Brandt JE, Bedja D, Kim JB, Georgakopoulos D, Gabrielson KL, Mitzner W, Fairweather D. IL-33 independently induces eosinophilic pericarditis and cardiac dilation: ST2 improves cardiac function. Circ Heart Fail. 2012;5:366–375. doi: 10.1161/CIRCHEARTFAILURE.111.963769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gupta M, Lent RW, Kaplan EL, Zabriskie JB. Serum cardiac troponin I in acute rheumatic fever. Am J Cardiol. 2002;89:779–782. doi: 10.1016/s0002-9149(01)02358-x. [DOI] [PubMed] [Google Scholar]
- 53.Quinn A, Kosanke S, Fischetti VA, Factor SM, Cunningham MW. Induction of autoimmune valvular heart disease by recombinant streptococcal M protein. Infect Immun. 2001;69:4072–4078. doi: 10.1128/IAI.69.6.4072-4078.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li Y, Heuser JS, Kosanke SD, Hemric M, Cunningham MW. Cryptic epitope identified in rat and human cardiac myosin S2 region induces myocarditis in the Lewis rat. J Immunol. 2004;172:3225–3234. doi: 10.4049/jimmunol.172.5.3225. [DOI] [PubMed] [Google Scholar]
- 55.Badorff C, Knowlton KU. Dystrophin disruption in enterovirus-induced myocarditis and dilated cardiomyopathy: from bench to bedside. Med Microbiol Immunol. 2004;193:121–126. doi: 10.1007/s00430-003-0189-7. [DOI] [PubMed] [Google Scholar]
- 56.Borghans JA, De Beor RJ. A minimal model for T-cell vaccination. Proc Biol Sci. 1995;259:173–178. doi: 10.1098/rspb.1995.0025. [DOI] [PubMed] [Google Scholar]
- 57.Lehmann PV, Forsthuber T, Miller A, Sercarz EE. Spreading of T cell autoimmunity to cryptic determinants of an autoantigen. Nature. 1992;358:155–157. doi: 10.1038/358155a0. [DOI] [PubMed] [Google Scholar]
- 58.Powell AM, Black MM. Epitope spreading: protection from pathogens, but propagation of autoimmunity? Clin Exp Dermatol. 2001;26:427–432. doi: 10.1046/j.1365-2230.2001.00852.x. [DOI] [PubMed] [Google Scholar]
- 59.Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol. 2002;2:85–95. doi: 10.1038/nri724. [DOI] [PubMed] [Google Scholar]
- 60.Notkins AL. Pathogenic mechanisms in autoimmune disease. Autoimmun Rev. 2004;3(Suppl 1):S7–S9. [PubMed] [Google Scholar]
- 61.Takata S, Nakamura H, Umemoto S, et al. Identification of autoantibodies with the corresponding antigen for repetitive coxsackievirus infection-induced cardiomyopathy. Circ J. 2004;68:677–682. doi: 10.1253/circj.68.677. [DOI] [PubMed] [Google Scholar]
- 62.Yung RL, Julius A. Epigenetics, aging, and autoimmunity. Autoimmunity. 2008;41:329–335. doi: 10.1080/08916930802024889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Denic A, Johnson AJ, Bieber AJ, et al. The relevance of animal models in multiple sclerosis research. Pathophysiology. 2011;18:21–29. doi: 10.1016/j.pathophys.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miller SD, Katz-Levy Y, Neville KL, Vanderlugt CL. Virus-induced autoimmunity: epitope spreading to myelin autoepitopes in Theiler’s virus infection of the central nervous system. Adv Virus Res. 2001;56:199–217. doi: 10.1016/s0065-3527(01)56008-x. [DOI] [PubMed] [Google Scholar]
- 65.Matsumoto Y, Park IK, Kohyama K. B-cell epitope spreading is a critical step for the switch from C-protein-induced myocarditis to dilated cardiomyopathy. Am J Pathol. 2007;170:43–51. doi: 10.2353/ajpath.2007.060544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Neu N, Craig SW, Rose NR, et al. Coxsackievirus induced myocarditis in mice: cardiac myosin autoantibodies do not cross-react with the virus. Clin Exp Immunol. 1987;69:566–574. [PMC free article] [PubMed] [Google Scholar]
- 67.Paque RE, Miller R. Autoanti-idiotypes exhibit mimicry of myocyte antigens in virus-induced myocarditis. J Virol. 1991;65:16–22. doi: 10.1128/jvi.65.1.16-22.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wolff PG, Kuhl U, Schultheiss HP. Laminin distribution and autoantibodies to laminin in dilated cardiomyopathy and myocarditis. Am Heart J. 1989;117:1303–1309. doi: 10.1016/0002-8703(89)90410-9. [DOI] [PubMed] [Google Scholar]
- 69.Magnusson Y, Marullo S, Hoyer S, et al. Mapping of a functional autoimmune epitope on the beta-adrenergic receptor in patients with idiopathic dilated cardiomyopathy. J Clin Invest. 1990;86:1658–1663. doi: 10.1172/JCI114888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wallukat G, Morwinski M, Kowal K, et al. Antibodies against the beta-adrenergic receptor in human myocarditis and dilated cardiomyopathy: beta-adrenergic agonism without desensitization. Eur Heart J. 1991;12(Suppl D):178–181. doi: 10.1093/eurheartj/12.suppl_d.178. [DOI] [PubMed] [Google Scholar]
- 71.Fu LX, Magnusson Y, Bergh CH, et al. Localization of a functional autoimmune epitope on the muscarinic acetylcholine receptor-2 in patients with idiopathic dilated cardiomyopathy. J Clin Invest. 1993;91:1964–1968. doi: 10.1172/JCI116416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ansari AA, Neckelmann N, Villinger F, et al. Epitope mapping of the branched chain a-ketoacid dehydrogenase dihydrolipoyl transacylase (BCKD-E2) protein that reacts with sera from patients with idiopathic dilated cardiomyopathy. J Immunol. 1994;153:4754–4765. [PubMed] [Google Scholar]
- 73.Schultheiss HP, Schulze K, Dorner A. Significance of the adenine nucleotide translocator in the pathogenesis of viral heart disease. Mol Cell Biochem. 1996;163/164:319–327. doi: 10.1007/BF00408672. [DOI] [PubMed] [Google Scholar]
- 74.Plotz PH. Autoantibodies are anti-idiotype antibodies to antiviral antibodies. Lancet ii. 1983:824–826. doi: 10.1016/s0140-6736(83)90740-7. [DOI] [PubMed] [Google Scholar]
- 75.Root-Bernstein R, Fairweather D. Complexities in the relationship between infection and autoimmunity. Curr Allergy Asthma Rep. 2014;14:407. doi: 10.1007/s11882-013-0407-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Paque RE, Miller R. Adoptively transferred anti-idiotype pulsed B cells mediate autoimmune myocarditis. Infect Immun. 1992;60:3396–3404. doi: 10.1128/iai.60.8.3396-3404.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Paque RE, Miller R. Modulation of murine coxsackievirus-induced myocarditis utilizing anti-idiotypes. Viral Immunol. 1988;1:207–224. doi: 10.1089/vim.1987.1.207. [DOI] [PubMed] [Google Scholar]
- 78.Paque RE, Miller R. Polyclonal anti-idiotypes influence macrophage chemotaxis in coxsackievirus-induced murine myocarditis. J Leukocyte Biol. 1988;45:79–86. doi: 10.1002/jlb.45.1.79. [DOI] [PubMed] [Google Scholar]
- 79.Paque RE, Miller R. Monoclonal anti-idiotypic antibodies regulate the expression of virus-induced murine myocarditis. Infect Immun. 1989;57:2864–2871. doi: 10.1128/iai.57.9.2864-2871.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Root-Bernstein R. Rethinking molecular mimicry in rheumatic heart disease and autoimmune myocarditis: laminin, collagen IV, CAR and B1AR as initial targets of disease. Front Pediatr. 2014;2:85. doi: 10.3389/fped.2014.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Root-Bernstein RS, Vonck J, Podufaly A. Antigenic complementarity between coxsackie virus and streptococci in rheumatic heart disease and myocarditis. Autoimmunity. 2009;22:177–187. doi: 10.1080/08916930802208540. [DOI] [PubMed] [Google Scholar]
- 82.Tandon R, Sharma M, Chandrasekhar Y, Kotb M, Yacoub MH, Narula J. Revisiting the pathogenesis of rheumatic fever and carditis. Nat Rev Cardiol. 2013;10:171–177. doi: 10.1038/nrcardio.2012.197. [DOI] [PubMed] [Google Scholar]
- 83.Selinka HC, Wolde A, Sauter M, Kandolf R, Klingel K. Virus-receptor interactions of coxsackie B viruses and their putative influence on cardiotropism. Med Microbiol Immunol. 2004;193:127–131. doi: 10.1007/s00430-003-0193-y. [DOI] [PubMed] [Google Scholar]
- 84.Shi Y, Chen C, Lisewski U, et al. Cardiac deletion of the coxsackievirus-adenovirus receptor abolishes coxsackievirus B3 infection and prevents myocarditis in vivo. J Am Coll Cardiol. 2009;53:1219–1226. doi: 10.1016/j.jacc.2008.10.064. [DOI] [PubMed] [Google Scholar]
- 85.Roberts BJ, Dragon JA, Moussawi M, Huber SA. Sex-specific signaling through Toll-like receptors 2 and 4 contributes to survival outcome of coxsackievirus B3 infection in C57BL/6 mice. Biol Sex Differ. 2012;3:25. doi: 10.1186/2042-6410-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fairweather D, Stafford KA, Sung YK. Update on coxsackievirus B3 myocarditis. Curr Opin Rheumatol. 2012;24:401–407. doi: 10.1097/BOR.0b013e328353372d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Orthopoulos G, Triantafilou K, Triantafilou M. Coxsackie B viruses use multiple receptors to infect human cardiac cells. J Med Virol. 2004;74:291–299. doi: 10.1002/jmv.20184. [DOI] [PubMed] [Google Scholar]
- 88.Cifuente JO, Ferrer MF, Jaquenod de Giusti C, et al. Molecular determinants of disease in coxsackievirus B1 murine infection. J Med Virol. 2011;83:1571–1581. doi: 10.1002/jmv.22133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Weremeichik H, Moraska A, Herzum M, Weller A, Huber SA. Naturally occurring anti-idiotypic antibodies-mechanisms for autoimmunity and immunoregulation? Eur Heart J. 1991;12(Suppl D):154–157. doi: 10.1093/eurheartj/12.suppl_d.154. [DOI] [PubMed] [Google Scholar]
- 90.Weller AH, Hall M, Huber SA. Polyclonal immunoglobulin therapy protects against cardiac damage in experimental coxsackievirus-induced myocarditis. Eur Heart J. 1992;13:115–119. doi: 10.1093/oxfordjournals.eurheartj.a060030. [DOI] [PubMed] [Google Scholar]
- 91.Newkirk MM. Rheumatoid factors: host resistance or autoimmunity? Clin Immunol. 2002;104:1–13. doi: 10.1006/clim.2002.5210. [DOI] [PubMed] [Google Scholar]
- 92.Cohen IR, Atlan H. Network regulation of autoimmunity: an automation model. J Autoimmun. 1989;2:613–625. doi: 10.1016/s0896-8411(89)80001-0. [DOI] [PubMed] [Google Scholar]
- 93.Segel LA, Jager E. Reverse engineering: a model for T-cell vaccination. Bull Math Biol. 1994;56:687–721. doi: 10.1007/BF02460717. [DOI] [PubMed] [Google Scholar]
- 94.Borghans JA, De Boer RJ, Sercarz E, Kumar V. T cell vaccination in experimental autoimmune encephalomyelitis: a mathematical model. J Immunol. 1998;161:1087–1093. [PubMed] [Google Scholar]
- 95.De Boer RJ, Hogeweg P. Idiotypic networks incorporating T-B cell co-operation. The conditions for percolation. J Theor Biol. 1989;139:17–38. doi: 10.1016/s0022-5193(89)80055-4. [DOI] [PubMed] [Google Scholar]
- 96.Sulzer B, Weisbuch G. Idiotypic regulation of B cell differentiation. Bull Math Biol. 1995;57:841–864. doi: 10.1007/BF02458297. [DOI] [PubMed] [Google Scholar]
- 97.Damian RT. A theory of immunoselection for eclipsed antigens of parasites and its implications for the problem of antigenic polymorphism in man. J Parasitol. 1962;48:16. [Google Scholar]
- 98.Damian RT. Molecular mimicry: antigen sharing by parasite and host and its consequences. Am Natur. 1964;98:129–149. [Google Scholar]
- 99.Lane D, Koprowski H. Molecular recognition and the future of monoclonal antibodies. Nature. 1982;296:200–202. doi: 10.1038/296200a0. [DOI] [PubMed] [Google Scholar]
- 100.Fujinami RS, Oldstone MBA, Wroblewska Z, Frankel ME, Koprowski H. Molecular mimicry in virus infection: crossreaction of measles virus phosphoprotein or of herpes simplex virus protein with human intermediate filaments. Proc Natl Acad Sci USA. 1983;80:2346–2350. doi: 10.1073/pnas.80.8.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fujinami RS, Oldstone MBA. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: mechanism for autoimmunity. Science. 1985;230:1043–1045. doi: 10.1126/science.2414848. [DOI] [PubMed] [Google Scholar]
- 102.Oldstone MBA. Molecular mimicry and autoimmune disease. Cell. 1987;50:819–820. doi: 10.1016/0092-8674(87)90507-1. [DOI] [PubMed] [Google Scholar]
- 103.Srinivasappa J, Saegusa J, Prabhakar BS, et al. Molecular mimicry: frequency of reactivity of monoclonal antiviral antibodies with normal tissues. J Virol. 1986;57:397–401. doi: 10.1128/jvi.57.1.397-401.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kaplan MH, Meyeserian M. An immunological cross-reaction between group A streptococcal cells and human heart tissue. Lancet. 1962;1:706. doi: 10.1016/s0140-6736(62)91653-7. [DOI] [PubMed] [Google Scholar]
- 105.Kaplan MH. Immunologic relation of streptococcal and tissue antigens. I. Properties of an antigen in certain strains of group A streptococci exhibiting an immunologic cross-reaction with human heart tissue. J Immunol. 1963;90:595. [PubMed] [Google Scholar]
- 106.Kaplan MH, Suchy ML. Immunologic relation of streptococcal and tissue antigens. II. Cross-reaction of antisera to mammalian heart tissue with a cell wall constituent of certain strains of Group A streptococci. J Exp Med. 1964;119:643. doi: 10.1084/jem.119.4.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kaplan MH. Rheumatic fever, rheumatic heart disease, and the streptococcal connection: the role of streptococcal antigens cross-reactive with heart tissue. Rev Infect Dis. 1979;1:988–986. doi: 10.1093/clinids/1.6.988. [DOI] [PubMed] [Google Scholar]
- 108.Wood JN, Hudson L, Jessell TM, Yamamoto M. A monoclonal antibody defining antigenic determinants on subpopulations of mammalian neurons and Trypanosoma cruzi parasites. Nature. 1982;296:34–38. doi: 10.1038/296034a0. [DOI] [PubMed] [Google Scholar]
- 109.Williams RC., Jr Rheumatic fever and the streptococcus: another look at molecular mimicry. Am J Med. 1983;75:727–730. doi: 10.1016/0002-9343(83)90399-6. [DOI] [PubMed] [Google Scholar]
- 110.Froude J, Gibofsky A, Buskirk DR, Khanna A, Zabriskie JB. Cross-reactivity between Streptococcus and human tissue: a model of molecular mimicry and autoimmunity. Curr Top Microbiol Immunol. 1989;145:5–26. doi: 10.1007/978-3-642-74594-2_2. [DOI] [PubMed] [Google Scholar]
- 111.Krisher K, Cunningham MW. Myosin: a link between streptococci and heart. Science. 1985;227:413–415. doi: 10.1126/science.2578225. [DOI] [PubMed] [Google Scholar]
- 112.Cunningham MW, McCormack JM, Fenderson PG, Ho MK, Beachey EH, Dale JB. Human and murine antibodies cross-reactive with streptococcal M protein and myosin recognize the sequence GLN-LYS-SER-LYS-GLN in M protein. J Immunol. 1989;143:2677–2683. [PubMed] [Google Scholar]
- 113.Cunningham MW, Antone SM, Gulizia JM, McManus BM, Fischetti VA, Gauntt CJ. Cytotoxic and viral neutralizing antibodies cross-react with streptococcal M protein, enteroviruses and human cardiac myosin. Proc Natl Acad Sci USA. 1992;89:1320–1324. doi: 10.1073/pnas.89.4.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Huber SA, Cunningham MW. Streptococcal M protein peptide with similarity to myosin induces CD4+ T cell-dependent myocarditis in MRL/++ mice and induces partial tolerance against coxsakieviral myocarditis. J Immunol. 1996;156:3528–3534. [PubMed] [Google Scholar]
- 115.Massilamany C, Gangaplara A, Steffen D, Reddy J. Identification of novel mimicry epitopes for cardiac myosin heavy chain-α that induce autoimmune myocarditis in A/J mice. Cell Immunol. 2011;271:438–449. doi: 10.1016/j.cellimm.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 116.Beisel KW, Srinivasappa J, Olsen MR, Stiff AC, Essani K, Prabhakar BS. A neutralizing monoclonal antibody against coxsackievirus B4 cross-reacts with contractile muscle proteins. Microb Pathog. 1990;8:151–156. doi: 10.1016/0882-4010(90)90079-6. [DOI] [PubMed] [Google Scholar]
- 117.Huber S, Polgar J, Moraska A, Cunningham MW, Schwimmbeck P, Schultheiss P. T lymphocyte responses in CVB3-induced murine myocarditis. Scand J Infect Dis. 1993;88(Suppl):67–78. [PubMed] [Google Scholar]
- 118.Gauntt CJ, Higdon AL, Arizpe HM, et al. Epitopes shared between coxsackievirus B3 (CVB3) and normal heart tissue contribute to CVB3-induced murine myocarditis. Clin Immunol Immunopathol. 1993;68:129. doi: 10.1006/clin.1993.1108. [DOI] [PubMed] [Google Scholar]
- 119.Wolfgram LJ, Beisel KW, Rose NR. Heart-specific autoantibodies following murine coxsackievirus B3 myocarditis. J Exp Med. 1985;161:1112. doi: 10.1084/jem.161.5.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Neumann DA, Rose NR, Ansari AA, Herskowitz A. Induction of multiple heart autoantibodies in mice with coxsackievirus B3- and cardiac myosin-induced autoimmune myocarditis. J lmmunol. 1994;152:343. [PubMed] [Google Scholar]
- 121.Ciháková D, Sharma RB, Fairweather D, Afanasyeva M, Rose NR. Animal models for autoimmune myocarditis and autoimmune thyroiditis. Methods Mol Med. 2004;102:175–193. doi: 10.1385/1-59259-805-6:175. [DOI] [PubMed] [Google Scholar]
- 122.Galvin JE, Hemric ME, Ward K, Cunningham MW. Cytotoxic mAb from rheumatic carditis recognizes heart valves and laminin. J Clin Invest. 2000;106:217–224. doi: 10.1172/JCI7132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shikhman AR, Greenspan NS, Cunningham MW. A subset of mouse monoclonal antibodies cross-reactive with cytoskeletal proteins and group A streptococcal M proteins recognizes N-acetyl-beta-D-glucosamine. J Immunol. 1993;151:3902–3913. [PubMed] [Google Scholar]
- 124.Guilherme L, Kalil J. Rheumatic fever: from sore throat to autoimmune heart lesions. Int Arch Allergy Immunol. 2004;134:56–64. doi: 10.1159/000077915. [DOI] [PubMed] [Google Scholar]
- 125.Guilherme L, Köhler KF, Kalil J. Rheumatic heart disease: mediation by complex immune events. Adv Clin Chem. 2011;53:31–50. [PubMed] [Google Scholar]
- 126.Rose NR. The role of infection in the pathogenesis of autoimmune disease. Semin Immunol. 1998;10:5–13. doi: 10.1006/smim.1997.0100. [DOI] [PubMed] [Google Scholar]
- 127.Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 128.Higgins PM. Splenomegaly in acute infections due to group A streptococci and viruses. Epidemiol Infect. 1992;109:199–209. doi: 10.1017/s0950268800050160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.De Rosa GL, Pardeo M, Stabile A, Rigante D. Rheumatic heart disease in children: from clinical assessment to therapeutical management. Eur Rev Med Pharmacol Sci. 2006;10:107–110. [PubMed] [Google Scholar]
- 130.Rose NR. Predictors of autoimmune disease: autoantibodies and beyond. Autoimmunity. 2008;41:419–428. doi: 10.1080/08916930802031686. [DOI] [PubMed] [Google Scholar]
- 131.Davies AM. Molecular mimicry: can epitope mimicry induce autoimmune disease? Immunol Cell Biol. 1997;75:113–126. doi: 10.1038/icb.1997.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rose NR, Mackay IR. Molecular mimicry: a critical look at exemplary instances in human diseases. Cell Mol Life Sci. 2000;57:542–551. doi: 10.1007/PL00000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Benoist CL, Mathis D. Autoimmunity provoked by infection: how good is the case for T cell epitope mimicry? Nat Immunol. 2001;2:797–801. doi: 10.1038/ni0901-797. [DOI] [PubMed] [Google Scholar]
- 134.Fourneau J-M, Bach J-M, van Endert PM, Bach J-F. The elusive case for a role of molecular mimicry in autoimmune diseases. Mol Immunol. 2004;40:1095–1102. doi: 10.1016/j.molimm.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 135.Fujinami RS, von Herrath MG, Christen U, Whitton JL. Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin Microbiol Rev. 2006;19:80. doi: 10.1128/CMR.19.1.80-94.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Massilamany C, Huber SA, Cunningham MW, Reddy J. Relevance of molecular mimicry in the mediation of infectious myocarditis. J Cardiovasc Trans Res. 2013;7:165–171. doi: 10.1007/s12265-013-9519-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gonnella PA, Del Nido PJ, McGowan FX. Oral tolerization with cardiac myosin peptide (614–629) ameliorates experimental autoimmune myocarditis: role of STAT 6 genes in BALB/CJ mice. J Clin Immunol. 2009:434–443. doi: 10.1007/s10875-009-9290-z. [DOI] [PubMed] [Google Scholar]
- 138.Geluk AL, van Meijgaarden KE, Ottenhoff TH. Flexibility in T-cell receptor ligand repertoires depends on MHC and T-cell receptor clonotype. Immunology. 1997;90:370–375. doi: 10.1111/j.1365-2567.1997.00370.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.de Haan EC, Wagenaar-Hilbers JPA, Liskamp RMJ, Moret EE, Wauben MHM. Limited plasticity in T cell recognition of modified T cell receptor contact residues in MHC class II bound peptides. Mol Immunol. 2005;42:355–364. doi: 10.1016/j.molimm.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 140.Hurford AL, Day T. Immune evasion and the evolution of molecular mimicry in parasites. Evolution. 2013;67:2889–2904. doi: 10.1111/evo.12171. [DOI] [PubMed] [Google Scholar]
- 141.Blyuss KB, Nicholson LB. The role of tunable activation thresholds in the dynamics of autoimmunity. J Theor Biol. 2012;308:45–55. doi: 10.1016/j.jtbi.2012.05.019. [DOI] [PubMed] [Google Scholar]
- 142.Pinto AA, Burroughs NJ, Ferreira M, Oliveira BM. Dynamics of immunological models. Acta Biotheor. 2010;58:391–404. doi: 10.1007/s10441-010-9117-6. [DOI] [PubMed] [Google Scholar]
- 143.Kristóf K, Madách K, Czaller I, Bajtay Z, Erdei A. Mathematical analysis of clinical data reveals a homunculus of bacterial mimotopes protecting from autoimmunity via oral tolerance in human. Mol Immunol. 2009;46:1673–1678. doi: 10.1016/j.molimm.2009.02.017. [DOI] [PubMed] [Google Scholar]
- 144.Dinkla K, Rohde M, Jansen WT, Kaplan EL, Chhatwal GS, Talay SR. Rheumatic fever-associated Streptococcus pyogenes isolates aggregate collagen. J Clin Invest. 2003;111:1905–1912. doi: 10.1172/JCI17247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dinkla K, Nitsche-Schmitz DP, Barroso V, et al. Identification of a streptococcal octapeptide motif involved in acute rheumatic fever. J Biol Chem. 2007;282:18686–18693. doi: 10.1074/jbc.M701047200. [DOI] [PubMed] [Google Scholar]
- 146.Dinkla K, Talay SR, Mörgelin M, et al. Crucial role of the CB3-region of collagen IV in PARF-induced acute rheumatic fever. PLoS One. 2009;4:e4666. doi: 10.1371/journal.pone.0004666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Henao-Martínez AF, Schwartz DA, Yang IV. Chagasic cardiomyopathy, from acute to chronic: is this mediated by host susceptibility factors? Trans R Soc Trop Med Hyg. 2012;106:521–527. doi: 10.1016/j.trstmh.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 148.Halsell JS, Riddle JR, Atwood JE, et al. Myopericarditis following smallpox vaccination among vaccinia-naive US military personnel. J Am Med Assoc. 2003;289:3283–3289. doi: 10.1001/jama.289.24.3283. [DOI] [PubMed] [Google Scholar]
- 149.Abrams D, Derrick G, Penny DJ, et al. Cardiac complications in children following infection with varicella zoster virus. Cardiol Young. 2001;11:647–652. doi: 10.1017/s1047951101001019. [DOI] [PubMed] [Google Scholar]
- 150.Bowles NE, Ni J, Kearney DL, et al. Detection of viruses in myocardial tissues by polymerase chain reaction: evidence of adenovirus as a common cause of myocarditis in children and adults. J Am Coll Cardiol. 2003;42:466–472. doi: 10.1016/s0735-1097(03)00648-x. [DOI] [PubMed] [Google Scholar]
- 151.Sato Y, Yamada T, Matsumori A. Hepatitis C virus and cardiomyopathy. In: Matsumori A, editor. Cardiomyopathies and Heart Failure: Biomolecular, Infectious, and Immune Mechanisms. Kluwer Academic Publishers; Boston, Mass: 2003. pp. 325–339. [Google Scholar]
- 152.Pauschinger M, Bowles NE, Fuentes-Garcia FJ, et al. Detection of adenoviral genome in the myocardium of adult patients with idiopathic left ventricular dysfunction. Circulation. 1999;99:1348–1354. doi: 10.1161/01.cir.99.10.1348. [DOI] [PubMed] [Google Scholar]
- 153.Pankoweit S, Lamparter S, Schoppet M, Maisch B. Parvovirus B19 genome in endomyocardial biopsy specimens. Circulation. 2004;109:e179. doi: 10.1161/01.CIR.0000124881.00415.59. [DOI] [PubMed] [Google Scholar]
- 154.Andréoletti L, Lévêque N, Boulagnon C, et al. Viral causes of human myocarditis. Arch Cardiovasc Dis. 2009;102:559–568. doi: 10.1016/j.acvd.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 155.Mahfoud F, Gärtner B, Kindermann M, et al. Virus serology in patients with suspected myocarditis: utility or futility? Eur Heart J. 2011;32:897–903. doi: 10.1093/eurheartj/ehq493. [DOI] [PubMed] [Google Scholar]
- 156.Cunha-Neto E, Bilate AM, Hyland KV, et al. Induction of cardiac autoimmunity in Chagas heart disease: a case for molecular mimicry. Autoimmunity. 2006;39:41–54. doi: 10.1080/08916930500485002. [DOI] [PubMed] [Google Scholar]
- 157.Gironès N, Cuervo H, Fresno M. Trypanosoma cruzi-induced molecular mimicry and Chagas’ disease. Curr Top Microbiol Immunol. 2005;296:89–123. doi: 10.1007/3-540-30791-5_6. [DOI] [PubMed] [Google Scholar]
- 158.Rose NR. Infection, mimics, and autoimmune disease. J Clin Invest. 2001;107:943–944. doi: 10.1172/JCI12673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lawson CM, O’Donoghue HL, Reed WD. Mouse cytomegalovirus infection induces antibodies which cross-react with virus and cardiac myosin: a model for the study of molecular mimicry in the pathogenesis of viral myocarditis. Immunology. 1992;75:513–519. [PMC free article] [PubMed] [Google Scholar]
- 160.O’Donoghue HL, Lawson CM, Reed WD. Autoantibodies to cardiac myosin in mouse cytomegalovirus myocarditis. Immunology. 1990;71:20–28. [PMC free article] [PubMed] [Google Scholar]
- 161.Teixeira-Coelho M, Cruz A, Carmona J, et al. TLR2 deficiency by compromising p19 (IL-23) expression limits Th17 cell responses to Mycobacterium tuberculosis. Int Immunol. 2011;23:89–96. doi: 10.1093/intimm/dxq459. [DOI] [PubMed] [Google Scholar]
- 162.Tough DF, Borrow P, Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science. 1996;272:1947–1950. doi: 10.1126/science.272.5270.1947. [DOI] [PubMed] [Google Scholar]
- 163.Tough DF, Sun S, Sprent J. T cell stimulation in vivo by lipopolysaccharide (LPS) J Exp Med. 1997;185:2089–2094. doi: 10.1084/jem.185.12.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Theil DJ, Tsunoda I, Rodriguez F, Whitton JL, Fujinami RS. Viruses can silently prime for and trigger central nervous system autoimmune disease. J Neurovirol. 2001;7:220–227. doi: 10.1080/13550280152403263. [DOI] [PubMed] [Google Scholar]
- 165.von Herrath MG, Fujinami RS, Whitton JL. Microorganisms and autoimmunity: making the barren field fertile? Nat Rev Microbiol. 2003;1:151–157. doi: 10.1038/nrmicro754. [DOI] [PubMed] [Google Scholar]
- 166.McCoy L, Tsunoda I, Fujinami RS. Multiple sclerosis and virus induced immune responses: autoimmunity can be primed by molecular mimicry and augmented by bystander activation. Autoimmunity. 2006;39:9–19. doi: 10.1080/08916930500484799. [DOI] [PubMed] [Google Scholar]
- 167.Gorton D, Blyth S, Gorton JG, Govan B, Ketheesan N. An alternative technique for the induction of autoimmune valvulitis in a rat model of rheumatic heart disease. J Immunol Methods. 2010;355:80–85. doi: 10.1016/j.jim.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 168.Fohlman J, Pauksen KK, Borein B, et al. High yield production of an inactivated coxsackievirus B3 adjuvant vaccine with protective effect against experimental myocarditis. Scand J Infect Dis. 1993;88(Suppl):103–108. [PubMed] [Google Scholar]
- 169.Vera-Lastra O, Medina G, Cruz-Dominguez P, Jara LJ, Shoenfeld Y. Autoimmune/inflammatory syndrome induced by adjuvants (Shoenfeld’s syndrome): clinical and immunological spectrum. Expert Rev Clin Immunol. 2013;9:361–371. doi: 10.1586/eci.13.2. [DOI] [PubMed] [Google Scholar]
- 170.Kasahara H, Ito M, Sugiyama T, et al. Autoimmune myocarditis induced in mice by cardiac c-protein. J Clin Invest. 1994;94:1026–1036. doi: 10.1172/JCI117416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kato N, Fujii Y, Agata N, et al. Experimental murine model for autoimmune myocarditis using Klebsiella pneunoniae O-3 lipopolysaccharide as a potent immunological adjuvant. Autoimmunity. 1993;14:231–236. doi: 10.3109/08916939309077370. [DOI] [PubMed] [Google Scholar]
- 172.Sutterwala FS, Haasken S, Cassel SL. Mechanism of NLRP3 inflammasome activation. Ann NY Acad Sci. 2014;1319:82–95. doi: 10.1111/nyas.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity. 2013;39:1003–1018. doi: 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Masedunskas A, Porat-Shliom N, Weigert R. Linking differences in membrane tension with the requirement for a contractile actomyosin scaffold during exocytosis in salivary glands. Commun Integr Biol. 2012;5:84–87. doi: 10.4161/cib.18258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Tsunoda I, Libbey JE, Fujinami RS. Sequential polymicrobial infections lead to CNS inflammatory disease: possible involvement of bystander activation in heterologous immunity. J Neuroimmunol. 2007;188:22–33. doi: 10.1016/j.jneuroim.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Burroughs NJ, Oliveira BMPM, Pinto AA. Autoimmunity arising from bystander proliferation of T cells in an immune response model. Math Comput Model. 2011;53:1389–1393. [Google Scholar]
- 177.Padovan E, Casorati G, Dellabona P, et al. Expression of two T cell receptor alpha chains: dual receptor T cells. Science. 1993;262:422–424. doi: 10.1126/science.8211163. [DOI] [PubMed] [Google Scholar]
- 178.Padovan E, Casorati G, Dellabona P, et al. Dual receptor T-cells: implications for allocreactivity and autoimmunity. Ann NY Acad Sci. 1995;756:66–70. doi: 10.1111/j.1749-6632.1995.tb44482.x. [DOI] [PubMed] [Google Scholar]
- 179.Davodeau F, Peyrat M-A, Romagné F, et al. Dual T cell receptor β chain expression on human T lymphocytes. J Exp Med. 1995;181:1391–1398. doi: 10.1084/jem.181.4.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Padovan E, Giachino C, Cella M, et al. Normal T lymphocytes can express two different T cell receptor β chains: implications for the mechanism of allelic exclusion. J Exp Med. 1995;181:1587–1591. doi: 10.1084/jem.181.4.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kekäläinen E, Hänninen A, Maksimow M, Arstila TP. T cells expressing two different T cell receptors form a heterogeneous population containing autoreactive clones. Mol Immunol. 2010;48:211–218. doi: 10.1016/j.molimm.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 182.Auger JL, Haasken S, Steinert EM, Binstadt BA. Incomplete TCR-β allelic exclusion accelerates spontaneous autoimmune arthritis in K/BxN TCR transgenic mice. Eur J Immunol. 2012:2354–2362. doi: 10.1002/eji.201242520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Elliott JI, Altmann DM. Dual T cell receptor alpha chain T cells in autoimmunity. J Exp Med. 1995;182:953–959. doi: 10.1084/jem.182.4.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Corthay A, Nandakumar KS, Holmdahl R. Evaluation of the percentage of peripheral T cells with two different T cell receptor alpha-chains and of their potential role in autoimmunity. J Autoimmun. 2001;16:423–429. doi: 10.1006/jaut.2001.0504. [DOI] [PubMed] [Google Scholar]
- 185.Westall FC, Root Bernstein RS. An explanation of prevention and suppression of experimental allergic encephalomyelitis. Mol Immunol. 1983;20:169–177. doi: 10.1016/0161-5890(83)90128-1. [DOI] [PubMed] [Google Scholar]
- 186.Westall FC, Root Bernstein RS. The cause and prevention of post infectious and post vaccinal encephalopathies in light of a new theory of autoimmunity. Lancet. 1986;2:251–252. doi: 10.1016/s0140-6736(86)92073-8. [DOI] [PubMed] [Google Scholar]
- 187.Pendergraft WF, 3rd, Preston GA, Shah RR, et al. Autoimmunity is triggered by cPR-3(105–201), a protein complementary to human autoantigen proteinase-3. Nat Med. 2004;10:72–79. doi: 10.1038/nm968. [DOI] [PubMed] [Google Scholar]
- 188.Pendergraft WF, 3rd, Pressler BM, Jennette JC, Falk RJ, Preston GA. Autoantigen complementarity: a new theory implicating complementary proteins as initiators of autoimmune disease. J Mol Med (Berl) 2005;83:12–25. doi: 10.1007/s00109-004-0615-3. [DOI] [PubMed] [Google Scholar]
- 189.Preston GA, Pendergraft WF, 3rd, Falk RJ. New insights that link microbes with the generation of antineutrophil cytoplasmic autoantibodies: the theory of autoantigen complementarity. Curr Opin Nephrol Hypertens. 2005;14:217–222. doi: 10.1097/01.mnh.0000165886.93427.b1. [DOI] [PubMed] [Google Scholar]
- 190.Preston G, Falk R. Autoimmunity: does autoantigen complementarity underlie PR3-ANCA AAV? Nat Rev Rheumatol. 2011;7:439–440. doi: 10.1038/nrrheum.2011.86. [DOI] [PubMed] [Google Scholar]
- 191.McGuire KL, Holmes DS. Role of complementary proteins in autoimmunity: an old idea re-emerges with new twists. Trends Immunol. 2005;26:367–372. doi: 10.1016/j.it.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 192.Root-Bernstein RS, Holsworth DD. Antisense peptides: a critical mini-review. J Theor Biol. 1998;190:107–119. doi: 10.1006/jtbi.1997.0544. [DOI] [PubMed] [Google Scholar]
- 193.Siemion IZ, Cebrat M, Kluczyk A. The problem of amino acid complementarity and antisense peptides. Curr Protein Pept Sci. 2004;5:507–527. doi: 10.2174/1389203043379413. [DOI] [PubMed] [Google Scholar]
- 194.Root-Bernstein RS, Yurochko F, Westall FC. Clinical suppression of experimental allergic encephalomyelitis by muramyl dipeptide “adjuvant”. Brain Res Bull. 1986;17:473–476. doi: 10.1016/0361-9230(86)90213-3. [DOI] [PubMed] [Google Scholar]
- 195.Root-Bernstein RS, Westall FC. Serotonin binding sites. II. Muramyl dipeptide binds to serotonin binding sites on myelin basic protein, LHRH, and MSH-ACTH 4-10. Brain Res Bull. 1990;25:827–841. doi: 10.1016/0361-9230(90)90178-3. [DOI] [PubMed] [Google Scholar]
- 196.Takeuchi Y, Root-Bernstein RS, Shih JC. Peptide displacement of [3H]5-hydroxytryptamine binding to bovine cortical membranes. Brain Res Bull. 1990;25:817–820. doi: 10.1016/0361-9230(90)90176-z. [DOI] [PubMed] [Google Scholar]
- 197.Root-Bernstein RS. Multiple antigen mediated autoimmunity (MAMA) in AIDS: a possible model for post infectious autoimmunity. Res Immunol. 1990;141:321–339. doi: 10.1016/0923-2494(90)90024-s. [DOI] [PubMed] [Google Scholar]
- 198.Root-Bernstein RS, Dobbelstein C. Insulin binds to glucagon forming a complex that is hyper-antigenic and inducing complementary antibodies having an idiotype-antiidiotype relationship. Autoimmunity. 2001;33:153–169. doi: 10.3109/08916930109008044. [DOI] [PubMed] [Google Scholar]
- 199.Root-Bernstein R. Autoreactive T-cell receptor (Vbeta/D/Jbeta) sequences in diabetes are homologous to insulin, glucagon, the insulin receptor, and the glucagon receptor. J Mol Recognit. 2009;2:177–187. doi: 10.1002/jmr.930. [DOI] [PubMed] [Google Scholar]
- 200.Root-Bernstein R, Podufaly A. T cell receptor variable regions in diabetes bind to each other, to insulin, glucagon or insulin receptor, and to their antibodies. Open Autoimmun J. 2012;4:10–22. [Google Scholar]
- 201.Root-Bernstein RS, Couturier J. Antigenic complementarity in the origins of autoimmunity: a general theory illustrated with a case study of idiopathic thrombocytopenia purpura. Clin Dev Immunol. 2006;13:49–65. doi: 10.1080/17402520600578731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Root-Bernstein RS. Antigenic complementarity in the induction of autoimmunity: a general theory and review. Autoimmun Rev. 2007;6:272–277. doi: 10.1016/j.autrev.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 203.Pearce JM. Effect of hemolytic toxin of streptococcus pyogenes on viral myocarditis in rabbit. AMA Arch Pathol. 1953;56:113–122. [PubMed] [Google Scholar]
- 204.Kogut EP, Livashova NV, Bondarenk AP, Zherdeva AI, Shuvalova IA. Eksperimental’noe izuchenie koksaki-streptokokkovoi infektsii [Experimental study of coxsackie-streptococcal infection] Vopr Virusol. 1978;6:690–695. [PubMed] [Google Scholar]
- 205.Dörner A, Grunert HP, Lindig V, et al. Treatment of coxsackievirus-B3-infected BALB/c mice with the soluble coxsackie adenovirus receptor CAR4/7 aggravates cardiac injury. J Mol Med (Berl) 2006;84:842–851. doi: 10.1007/s00109-006-0076-y. [DOI] [PubMed] [Google Scholar]
- 206.Goodfellow IG, Evans DJ, Blom AM, et al. Inhibition of coxsackie B virus infection by soluble forms of its receptors: binding affinities, altered particle formation, and competition with cellular receptors. J Virol. 2005;79:12016–12024. doi: 10.1128/JVI.79.18.12016-12024.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.De Boer RJ, Hogeweg P. Memory but no suppression in low-dimensional symmetric idiotypic networks. Bull Math Biol. 1989;51:223–246. doi: 10.1007/BF02458444. [DOI] [PubMed] [Google Scholar]
- 208.Suresh L, Chandrasekar S, Rao RS, Ravi V, Badrinath S. Coxsackie virus and rheumatic fever: a correlative study. J Assoc Physicians India. 1989;37:582–585. [PubMed] [Google Scholar]
- 209.Zaher SR, Kassem AS, Hughes JJ. Coxsackie virus infections in rheumatic fever. Indian J Pediatr. 1993;60:289–298. doi: 10.1007/BF02822194. [DOI] [PubMed] [Google Scholar]
- 210.Vikerfors T, Stjerna A, Olcen P, Malmcrona R, Magnius L. Acute myocarditis: serologic diagnosis, clinical findings and follow-up. Acta Med Scand. 1988;223:45–52. [PubMed] [Google Scholar]
- 211.Novikov I. O dignostike nervmaticheskikh miokarditov [Diagnosis of nonrheumatic myocarditis] Kardiologiia. 1983;23:50–55. [PubMed] [Google Scholar]
- 212.Kuhl U, Pauschinger M, Noutsias M, et al. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation. 2005;111:887–893. doi: 10.1161/01.CIR.0000155616.07901.35. [DOI] [PubMed] [Google Scholar]
- 213.Jacobson DL, Gange SJ, Rose NR, Graham NM. Epidemiology and estimated population burden of selected autoimmune disease in the United States. Clin Immunol Immunopathol. 1997;84:223–243. doi: 10.1006/clin.1997.4412. [DOI] [PubMed] [Google Scholar]
- 214.Whitacre CC. Sex differences in autoimmune disease. Nat Immunol. 2001;2:777–780. doi: 10.1038/ni0901-777. [DOI] [PubMed] [Google Scholar]
- 215.Zandman-Goddard G, Peeva E, Shoenfeld YY. Gender and autoimmunity. Autoimm Rev. 2007;6:366–372. doi: 10.1016/j.autrev.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 216.Fairweather D, Frisancho-Kiss S, Rose NR. Sex differences in autoimmune disease from a pathologic perspective. Am J Pathol. 2008;173:600–609. doi: 10.2353/ajpath.2008.071008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Miller VM. Why are sex and gender important to basic physiology and translational and individualized medicine? Am J Physiol Heart Circ Physiol. 2014;306:H781–H788. doi: 10.1152/ajpheart.00994.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Styrt B, Sugarman B. Estrogens and infection. Rev Infect Dis. 1991;13:1139–1150. doi: 10.1093/clinids/13.6.1139. [DOI] [PubMed] [Google Scholar]
- 219.Lang TJ. Estrogen as an immunomodulator. Clin Immunol. 2004;113:224–230. doi: 10.1016/j.clim.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 220.Cook IF. Sexual dimorphism of humoral immunity with vaccines. Vaccine. 2008;26:3551–3555. doi: 10.1016/j.vaccine.2008.04.054. [DOI] [PubMed] [Google Scholar]
- 221.Flanagan KL, Klein SL, Skakkebaek NE, et al. Sex differences in the vaccine-specific and non-targeted effects of vaccines. Vaccine. 2011;16:2349–2354. doi: 10.1016/j.vaccine.2011.01.071. [DOI] [PubMed] [Google Scholar]
- 222.Straub RH. The complex role of estrogens in inflammation. Endocrine Rev. 2007;28:521–574. doi: 10.1210/er.2007-0001. [DOI] [PubMed] [Google Scholar]
- 223.Rubtsov A, Rubtsova K, Kappler JW, Marrack P. Genetic and hormonal factors in female-biased autoimmunity. Autoimm Rev. 2010;9:494–498. doi: 10.1016/j.autrev.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Lahita RG. Sex hormones and immune function. In: Legato MJ, editor. Principles of Gender-Specific Medicine. 2. Elsevier; Massachusetts: 2010. pp. 615–626. [Google Scholar]
- 225.Wilder RL. Neuroendocrine-immune system interactions and autoimmunity. Annu Rev Immunol. 1995;13:307–338. doi: 10.1146/annurev.iy.13.040195.001515. [DOI] [PubMed] [Google Scholar]
- 226.Gharaee-Kermani M, Hatano K, Nozaki Y, Phan SH. Gender-based differences in bleomycin-induced pulmonary fibrosis. Am J Pathol. 2005;166:1593–1606. doi: 10.1016/S0002-9440(10)62470-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Pennell LM, Galligan CL, Fish EN. Sex affects immunity. J Autoimm. 2012;38:J282–J291. doi: 10.1016/j.jaut.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 228.Fairweather D, Petri MA, Coronado MJ, Cooper LT., Jr Autoimmune heart disease: role of sex hormones and autoantibodies in disease pathogenesis. Expert Rev Clin Immunol. 2012;8:269–284. doi: 10.1586/eci.12.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Koenig A, Sateriale A, Budd RC, Huber SA, Buskiewicz IA. The role of sex differences in autophagy in the heart during coxsackievirus B3-induced myocarditis. J Cardiovasc Transl Res. 2014;7:182–191. doi: 10.1007/s12265-013-9525-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Olgunturk R, Okur I, Cirak MY, et al. The role of viral agents in aetiopathogenesis of acute rheumatic fever. Clin Rheumatol. 2011;30:15–20. doi: 10.1007/s10067-010-1447-x. [DOI] [PubMed] [Google Scholar]
- 231.Burch GE, Sun SC, Colcolough HL, Sohal RS, De Pasquale NP. Coxsackie B viral myocarditis and valvulitis identified in routine autopsy specimens by immunofluorescent techniques. Am Heart J. 1967;74:13–23. doi: 10.1016/0002-8703(67)90035-x. [DOI] [PubMed] [Google Scholar]
- 232.Burch GE, Giles TD, Colcolough HL. Pathogenesis of “rheumatic” heart disease: critique and theory. Am Heart J. 1970;80:556–561. doi: 10.1016/0002-8703(70)90205-x. [DOI] [PubMed] [Google Scholar]
- 233.Burch GE, Giles TD. The role of viruses in the production of heart disease. Am J Cardiol. 1972;29:231–240. doi: 10.1016/0002-9149(72)90634-0. [DOI] [PubMed] [Google Scholar]
- 234.Pongpanich B, Suthas NA, Ayuthya P, Jayavasu J, Sangkavibha N. Coxsackie group B virus and acquired valvular heart disease in children. J Med Assoc Thai. 1976;59:452–456. [PubMed] [Google Scholar]
- 235.Chandy KG, John TJ, Mukundan P, Cherian G. Coxsackie B antibodies in “rheumatic” valvular heart-disease. Lancet. 1979;1:381. doi: 10.1016/s0140-6736(79)92918-0. [DOI] [PubMed] [Google Scholar]
- 236.Chandy KG, John TJ, Cherian G. Coxsackieviruses and chronic valvular heart disease. Am Heart J. 1980;100:578–580. doi: 10.1016/0002-8703(80)90674-2. [DOI] [PubMed] [Google Scholar]
- 237.Górska A, Urban M, Głowińska B, Kowalewski M. Is infection with group A streptococcus the only reason for rheumatic fever? A case report of rheumatic fever coexisting with Coxsackie B1 virus infection. Przegl Lek. 1998;55:418–419. [PubMed] [Google Scholar]
- 238.Limson BM, Chan VF, Guzman SV, Maaba MR, Mendoza MT. Occurrence of infection with group B coxsackievirus in rheumatic and non-rheumatic Filipino children. J Infect Dis. 1979;140:415–418. doi: 10.1093/infdis/140.3.415. [DOI] [PubMed] [Google Scholar]
- 239.Pearce JM. The effect of hemolytic streptococcus toxin on experimental viral carditis. Am J Pathol. 1951;27:699–700. [PubMed] [Google Scholar]
- 240.Mallampalli MP, Davies E, Wood D, Robertson H, Polato F, Carter CL. Role of environmental and sex differences in the development of autoimmune disease: a roundtable meeting report. J Women’s Health. 2013;22:578–586. doi: 10.1089/jwh.2013.4462. [DOI] [PMC free article] [PubMed] [Google Scholar]