

Abstract
Depression is a common cause of mortality and morbidity, but the biological bases of the deficits in emotional and cognitive processing remain incompletely understood. Current antidepressant therapies are effective in only some patients and act slowly. We propose an excitatory synapse hypothesis of depression in which chronic stress and genetic susceptibility cause changes in the strength of subsets of glutamatergic synapses at multiple locations, including the prefrontal cortex, hippocampus and nucleus accumbens, leading to a dysfunction of cortico-mesolimbic reward circuitry that underlies many of the symptoms of depression. This hypothesis accounts for current depression treatments and suggests an updated framework for the development of better therapeutic compounds.
Keywords: glutamate, stress, ketamine, reward, hippocampus, nucleus accumbens
Major depressive disorder (MDD) is one of the most common and costly of neuropsychiatric syndromes, with a lifetime prevalence of 7–12% in men and 20–25% in women, and a multi-billion dollar annual economic burden in the US [1,2].
The most tragic consequence of untreated depression is suicide, attempted by as many as 8% of severely depressed patients. According to the Centers for Disease Control and Prevention, nearly half a million patients receive emergency care for suicide attempts each year in the United States and over 38,000 individuals die by intentional self-inflicted injuries - twice as many lives as are lost to homicide. Shockingly, 23% of suicide victims were being treated with antidepressants at the time [3]. In fact, only half of patients with major depression respond to standard-of-care antidepressants (ADs), such as selective serotonin reuptake inhibitors (SSRIs)[4], with 70% failing to achieve full remission [5]. A better understanding of the nature of the changes in the brain driving the wide-range of behavioral symptoms that characterize depression is essential for developing more effective treatments for this disorder and preventing suicide.
In the search for ways to prevent, treat, and cure diseases, a strong hypothesis can prove invaluable if it accounts for known etiologies, is consistent with existing human and preclinical data gathered across a wide range of modalities, makes clearly testable predictions, explains the effectiveness of existing therapies mechanistically, and offers guidance for the development of novel prophylactic and therapeutic approaches.
The serotonin hypothesis of depression, formulated in the early 1960’s, provides an excellent example, driving the field of biological psychiatry forward and leading to the development of SSRIs (Box 1). More recently, considerable evidence of the involvement of neurotrophins in depression and antidepressant action has accumulated, leading to a neurotrophin hypothesis of depression (Box 2). Nevertheless, these hypotheses leave many unanswered questions about both the causes and treatment of depression.
BOX 1. The serotonin hypothesis of depression.
The monoamine neurotransmitter serotonin (5-hydroxytryptamine, 5-HT) is synthesized by neurons in the dorsal raphe (DR) nucleus. These neurons integrate inputs from multiple brain regions, including the nucleus accumbens (NAc), amygdala, lateral habenula (LHb), and PFC, and send projections throughout the brain, including to the hippocampus, PFC, substantia nigra, and NAc.
Chance observations of mood-altering compounds more than 50 years ago led to the first coherent theory of depression and opened the door for current first-line treatments. Evidence that these agents alter the concentration of monoamine neurotransmitters, such as serotonin, dopamine, and norepinephrine (e.g. [136]), led to the hypothesis that depression is caused by a deficiency of monoamines [137–139]. Because the pharmacological profile of many AD drugs is more consistent with changes in serotonin levels than other monoamine neurotransmitters, it is now more common to speak of a serotonin hypothesis of depression, although inhibitors of norepinephrine uptake are also effective ADs, including the tricyclics. Most statements of the serotonin hypothesis fail to offer mechanistic explanations, nevertheless this theory became a foundation for research in biological psychiatry, and the central dogma in the field of major depression for many decades.
Although deficits in serotonin levels or release are a central prediction of this hypothesis, the relationship between human depression and the levels of serotonin or its metabolite 5-hydroxyindoleacetic acid (5-HIAA) remains unclear, with early enthusiasm giving way to mixed results [140]. Experimental manipulations of serotonin levels in humans by depleting or enhancing the precursor for its synthesis, tryptophan, also remain inconclusive [141–144]. In general, acute manipulation of tryptophan levels is more likely to affect mood in patients that are depressed than in otherwise healthy individuals, perhaps because it impairs the beneficial actions of their SSRIs.
One prediction of this hypothesis is that elevation of serotonin levels would relieve the symptoms of depression and testing this prediction led directly to the development of SSRIs, which produce a rapid elevation of extracellular serotonin concentration [145]. With fewer side effects than the less-specific tricyclics, SSRIs were prescribed to tens of millions in their first decade.
SSRI-induced elevation of serotonin increases activation of serotonin receptors, of which there are 14 subtypes, each having a unique pattern of expression and localization. Alterations in a single population, or subpopulation, of serotonin receptors might also be responsible for the symptoms of depression, rather than a global dysfunction of the neurotransmitter system, but evidence of such changes remains inconclusive [146–148].
50 years later, considerable debate about the validity of the serotonin hypothesis thus remains, with only inconsistent and inconclusive evidence supporting it. It is unlikely that depression is caused by a simple decrease in serotonin synthesis, release or receptor expression. Nevertheless, there is clear evidence that elevated levels of monoamines can relieve the symptoms of depression.
Box 2. The neurotrophin hypothesis of depression.
Depression has been hypothesized to result when environmental factors, chronic stress, and genetic susceptibility interact to impair neurotrophin signaling [15,98,149]. The consequences of inadequate neurotrophin signaling that might contribute to the genesis of depression include impaired neurogenesis in the dentate gyrus, atrophy of distal dendrites, and impaired neuronal plasticity. It has been further postulated that ADs alleviate the symptoms of depression by reversing or blocking this effect, so as to restore neurotrophin signaling. Because of their predominance in the neocortex and hippocampus, the most data concerns brain-derived neurotrophic factor (BDNF) and its receptor, tropomyosin-related kinase type B (trkB)[150,151].
This hypothesis predicts that BDNF signaling is decreased in depressed patients and restored with successful AD treatment and there is some evidence of decreased BDNF-trkB signaling in human depression. First, serum BDNF levels are decreased in depressed patients [152] and recover with AD treatment [153]. Additionally, BDNF and trkB mRNA levels are decreased in postmortem tissue from suicide victims [154]. Patients treated with ADs have a higher level of BDNF than untreated patients, specifically in the dentate gyrus, hilus, and supragranular regions of the hippocampus [155], and increased hippocampal neurogenesis [156]. Furthermore, a polymorphism (Val66Met) in the BDNF gene is correlated with various aspects of depression and AD action (e.g. [157,158]), including reduced hippocampal volume, depression, and suicidality [159–161].
The hypothesis also predicts that impairing BDNF signaling should induce a depressive-like phenotype and interfere with AD responses in animal models. Some mice with decreased BDNF-trkB signaling display a depression-like phenotype, including decreased neural proliferation in the dentate gyrus [94] and reduced dendritic spine density in CA1 cells [162], as well as reduced responses to acute ADs [163]. On the other hand, heterozygous BDNF knockout mice do not differ from wild types in anxiety and behavioral despair measures in some [164,165], but not all [166], studies. Monteggia et al. report that only female conditional forebrain BDNF knockout mice have a depressed phenotype and fail to respond acutely to ADs [167]. Pyramidal cells in the prefrontal cortex of transgenic mice with the BDNF Met allele knocked in display atrophy of distal apical dendrites and dendritic spines, as well as a depression of excitatory synaptic responses [168]. Unfortunately, depression-related behavioral assays with construct and face validity, such as the sucrose preference and novelty suppressed feeding tests, have not yet been tested in these mice.
Such experiments are complicated for two reasons. First, BDNF-trkB signaling is crucial for brain development, rendering interpretation of results from knock-out animals difficult. Second, BDNF-trkB signaling does not exert the same actions in all brain regions. In the hippocampus and cortex, BDNF-trkB signaling promotes resilience to stress, whereas in the NAc BDNF promotes susceptibility [38]. For example, disrupted BDNF signaling in the NAc renders mice more resistant to chronic social defeat [169].
Altogether, it appears that the neurotrophin hypothesis of depression is incomplete. In addition to the conflicting data in the literature, there is no established mechanism linking serotonin elevation to subsequent increases in BDNF transcription, and therefore no explanation as to how ADs such as SSRIs activate this pathway. Nevertheless, these findings highlight the possibility that changes in neuronal strength and plasticity may underlie depression and the action of ADs.
In this review, we highlight emerging evidence of dysfunctions of excitatory synaptic transmission [6], and suggest how this defect may correlate with the genesis of depression and the evidence that serotonin and neurotrophins play a role in depression. We are now moving beyond a symptom-based description of depression as a single disease entity and beginning to identify biological phenotypes that can be compared across species and across classically defined labels, as encouraged by the NIMH’s Research Domain Criteria. A significant goal of basic research is thus to identify the neurobiological consequences of disease-promoting conditions at the level of alterations in gene products, synapses, cells, and circuits and then, in a bottom up manner, map these discoveries to the specific behavioral deficits characterizing human neuropsychiatric conditions. Here we focus on the circuits mediating reward behavior, which underlies many of the symptoms of human depression, such as anhedonia and aberrant reward-associated perception and memory. We will also highlight how this excitatory synapse hypothesis of depression offers a new framework with which to approach the treatment of patients with depression.
The etiology of depression
Despite its high incidence and its socioeconomic impact, the causes of depression remain poorly understood. Depression involves a combination of genetic and epigenetic susceptibility together with environmental risk factors, such as stress, emotional trauma, or traumatic head injury [7], with heritable factors contributing slightly less than half of the risk. Many single nucleotide polymorphisms and epigenetic differences are linked to increased risk for depression, but no single gene candidate produces a strong enough effect to provide convincing mechanistic hypotheses [8]. This reinforces the fact that MDD is a complex and heterogeneous collection of symptoms caused by variations in multiple genes, each responsible for a small effect on risk, that ultimately converge onto common circuit, cellular, and molecular pathways.
Stress and depression
Stressful life events are a key environmental risk factor for depressive disorders in genetically susceptible individuals [9] and are suspected to be causal in many patients. Depressed patients report more stressful life events and have fewer social resources than non-depressed subjects [10]. Personality traits, in particular neuroticism and lack of a confidence, have also been linked to the etiology of depression and may increase risk in response to stress [11].
McEwen, Sapolsky, and others have emphasized the importance of allostatic overload as an explanation for many chronic illnesses of modern human life, including depression [12,13]. These stressors may be physical or psychosocial, the latter including low self-esteem, loneliness, deficient social skills, excessive anxiety, rumination, and negative thinking. All stressors activate the sympathetic nervous system and the hypothalamus-pituitary-adrenal (HPA) axis, causing elevation of glucocorticoids (GC) and other stress hormones. Normally, the physiological stress-response is self-terminating, due to negative feedback, directly via the hypothalamus and pituitary and indirectly via several brain areas enriched in glucocorticoid receptors (GRs), including the prefrontal cortex (PFC) and hippocampus.
A subset of depressed patients display abnormal HPA axis activity, resulting in increased basal levels of GCs and a blunted circadian rhythm [14]. GCs regulate neuronal survival, excitability, proliferation, and metabolism, and allostatic overload may promote depression by impairing these processes [15]. Because of their role in negative feedback, a dysfunction of GRs in areas like the hippocampus can cause HPA dysregulation and failure to limit the stress response [16]. Indeed, some patients with depression display an inability to suppress the axis in response to a challenge [17]. However, HPA challenges lack sensitivity and specificity as a diagnostic biomarker. Interestingly, patients treated chronically with corticosteroids and patients with Cushing’s disease, both hypercortisolemic states, are more likely to have depression and experience a range of depression-related cognitive and memory deficits [18,19].
The structure of the depressed brain is different from the healthy brain, likely representing one visible manifestation of pathophysiology. Postmortem studies of suicide completers and severely depressed patients have revealed a reduction in the volume of the PFC and hippocampus, regions thought to play a role in the cognitive aspects of depression [20]. An impairment of normal, ongoing neurogenesis in the dentate gyrus of the hippocampus, as observed in several models of chronic stress [21], and atrophy of dendrites [22] might underlie this decrease in hippocampal volume. Successful AD treatment reverses these decreases in hippocampal volume (i.e. [23]). As reviewed elsewhere, the size of other brain regions involved in regulation of reward and emotion may also be affected in depressed patients, including the amygdala [24] and striatum [25].
Changes in excitatory synapses in depression
A common element linking stress, serotonin, and neurotrophins is their effects on excitatory synaptic transmission [26]. In preclinical studies, there is increasing evidence that chronic stress exerts deleterious effects on excitatory synaptic structure and function in multiple brain regions associated with cognitive and emotional control of reward behaviors which resemble those seen in human depression. Conversely, serotonin and neurotrophins exert an opposing action, generally promoting excitatory synaptic transmission in the same brain regions. Many symptoms of depression seem to result from a dysfunction in the valuation of stimuli, with decreases in positive valuation (i.e. anhedonia) occurring in parallel with increases in negative valuation (disappointment, fear, expectation of punishment). Changes in the strength of various excitatory synapses in the distinct circuits mediating both of these processes have been described.
Nucleus Accumbens
The NAc is critical for integrating cortical and hippocampal inputs and regulating the firing of neurons in the ventral tegmental area (VTA) [27], thereby influencing the motivation to seek rewarding stimuli [28], as well as motor behavior. Lim et al. [29] made a particularly significant advance in our understanding of the neurobiology of depression with their study of the effects of chronic stress on excitatory synapses in the NAc (Fig. 1). They observed that excitatory synapses in medium spiny neurons (MSNs) in the NAc that express the D1 dopamine receptor (D1R) displayed a selective decrease in AMPAR-mediated excitation after five days of chronic restraint stress. The decrease in excitation occurred in parallel with anhedonia in the sucrose preference test and depressive-like changes in the tail suspension and forced swim tests. Because D1R-expressing MSNs promote activation of dopaminergic cells [30], decreased excitation of D1R cells should lower dopamine release, a key regulator of reward-seeking and motivated behavior.
Figure 1. Stress induced internalization of AMPARs in D1R-expressing medium spiny neurons in the NAc underlies some stress-induced depression-like behavioral changes.
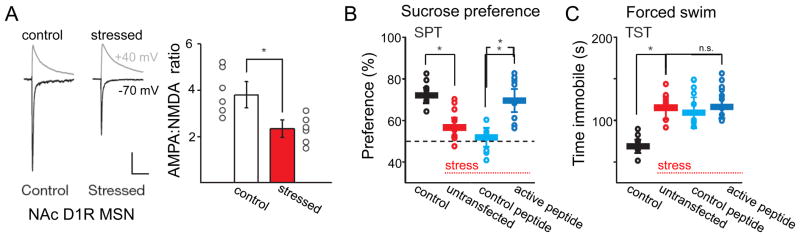
A. Whole-cell recording of evoked excitatory synaptic currents from D1R-expressing MSNs at −70 mV and +40 mV for analysis of AMPAR- and NMDAR-mediated components in slices from control and chronically stressed mice. Chronic stress decreased the AMPAR component, but not the NMDAR component. B,C. Transfection of D1R-expressing cells with a peptide that prevents stress-induced AMPAR internalization prevented the stress-induced loss of sucrose preference (B), but not stress-induced immobility in the forced swim test (C). Modified with permission from Lim et al., 2012 [29].
Experimental manipulations that decreased AMPAR-mediated excitation in D1R-expressing cells mimicked the stress-induced behavioral changes in unstressed animals, and manipulations that prevented stress-induced decreases in AMPAR-mediated excitation blocked some behavioral changes (sucrose preference test) in stressed animals[29]. Interestingly, other behaviors that are widely assayed in depression studies, such as the tail suspension test and the forced swim test, were not rescued by preventing AMPAR downregulation in the NAc. The authors conclude that the stress-induced decrease in excitation is sufficient and necessary for many behavioral changes that resemble human depression symptoms, specifically anhedonia, highlighting the central importance of these synaptic circuits to the genesis of anhedonia. The source of the axons forming these excitatory synapses was not identified, but is likely to include afferents from the hippocampus, prefrontal cortex, and amygdala. In addition to these postsynaptic changes in excitatory synaptic strength, decreases in the frequency of miniature excitatory postsynaptic currents (EPSCs), presumably indicative of presynaptic changes in release probability, have also been reported in D1R MSNs [31,32]. Accompanying these physiological changes, modest increases in the density of stubby dendritic spines are observed in susceptible mice subjected to chronic social defeat [31,33], but the identity of the MSNs was not determined.
Ventral Tegmental Area
The cell bodies of dopamine-releasing cells enervating the forebrain are located in the VTA, intermingled with a population of local GABAergic interneurons, both of which receive input from NAc MSNs and the PFC [27]. Changes in the electrophysiological responses of dopaminergic VTA neurons in chronic stress models remain controversial. In one study, optogenetic activation of dopamine-releasing VTA neurons relieved depression-like symptoms in chronically stressed mice and, conversely, inhibition of VTA neurons triggered a depression-like behavioral phenotype [34]. The decrease in the ability of cortical and hippocampal inputs to excite D1R cells in the NAc after chronic stress, described above, could thus underlie the reduced firing of VTA neurons [35,36], reduced dopamine release [37], and the loss of the rewarding properties of various stimuli in these models. Changes in synaptic strength within the VTA itself could also contribute [34]. We predict that restoration of normal behavior by ADs should be accompanied by a restoration of synaptic strength, but this has not yet been tested. Other studies, however, report that VTA discharge is increased in chronic stress models [38–40] and that optogenetic activation of VTA neurons promotes depression-like behavior [41]. Given that 1) drugs that elevate dopamine, like cocaine, are clearly not pro-depressive, 2) optogenetic stimulation of the PFC exerts antidepressive effects [42,43], and 3) that optogenetic activation of D1R MSNs promotes resilience to social stress-induced depression [32], it is unclear how the observed increases in VTA firing are generated and why they should be pro-depressive. Further work is needed to reconcile these differences, perhaps by identifying subpopulations of inputs, synapses, or neurons within the NAc.
Prefrontal Cortex
[There are many subdivisions of the PFC (infralimbic, prelimbic, orbital, dorsolateral, ventromedial) and, for simplicity, we omit any distinction of these regions.] The PFC is an important site at which cognitive evaluations, such as the controllability of a stressor [44] or the pleasantness of a stimulus [45], can influence affect and reward. Depressed patients display evidence of reduced activity in the PFC and SSRI treatment restores normal activity levels [23,46]. In animal models, chronic stress results in atrophy of distal dendrites of pyramidal cells in the PFC and loss of dendritic spines [47,48]. Yuen et al. [49] have reported that seven days of chronic restraint stress produces a decrease in synaptic excitation in layer V pyramidal cells in the PFC. Both AMPAR- and NMDAR- components of the EPSP were equally affected, unlike in the NAc [29] and hippocampus (see below), and levels of GluA1 and GluN1 protein were down-regulated in parallel (Fig. 2). The decreased expression of GluA1 subunits was mediated by ubiquitination and proteosomal degradation. In addition to this postsynaptic mechanism, there may also have been a decrease in presynaptic release probability (decreases in mEPSC frequency, albeit no change in the paired-pulse ratio of evoked EPSPs). No change in PSD95 expression was detected, leaving uncertain the question of whether changes in synapse number and presynaptic function accompany the observed postsynaptic changes. Yuen et al. also observed no change in GluA1 function or expression in the hippocampus with seven days of chronic restraint stress, a time at which GluA1 and GluN1 are decreased in PFC, suggesting that the PFC may be more susceptible to stress. Consistent with the decrease in excitation, expression of several activity-dependent genes is decreased in the PFC of depressed humans and in rodent models [42].
Figure 2. Stress-induced internalization of AMPARs and NMDARs in the prefrontal cortex is mediated by ubiquitination and proteosomal degradation.

AMPAR-mediated (A, −70 mV, representative currents at right) and NMDAR-mediated (B, +60 mV) excitatory currents recorded from layer V pyramidal cells in response to a range of stimulation intensities in slices from unstressed control rats (open circle) and rats subjected to chronic restraint stress (filled triangle) or chronic unpredictable stress (filled square). Stress depressed both components regardless of stimulation intensity. C. Chronic restraint stress decreased both expression and plasma membrane surface insertion of GluA1 and GluN1 receptor subunits. D. Pharmacological inhibition of proteosomal degradation prevented decreases in AMPAR-mediated synaptic currents in response to chronic stress, but had no effect in unstressed animals. Representative currents shown at right. Modified with permission from Yuen et al., 2012 [49].
Activation of 5-HT2ARs triggers a rapid increase in the frequency of spontaneous glutamatergic excitatory postsynaptic responses in pyramidal cells in the PFC that is mediated by the excitation of a subset of pyramidal cells in layer V–VI, while evoked EPSCs are depressed simultaneously [50–52]. The effect of serotonin on mEPSC frequency is diminished after chronic stress [53], although the authors failed to report whether there is any change in the basal frequency or amplitude after stress or whether serotonin is less effective at exciting layer V–VI cells. One potential explanation is that serotonin’s effects are weaker because the intracortical excitatory synapses formed by the deep layer cells are weakened by stress, although this has not been tested.
Lateral Habenula
The LHb has received increased attention in the context of depression because of behavioral studies indicating that it signals negative reward (i.e. disappointment)[54,55]. The LHb forms a glutamatergic monosynaptic excitatory projection directly to both dopaminergic neurons in the VTA and serotonergic neurons in the DR, as well to the mesopontine rostromedial tegmental nucleus (RMTg), which then sends substantial GABAergic projections to the VTA and DR [56,57]. Stimulation of the LHb inhibits discharge of VTA neurons [58] and presumably DR cells, as well. When animals perform tasks with various rewarding, neutral or punishing stimuli, neurons in the LHb are excited by both the absence of an expected reward or the presence of a punishment, and they are inhibited by the rewarding stimulus, especially when the stimuli are unpredictable [59]. Induction of LHb discharge by negative reward would thus presumably inhibit both dopamine and serotonin secretion.
In the learned helplessness model, LHb neurons displayed an increased frequency of mEPSCs compared to naïve controls with no change in their mean amplitude, reflecting an increased probability of glutamate release from the nerve terminals of some population of presynaptic neurons [60]. Consistent with this increased excitation, the spontaneous action potential discharge rate of LHb neurons was significantly higher after learned helplessness. These changes could thus suggest a physiological basis for excessive disappointment or negative thinking in depression. ON the other hand, depressed patients subjected to acute tryptophan depletion in order to induce depressive symptoms displayed significant decreases in activity-dependent PET signals in the LHb that were closely correlated with the occurrence and severity of the depressive symptoms [61].
Amygdala
The amygdala is critical for learning to fear aversive stimuli and learning that previously aversive stimuli are no longer dangerous [62]. Unlike the hippocampus, the amygdala displays increases in volume in depressed patients [24] and is hyperactive [63]. Unlike the hippocampus and PFC, chronic stress induces dendritic hypertrophy in the amygdala [64]. Relatively little is known about changes in synaptic function to, from, and within the amygdala in chronic stress models, but optogenetic activation of amygdala inputs to the ventral hippocampus can impair normal social behaviors [65].
Hippocampus
Chronic stress induces changes in synaptic function that occur in parallel with the atrophy of distal apical dendritic branches in pyramidal cells and decreases in the size and number of dendritic spines [66,67].
While stress has repeatedly been shown to affect excitatory synapses in the hippocampus, published results are somewhat conflicting. At Schaffer collateral (SC) synapses onto CA1 cells, chronic stress is reported to have no effect on basal synaptic strength, although it does impair long-term potentiation (LTP), decreases GluA1 mRNA [68–70], and enhances synaptic currents mediated by NMDARs, while leaving AMPAR-mediated excitation unaltered in area CA3 [71].
One factor that could reconcile these discrepancies is the dendritic location of the synapses. We have recently described stress-induced changes in synaptic function at the synapses in distal apical dendrites of CA1 pyramidal cells formed by temporoammonic (TA) inputs from entorhinal cortex [72](Fig. 3). We observed that 3–6 weeks of chronic unpredictable stress (CUS), which is sufficient to produce depression-like behavior in the sucrose preference test and other behavioral assays, resulted in a 50% decrease in AMPAR-mediated excitation at TA-CA1 synapses. NMDAR-mediated excitation at TA-CA1 synapses was unaffected, as was AMPAR-mediated excitation at neighboring Schaffer collateral-CA1 synapses. Accompanying the decreased excitation was a corresponding layer-specific decrease in the expression of GluA1 and PSD-95 protein. No changes in the levels of GluA2 or GluN1 protein were detected. This synaptic pathology had deleterious behavioral consequences, as shown by the ability of CUS to disrupt long-term memory consolidation in a spatially-cued Morris water maze task, a function of TA-CA1 synapses [73]. Further strengthening the relationship between this synaptic pathology and the behavioral alterations produced by CUS, we observed that changes in both AMPAR-mediated excitation and GluA1 protein levels were restored to normal levels in stressed animals treated chronically, but not acutely, with fluoxetine, just like the restoration of normal reward behavior.
Figure 3. Stress decreases AMPAR-mediated excitation and GluA1 expression at temporoammonic to CA1 cell synapses and chronic fluoxetine reverses these effects.

A. Field excitatory postsynaptic potentials (fEPSPs) are recorded in stratum lacunosum moleculare in response to stimulation of the temporoammonic (TA) pathway in Mg2+-free saline, allowing dissection of AMPAR- and NMDAR-mediated components. AMPAR-mediated transmission was reduced over a range of stimulation intensities after chronic unpredictable stress (red) compared to unstressed rats (blue). Quantification of AMPA:NMDA ratios (B) and GluA1 protein (C) reveals that the selective decrease in AMPAR-mediated transmission is due to decreased GluA1 expression. Administration of fluoxetine for three weeks to stressed rats restored both AMPA:NMDA ratios and GluA1 expression. Modified with permission from Kallarackal et al., 2013 [72].
Differences in the ability of allostatic load to affect excitatory synaptic function may play a role in the determination of susceptibility to becoming depressed. Schmidt et al. [70] examined the difference between mice that were vulnerable to behavioral changes after chronic social stress and those that were resilient. Vulnerable mice exhibited a significant decrease in GluA1 and increase in GluA2 subunits in area CA1, compared to resilient mice. The authors further examined genetic polymorphisms in genes encoding GluA1 subunits and found a single nucleotide polymorphism that correlated with vulnerability to stress. How this compares to human GluA1 gene SNPs was not described, but this evidence converges with human data implicating AMPARs [74,75].
We have also observed that chronic stress quantitatively and qualitatively changes the way TA-CA1 excitatory synapses respond to serotonergic stimulation [76]. Whereas activation of postsynaptic 5-HT1BRs produces a doubling of AMPAR-mediated excitation in control animals that recovers within ca. 30 min of washing out the agonist, activation of 5-HT1BRs in chronically stressed animals produces a potentiation that is both significantly greater in magnitude and more persistent (>1–2 hours) after removal of the agonist (Fig. 4). Normal transient responses to 5-HT1BR activation are restored in CUS animals administered fluoxetine chronically, but not acutely. We have not yet determined the molecular changes in the signaling pathways that underlie 5-HT1BR-induced potentiation that underlie this effect, but they must be confined to excitatory synapses in the postsynaptic cell where this potentiation is induced and expressed in order to account for the stress-induced changes we observe.
Figure 4. Serotonin causes a 5-HT1BR-dependent potentiation of TA-CA1 synapses and this potentiation is altered by chronic stress.
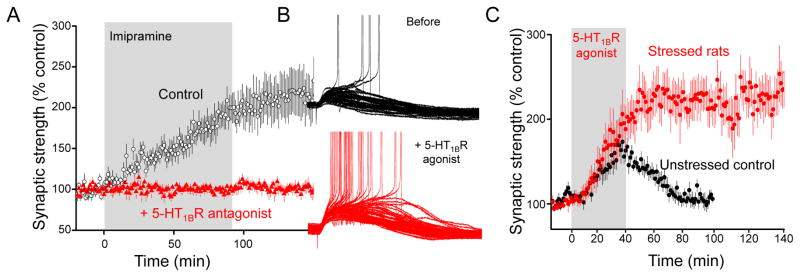
A. fEPSPs are recorded in stratum lacunosum moleculare in response to stimulation of the TA pathway during application of the tricyclic antidepressant imipramine in control saline (black) or saline containing the 5-HT1BR antagonist isamoltane. Elevation of endogenous serotonin produces a doubling of synaptic strength. B. Activation of 5-HT1BRs with the selective agonist anpirtoline promotes action potential discharge in CA1 cells. C. Anpirtoline produces a robust and reversible potentiation of TA-CA1 excitatory postsynaptic currents in slices from unstressed control animals, but produces an enhanced and persistent potentiation in slices from rats subjected to chronic unpredictable stress. Modified with permission from Cai et al., 2013 [76].
Not only do chronic stress models produce changes in glutamatergic receptor function and expression, but these changes may be sufficient to cause changes in behaviors related to depression. For example, Chourbaji et al. [77] found that knocking out the GluA1 gene globally resulted in a depression-like phenotype in the learned helplessness test. Similarly, we have reported that mice in which the GluA1 gene is mutated so that it can no longer be phosphorylated by CaM kinase display a depression-like phenotype in the sucrose preference and novelty suppressed feeding tests, but not in the tail suspension test [76]. Together these data make a strong argument that disruption of hippocampal glutamatergic transmission, specifically AMPAR function, can accompany depression-like behavioral changes.
Human studies
Although far less extensive than the literature on serotonin and its receptors, there is some evidence for changes in glutamatergic function in depressed patients (Table 1); much of it mixed. Plasma levels of glutamate are decreased in some studies [78,79], but not others [80]. A considerable difficulty in studies of this type is that glutamate is present in a very large metabolic pool, rendering it impossible to know what fraction of the measured glutamate is synaptic in origin. Postmortem studies have revealed decreases in multiple glutamate receptor subunits in depressed patients, including the GluA1 subunit [81,82], as well as decreased AMPAR binding in the striatum of suicide completers [74,75]. Hopefully, reliable biomarkers can be found with which to more directly assay glutamatergic function in depressed patients.
TABLE 1.
| Study | Protein or gene | Major finding |
|---|---|---|
| Proteins and genes | ||
| [181] | NMDAR | Abnormal binding in brains of suicide completers |
| [182] | 5-HTT gene | Promoter short allele variant promotes depression and suicidality after early life stress |
| [183] | BDNF | Val66Met variant interacts with stress to promote depression |
| [184] | BDNF (review) | Lower serum BDNF in depressed patients, normalized with AD treatment |
| [185] | 5HT1AR (review) | C(-1019)G SNP correlates with MDD, suicidality, and decreased response to SSRIs |
| [186] | mGluR5 | Decreased in PFC in unmedicated depressed post-mortem samples |
| [187] | Multiple synaptic proteins, 5- HTRs | Decreased SNAP25, GLUR1, GLUR3; increased HTR2C, decreased HTR4 and HTR7 in hippocampal subfields of depressed |
| PET findings in depression patients in vivo | ||
| [186] | mGluR5 | Reduction in several regions, including HC and PFC |
| [188] | 5HT1AR (review) | Mixed results, with clear trend for decrease in many brain areas |
| [189] | 5HT1BR | Decreased binding in ventral striatum and pallidum |
| [190] | 5HT2R | Reduction correlates with ECT effect |
| [191] | 5-HTT (review) | Mixed results: Less binding or no effect |
| [192] | MAO type A | Greater binding in many areas including PFC, ACC, ventral striatum, hippocampus |
| Metabolism and activity (fMRI) | ||
| [193] | Hypoactive areas include frontal and temporal cortices; select subcortical and limbic structures hyperactive (meta-analysis) | |
| [194] | Disrupted resting state interhemispheral coherence | |
| [195] | Increased baseline metabolism of OFC, amygdala, posterior cingulate; decreased metabolism subgenual ACC and dlPFC | |
| [196] | Subgenual ACC more active, dorsal ACC less active in response to reward-related feedback in anhedonic individuals | |
| [46] | Successful SSRI treatment reverses metabolism deficits in dlPFC, mPFC, dACC | |
| [197] | Reduced activation of temporal and occipital cortices during emotional task | |
Formal statement of the excitatory synapse hypothesis
Major depression is caused by a weakening of specific subsets of excitatory synapses in multiple brain regions that are critical in the determination of affect and reward. Chronic hyperactivity of the HPA axis in response to excessive stress, known to be important allostatic risk factor in the gene x environmental axis determining susceptibility to depression, is one potential mediator of these changes. High levels of GRs in cells in these regions contribute to their vulnerability.
Many of the characteristic changes in behavior that define the symptomatology of human depression, such as anhedonia and depressed mood, result because impaired excitatory synaptic transmission leads to reduced activity in the mesolimbic reward circuitry. Other circuits that are important in the mediation of other behaviors that are altered in depression, such as sleep, sex drive, sociality, working memory, and attention are similarly affected.
Restoration of excitatory synaptic strength is the critical action of effective antidepressants, including both conventional agents, such as SSRIs and ECT, and newer compounds, such as ketamine.
Several authors have highlighted stress-induced pathophysiological changes in reward circuits as the biological underpinning of anhedonia and other symptoms of depression [83–85]. There is a strong correlation between anhedonia and suicide [86,87]. Indeed, anhedonia has been found to be a more significant predictor of imminent suicide than any other psychiatric variable [88]. Social anhedonia (e.g., loss of interest in social interaction) appears especially predictive of suicidal ideation [87]. We can predict how changes in excitatory synapses in regions such as the hippocampus and prefrontal cortex might interact with reward circuits in the mesolimbic system to change affective behaviors and produce a depression-like behavioral phenotype (Fig. 5).
Figure 5. The hypothesized impact of stress-induced changes in excitatory synaptic transmission on the reward circuitry and the mechanisms of antidepressant action.

The hippocampus and PFC send glutamatergic projections (red) to the NAc where they excite D1R-expressing GABAergic MSNs. These cells inhibit a population of interneurons within the VTA that form GABAergic inhibitory synapses (blue) onto dopaminergic VTA cells (green) projecting back to the NAc, hippocampus, and PFC. When the hippocampus or PFC are active, they excite D1R cells, thereby disinhibiting VTA dopaminergic cells and promoting dopamine release (left panel). There is a distinct circuit mediating responses to negative stimuli in which glutamatergic cells in the LHb project to GABAergic neurons in the rostromedial tegmentum (RMTg), which in turn inhibit dopaminergic neurons in the VTA. Chronic stress induces anhedonia and other reward-related symptoms of depression because of a weakening of excitatory synapses in the hippocampus and PFC and because of a weakening of excitatory synapses on D1R MSNs. There is also a potentiation of excitatory inputs onto LHb neurons, resulting in an increase in the excitation of RMTg neurons and subsequent increase in the inhibition of VTA cells. These changes synergistically increase the inhibition of VTA dopaminergic cells thereby impairing dopamine release (center panel). Effective antidepressant therapies (SSRIs, ketamine, DBS, or ECT) can all trigger an activity- and/or serotonin-dependent strengthening of excitatory synapses in the hippocampus, PFC, and NAc, thereby restoring the normal release of dopamine in response to rewarding stimuli (right panel). Evidence of changes in synaptic excitation: 1. (Lim et al., 2012 [29]); 2. (Yuen et al., 2012; Li et al., 2011; Liston et al., 2006; Radley et al., 2008; Liu and Aghajanian, 2008 [47–49,53,104]); 3. (Kallarackal et al., 2013; Alfarez et al., 2003; Schmidt et al., 2010; Watanabe et al., 1992; Sousa et al., 2000 [69,70,72,172,178]). 4. (Li et al., 2011 [60]); 5. (Stamatkis and Stuber, 2012 [179]). Evidence of synaptic strengthening by ADs: 6. (Cai et al., 2013, SSRI [76]; Conrad et al., 1996, tricyclic [180]). 7. (Li et al., 2011, ketamine [104]); 8. (Covington et al., 2010, LTP? [42]).
One important target of outputs from both the hippocampus and prefrontal cortex is the NAc, and activation of either brain region is positively correlated with NAc activity [e.g. 84]. One important target of the NAc is the VTA. Numerous studies have shown that increased dopamine release is critical for appetitive motivational processes such as initiation of behavior, exertion of effort, sustained task engagement, and instrumental learning [90]- precisely the behaviors that are disturbed in depression. MSNs in the NAc are GABAergic and the D1R-expressing subset of MSNs activates dopaminergic cells by inhibiting intrinsic VTA GABAergic cells [30]. The net effect of activation of D1R cells in response to inputs from the hippocampus and PFC is thus to promote dopamine release. Stress-induced weakening of excitatory synapses onto D1R cells [29] would make it harder for rewarding stimuli to trigger the normal activation of dopaminergic VTA neurons and the release of dopamine. D1R MSNs would hence be less effective at eliciting positive reinforcement or motivated behavior. Similarly, weakened excitatory synapses in the hippocampus and PFC would decrease the afferent drive that the NAc receives from these regions [91] and also diminish the release of dopamine by reward reinforcing stimuli (e.g. [37,92]). Decreased drive to the amygdala from the PFC and hippocampus may also be important. These changes occur in parallel to, and are synergistic with, the increased inhibition of VTA cells mediated by changes in the LHb - RMTg circuit, as discussed above.
It is important to note that we do not predict that every excitatory synapse is affected by stress, even in the same region. In the NAc, synapses on D1R cells are affected, but not those on D2R cells [29,32]. In the hippocampus, TA-CA1 synapses are weakened by chronic stress, but Schaffer collateral synapses are not [72]. Second, not every stress-sensitive excitatory synapse responds in the same manner. For example, synapses involved with positive valuation are more likely to be weakened by stress, whereas synapses involved in negative valuation become strengthened. More synapses need to be examined to clarify these issues. Finally, the wide range of behaviors that are altered in stress and depression provides clear evidence that there are likely to be multiple synapses that are adversely affected by stress, with no one synapse likely to be uniquely responsible for generating the depressed phenotype.
Restoration of excitatory synaptic strength within the hippocampus and PFC, as well as restoration of the strength of excitatory synapses onto D1R cells, would restore the ability of rewarding stimuli to promote dopamine secretion. The hypothesis thus predicts that any agent or manipulation that strengthens key stress-sensitive excitatory synapses in the reward system may be an effective AD. Indeed, optogenetic stimulation of the PFC exerts AD-like effects in the sucrose preference and social interaction tests in rodent models [42,43], equivalent to the effects of chronic SSRIs. Furthermore, direct optogenetic activation of D1R MSNs promotes resilience to social stress-induced depression [32].
The excitatory synapse is also a place where serotonin and BDNF - trkB receptor signaling converge. BDNF secretion is promoted by glutamatergic excitation. Considerable evidence ties BDNF and trkB to synapse strengthening processes such as LTP. Application of BDNF also results in morphological and functional changes that parallel LTP, including increased spine formation and higher amplitudes and frequency of miniature EPSCs in CA1 neurons in the hippocampus [93]. Finally, there is considerable evidence that BDNF - trkB signaling promotes the generation and survival of granule cells in the dentate gyrus [94]. Proliferation of these cells and their incorporation into the hippocampal circuitry should also promote activity within hippocampal circuits, thereby promoting hippocampal throughput and output. BDNF - trkB signaling is thus both promoted by excitation and promotes excitation. Similarly, insufficient serotonin (Box 1) may lead to loss of on-going maintenance of synaptic strength (see below). Conversely, insufficient synaptic strength may weaken the drive of neurons in the dorsal raphe from the limbic cortices [95] and diminish serotonin release.
Therapeutic implications of the excitatory synapse hypothesis
Although the development of SSRIs has improved the treatment of major depression, there is still considerable need for better therapeutic options. Can the excitatory synapse hypothesis account for the actions of existing therapies and point towards new strategies?
SSRIs
Our understanding of how SSRIs relieve depression depends upon understanding how serotonin affects brain function. Two effects appear central to understanding the beneficial actions of SSRIs: promoting neurogenesis and promoting excitatory interactions at specific loci. There is strong evidence that SSRIs promote the proliferation of progenitor cells in the subgranular zone of the dentate gyrus, leading to the increased genesis and survival of dentate granule cells [96]. Given the evidence that chronic stress impairs neurogenesis of these same cells [97], it has been proposed that restoration of neurogenesis is a critical factor in the AD action of SSRIs [98], presumably by increasing hippocampal output, although this is rarely discussed.
There is also considerable evidence that serotonin regulates excitatory synaptic transmission in various brain regions through actions at multiple serotonin receptor subtypes. In the hippocampus, elevation of serotonin levels with SSRIs potentiates excitation at both mossy fiber – CA3 cell synapses, via an action at 5-HT4Rs [99], and TA-CA1 cell synapses, via an action at 5-HT1BRs [76]. Furthermore, the ability of fluoxetine to restore normal sucrose preference and novelty suppressed feeding behaviors is compromised when 5-HT1BRs are blocked pharmacologically or deleted genetically, suggesting that this potentiation is required for the therapeutic actions of SSRIs [76]. As discussed above, serotonin promotes excitatory synaptic transmission in the PFC, as well.
Ketamine and NMDAR antagonists
One of the greatest recent advances in our understanding of depression was the finding that antagonists of NMDA receptors, particularly ketamine, have rapid, robust, and sustained (7–10 days) AD-like effects in humans [100,101]. Multiple studies have shown that acute administration of ketamine, at doses that are said to be lower than those that produce psychotomimetic responses, produces improvement in mood and reduces depressive symptoms, including suicidal ideation, within 1–2 hours; these improvements persist for up to two weeks. Rodents treated acutely with ketamine exhibit an AD-like phenotype in the forced swim [102], novelty suppressed feeding, and learned helplessness tests [103]. Furthermore, ketamine restores sucrose preference and novelty suppressed feeding behaviors rapidly in chronically stressed animals [104].
Although an area of on-going investigation, the generally accepted model of ketamine’s therapeutic effect is consistent with the hypothesized involvement of excitatory synapses in depression. Ketamine’s actions can be divided into an induction phase, during which it is present in the brain at sufficient concentrations to inhibit NMDARs, and an expression phase, in which symptoms are relieved for 1–2 weeks after washout of the drug from the brain.
There are two hypotheses about how ketamine acts during the induction phase. Ketamine may exert a preferential inhibition of NMDARs on GABAergic inhibitory interneurons [105–107]. This could be due to differences in subunit composition or because interneurons are more depolarized than pyramidal cells, relieving ion channel block by Mg2+ and allowing NMDARs to contribute more to their overall excitation. Ketamine thus produces a mild disinhibition of the neuronal population, an increase in high frequency oscillatory activity in rats [108,109] and humans [110,111], and a neurochemically detectable surge of glutamate release in the PFC and NAc [105,112,113]. Coincident with the washout of ketamine at the end of the induction phase, glutamate concentrations return to normal [114]. Ketamine has recently been shown to exert an antidepressant-like action in the forced swim test in mice lacking NMDARs in a subset of GABAergic interneurons [115], which could indicate that another population of interneurons is critical or that its actions do not depend on blocking interneuron NMDARs. Unfortunately, the authors did not determine whether ketamine was or was not able to elicit increased activity in these animals.
The alternative hypothesis states that ketamine blocks on-going activation of NMDARs mediated by spontaneous transmitter release [116]. This on-going activity is postulated to produce constitutive suppression of signaling pathways that promote excitatory synaptic transmission. By relieving this suppression, ketamine may promote expression of several proteins that potentiate excitatory synapses, including AMPARs [104,116].
Regardless of which hypothesis is true, both share the strengthening of excitatory synapses, persisting throughout the expression phase, as their common effector mechanism. Several activity-dependent consequences of a period of increased network activity have been implicated, such as activation of the mTOR signaling pathway or deactivation of the eEF2 pathway, and rapid translation of proteins from pre-existing mRNAs, as well as induction of the immediate early gene FosB and its downstream genes. All three mechanisms ultimately lead to direct potentiation of synapses, the induction of synapse-related genes, and increased synthesis of synaptic proteins within hours [103,116], thereby strengthening pathologically weakened excitatory synapses [104]. Ketamine appears to induce LTP-like processes, as suggested by the finding that ketamine triggers an increase in surface expression of AMPARs [117]. Indeed, ketamine loses its AD effect in the absence of functional AMPARs [117,118]. Ketamine also promotes BDNF synthesis and release, which may be required for its actions [116].
Unfortunately, ketamine’s side effects have hindered its clinical usefulness. Considerable effort is now being made to develop other inhibitors of NMDAR function that might preserve ketamine’s rapid AD actions without its side effects. Alternatively, the hypothesis predicts that any compound that either promotes LTP-like processes or promotes ketamine-like increases in oscillatory activity by any other means would also strengthen excitatory transmission and exert an AD action. Indeed, Kumar et al. [119] have shown that rhythmic optogenetic activation of the PFC exerts an AD-like effect in the forced swim test and increases oscillatory activity throughout the mesolimbic reward circuitry, including the VTA and NAc [119].
Scopolamine
The nonselective antagonist of muscarinic acetylcholine receptors, scopolamine, exerts an antidepressant action that is slower than ketamine, but faster than SSRIs (ca. 3–5 days) [120]. Like ketamine, scopolamine increases signaling via the mTOR pathway in the PFC and increases the number and size of dendritic spines on layer V pyramidal cells, as well as the ability of serotonin to increase the frequency and amplitude of spontaneous glutamatergic synaptic currents [121]. These responses are probably triggered by a short burst of neuronal activity, as suggested by detection of elevated glutamate levels with microdialysis, like ketamine. Indeed, blocking PFC AMPARS was sufficient to prevent the ability of scopolamine to produce and AD-like response in the forced swim test.
Positive allosteric modulators of AMPARs
Drugs that block AMPAR desensitization and/or deactivation are one promising way to exert AD action by directly potentiating excitatory synapses. The AMPAR potentiator LY392098, for example, exerted AD-like actions in the tail suspension and forced swim tests, although it did not restore sucrose preference after chronic stress [122,123]. In addition to their direct effects on excitation, AMPAR potentiators increase BDNF mRNA in the hippocampus [124]. Finally, AMPAR potentiators may be useful in accelerating the therapeutic response to SSRIs [125].
Deep brain stimulation (DBS)
DBS has attracted considerable attention as a means to alleviate treatment-resistant depression, although the mechanisms underlying its therapeutic effects and specific loci of action remain poorly understood. In humans, repetitive stimulation of the subcallosal cingulate gyrus has been shown to be therapeutic in some patients [126]. Similarly, in preclinical models, optogenetic and electrical stimulation of the medial PFC has an AD effect [42]. DBS of the rat PFC gray matter also causes an AD-like response in the forced swim, novelty suppressed feeding, and sucrose preference tests [127].
The hypothesis described here predicts that repetitive stimulation, particularly of PFC and hippocampal inputs to the NAc, may induce activity-dependent synaptic potentiation or induce activity-dependent genes and transcription factors, much like ketamine. Whether and where in the brain these processes occur remains to be determined. Interestingly, direct stimulation of the ventral striatal reward circuitry may be equally efficacious in depressed humans [128,129]. Finally, preclinical studies have suggested that DBS may act indirectly by promoting the release of serotonin [127].
Electroconvulsive Therapy (ECT)
There is a compelling overlap between direct methods of exciting the brains of depressed patients to improve mood and the high-frequency stimuli that induce LTP at excitatory synapses within and between many of the brain areas discussed above. Electroconvulsive therapy (ECT), in which a generalized seizure is induced by delivering 20–120 Hz stimulation, is one of the oldest treatments for depression still in use, although typically reserved for patients resistant to pharmacological treatment, or where such treatment is contraindicated, such as many elderly patients.
ECT-like stimulation in rats is known to induce an LTP-like potentiation of excitatory synapses in the dentate gyrus that persists for up to 40 days [130]. Like conventional LTP, this ECT-induced potentiation is inhibited by an NMDAR antagonist [131] and is accompanied by an increase in phosphorylation of serine 831 of GluA1 and S1301 of GluN2B. Changes in overall receptor expression were observed in some studies [132], but not others [133]. Interestingly, ECT also promotes an NMDAR-dependent increase in BDNF expression in animal models [134,135].
In conclusion, although there are many important questions that remain unanswered (Box 3), there is considerable evidence that dysfunction of excitatory synapses contributes to the pathology of depression and that many effective antidepressants act to restore normal synaptic strength. We recognize that the evidence in support of the hypothesis is far from complete and that the circuits are far more complex than we have indicated here. Our intent in formulating this hypothesis is to stimulate further work and to provide a conceptual framework with which to interpret the results. We hope that these efforts lead to better understanding of the causes of depression and thereby to better therapeutic options that increase the number of patients that can be treated effectively and that their symptoms can be relieved more quickly.
Box 3. Unanswered questions.
How does chronic stress impair synaptic structure and function? Changes associated with stress have been attributed to a variety of mediators, including corticosteroids, peptide hormones (e.g. CRH), opioid peptides, monoamines (e.g. serotonin, dopamine), inflammatory cytokines, endocannabinoids, and neurosteroids, to name a few [170]. Glucocorticoids are by no means singly important but are likely the best known. In one example, chronic administration of corticosterone to naïve rats and mice is sufficient to produce depressive-like behavior and recapitulate several molecular markers of chronic stress models [171], presumably via glucocorticoid receptors. Neurons throughout the cortex, hippocampus, and ventral striatum express GRs at high levels, rendering them vulnerable to chronic stress. The downstream mediators of persistent GR activation are unknown but ultimately alter expression of synaptic proteins through altered synthesis, turnover, and degradation [49]. What makes some synapses selectively vulnerable? Distal dendrites are particularly sensitive to the atrophic effects of chronic stress and contributes, at least in part, for the localization of the altered synaptic composition and function [72,172]. Studies continue to unravel new roles for all the known mediators of stress, sometimes with differing short- and long-term actions, and continue to elucidate the molecular mediators between stress and impaired synaptic function.
How do SSRIs act in the NAc and LHb? Activation of 5-HT1BRs triggers a persistent inhibition of excitatory synaptic transmission in the NAc in unstressed animals [173,174]. The hypothesis predicts that this action of serotonin would promote, not relieve, the symptoms of depression. It remains to be determined what the effects of chronic SSRI or acute ketamine administration are on these synapses. Similarly, do SSRIs re-normalize plastic changes in synapses in the LHb and RMTg? This will be particularly informative because the direction of the depression-associated synaptic plasticity in this circuit (strengthening) appears opposite to that seen in the hippocampus, PFC, and NAc (weakening).
Why do SSRIs induce immediate elevation of serotonin levels [145] and rapid potentiation of excitatory synapses [76], but a delayed therapeutic response? The hypothesis predicts that an immediate strengthening of stress-weakened synapses should relieve symptoms rapidly. Indeed, cocaine produces a rapid strengthening of excitatory synapses in the NAc [175] and an immediate euphoric response. It may be that multiple bouts of serotonin-induced potentiation are necessary to render them persistent, perhaps in conjunction with slower changes in neurotrophin signaling, for the effects to be sustained. Only repeated administration of cocaine, for example, leads to a lasting disinhibition [30] or increases in mEPSC frequency [176] in VTA dopamine neurons. Alternatively, SSRIs may elicit acute actions in other brain regions, such as the amygdala, that may oppose their actions in reward circuits. Not enough is known about the actions of SSRIs in mesolimbic reward circuits. Finally, it will be important to determine whether ADs that affect NE and DA levels also produce changes in these circuits.
How and where does DBS work? The effects described in some patients offer encouragement that DBS may offer sorely needed relief for the most severely depressed, and for treatment resistant patients [126], but the low rate of surgical success suggests that we do not yet know enough about what structures or pathways to target and perhaps what stimulus parameters to use. Preclinical studies are likely to suggest the best places to re-normalize reward circuitry.
Can compounds be developed that retain ketamine’s beneficial rapid antidepressant action without the negative side effects? Current efforts to develop NMDAR antagonists with unique pharmacological properties and subunit selectivity appear promising [177], although the side-effect profile is not yet clear. If our understanding of how ketamine works is correct, then any drug that promotes synchronized oscillatory activity in the PFC and hippocampus should also have an AD action, like ketamine.
Highlights.
We review evidence of dysfunction of excitatory synapses in reward circuits in depression.
We suggest how these defects arise and how they negatively impact circuit function.
We review evidence that many antidepressant drugs and treatments reverse these changes.
Acknowledgments
We thank our colleagues Drs. Mary Kay Lobo, Brian Mathur, Todd Gould, and Robert Schwarcz and Mr. T. Chase Francis for their comments on the manuscript. SMT and XC were supported by grant R01 MH086828, AJK and AMV were supported by T32 GM008181, MDK was supported by T32 NS063391, and TAL was supported by T32 NS007375.
GLOSSARY OF TERMS USED
- Allostatic load/overload
Allostasis is the concept that homeostatic setpoints can be differentially regulated to meet different demands in the internal and external environment (e.g. physical or psychological stress). Repeated allostatic adaptation exacts a cost on the brain.
Behavioral measures of ‘helplessness’ (All sensitive to acute SSRIs.)
- Tail suspension test
Mice are dangled by their tails until they cease to struggle. Naïve, unstressed animals have longer latencies to immobility than stressed animals
- Forced swim test
Animals are placed in a tank of water until they cease struggling. Naïve, unstressed animals have longer latencies to cease struggling than stressed animals
- Learned helplessness test
Animals are placed in a chamber where they previously received inescapable foot shocks paired with a conditioned stimulus. Latency to escape from the same chamber is later measured in response to the conditioned stimulus.
Behavioral measures of hedonic state (All sensitive to chronic, but not acute, SSRIs.)
- Sucrose preference test
A two bottle choice between plain water and dilute (1%) sucrose solution. Naïve, unstressed animals drink about 80–90% of their liquid from the sucrose solution, whereas chronically stressed animals drink nearly equally from both bottles, presumably because the sucrose solution is no longer rewarding
- Social interaction test
Animals are placed in an arena with a novel juvenile animal held in a small cage. Time spent in the vicinity of the cage is compared in the presence and absence of the juvenile. Naïve, unstressed animals spend more time in the vicinity of the cage when the juvenile is present, whereas chronically stressed animals do not, presumably because the social interaction is no longer rewarding
- Novelty suppressed feeding test
Animals are food deprived then placed in a brightly lit arena with food in the center. The latency until they venture into the center and eat is measured. Naïve, unstressed animals display a shorter latency than stressed animals. This test may reveal more about anxiety than hedonic state.
Models of depression-like behavioral changes
- Chronic social defeat
Animals are typically placed in the home cage of a novel aggressor for 30 min, once/day for three weeks
- Chronic unpredictable stress
Animals experience two bouts of one of many mild stressors twice per day for three weeks
- Chronic restraint stress
Animals are placed in restraint tubes for several hours daily, repeated over several days (e.g. four hours/day for 10–14 days)
- Learned helplessness
Animals receive a single session with repeated unpredictable and uncontrollable stressors (foot shocks)
Footnotes
The authors declare they have no conflicting financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kessler RC, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:593–602. doi: 10.1001/archpsyc.62.6.593. [DOI] [PubMed] [Google Scholar]
- 2.Belmaker RH, Agam G. Major depressive disorder. N Engl J Med. 2008;358:55–68. doi: 10.1056/NEJMra073096. [DOI] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention. Suicide Facts at a Glance. 2009 Retrieved from http://www.cdc.gov/violenceprevention/pdf/Suicide_DataSheet-a.pdf.
- 4.Gartlehner G, et al. Second-Generation Antidepressants in the Pharmacologic Treatment of Adult Depression: An Update of the 2007 Comparative Effectiveness Review. AHRQ Comp Eff Rev. 2011 [PubMed] [Google Scholar]
- 5.Gaynes B, Warden D. What did STAR* D teach us? Results from a large-scale, practical, clinical trial for patients with depression. Psychiatry Serv. 2009;60:1439–45. doi: 10.1176/ps.2009.60.11.1439. [DOI] [PubMed] [Google Scholar]
- 6.Duman RS, Aghajanian GK. Synaptic dysfunction in depression: potential therapeutic targets. Science. 2012;338:68–72. doi: 10.1126/science.1222939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nestler E, et al. Neurobiology of depression. Neuron. 2002;34:13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- 8.Flint J, Kendler KS. The genetics of major depression. Neuron. 2014;81:484–503. doi: 10.1016/j.neuron.2014.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kendler K. Anna-Monika-Prize paper. Major depression and the environment: a psychiatric genetic perspective. Pharmacopsychiatry. 1998;31:5–9. doi: 10.1055/s-2007-979287. [DOI] [PubMed] [Google Scholar]
- 10.Billings AG, et al. Social-environmental factors in unipolar depression: comparisons of depressed patients and nondepressed controls. J Abnorm Psychol. 1983;92:119–33. doi: 10.1037//0021-843x.92.2.119. [DOI] [PubMed] [Google Scholar]
- 11.Korte SM, et al. The Darwinian concept of stress: benefits of allostasis and costs of allostatic load and the trade-offs in health and disease. Neurosci Biobehav Rev. 2005;29:3–38. doi: 10.1016/j.neubiorev.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 12.Sapolsky R. The stress of Gulf War syndrome. Nature. 1998;393:308–9. doi: 10.1038/30606. [DOI] [PubMed] [Google Scholar]
- 13.McEwen BS. Allostasis and allostatic load: Implications for neuropsychopharmacology. Neuropsychopharmacology. 2000;22:108–124. doi: 10.1016/S0893-133X(99)00129-3. [DOI] [PubMed] [Google Scholar]
- 14.Pariante CM. Risk factors for development of depression and psychosis. Glucocorticoid receptors and pituitary implications for treatment with antidepressant and glucocorticoids. Ann N Y Acad Sci. 2009;1179:144–52. doi: 10.1111/j.1749-6632.2009.04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23:477–501. doi: 10.1016/S0893-133X(00)00159-7. [DOI] [PubMed] [Google Scholar]
- 16.Anacker C, et al. The glucocorticoid receptor: pivot of depression and of antidepressant treatment? Psychoneuroendocrinology. 2011;36:415–25. doi: 10.1016/j.psyneuen.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spijker A, van Rossum E. Glucocorticoid sensitivity in mood disorders. Neuroendocrinology. 2012;95:179–86. doi: 10.1159/000329846. [DOI] [PubMed] [Google Scholar]
- 18.Brown ES. Effects of glucocorticoids on mood, memory, and the hippocampus. Treatment and preventive therapy. Ann N Y Acad Sci. 2009;1179:41–55. doi: 10.1111/j.1749-6632.2009.04981.x. [DOI] [PubMed] [Google Scholar]
- 19.Frol AB, et al. A comparison of clinician-rated neuropsychological and self-rated cognitive assessments in patients with asthma and rheumatologic disorders. Allergy asthma Proc. 2013;34:170–5. doi: 10.2500/aap.2013.34.3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McEwen BS. Brain on stress: how the social environment gets under the skin. Proc Natl Acad Sci USA. 2012;109(Suppl):17180–5. doi: 10.1073/pnas.1121254109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yun J, et al. Chronic restraint stress impairs neurogenesis and hippocampus-dependent fear memory in mice: Possible involvement of a brain-specific transcription factor Npas4. J Neurochem. 2010;114:1840–1851. doi: 10.1111/j.1471-4159.2010.06893.x. [DOI] [PubMed] [Google Scholar]
- 22.Magariños AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: comparison of stressors. Neuroscience. 1995;69:83–8. doi: 10.1016/0306-4522(95)00256-i. [DOI] [PubMed] [Google Scholar]
- 23.Fales CL, et al. Antidepressant treatment normalizes hypoactivity in dorsolateral prefrontal cortex during emotional interference processing in major depression. J Affect Disord. 2009;112:206–11. doi: 10.1016/j.jad.2008.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frodl T, et al. Larger amygdala volumes in first depressive episode as compared to recurrent major depression and healthy control subjects. Biol Psychiatry. 2003;53:338–44. doi: 10.1016/s0006-3223(02)01474-9. [DOI] [PubMed] [Google Scholar]
- 25.Lorenzetti V, et al. Structural brain abnormalities in major depressive disorder: a selective review of recent MRI studies. J Affect Disord. 2009;117:1–17. doi: 10.1016/j.jad.2008.11.021. [DOI] [PubMed] [Google Scholar]
- 26.Pittenger C, Duman RS. Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology. 2008;33:88–109. doi: 10.1038/sj.npp.1301574. [DOI] [PubMed] [Google Scholar]
- 27.Sesack SR, Grace Aa. Cortico-Basal Ganglia reward network: microcircuitry. Neuropsychopharmacology. 2010;35:27–47. doi: 10.1038/npp.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith KS, et al. Disentangling pleasure from incentive salience and learning signals in brain reward circuitry. Proc Natl Acad Sci USA. 2011;108:E255–E264. doi: 10.1073/pnas.1101920108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lim BK, et al. Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature. 2012;487:183–9. doi: 10.1038/nature11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bocklisch C, et al. Cocaine disinhibits dopamine neurons by potentiation of GABA transmission in the ventral tegmental area. Science. 2013;341:1521–5. doi: 10.1126/science.1237059. [DOI] [PubMed] [Google Scholar]
- 31.Christoffel DJ, et al. IκB kinase regulates social defeat stress-induced synaptic and behavioral plasticity. J Neurosci. 2011;31:314–21. doi: 10.1523/JNEUROSCI.4763-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Francis TC, et al. Nucleus accumbens medium spiny neuron subtypes mediate depression-related outcomes to social defeat stress. Biol Psychiatry. 2015;77:212–22. doi: 10.1016/j.biopsych.2014.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Golden SA, et al. Epigenetic regulation of RAC1 induces synaptic remodeling in stress disorders and depression. Nat Med. 2013;19:337–44. doi: 10.1038/nm.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tye KM, et al. Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature. 2013;493:537–41. doi: 10.1038/nature11740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valenti O, et al. Different stressors produce excitation or inhibition of mesolimbic dopamine neuron activity: response alteration by stress pre-exposure. Eur J Neurosci. 2012;35:1312–21. doi: 10.1111/j.1460-9568.2012.08038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Belujon P, Grace Aa. Restoring mood balance in depression: ketamine reverses deficit in dopamine-dependent synaptic plasticity. Biol Psychiatry. 2014;76:927–36. doi: 10.1016/j.biopsych.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cabib S, Puglisi-Allegra S. The mesoaccumbens dopamine in coping with stress. Neurosci Biobehav Rev. 2012;36:79–89. doi: 10.1016/j.neubiorev.2011.04.012. [DOI] [PubMed] [Google Scholar]
- 38.Krishnan V, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 39.Cao JL, et al. Mesolimbic dopamine neurons in the brain reward circuit mediate susceptibility to social defeat and antidepressant action. J Neurosci. 2010;30:16453–8. doi: 10.1523/JNEUROSCI.3177-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barik J, et al. Chronic stress triggers social aversion via glucocorticoid receptor in dopaminoceptive neurons. Science. 2013;339:332–5. doi: 10.1126/science.1226767. [DOI] [PubMed] [Google Scholar]
- 41.Chaudhury D, et al. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature. 2013;493:532–6. doi: 10.1038/nature11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Covington HE, et al. Antidepressant effect of optogenetic stimulation of the medial prefrontal cortex. J Neurosci. 2010;30:16082–16090. doi: 10.1523/JNEUROSCI.1731-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vialou V, et al. Prefrontal cortical circuit for depression- and anxiety-related behaviors mediated by cholecystokinin: role of ΔFosB. J Neurosci. 2014;34:3878–87. doi: 10.1523/JNEUROSCI.1787-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amat J, et al. Medial prefrontal cortex determines how stressor controllability affects behavior and dorsal raphe nucleus. Nat Neurosci. 2005;8:365–371. doi: 10.1038/nn1399. [DOI] [PubMed] [Google Scholar]
- 45.Berridge KC, Kringelbach ML. Neuroscience of affect: brain mechanisms of pleasure and displeasure. Curr Opin Neurobiol. 2013;23:294–303. doi: 10.1016/j.conb.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kennedy SH, et al. Changes in regional brain glucose metabolism measured with positron emission tomography after paroxetine treatment of major depression. Am J Psychiatry. 2001;158:899–905. doi: 10.1176/appi.ajp.158.6.899. [DOI] [PubMed] [Google Scholar]
- 47.Liston C, et al. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J Neurosci. 2006;26:7870–4. doi: 10.1523/JNEUROSCI.1184-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Radley JJ, et al. Repeated stress alters dendritic spine morphology in the rat medial prefrontal cortex. J Comp Neurol. 2008;507:1141–1150. doi: 10.1002/cne.21588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuen EY, et al. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron. 2012;73:962–77. doi: 10.1016/j.neuron.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aghajanian GK, Marek GJ. Serotonin induces excitatory postsynaptic potentials in apical dendrites of neocortical pyramidal cells. Neuropharmacology. 1997;36:589–599. doi: 10.1016/s0028-3908(97)00051-8. [DOI] [PubMed] [Google Scholar]
- 51.Lambe EK, et al. Serotonin induces EPSCs preferentially in layer V pyramidal neurons of the frontal cortex in the rat. Cereb Cortex. 2000;10:974–80. doi: 10.1093/cercor/10.10.974. [DOI] [PubMed] [Google Scholar]
- 52.Béïque JC, et al. Mechanism of the 5-hydroxytryptamine 2A receptor-mediated facilitation of synaptic activity in prefrontal cortex. Proc Natl Acad Sci USA. 2007;104:9870–9875. doi: 10.1073/pnas.0700436104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu RJ, Aghajanian GK. Stress blunts serotonin- and hypocretin-evoked EPSCs in prefrontal cortex: role of corticosterone-mediated apical dendritic atrophy. Proc Natl Acad Sci USA. 2008;105:359–64. doi: 10.1073/pnas.0706679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsumoto M, Hikosaka O. Lateral habenula as a source of negative reward signals in dopamine neurons. Nature. 2007;447:1111–5. doi: 10.1038/nature05860. [DOI] [PubMed] [Google Scholar]
- 55.Proulx CD, et al. Reward processing by the lateral habenula in normal and depressive behaviors. Nat Neurosci. 2014;17:1146–52. doi: 10.1038/nn.3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gonçalves L, et al. Differential projections from the lateral habenula to the rostromedial tegmental nucleus and ventral tegmental area in the rat. J Comp Neurol. 2012;520:1278–1300. doi: 10.1002/cne.22787. [DOI] [PubMed] [Google Scholar]
- 57.Sego C, et al. Lateral habenula and the rostromedial tegmental nucleus innervate neurochemically distinct subdivisions of the dorsal raphe nucleus in the rat. J Comp Neurol. 2014;522:1454–1484. doi: 10.1002/cne.23533. [DOI] [PubMed] [Google Scholar]
- 58.Christoph GR, et al. Stimulation of the lateral habenula inhibits dopamine-containing neurons in the substantia nigra and ventral tegmental area of the rat. J Neurosci. 1986;6:613–9. doi: 10.1523/JNEUROSCI.06-03-00613.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matsumoto M, Hikosaka O. Representation of negative motivational value in the primate lateral habenula. Nat Neurosci. 2009;12:77–84. doi: 10.1038/nn.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li B, et al. Synaptic potentiation onto habenula neurons in the learned helplessness model of depression. Nature. 2011;470:535–9. doi: 10.1038/nature09742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morris JS, et al. Covariation of activity in habenula and dorsal raphé nuclei following tryptophan depletion. Neuroimage. 1999;10:163–172. doi: 10.1006/nimg.1999.0455. [DOI] [PubMed] [Google Scholar]
- 62.Duvarci S, Pare D. Amygdala microcircuits controlling learned fear. Neuron. 2014;82:966–80. doi: 10.1016/j.neuron.2014.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Drevets WC. Neuroimaging abnormalities in the amygdala in mood disorders. Ann N Y Acad Sci. 2003;985:420–44. doi: 10.1111/j.1749-6632.2003.tb07098.x. [DOI] [PubMed] [Google Scholar]
- 64.Vyas A, et al. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci. 2002;22:6810–8. doi: 10.1523/JNEUROSCI.22-15-06810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Felix-Ortiz AC, Tye KM. Amygdala inputs to the ventral hippocampus bidirectionally modulate social behavior. J Neurosci. 2014;34:586–95. doi: 10.1523/JNEUROSCI.4257-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Magariños AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience. 1995;69:89–98. doi: 10.1016/0306-4522(95)00259-l. [DOI] [PubMed] [Google Scholar]
- 67.Pawlak R, et al. Tissue plasminogen activator and plasminogen mediate stress-induced decline of neuronal and cognitive functions in the mouse hippocampus. Proc Natl Acad Sci USA. 2005;102:18201–6. doi: 10.1073/pnas.0509232102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bartanusz V, et al. Stress-induced changes in messenger RNA levels of N-methyl-D-aspartate and AMPA receptor subunits in selected regions of the rat hippocampus and hypothalamus. Neuroscience. 1995;66:247–52. doi: 10.1016/0306-4522(95)00084-v. [DOI] [PubMed] [Google Scholar]
- 69.Alfarez DN, et al. Chronic unpredictable stress impairs long-term potentiation in rat hippocampal CA1 area and dentate gyrus in vitro. Eur J Neurosci. 2003;17:1928–1934. doi: 10.1046/j.1460-9568.2003.02622.x. [DOI] [PubMed] [Google Scholar]
- 70.Schmidt MV, et al. Individual stress vulnerability is predicted by short-term memory and AMPA receptor subunit ratio in the hippocampus. J Neurosci. 2010;30:16949–58. doi: 10.1523/JNEUROSCI.4668-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kole MHP, et al. The antidepressant tianeptine persistently modulates glutamate receptor currents of the hippocampal CA3 commissural associational synapse in chronically stressed rats. Eur J Neurosci. 2002;16:807–816. doi: 10.1046/j.1460-9568.2002.02136.x. [DOI] [PubMed] [Google Scholar]
- 72.Kallarackal AJ, et al. Chronic stress induces a selective decrease in AMPA receptor-mediated synaptic excitation at hippocampal temporoammonic-CA1 synapses. J Neurosci. 2013;33:15669–74. doi: 10.1523/JNEUROSCI.2588-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Remondes M, Schuman EM. Role for a cortical input to hippocampal area CA1 in the consolidation of a long-term memory. Nature. 2004;431:699–703. doi: 10.1038/nature02965. [DOI] [PubMed] [Google Scholar]
- 74.Freed WJ, et al. Properties of [3H]AMPA binding in postmortem human brain from psychotic subjects and controls: increases in caudate nucleus associated with suicide. Exp Neurol. 1993;121:48–56. doi: 10.1006/exnr.1993.1070. [DOI] [PubMed] [Google Scholar]
- 75.Meador-Woodruff JH, et al. Striatal ionotropic glutamate receptor expression in schizophrenia, bipolar disorder, and major depressive disorder. Brain Res Bull. 2001;55:631–40. doi: 10.1016/s0361-9230(01)00523-8. [DOI] [PubMed] [Google Scholar]
- 76.Cai X, et al. Local potentiation of excitatory synapses by serotonin and its alteration in rodent models of depression. Nat Neurosci. 2013;16:464–72. doi: 10.1038/nn.3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chourbaji S, et al. AMPA receptor subunit 1 (GluR-A) knockout mice model the glutamate hypothesis of depression. FASEB J. 2008;22:3129–34. doi: 10.1096/fj.08-106450. [DOI] [PubMed] [Google Scholar]
- 78.Auer DP, et al. Reduced glutamate in the anterior cingulate cortex in depression: an in vivo proton magnetic resonance spectroscopy study. Biol Psychiatry. 2000;47:305–13. doi: 10.1016/s0006-3223(99)00159-6. [DOI] [PubMed] [Google Scholar]
- 79.Pfleiderer B, et al. Effective electroconvulsive therapy reverses glutamate/glutamine deficit in the left anterior cingulum of unipolar depressed patients. Psychiatry Res. 2003;122:185–92. doi: 10.1016/s0925-4927(03)00003-9. [DOI] [PubMed] [Google Scholar]
- 80.Mitani H, et al. Correlation between plasma levels of glutamate, alanine and serine with severity of depression. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:1155–8. doi: 10.1016/j.pnpbp.2006.03.036. [DOI] [PubMed] [Google Scholar]
- 81.Choudary PV, et al. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc Natl Acad Sci USA. 2005;102:15653–8. doi: 10.1073/pnas.0507901102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Beneyto M, et al. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology. 2007;32:1888–902. doi: 10.1038/sj.npp.1301312. [DOI] [PubMed] [Google Scholar]
- 83.Nestler EJ, Carlezon WA. The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59:1151–9. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 84.Akiskal HS, McKinney WT. Depressive disorders: toward a unified hypothesis. Science. 1973;182:20–9. doi: 10.1126/science.182.4107.20. [DOI] [PubMed] [Google Scholar]
- 85.Leuner B, Shors TJ. Neuroscience. Vol. 251. IBRO; 2013. Stress, anxiety, and dendritic spines: What are the connections? pp. 108–119. [DOI] [PubMed] [Google Scholar]
- 86.Spijker J, et al. Predictors of suicidality in depressive spectrum disorders in the general population: results of the Netherlands Mental Health Survey and Incidence Study. Soc Psychiatry Psychiatr Epidemiol. 2010;45:513–21. doi: 10.1007/s00127-009-0093-6. [DOI] [PubMed] [Google Scholar]
- 87.Winer ES, et al. Anhedonia predicts suicidal ideation in a large psychiatric inpatient sample. Psychiatry Res. 2014;218:124–8. doi: 10.1016/j.psychres.2014.04.016. [DOI] [PubMed] [Google Scholar]
- 88.Fawcett J, et al. Time-related predictors of suicide in major affective disorder. Am J Psychiatry. 1990;147:1189–94. doi: 10.1176/ajp.147.9.1189. [DOI] [PubMed] [Google Scholar]
- 89.Floresco SB, et al. Glutamatergic afferents from the hippocampus to the nucleus accumbens regulate activity of ventral tegmental area dopamine neurons. J Neurosci. 2001;21:4915–22. doi: 10.1523/JNEUROSCI.21-13-04915.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Salamone JD, Correa M. The mysterious motivational functions of mesolimbic dopamine. Neuron. 2012;76:470–85. doi: 10.1016/j.neuron.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.O’Donnell P, Grace AA. Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. J Neurosci. 1995;15:3622–3639. doi: 10.1523/JNEUROSCI.15-05-03622.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Legault M, et al. Chemical stimulation of the ventral hippocampus elevates nucleus accumbens dopamine by activating dopaminergic neurons of the ventral tegmental area. J Neurosci. 2000;20:1635–42. doi: 10.1523/JNEUROSCI.20-04-01635.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tyler WJ, Pozzo-Miller L. Miniature synaptic transmission and BDNF modulate dendritic spine growth and form in rat CA1 neurones. J Physiol. 2003;553:497–509. doi: 10.1113/jphysiol.2003.052639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sairanen M, et al. Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. J Neurosci. 2005;25:1089–94. doi: 10.1523/JNEUROSCI.3741-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weissbourd B, et al. Presynaptic partners of dorsal raphe serotonergic and GABAergic neurons. Neuron. 2014;83:645–62. doi: 10.1016/j.neuron.2014.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Malberg JE, et al. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20:9104–10. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gould E, et al. Proliferation of granule cell precursors in the dentate gyrus of adult monkeys is diminished by stress. Proc Natl Acad Sci USA. 1998;95:3168–3171. doi: 10.1073/pnas.95.6.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sahay A, Hen R. Adult hippocampal neurogenesis in depression. Nat Neurosci. 2007;10:1110–5. doi: 10.1038/nn1969. [DOI] [PubMed] [Google Scholar]
- 99.Kobayashi K, et al. Chronic fluoxetine bidirectionally modulates potentiating effects of serotonin on the hippocampal mossy fiber synaptic transmission. J Neurosci. 2008;28:6272–80. doi: 10.1523/JNEUROSCI.1656-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Berman RM, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 101.Zarate CA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–64. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 102.Garcia LSB, et al. Acute administration of ketamine induces antidepressant-like effects in the forced swimming test and increases BDNF levels in the rat hippocampus. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:140–4. doi: 10.1016/j.pnpbp.2007.07.027. [DOI] [PubMed] [Google Scholar]
- 103.Li N, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–64. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li N, et al. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. 2011;69:754–61. doi: 10.1016/j.biopsych.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Moghaddam B, et al. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–7. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dwyer JM, Duman RS. Activation of mammalian target of rapamycin and synaptogenesis: role in the actions of rapid-acting antidepressants. Biol Psychiatry. 2013;73:1189–98. doi: 10.1016/j.biopsych.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Carlén M, et al. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry. 2012;17:537–48. doi: 10.1038/mp.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kittelberger K, et al. Comparison of the effects of acute and chronic administration of ketamine on hippocampal oscillations: relevance for the NMDA receptor hypofunction model of schizophrenia. Brain Struct Funct. 2012;217:395–409. doi: 10.1007/s00429-011-0351-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Maksimow A, et al. Increase in high frequency EEG activity explains the poor performance of EEG spectral entropy monitor during S-ketamine anesthesia. Clin Neurophysiol. 2006;117:1660–1668. doi: 10.1016/j.clinph.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 111.Cornwell BR, et al. Synaptic potentiation is critical for rapid antidepressant response to ketamine in treatment-resistant major depression. Biol Psychiatry. 2012;72:555–61. doi: 10.1016/j.biopsych.2012.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lorrain DS, et al. Effects of ketamine and N-methyl-D-aspartate on glutamate and dopamine release in the rat prefrontal cortex: Modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neuroscience. 2003;117:697–706. doi: 10.1016/s0306-4522(02)00652-8. [DOI] [PubMed] [Google Scholar]
- 113.Razoux F, et al. Ketamine, at a dose that disrupts motor behavior and latent inhibition, enhances prefrontal cortex synaptic efficacy and glutamate release in the nucleus accumbens. Neuropsychopharmacology. 2007;32:719–727. doi: 10.1038/sj.npp.1301057. [DOI] [PubMed] [Google Scholar]
- 114.Aan Het Rot M, et al. Ketamine for depression: where do we go from here? Biol Psychiatry. 2012;72:537–47. doi: 10.1016/j.biopsych.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pozzi L, et al. Mice lacking NMDA receptors in parvalbumin neurons display normal depression-related behavior and response to antidepressant action of NMDAR antagonists. PLoS One. 2014;9:e83879. doi: 10.1371/journal.pone.0083879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Autry AE, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475:91–95. doi: 10.1038/nature10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nosyreva E, et al. Acute suppression of spontaneous neurotransmission drives synaptic potentiation. J Neurosci. 2013;33:6990–7002. doi: 10.1523/JNEUROSCI.4998-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Maeng S, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63:349–52. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 119.Kumar S, et al. Cortical control of affective networks. J Neurosci. 2013;33:1116–29. doi: 10.1523/JNEUROSCI.0092-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Furey ML, Drevets WC. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch Gen Psychiatry. 2006;63:1121–9. doi: 10.1001/archpsyc.63.10.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Voleti B, et al. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry. 2013;74:742–9. doi: 10.1016/j.biopsych.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li X, et al. Antidepressant-like actions of an AMPA receptor potentiator (LY392098) Neuropharmacology. 2001;40:1028–33. doi: 10.1016/s0028-3908(00)00194-5. [DOI] [PubMed] [Google Scholar]
- 123.Farley S, et al. Antidepressant-like effects of an AMPA receptor potentiator under a chronic mild stress paradigm. Int J Neuropsychopharmacol. 2010;13:1207–1218. doi: 10.1017/S1461145709991076. [DOI] [PubMed] [Google Scholar]
- 124.Mackowiak M, et al. An AMPA receptor potentiator modulates hippocampal expression of BDNF: an in vivo study. Neuropharmacology. 2002;43:1–10. doi: 10.1016/s0028-3908(02)00066-7. [DOI] [PubMed] [Google Scholar]
- 125.Li X, et al. Enhancement of antidepressant potency by a potentiator of AMPA receptors. Cell Mol Neurobiol. 2003;23:419–30. doi: 10.1023/A:1023648923447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mayberg HS, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45:651–60. doi: 10.1016/j.neuron.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 127.Hamani C, et al. Antidepressant-Like Effects of Medial Prefrontal Cortex Deep Brain Stimulation in Rats. Biol Psychiatry. 2010;67:117–124. doi: 10.1016/j.biopsych.2009.08.025. [DOI] [PubMed] [Google Scholar]
- 128.Malone DA, et al. Deep brain stimulation of the ventral capsule/ventral striatum for treatment-resistant depression. Biol Psychiatry. 2009;65:267–75. doi: 10.1016/j.biopsych.2008.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schlaepfer TE, et al. Deep brain stimulation of the human reward system for major depression--rationale, outcomes and outlook. Neuropsychopharmacology. 2014;39:1303–14. doi: 10.1038/npp.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Stewart CA, et al. LTP-like synaptic efficacy changes following electroconvulsive stimulation. Neuroreport. 1994;5:1041–4. doi: 10.1097/00001756-199405000-00006. [DOI] [PubMed] [Google Scholar]
- 131.Stewart CA, Reid IC. Ketamine prevents ECS-induced synaptic enhancement in rat hippocampus. Neurosci Lett. 1994;178:11–4. doi: 10.1016/0304-3940(94)90277-1. [DOI] [PubMed] [Google Scholar]
- 132.Naylor P, et al. Repeated ECS induces GluR1 mRNA but not NMDAR1A-G mRNA in the rat hippocampus. Brain Res Mol Brain Res. 1996;35:349–53. doi: 10.1016/0169-328x(95)00264-s. [DOI] [PubMed] [Google Scholar]
- 133.Fumagalli F, et al. Repeated electroconvulsive shock (ECS) alters the phosphorylation of glutamate receptor subunits in the rat hippocampus. Int J Neuropsychopharmacol. 2010;13:1255–60. doi: 10.1017/S1461145710000544. [DOI] [PubMed] [Google Scholar]
- 134.Nibuya M, et al. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–47. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Altar CA, et al. Effects of electroconvulsive seizures and antidepressant drugs on brain-derived neurotrophic factor protein in rat brain. Biol Psychiatry. 2003;54:703–9. doi: 10.1016/s0006-3223(03)00073-8. [DOI] [PubMed] [Google Scholar]
- 136.Marshall EF, et al. The effect of iproniazid and imipramine on the blood platelet 5-hydroxytrptamine level in man. Br J Pharmacol Chemother. 1960;15:35–41. doi: 10.1111/j.1476-5381.1960.tb01207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Woolley DW, Shaw E. A biochemical and pharmacological suggestion about certain mental disorders. Proc Natl Acad Sci USA. 1954;40:228–31. doi: 10.1073/pnas.40.4.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Schildkraut JJ. The catecholamine hypothesis of affective disorders: a review of supporting evidence. Am J Psychiatry. 1965;122:509–22. doi: 10.1176/ajp.122.5.509. [DOI] [PubMed] [Google Scholar]
- 139.Lapin IP, Oxenkrug GF. Intensification of the central serotoninergic processes as a possible determinant of the thymoleptic effect. Lancet. 1969;1:132–6. doi: 10.1016/s0140-6736(69)91140-4. [DOI] [PubMed] [Google Scholar]
- 140.Jacobsen JPR, et al. The 5-HT deficiency theory of depression: perspectives from a naturalistic 5-HT deficiency model, the tryptophan hydroxylase 2Arg439His knockin mouse. Philos Trans R Soc Lond B Biol Sci. 2012;367:2444–59. doi: 10.1098/rstb.2012.0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mendels J, et al. Amine precursors and depression. Arch Gen Psychiatry. 1975;32:22–30. doi: 10.1001/archpsyc.1975.01760190024002. [DOI] [PubMed] [Google Scholar]
- 142.Delgado PL, et al. Serotonin and the neurobiology of depression. Effects of tryptophan depletion in drug-free depressed patients. Arch Gen Psychiatry. 1994;51:865–74. doi: 10.1001/archpsyc.1994.03950110025005. [DOI] [PubMed] [Google Scholar]
- 143.Salomon RM, et al. Lack of behavioral effects of monoamine depletion in healthy subjects. Biol Psychiatry. 1997;41:58–64. doi: 10.1016/0006-3223(95)00670-2. [DOI] [PubMed] [Google Scholar]
- 144.Moreno FA, et al. Tryptophan depletion and depressive vulnerability. Biol Psychiatry. 1999;46:498–505. doi: 10.1016/s0006-3223(99)00095-5. [DOI] [PubMed] [Google Scholar]
- 145.Guan XM, McBride WJ. Fluoxetine increases the extracellular levels of serotonin in the nucleus accumbens. Brain Res Bull. 1988;21:43–46. doi: 10.1016/0361-9230(88)90118-9. [DOI] [PubMed] [Google Scholar]
- 146.Cheetham SC, et al. Brain 5-HT1 binding sites in depressed suicides. Psychopharmacology. 1990;102:544–8. doi: 10.1007/BF02247138. [DOI] [PubMed] [Google Scholar]
- 147.López-Figueroa AL, et al. Serotonin 5-HT1A, 5-HT1B, and 5-HT2A receptor mRNA expression in subjects with major depression, bipolar disorder, and schizophrenia. Biol Psychiatry. 2004;55:225–33. doi: 10.1016/j.biopsych.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 148.Savitz JB, et al. 5-HT(1A) receptor function in major depressive disorder. Prog Neurobiol. 2009;88:17–31. doi: 10.1016/j.pneurobio.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–27. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 150.Duman RS, et al. A molecular and cellular theory of depression. Arch Gen Psychiatry. 1997;54:597–606. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- 151.Chourbaji S, et al. Altering BDNF expression by genetics and/or environment: impact for emotional and depression-like behaviour in laboratory mice. Neurosci Biobehav Rev. 2011;35:599–611. doi: 10.1016/j.neubiorev.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 152.Karege F, et al. Decreased serum brain-derived neurotrophic factor levels in major depressed patients. Psychiatry Res. 2002;109:143–8. doi: 10.1016/s0165-1781(02)00005-7. [DOI] [PubMed] [Google Scholar]
- 153.Shimizu E, et al. Alterations of serum levels of brain-derived neurotrophic factor (BDNF) in depressed patients with or without antidepressants. Biol Psychiatry. 2003;54:70–5. doi: 10.1016/s0006-3223(03)00181-1. [DOI] [PubMed] [Google Scholar]
- 154.Dwivedi Y, et al. Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry. 2003;60:804–15. doi: 10.1001/archpsyc.60.8.804. [DOI] [PubMed] [Google Scholar]
- 155.Chen B, et al. Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol Psychiatry. 2001;50:260–5. doi: 10.1016/s0006-3223(01)01083-6. [DOI] [PubMed] [Google Scholar]
- 156.Boldrini M, et al. Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology. 2009;34:2376–89. doi: 10.1038/npp.2009.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jessen F, et al. No association of the Val66Met polymorphism of the brain-derived neurotrophic factor with hippocampal volume in major depression. Psychiatr Genet. 2009;19:99–101. doi: 10.1097/YPG.0b013e32832080ce. [DOI] [PubMed] [Google Scholar]
- 158.Schenkel LC, et al. The BDNF Val66Met polymorphism is an independent risk factor for high lethality in suicide attempts of depressed patients. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:940–4. doi: 10.1016/j.pnpbp.2010.04.023. [DOI] [PubMed] [Google Scholar]
- 159.Verhagen M, et al. Meta-analysis of the BDNF Val66Met polymorphism in major depressive disorder: effects of gender and ethnicity. Mol Psychiatry. 2010;15:260–71. doi: 10.1038/mp.2008.109. [DOI] [PubMed] [Google Scholar]
- 160.Hajek T, et al. Reduced hippocampal volumes in healthy carriers of brain-derived neurotrophic factor Val66Met polymorphism: meta-analysis world. J Biol psychiatry. 2012;13:178–87. doi: 10.3109/15622975.2011.580005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zai CC, et al. The brain-derived neurotrophic factor gene in suicidal behaviour: a meta-analysis. Int J Neuropsychopharmacol. 2012;15:1037–42. doi: 10.1017/S1461145711001313. [DOI] [PubMed] [Google Scholar]
- 162.Magariños AM, et al. Effect of brain-derived neurotrophic factor haploinsufficiency on stress-induced remodeling of hippocampal neurons. Hippocampus. 2011;21:253–64. doi: 10.1002/hipo.20744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Saarelainen T, et al. Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J Neurosci. 2003;23:349–57. doi: 10.1523/JNEUROSCI.23-01-00349.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.MacQueen GM, et al. Performance of heterozygous brain-derived neurotrophic factor knockout mice on behavioral analogues of anxiety, nociception, and depression. Behav Neurosci. 2001;115:1145–53. doi: 10.1037//0735-7044.115.5.1145. [DOI] [PubMed] [Google Scholar]
- 165.Chourbaji S, et al. Mice with reduced brain-derived neurotrophic factor expression show decreased choline acetyltransferase activity, but regular brain monoamine levels and unaltered emotional behavior. Brain Res Mol Brain Res. 2004;121:28–36. doi: 10.1016/j.molbrainres.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 166.Chan JP, et al. Examination of behavioral deficits triggered by targeting BDNF in fetal or postnatal brains of mice. Neuroscience. 2006;142:49–58. doi: 10.1016/j.neuroscience.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 167.Monteggia LM, et al. Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol Psychiatry. 2007;61:187–97. doi: 10.1016/j.biopsych.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 168.Goldwater DS, et al. Structural and functional alterations to rat medial prefrontal cortex following chronic restraint stress and recovery. Neuroscience. 2009;164:798–808. doi: 10.1016/j.neuroscience.2009.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Berton O, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–8. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- 170.Joëls M, Baram TZ. The neuro-symphony of stress. Nat Rev Neurosci. 2009;10:459–66. doi: 10.1038/nrn2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gourley SL, Taylor JR. Recapitulation and reversal of a persistent depression-like syndrome in rodents. Current Protocols in Neuroscience. 2009 Oct;Chapter 9(Unit 9.32) doi: 10.1002/0471142301.ns0932s49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Watanabe Y, et al. Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Res. 1992;588:341–5. doi: 10.1016/0006-8993(92)91597-8. [DOI] [PubMed] [Google Scholar]
- 173.Mathur BN, et al. Serotonin induces long-term depression at corticostriatal synapses. J Neurosci. 2011;31:7402–11. doi: 10.1523/JNEUROSCI.6250-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Dölen G, et al. Social reward requires coordinated activity of nucleus accumbens oxytocin and serotonin. Nature. 2013;501:179–84. doi: 10.1038/nature12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Pascoli V, et al. Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature. 2012;481:71–5. doi: 10.1038/nature10709. [DOI] [PubMed] [Google Scholar]
- 176.Chen BT, et al. Cocaine but Not Natural Reward Self-Administration nor Passive Cocaine Infusion Produces Persistent LTP in the VTA. Neuron. 2008;59:288–297. doi: 10.1016/j.neuron.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sanacora G, et al. Lanicemine: a low-trapping NMDA channel blocker produces sustained antidepressant efficacy with minimal psychotomimetic adverse effects. Mol Psychiatry. 2014;19:978–85. doi: 10.1038/mp.2013.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Sousa N, et al. Reorganization of the morphology of hippocampal neurites and synapses after stress-induced damage correlates with behavioral improvement. Neuroscience. 2000;97:253–266. doi: 10.1016/s0306-4522(00)00050-6. [DOI] [PubMed] [Google Scholar]
- 179.Stamatakis AM, Stuber GD. Activation of lateral habenula inputs to the ventral midbrain promotes behavioral avoidance. Nat Neurosci. 2012;15:1105–1107. doi: 10.1038/nn.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Conrad CD, et al. Chronic stress impairs rat spatial memory on the Y maze, and this effect is blocked by tianeptine pretreatment. Behav Neurosci. 1996;110:1321–1334. doi: 10.1037//0735-7044.110.6.1321. [DOI] [PubMed] [Google Scholar]
- 181.Nowak G, et al. Alterations in the N-methyl-D-aspartate (NMDA) receptor complex in the frontal cortex of suicide victims. Brain Res. 1995;675:157–64. doi: 10.1016/0006-8993(95)00057-w. [DOI] [PubMed] [Google Scholar]
- 182.Caspi A, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–9. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- 183.Hosang GM, et al. Interaction between stress and the BDNF Val66Met polymorphism in depression: a systematic review and meta-analysis. BMC Med. 2014;12:7. doi: 10.1186/1741-7015-12-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sen S, et al. Serum brain-derived neurotrophic factor, depression, and antidepressant medications: meta-analyses and implications. Biol Psychiatry. 2008;64:527–32. doi: 10.1016/j.biopsych.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Albert PR, Le François B. Modifying 5-HT1A Receptor Gene Expression as a New Target for Antidepressant Therapy. Front Neurosci. 2010;4:35. doi: 10.3389/fnins.2010.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Deschwanden A, et al. Reduced metabotropic glutamate receptor 5 density in major depression determined by [(11)C]ABP688 PET and postmortem study. Am J Psychiatry. 2011;168:727–34. doi: 10.1176/appi.ajp.2011.09111607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Duric V, et al. Altered expression of synapse and glutamate related genes in postmortem hippocampus of depressed subjects. Int J Neuropsychopharmacol. 2013;16:69–82. doi: 10.1017/S1461145712000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Shrestha S, et al. Serotonin-1A receptors in major depression quantified using PET: controversies, confounds, and recommendations. Neuroimage. 2012;59:3243–51. doi: 10.1016/j.neuroimage.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Murrough JW, et al. Reduced ventral striatal/ventral pallidal serotonin1B receptor binding potential in major depressive disorder. Psychopharmacology. 2011;213:547–53. doi: 10.1007/s00213-010-1881-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Yatham LN, et al. Effect of electroconvulsive therapy on brain 5-HT(2) receptors in major depression. Br J psychiatry. 2010;196:474–9. doi: 10.1192/bjp.bp.109.069567. [DOI] [PubMed] [Google Scholar]
- 191.Smith DF, Jakobsen S. Molecular Neurobiology of Depression: PET Findings on the Elusive Correlation with Symptom Severity. Front psychiatry. 2013;4:8. doi: 10.3389/fpsyt.2013.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Meyer JH, et al. Brain monoamine oxidase A binding in major depressive disorder: relationship to selective serotonin reuptake inhibitor treatment, recovery, and recurrence. Arch Gen Psychiatry. 2009;66:1304–12. doi: 10.1001/archgenpsychiatry.2009.156. [DOI] [PubMed] [Google Scholar]
- 193.Fitzgerald PB, et al. A meta-analytic study of changes in brain activation in depression. Hum Brain Mapp. 2008;29:683–95. doi: 10.1002/hbm.20426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wang L, et al. Interhemispheric functional connectivity and its relationships with clinical characteristics in major depressive disorder: a resting state fMRI study. PLoS One. 2013;8:e60191. doi: 10.1371/journal.pone.0060191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Drevets WC, et al. Functional anatomical correlates of antidepressant drug treatment assessed using PET measures of regional glucose metabolism. Eur Neuropsychopharmacol. 2002;12:527–44. doi: 10.1016/s0924-977x(02)00102-5. [DOI] [PubMed] [Google Scholar]
- 196.Mies GW, et al. Neurophysiological correlates of anhedonia in feedback processing. Front Hum Neurosci. 2013;7:96. doi: 10.3389/fnhum.2013.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Townsend JD, et al. fMRI activation in the amygdala and the orbitofrontal cortex in unmedicated subjects with major depressive disorder. Psychiatry Res. 2010;183:209–17. doi: 10.1016/j.pscychresns.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]