

Abstract
The potential utility of synthetic macrocycles as drugs, particularly against low druggability targets such as protein-protein interactions, has been widely discussed. There is little information, however, to guide the design of macrocycles for good target protein-binding activity or bioavailability. To address this knowledge gap we analyze the binding modes of a representative set of macrocycle-protein complexes. The results, combined with consideration of the physicochemical properties of approved macrocyclic drugs, allow us to propose specific guidelines for the design of synthetic macrocycles libraries possessing structural and physicochemical features likely to favor strong binding to protein targets and also good bioavailability. We additionally provide evidence that large, natural product derived macrocycles can bind to targets that are not druggable by conventional, drug-like compounds, supporting the notion that natural product inspired synthetic macrocycles can expand the number of proteins that are druggable by synthetic small molecules.
Keywords: druglikeness, druggability, ligand efficiency, binding mode, macrocyclic drugs
INTRODUCTION
It has been estimated that, of the ~3000 human proteins with potential utility as drug targets, only a minority are addressable using current approaches for small molecule drug discovery1. There is consequently great interest in developing approaches for identifying therapeutically useful inhibitors for the large number of targets that are not conventionally “druggable”, such as protein-protein interfaces (PPI)2–5. Two decades of experience applying fragment-based lead identification methods has shown that the difficulty with PPI targets is not simply that drug-like compounds that can bind them are rare and require more powerful screening strategies, but rather reflects an intrinsically low propensity to bind small druglike molecules with high affinity4,6,7. Thus, although a number of druglike inhibitors of PPI targets have been reported2,8,9, attention is increasingly turning to exploration of compound classes that fall outside conventional definitions of druglikeness10–13. The search for suitable new chemotypes is complicated by the need to consider not only their potential to bind strongly and specifically to their intended target, but also whether they are likely to possess the properties required for pharmacological activity in vivo, such as good solubility, the ability to penetrate cell membranes to access intracellular targets, and resistance to metabolic degradation.
Synthetic macrocycles have received growing attention for their potential as drugs, due in large part to their proposed utility against low druggability targets14–19. This interest was initially sparked by the observation that natural product-derived drugs often violate conventional definitions of druglikeness, suggesting that they identify distinct regions of chemical space that represent alternative solutions to the challenge of achieving both potent target modulation and good pharmaceutical properties20. The high prevalence of macrocycles among these drugs strongly suggests that a macrocyclic structure helps confer these favorable properties. The notion that cyclization of a ligand can increase its binding affinity by eliminating unproductive conformations is well established14,16,17, but how a macrocyclic structure might help confer drug-like pharmaceutical properties is less clear. Lipinski21 and others20 have speculated that natural products might be substrates for transporter proteins that mediate their active uptake through cell membranes. Moreover, there is evidence that the constrained flexibility of macrocycles also contributes to increased passive permeation through biological membranes22–24 and thus to improved oral bioavailability25. The ability of macrocycles to display “chameleonic” properties by adopting conformations that partially bury hydrophilic or hydrophobic functionality from the solvent environment, for example by formation of intramolecular hydrogen bonds to bury polar groups during permeation through biological membranes, has also been invoked as a potential contributor to solubility and membrane permeability14,22–24. It is also proposed that some cyclic peptides might permeate membranes by pore formation, chelation of divalent metals, or direct binding to specific phospholipids14,24.
The term “macrocycle” encompasses an enormous range of chemical structures, only a tiny fraction of which are likely to have the characteristics required for good pharmacological activity. To aid in the design of MC compound libraries for drug discovery it would be helpful to know what structural and physicochemical features render MCs most likely to bind strongly and specifically to a targeted protein, and to have good solubility, good cell permeability and high metabolic stability. For example, it would be useful to know whether protein binding is better achieved by considering the MC ring merely as a scaffold that presents a set of substituents that engage the protein, or whether a better approach is to design compounds with the expectation that some ring atoms will directly participate in binding. The physicochemical balance of the compounds is also an important question. Conventional metrics for druglikeness are of limited value in addressing this question, as generally these have been constructed by empirically evaluating conventional drug candidates, which are dominated by non-macrocyclic compounds. New, MC-specific design guidelines are thus needed.
One approach to filling this information gap is to analyze how proteins bind pharmacologically active MC natural products, with the goal of identifying common features that might inform the design of synthetic MCs intended to target proteins. Surprisingly, no systematic structural survey of MC binding modes has previously been reported. Here we identify a representative set of MC-protein complexes for which co-crystal structures have been reported, and examine these structures to establish the key characteristics of their binding modes in comparison to complexes with conventional, drug-like ligands. We also develop a refinement to the FTMap algorithm for computational fragment mapping26–28, and use this method to evaluate the number, strength and spatial distribution of druggable subsites (a.k.a. binding energy “hot spots”) within each MC binding site, to assess how MC binding sites differ from sites that bind conventional drugs. Based on our analysis we propose specific guidelines for the design of synthetic MC libraries that possess structural and physicochemical features likely to be favorable for binding to protein targets and also for good bioavailability. We additionally provide evidence that large, natural product derived MCs can bind targets that are not druggable by conventional, drug-like compounds, giving concrete support to the notion that natural product inspired MCs can expand the range of proteins that can be targeted with pharmaceutically relevant synthetic small molecules.
RESULTS
To identify a representative, non-redundant set of protein-MC X-ray co-crystal structures for analysis, we began by identifying all entries in the Protein Data Bank (PDB) that contained natural products or natural product-derived MC ligands with ring sizes of 14 or more atoms. We filtered the structures in this set using several additional criteria, detailed in Methods, to exclude low quality structures and also certain chemotypes, such as cyclic nucleotides, that we considered of low relevance for drug discovery. Most importantly, where the database contained complexes of the same protein bound to close structural analogues of a given MC, or complexes of a given MC bound to paralogs or orthologs of the same protein target, we selected one representative example to avoid biasing our data set with multiple copies of essentially the same complex. This selection process resulted in the surprisingly small number of 22 distinct MC-protein complexes, encompassing 19 distinct MCs and 13 distinct proteins (Supplementary Results, Supplementary Fig. 1). Although this set represents a relatively small number of complexes, it contains all non-redundant examples we could find that met our quality criteria, and thus represents the most complete data set available on which to base such an analysis.
Characteristics of the Test Set
The MCs in the test set range in size from 14–35 ring atoms, with molecular weights ranging from 365–1291 Da. For the subsequent analysis we found it useful to divide these compounds into two size categories: “small macrocycles”, which include the six MCs in the test set with molecular weights <600 Da. – i.e. within or close to the accepted upper limit for conventional drugs21 – and “large macrocycles”, which comprise the 13 MCs with m.w. >600 Da, which thus substantially violate this key descriptor of conventional druglikeness. To determine whether the limited number of non-redundant MCs for which complex structures exist are representative of natural product MCs in general, and of MC drugs in particular, we compared our test set against 3747 natural product MCs15, and also against 44 MC compounds identified as approved drugs, primarily taken from the CHEMBL database29 (Supplementary Table 1). In terms of ring size, our test set is fairly representative of all natural product MCs, and closely mirrors the size distribution found for approved MC drugs even though only three compounds are common to both sets (Supplementary Fig. 2). In terms of lipophilicity (clogP), polar surface area (PSA), number of hydrogen bond donors (HBD) and acceptors (HBA), and number of rotatable bonds (NRB), the large MCs in the test set closely match the oral MC drugs, with an average MW that is ~3-fold higher than that for conventional oral drugs, and a substantially higher number of hydrogen bond donors and acceptors, but with comparable lipophilicity as measured by clogP (Fig 1a–d, Supplementary Table 2). Both the large MCs in the test set and the oral MC drugs additionally have a greater number of rotatable bonds compared to the set of all oral drugs. In contrast, the small MCs in the test set possess structural and physicochemical properties that fall within the ranges observed for conventional oral drugs, aside from having a slightly higher average molecular weight. From these results it is clear that (i) approved MC drugs occupy a region of chemical space that is quite distinct from that defined by conventional druglike ligands, and (ii) the MCs in our test set appear to resemble the subset of MC drugs that can be taken orally.
Figure 1.

Properties of MCs in the test set, compared to MC drugs and to all oral drugs. (a)–(d) Physicochemical properties relevant to druglikeness for the MCs from the test set, in comparison to the 18 oral MC drugs and the 26 non-oral MC drugs from Supplementary Table 1, and also 1193 oral drugs described previously50. The bold horizontal lines indicate the mean value, and the vertical bars show the 10–90% value range. An asterisk (*) indicates that the mean value differs from that for all oral drugs at the P < 0.05 significance level, calculated using classical (non-paired) t-tests after establishing sample normality using the Anderson-Darling test (see Methods). Numerical values for these and other properties are collected in Supplementary Table 2. (e) Conventional drugs and MC drugs occupy distinct regions of chemical space. The spheroids represent approximately the 10th–90th percentile range of values observed for molecular weight, polar surface area and number of rotatable bonds (NRB). The colored “X” symbols show the mean values for each compound class, and the dashed lines show the projection of the mean values on the MW versus NRB axes that represent the floor of the plot. The transparent blue box shows the range of property values encompassed by Lipinski’s “Rule of Five” (MW ≤ 500 Da)21 and Veber’s Rules (PSA ≤ 140 Å2NRB ≤10)48.
Protein-MC binding geometry and extent of interface
The MC-protein binding modes observed among the test set can be described in terms of three broadly distinct interaction geometries. Slightly more than half of the large MCs bind with the MC ring roughly perpendicular to the protein surface, such that one edge of the ring binds along the bottom of an extended groove or cleft on the protein, with substituents interacting with adjacent binding pockets, and the outer edge of the ring exposed to solvent. An example of this “edge-on” binding mode is shown in Figure 2a. The remaining large MCs adopt a different binding geometry in which the MC ring lies face-on to the protein surface, making contacts across a large area (Figure 2b). The MCs that display this face-on binding mode invariably have 1–2 large substituents that interact with substantial adjacent clefts or pockets on the protein. In contrast, most of the small MCs adopt a compact, roughly globular conformation and bind in a cleft or pronounced depression on the protein (Figure 2c).
Figure 2.
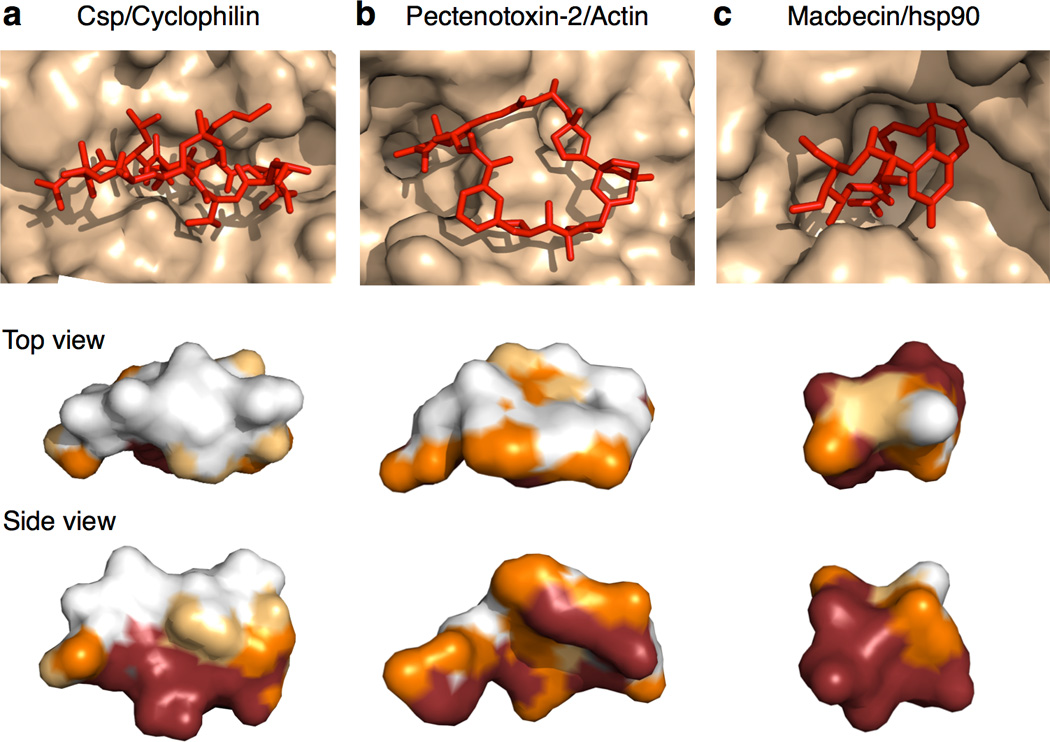
MC Binding Modes. (a) Edge-on binding mode, as exemplified by cyclosporin (Csp) binding to cyclophilin. MCs that bind edge-on typically adopt a conformation in which the ring is flattened and elongated, such that even substituents attached to the solvent-exposed edge of the ring can reach to make extensive contact with the protein. (b) Face-on binding mode, exemplified by the binding of Pectenotoxin-2 to actin. MCs that bind face-on typically project a large substituent into a substantial neighboring pocket or cleft. (c) Compact binding mode observed for most of the small MCs, exemplified by Macbecin bound to hsp90. Upper panel shows the conformation of the ligand (red) when bound to its protein target (wheat). The images below show surface representations of the MC ligands from the upper panels, viewed looking down on the exposed portion of the compound (upper image) and from the side (lower image), with the ligand atoms color-coded according to how much contact they make with the protein (Red ≥ 90% buried, orange = 50–90 % buried, Yellow = 25–50% buried, and White = <25% buried).
The small MCs tended to be almost fully enveloped within their protein binding sites, burying a quite uniform 82 ± 4 % of their total solvent accessible surface area (SASA) upon binding (Figure 3a; Supplementary Table 3), as exemplified by Macbecin in its complex with hsp90 (Figure 2c). In contrast, the large MCs appeared to bury a fairly uniform 630 ± 120 Å2 of surface upon binding, corresponding to an average of 57 ± 8 % of the compounds’ total SASA, with at most a modest dependence on MC size. This value is roughly twice the 300 ± 130 Å2 of SASA buried by a typical drug30, and approaches the 800 ± 200 Å2 of SASA buried on average by each binding partner at a protein-protein interface31.
Figure 3.

Extent and character of the protein-MC binding interface. (a) Plot of buried SASA versus total SASA. The dotted line represents the line of identity, corresponding to 100% of MC SASA buried in the complex. Small MCs (triangles) bury ~80% of their SASA upon binding, with the size of the binding interface being roughly proportional to the surface area of the MC ligand. The large MCs (circles) bury a roughly constant 630 ± 150 Å2 of SASA (dashed line), with only a small dependence on compound size. The solid curve is an arbitrary interpolation of the data. (b) Comparison of the fraction of MC atoms that make direct contact with the protein (defined as atoms burying >5 Å2 of MC SASA) that are polar versus nonpolar, versus the corresponding ratio for all MC atoms. (c) Example showing how MC heavy atoms can be categorized by region into ring atoms (black), substituent atoms (blue) and “peripheral” atoms (green). (d) Contributions to total MC buried surface by region. (e) Percentage of atoms from each region that make direct contact with the protein (defined as atoms burying >5 Å2 of MC SASA). (f) Average polar/nonpolar ratio for the atoms from each MC region that make contact with the protein. Error bars are standard deviations; an asterisk (*) indicates that the specified difference is statistically significant using the Mann-Whitney U (rank) test (see Methods).
Physicochemical Characteristics of Binding Regions of MCs
It has been proposed that the advantageous pharmacological properties seen for certain large natural product MCs arises because these compounds possess distinct structural domains whereby, for example, a predominantly hydrophobic target binding region is physicochemically balanced by the presence of polar functionality on other parts of the molecule14,15. To test this proposal we analyzed the polar/non-polar balance of the MC atoms that make contact with their protein targets, as defined by their burial of >5 Å2 of SASA in the complex. For the large MCs in our test set, on average 73 ± 9 % of the MC atoms that make contact with the protein are nonpolar (i.e. C, S or Cl) and 27 % are polar (N or O) (Fig. 3b). This non-polar/polar ratio is essentially identical to the 71%/29% (±7%) ratio we found for the MC atoms as a whole. Similar results are obtained if the ratio of non-polar/polar contacts is instead quantified in terms of MC atoms within 4.5 Å of the protein, or total buried surface area. Thus, for the complexes with large MCs for which X-ray structures exist, the physicochemical nature of the regions that participate directly in binding is similar to that for the compound as a whole.
Participation of Different MC Regions in Target Binding
To gain insight into the roles that different regions within the MC structure play in protein binding, we categorized MC heavy atoms into three different regional types (Fig. 3c). These are; (i) “ring atoms”, that comprise the contiguous, sigma-bonded ring by which the MC size is defined; (ii) “peripheral atoms”, which are small groups such as methyl, carbonyl, hydroxyl, and halogens that consist of a single heavy atom directly appended to the ring; and (iii) “substituent atoms”, comprising larger (i.e. two or more heavy atoms) structures connected to the ring. The large MCs in our test set, and also the oral MC drugs, contain an average of 4–5 of these larger substituents, together encompassing roughly half of the heavy atoms in the MC (Supplementary Table 4). Approximately 38% of the structure is made up of ring atoms, on average, while peripheral groups make up the remaining ~15%. Analyzing the extent to which different regions of MC structure participate in binding we found that, for the large MCs, ~60% of MC atoms that make direct contact with the protein are contributed by substituents, with ring atoms contributing only 15% and peripheral atoms – which are the least numerous regional atom type – contributing a surprisingly high 22% of the total contact atoms (Figure 3d). Normalizing for the different abundances of the regional atom types (Figure 3e) shows that, although peripheral atoms make up only a small fraction of the structure of a given MC, where present they are highly likely to directly participate in binding to the target. Specifically, among the large MCs 72 ± 17 % of peripheral atoms make direct contact with the protein, whereas only about half of substituent atoms do so, and about one quarter of ring atoms (Supplementary Table 4).
Examining the physicochemical composition of contact atoms from different sites on the MC ligands revealed the surprising finding that, although the physicochemical composition of contact atoms overall matched that of the whole MC molecule, significant biases exist for atoms in certain regions of the MC structure. Figure 3f shows the percentage of polar versus nonpolar contact atoms, enumerated separately for substituent, peripheral and ring atoms. It can be seen that the balance of polar versus nonpolar contact atoms in the substituents tends to match the composition of the compound as a whole. Contacts involving ring atoms are largely nonpolar, however. Peripheral atoms, in contrast, are much more likely to be polar compared to other regions of MC structure, and those peripheral atoms that directly contact the protein similarly are predominantly polar.
We additionally evaluated the occurrence and locations of intramolecular hydrogen bonds in the bound MCs. We found that in 12 of the 22 protein-MC complexes the bound MC showed no intramolecular hydrogen bonds, and all but one of the remaining complexes showed only one or two such interactions (Supplementary Table 5). The exception was argadin bound to chitinase, for which the 10 HBD and 11 HBA present on the MC engage in 5 intramolecular hydrogen bonds. The overall picture, however, is that among MCs in our test set only a small minority of HBD or HBA are internally complemented in the bound conformations. This finding suggests that if these compounds passively permeate cell membranes they likely adopt alternative conformations to do so.
Characteristics of MC binding sites on proteins
In addition to understanding how natural product MCs bind to their targets, it is also of interest to know whether the sites on proteins that bind MCs differ in measurable ways from sites that bind conventional drug-like small ligands (Fig. 4). Identifying distinctive properties of MC binding sites might give insights into how to design synthetic MCs to be complementary to such sites, and might additionally provide a basis for identifying which proteins are most suitable to target with natural product-inspired MCs. In addition to global binding site characteristics such as size, shape and physicochemical composition32, it has been shown that the ligand-binding properties of protein surfaces are governed by the number, strength and spatial distribution of binding energy “hot spots”33–35. Hot spots are local surface regions whose shape and physicochemical character gives them the potential to develop substantial binding energy through interaction with atoms from a binding partner.
Figure 4.
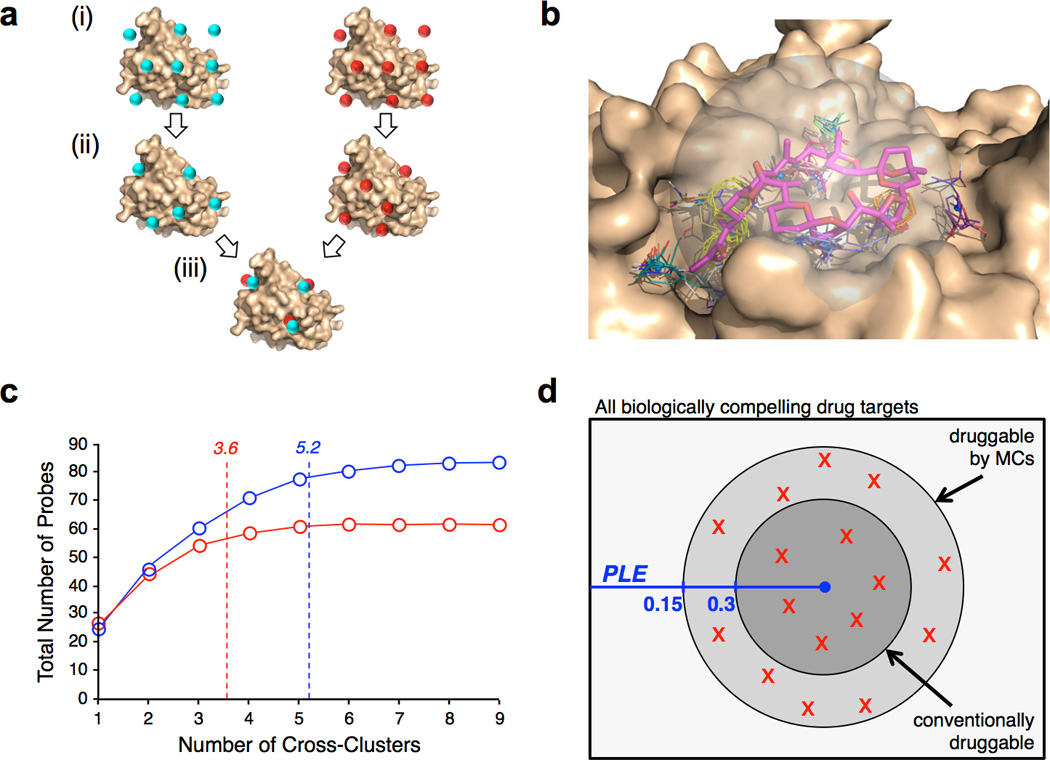
FTMap analysis of MC binding sites. (a) FTMap involves (i) placing probe molecules (represented by cyan or red spheres) on a dense grid around the protein, (ii) energy minimization and clustering to identify regions on the protein that interact most favorably with each probe type, and (iii) overlaying the results across all probe types to define “Cross-Clusters” (CCs) that identify binding energy hot spots26(b) Representative result of FTMap analysis for Pectenotoxin-2 (magenta) bound to actin (wheat). CCs are shown as colored sticks. (c) Number of CCs occupied by ligands from the MC test set (blue) or the drug-like ligand comparator set (red). The plot shows the total number of probes the ligands overlap with, starting from the most highly-populated of the occupied CCs (ranked number 1) to the least populated (highest CC number), averaged over the entire set of complexes. The average number of CCs occupied is 5.2 for the MC ligands versus 3.6 for the druglike-ligands (p < 0.01; see Table 1). (d) Venn diagram illustrating the proposal that MCs can bind conventionally druggable targets, and also additional targets whose Potential Ligand Efficiency (PLE) falls below 0.3 kcal.mol−1/HA. The distribution of druggabilities observed for the 16 large MC binding sites assessed using FTMap are shown by red “X” symbols.
To analyze the MC binding sites in the test set we developed a modification to the well-validated FTMap method for computational fragment mapping (Figure 4a). Validation of the FTMap algorithm across a large number of different systems has shown that the method can reliably identify the locations of binding energy hot spots that are exploited by known ligands26–28,36–39. It has also been shown that the number of probe clusters in a given CC provides a relative measure of the energetic importance of the hot spot for binding to small molecule ligands40 or to other proteins39,41. For the current work we constrained the initial mapping to a sphere of radius 10 Å centered on the ligand binding site (Figure 4b). This constraint restricted the analysis to a more or less uniform area of the protein surface, large enough to encompass the regions that accommodate even the largest MCs in our set, so that the number and strengths of the CCs could be compared between different proteins. For comparison to the MC-protein complexes, we assembled a diverse “comparator drug set” of 24 X-ray co-crystal structures of conventional drugs or drug-like ligands bound to their protein targets (Supplementary Table 6). When computationally mapping the comparator drug set the initial probe map was restricted to the same 10 Å sphere used for the MC complexes, so that the number of binding energy hot spots identified and the density of probes at each hot spot could be directly compared. The FTMap analysis was done using the protein from each complex structure after removing the ligand atoms, to characterize the binding site without reference to which portions of it are occupied by a particular ligand.
Figure 4b shows a typical result from FTMap analysis of a MC binding site, for the Pectenotoxin-2 binding site on actin. Multiple binding energy hot spots were identified. A subset of these – including the top ranking CCs that are predicted to be energetically most significant – trace out the regions of the protein surface that accommodate the MC ligand. Other CCs occur in nearby regions of the protein surface, indicating that these regions have the potential to interact favorably with ligands, but are not exploited by the particular ligand present in the complex. The results of analysis of all the MC binding sites, together with the comparator set of druglike ligand binding sites, are collected in Table 1. They show that the binding sites that accommodate the large MCs are about 15% larger than those for conventional drug-like ligands (p < 0.05), as measured by the distance between the two CCs that define the furthest extent of the site. This difference is derived from the locations of the binding energy hot spots alone, without regard to whether these hot spots are occupied by the ligand in the experimentally observed complex, and thus reflect the intrinsic properties of the binding sites. The MC binding sites and conventional drug binding sites on average contained the same number of hot spots, but in the large MC binding sites these are spaced significantly further apart (P < 0.01). In particular, the two top ranking hot spots, which are most important for binding26,28, are on average separated by 10.2 Å for the large MC binding sites versus only 7.5 Å for the drug-like comparator set (p < 0.01). The top two binding hot spots were not always widely separated in the large MC binding sites, but in most instances were >10 Å apart and in some cases were separated by as much as 18 Å. In contrast, for the binding sites for the drug-like ligands, in over 80% of cases the two most important CCs were within 9 Å of each other. In all of the above measures, the binding sites for the small MCs were essentially indistinguishable from sites that bind conventional drug-like ligands.
Table 1.
Binding site statistics for macrocycles versus conventional druglike ligandsa
| Binding Site Property | Macrocycles | Comparator Drug Set |
||
|---|---|---|---|---|
| All | Small (mw <600) |
Large (mw >600) |
||
| Longest dimension | 15.9 Å (P = 0.13) |
14.4 Å (P = 0.90) |
16.9 Å (P = 0.044) |
14.3 Å |
| Number of Hot Spots | 7.9 (P = 0.68) |
7.9 (P = 0.90) |
7.9 (P = 0.98) |
8.2 |
| Occupied Hot Spots | 5.1 (64%) (P= 0.0008) |
5.0 (60%) (P = 0.011) |
5.2 (66%) (P = 0.008) |
3.6 (44%) |
|
Average Separation of Hot Spots |
8.9 Å (P = 0.07) |
8.3 Å (P = 0.90) |
9.3 Å (P = 0.006) |
8.1 Å |
|
Separation of Top Two Hot Spots |
9.5 Å (P = 0.013) |
7.8 Å (P = 0.40) |
10.6 Å (P = 0.004) |
7.5 Å |
| Average CC Population | 15.7 (P = 0.81) |
16.1 (P = 0.90) |
15.4 (P = 0.85) |
15.7 |
| Top CC population | 26.5 (P = 0.21) |
26.3 (P = 0.50) |
26.7 (P = 0.23) |
28.5 |
| Top two CC population | 49.1 (P = 0.17) |
49.3 (P = 0.12) |
48.9 (P = 0.51) |
50.9 |
Values shown are mean values. P values are based on the Mann-Whitney U test, and indicate whether the mean for the set or subset of MCs is significantly different from the mean for the comparator drug set.
In addition to differences in the binding sites themselves, the MC and non-MC ligands also differed in how they exploited binding energy hot spots within their binding sites. Hot spots were designated as being occupied if both (i) the center of mass of the probes in a given CC fell within 2 Å of any non-hydrogen atom of the ligand, and (ii) at least 25% of the CC probe atoms were within 1.25 Å of the MC atoms. On average, the drug-like ligands in our comparator set occupied 3.6 CCs, representing 45% of the hot spots present at the binding site. In contrast, the large MCs utilized an average of 5.2 distinct hot spots, comprising some 66% of those available (Fig. 4c). This difference in hot spot utilization was statistically significant (P < 0.01; Table 1). Therefore, in addition to being somewhat larger than sites that bind conventional druglike ligands, the MC binding sites were also more fully occupied by their ligands. These two factors together lead to the very large protein-ligand contact area for the large MCs that was described above (Fig. 3a, Supplementary Table 3). Analyzing the degree to which different regions of the MC structure engage with hot spots in the binding site we found that ring, peripheral and substituent atoms that make contact with the protein were equally likely to interact at a hot spot versus a non hot spot region (Supplementary Fig. 4). This result suggests that contacts involving any portion of the MC structure can contribute to the generation of binding energy. Interestingly, the finding that MCs interact with a greater number of hot spots than conventional drug-like ligands was also seen for the small MCs, emphasizing the different binding modes of small MCs and drug-like ligands despite their rather similar dimensions and physicochemical properties.
Role of Target Site in Determining MC Interaction Mode
Comparing the binding of structurally different MCs that bind at the same site provides insight into the extent to which binding site structure dictates binding mode. The binding of small MC ligands such as geldanamycin and radicicol to the nucleotide binding site of HSP90 has been described42. The commonalities in the binding modes of Rapamycin and FK506 for FKBP have also been extensively discussed14, though this ligand pair represents a special case in that the chemical structures of the regions that make contact with the protein are essentially identical. Among the complexes in the test set, however, are several examples of MC pairs with quite distinct structures that bind at the same site on a common target protein.
Reidispongiolide A (RspA) and Kabiramide C (KabC) each bind to the same surface site on actin. Figure 5a shows that the macrocyclic portions of these two natural products employ different strategies to achieve binding to the target, finding alternative ways to position their large and structurally homologous substituents so they can exploit key binding hot spots in an adjacent cleft. Figure 5b shows that the 15-member MC argadin and the 17-member MC argifin bind to a common surface site on chitinase, but again exploit this site in different ways. The two ligands occupy roughly the same space and exploit many of the same hot spots, but the geometries with which the ring and substituent portions of these ligands interact with the site are quite different. The MC rings of the compounds do not significantly overlap, and the largest substituents project in opposite directions, leading to an almost “head to tail” relationship between the binding geometries. As a third example, Cyclosporin (Csp) and Sanglifehrin A (SfA) display an “edge on” binding mode with cyclophilin (Figure 5c), with the MC rings themselves occupying largely the same region of space and interacting with mostly the same binding hot spots. However, the two ligands exploit different hot spots with some of their substituents. Overall, these examples show that a given binding site can interact with different MC structures by employing quite distinct binding mechanisms.
Figure 5.
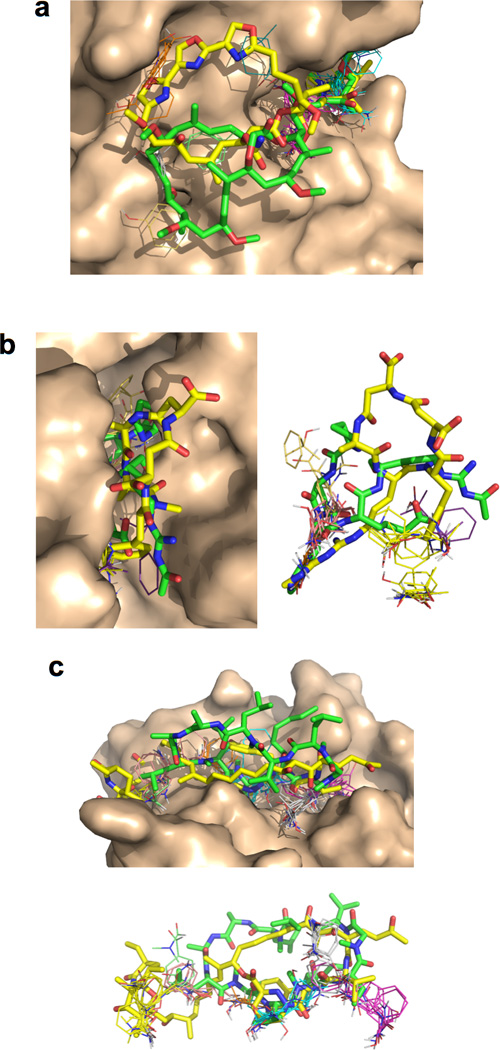
Comparison of binding modes for distinct MCs that bind at a common target site. (a) Reidispongiolide A (yellow) and Kabiramide C (green) bound to actin (wheat). The locations of the FTMap CCs are shown as colored sticks. Both compounds utilize the top two ranking hot spots, which line the site that accommodates their homologous large substituents, but the 26-member ring of RspA and the 25-atom ring of KabC exploit different sets of hot spots in their face-on interaction with the adjacent protein surface. (b) Argadin (green) and Argifin (yellow) bound to chitinase (wheat). Right panel is a superposition of the two ligands with the protein removed, to more clearly show the “head-to-tail” relationship between their binding modes, and the overlap with the FTMap CCs. (c) Cyclosporin A (green) and Sangliferhrin A (yellow) bound to cyclophilin (wheat). Lower panel is a superposition of the two ligands with the protein removed. The MC rings of these compounds bind edge-on, occupying largely similar sets of hot spots along the bottom of the binding cleft. But the large substituent of Sanglifehrin A reaches into a strong hot spot that is not exploited by Cyclosporin A, while an isobutyl substituent on the larger Cyclosporin A ring instead interacts with other hot spots not used by Sangliferhrin A.
Druggability of MC binding sites
Finally, we addressed the question of whether the MC binding sites in the test set can be considered druggable with respect to conventional small molecule ligands, or whether they represent conventionally undruggable sites that are uniquely targeted by MCs. The latter result would provide concrete support for the hypothesis that using natural product-inspired MCs might expand the range of proteins that can be targeted with pharmaceutically relevant synthetic molecules. It is well established that the hit rate achieved in an experimental fragment screen is a good predictor of druggability6,43, and that application of the same principle computationally using FTMap can distinguish druggable from non druggable targets with high reliability26,28,35,44. Specifically, published studies show that when a protein is globally mapped (i.e. with no constraints on where probes can lie on the protein surface) using the standard FTMap 16 probe library, druggable binding sites are characterized by the presence of a strong (>16 probe clusters) CC plus at least one other CC containing 5 or more probe clusters, located within 7 Å of the first26,28. We therefore used FTMap to globally map the 16 distinct MC binding proteins represented in the test set, to evaluate which sites would be rated as druggable using these previously established criteria. As was the case in the published FTMap druggability analyses, the protein targets were mapped using separate crystal structures of the unbound proteins, rather than using the protein structures from the bound complexes. The analysis showed that six of the MC binding sites were rated as druggable and ten as not druggable (Supplementary Table 7). Thus, a substantial proportion of the MC binding sites in the test set appear to be poor prospects to bind conventional drug-like ligands with high affinity.
DISCUSSION
The notion that MCs might provide a means to achieve pharmaceutically useful inhibitors of PPIs and other traditionally difficult targets rests principally upon two ideas: (i) being larger than conventional drugs, and also conformationally constrained, MCs can potentially make more extensive contact with the protein target, and can do so without excessive entropic penalty16; and (ii) a macrocyclic structure can promote good pharmaceutical properties despite a high compound molecular weight and other significant deviations from conventional definitions of druglikeness14,17,19,23,24,45. The validity of these ideas is supported by the existence of a modest number of PPI-targeting MC drugs that violate conventional druglikeness guidelines14,19,23. However, we currently lack any framework for assessing the utility of MCs as a general approach to inhibiting low druggability targets, or for predicting which types of macrocyclic molecular structures might be good for this purpose.
The premise that the larger size of MCs renders them good prospects to bind and inhibit PPI targets can be quantitatively framed in terms of Ligand Efficiency (LE), a concept in which the binding affinity of a ligand is normalized for differences in ligand size to provide a measure of how efficiently the ligand generates binding energy per heavy atom (HA) of its structure46. To achieve a pharmacologically relevant binding affinity of KD ≤10 nM, a ligand with a molecular weight within the Rule of Five threshold of 500 Da. – which on average corresponds to ~38 HA – must generate at least –RTln10−8/38 = ~0.3 kcal.mol−1 of binding energy per HA of ligand structure46,47. Supplementary Table 2 shows that oral MC drugs have an average MW of 920 Da., and range up to well over 1000 Da., corresponding to 75 or more heavy atoms, and that some non-oral MC drugs are even larger. Thus, for a MC to achieve a binding affinity of 10 nM would require only −RTln (10−8)/75 = 0.15 kcal.mol−1/HA, or even less for larger MCs. Thus, MCs of a size that demonstrably can have good pharmaceutical properties are potentially able to target proteins that are less than half as druggable as conventional drug targets, where druggability is defined as the potential of a site to generate binding energy with a small molecule ligand. Indeed, developing an earlier suggestion3 we propose that Potential Ligand Efficiency (PLE) represents a quantitative way to think about the druggability of PPI interfaces and other challenging targets that can give insight into the level of difficulty presented, and also into the prospects for compounds of a given size to achieve strong binding.
The above analysis suggests that natural product inspired MCs do indeed have broad potential to bind strongly to low druggability targets, provided that they can interact with a geometry that results in a substantially increased contact area compared to conventional drug-like ligands. Our analysis shows that this condition is invariably satisfied by the large MCs in the test set, whose average contact area is more than twice that seen for conventional drug-like ligands. Consistent with this idea, our analysis of the druggability of the MC binding sites in the test set revealed that the majority do not appear to be druggable as assessed by the FTMap benchmarks previously established for other target classes26,28,35,44. This result suggests that MCs can bind to target sites capable of generating as little as 0.1–0.15 kcal.mol−1 of binding energy per HA of ligand structure, which would be very far from druggable by conventional compounds. Overall, our results suggest that use of appropriately designed MCs can substantially expand the range of druggable targets, as illustrated in Figure 4d. The historical success of macrocyclic natural products and their derivatives as drugs14,19,20,23 suggests that such targets present a significant opportunity for drug discovery.
Exploiting the opportunity provided by natural product-inspired MCs would be greatly enabled by knowledge of what structural features of such compounds are likely to promote strong binding to protein targets and, separately, good pharmaceutical properties. To meet the latter need, we might envision a set of guidelines analogous to Lipinski’s Rule of Five21, Veber’s Rules48, or similar druglikeness guidelines8, but based on the behavior of macrocyclic chemotypes. Based on the value ranges observed for the molecular properties encompassed in the Rule of Five and Veber’s Rules for the 18 orally available MC drugs (Supplementary Table 2), we tentatively propose a modified set of property ranges that we believe is more appropriate for the design of synthetic MC libraries for use in the discovery of oral drugs (Table 2). The set of compounds on which these ranges are based is necessarily very small, though it includes all known examples of orally available macrocyclic drugs. Nonetheless, the results show that the oral MC drugs display quite consistent properties that in many cases are clearly distinct from those observed for conventional drugs.
Table 2.
Proposed physicochemical guidelines for the design of synthetic large MC libraries for use in the discovery of oral drugs
An enormous variety of MC structures might be envisioned that conform to the guidelines in Table 2. We therefore attempted to use our analysis of MC binding modes to devise more specific design guidelines for the kinds of MC structures likely to bind to proteins, and therefore likely to have useful pharmacological as well as pharmaceutical properties. The properties of the large MCs contained in our test set of protein-MC complexes coincide closely with the oral MC drugs, supporting the notion that analysis of the binding modes of the test set might return information relevant to the design of orally available MC compounds. This analysis led us to identify a number of structural features in the MCs that are common to these pharmacologically active MCs.
The oral MC drugs and the large MCs in our test set typically contain 1–2 large substituents, often totaling 20–30 heavy atoms or more, plus several much smaller substituents such as acetyl, methoxy or isobutyl groups. In natural product MCs the vast majority of substituent atoms, including small peripheral groups attached to ring atoms, participate directly in contact with the protein. Thus, structural diversity in these regions is an important consideration when designing MC libraries for drug discovery. Although only about one in three ring atoms contacts the protein, collectively these contribute ~25% of the contact area. Moreover, ring, peripheral and substituent atoms that contact the protein are equally likely to bind at a hot spot. Therefore, all regions of the MC must be considered potentially important for achieving good binding complementarity.
The disproportionate role in binding played by single HA substituents attached to the ring suggests that achieving an appropriate number and diversity of such peripheral groups is particularly important for good protein binding activity. Mimicking natural product MCs by including multiple polar atoms in these peripheral positions also provides a means to ensure adequate PSA, which is critical for good aqueous solubility and thus for pharmaceutical utility49.
The regions of the MC that interact with the protein target, and the MC structure as a whole, have a drug-like physicochemical balance of one polar (O or N) atom per 2–3 nonpolar (C, S, Cl) atoms. Consequently, the clogP for oral MC drugs is similar to that for conventional drugs, while absolute PSA scales with molecular weight and is much higher.
Whether a large MC adopts an edge-on or a face-on binding mode appears to be dictated by the topology of the protein surface. However, different MC structures can interact at a given binding site in otherwise quite distinct ways, by exploiting different sub-sets of the available binding energy hot spots. The face-on binders often bind such that a large – in some cases almost drug-sized – substituent can access a substantial neighboring pocket or cleft. The edge-on binders typically display a bound conformation in which the ring is flattened and elongated, such that even substituents attached to the solvent-exposed edge of the ring can reach to make extensive contact with the protein (see, for example Figure 5c). Thus, even for edge-on binders the substituents are typically not restricted to a single edge of the MC ring. These findings argue that a diverse, general purpose MC library with large and small substituents distributed around the ring might have utility across a wide range of different protein binding site topologies.
A common feature among the oral MC drugs and the MC test set is a significant degree of unsaturation in the ring, due to alkene or amide bonds or to cross-links or externally fused small rings. Unlike rotatable bonds in general, single bonds within an MC ring do not substantially mitigate against oral availability48. Nevertheless, the relatively high degree of unsaturation observed for these MCs suggests that substantial rigidification of the MC ring is a feature of pharmaceutically relevant MC chemotypes.
The MC structural features described above are summarized in Table 3, which, together with the property ranges in Table 2, we tentatively propose as a set of design guidelines for synthetic MCs intended as pharmaceutically useful binders or inhibitors of protein drug targets. These guidelines must be considered as provisional, requiring validation and further refinement based on prospective experimental tests. Nonetheless, the available evidence supports the notion that compounds conforming to these properties represent a useful class for the discovery of pharmacologically active synthetic MCs
Table 3.
Proposed structural guidelines for the design of synthetic large MC libraries for use in the discovery of oral drugs
| Property | Observed Rangea |
|---|---|
| Ring Size (R) | 14–38 |
| Number of Substituents | 4.4 (3–8) |
| Large substituents (≥5 HA)b | 1.9 (1–3) |
| Small Substituents (2–4 HA)b | 2.4 (1–6) |
| Proportion of HA that are in Substituents | 47% (40–59%) |
|
Number of
Peripheral Groupsc |
5–12 |
| Polar/Nonpolar balance, Substituents | ~30/70 |
|
Polar/Nonpolar balance,
Peripheral Groups |
~60/40 |
| Degrees of Unsaturation in Ring | ~0.4R – 4 (±3) |
| N:O ratio | 0.25:1 (0–0.4:1) |
| Chiral Centers | 15 (9–18) |
Mean (10–90% range).
HA = Heavy (i.e. non-hydrogen) atoms.
Peripheral groups are groups connected to the MC ring that contain only a single HA (see text).
Finally, our analysis also provides clues as to what features on a protein target might make it suitable for inhibition by a macrocyclic ligand. Specifically, the results of the FTMap analysis of the MC binding sites in our test set suggests that such sites typically contain at least 5 relatively strong binding energy hot spots, 1–2 more than is required for binding a conventional drug. These hot spots can be substantially further apart than is acceptable for a conventionally druggable binding site. In particular, the two strongest hot spots – which for conventional drug binding must be close together – for MCs can be separated by as much as 18 Å. These criteria potentially provide a means to identify protein targets that are particularly well suited to bind MC ligands, based only on computational analysis of the structures of the unbound proteins.
METHODS
Selection of the MC-binding protein set
Selection began by identifying Protein Data Bank (PDB) entries containing natural product MC compounds (or a close derivative), excluding compounds that were discovered via linking or tethering of acyclic leads. This search was done over a period of several months, and included PDB entries deposited up to December 2012. The resulting complexes were then subject to a series of selection criteria in order to ensure set consistency and remove redundancies. MC were included only if they contained a minimum of 14 atoms in the main ring and were documented to function as a protein inhibitor (no substrates or coordinated metal cofactors). “Ring atoms” were defined as atoms in the continuous, sigma-bonded chain of atoms that defines the macrocyclic scaffold. In cases of fused ring or multi-cyclic systems, where two or more sigma bonds are shared between the rings, the system was regarded as a fused ring – i.e. the macrocyclic chain encompass both rings, and thus comprises the longest continuous cyclic sigma-bonded chain of atoms. Exceptions to this latter rule were if the smaller fused ring was aromatic, in which case it was counted along the shorter continuous side of sigma-bonded atoms. Also, where the fused rings share only a single sigma bond, then the macrocyclic chain was considered to include only the larger of the two rings, with the smaller fused ring considered as a substituent.
To minimize redundancy, if a compound had a series of closely related analogs bound to the same protein, only one was selected. For cases where identical MCs bound orthologs of a protein, one was selected, prioritizing human when possible. Structures of the same MC complexed to distinct proteins, or the same protein bound to different MC ligands, were not considered to be duplicates, because a different binding partner requires a distinct binding mode. Complexes with a mutation in the binding site were excluded, as well as those with a binding site conformation known to be dependent on crystal contacts. The PDB codes for the 22 complexes in the final test set are as follows: cyclosporin/cyclophilin, 1cwa; sanglifehrin/cyclophilin, 1ynd; rapamycin/FKBP, 2dg3; FK506/FKBP, 2fke; scyptolin/pan-elastase, 1okx; ge2270a/ef-tu, 1d8t; nodularin R/PP1a, 3e7a; arylomycin/signal peptidase, 1t7d; argadin/chitinase, 1waw; argifin/chitinase, 1wb0; pectenotoxin/ actin, 2q0r; kabiramide C/actin, 1qz5; reidispongiolide A/actin, 2asm; latrunculin B/actin, 2q0u; sorephan A/acetylCoA carboxylase, 3gid; geldanamycin/hsp90 (hu), 1yet; pochoxime A/hsp90 (hu), 3inw; macbecin/hsp90 (yeast), 2vwc; radicicol/hsp90 (yeast), 1bgq; radicicol/topoisomerase VI-B, 2hkj; radiciol/PDK3, 2q8i; radicicol/PhoQ, 3cgy.
Defining Protein-MC Contact Regions
The interaction of the ligands with their respective proteins was assessed in two ways: (1) solvent accessible surface area (SASA) analysis and (2) determination of contact atoms. Changes in SASA were determined both for the ligand binding as a whole and for the individual ligand atoms using methods based on Lee and Richards51. For the individual ligand atom analysis, only those atoms burying more than 5 Å2 SASA upon binding were considered in order to exclude internal atoms that were unlikely to be able to interact with the protein. Contact atoms were defined as heavy atoms of the ligand within a 4.5 Å radius of a heavy atom of the protein.
Characterizing MC binding sites on proteins
To investigate the binding sites of proteins, a slightly modified version of the FTMap26 algorithm was used, to normalize the results and thereby allow direct comparison between different protein types. The modification constrained the initial placement of the probes to a 10 Å radius sphere around the center of mass of the ligand, so that mapping of each protein involved distribution of the probes across a similar extent of protein surface. The 10 Å distance constraint restricted only the starting positions for the probes, which through subsequent energy minimization could relax to locations slightly outside the initial placement perimeter.
The comparator set of complexes containing conventional druglike ligands was created from proteins found in the Astex Diverse Set 52 and the EMBL-EBI index of approved drugs (http://www.ebi.ac.uk/thornton-srv/databases/drugport/). Inhibitors with IC50 values of ≤ 1 µM or KD ≤ 100 nM were selected from these established sets. Only one protein from each family was used, to minimize bias. The complexes included in the comparator set are listed in Supplementary Table 6.
The analysis of which hot spots are occupied by the MC ligand was also done using the results of the constrained mapping. A consensus cluster, identifying a hot spot, was considered “occupied” by the ligand if any heavy atom of the ligand was within 2 Å of the geometric center of the cluster. For borderline cases an additional criterion was used, requiring a minimum of 25% of all probe atoms in the consensus cluster to be within 1.25 Å of a heavy atom of the ligand.
For the analysis of druggability, global (unconstrained) mapping of the ligand-free structures of MC binding proteins was performed using the FTMap algorithm26, implemented as the FTMap server (http://ftmap.bu.edu/). Based on previously published benchmarks26,53,54, a site was considered druggable only if it contained a consensus cluster containing at least 16 probe clusters, plus at least one other consensus cluster located within a 7 Å radius and containing 5 or more probe clusters.
Statistical Analysis
Table 1. The non-parametric Mann-Whitney U (rank) test55 was used to compare various properties of macrocycles to the same properties calculated for the comparator drug set. The P value shown is the probability that the two populations do not significantly differ.
Table S2. Here the statistical problem is to compare macrocycle properties to the same properties observed for all oral drugs. Since the latter sample is much larger than the others, non-parametric (rank-based) tests are not effective, and hence we had to use classical non-paired t-tests based on the sample estimates of mean and standard deviation. The test assumes that each of the two populations being compared follows a normal distribution. We used the Anderson-Darling test56 to check the normality based on the samples. Since the test requires at least 7 samples, no test was applied to the small macrocycles. We found that each property of the large macrocycles followed a normal distribution, as the hypothesis of normality cannot be rejected at the P=0.01 level. Similarly, the properties of the not orally available MC drugs followed normal distributions. For all oral drugs we had only the statistics and not the detailed data. However, this sample is so large that significant deviations from normality are very unlikely. The only data where deviations from normality occurs are the orally available MC drugs, although in most cases the hypothesis of normality still cannot be rejected at the P=0.001 level. In spite of this result, the P values shown in Table S2, comparing macrocycle properties to the properties of all oral drugs, are based on the t-test, since we do not have other viable options, and the t-test results are generally not very sensitive to deviations from normality. Table 2 shows the two-tailed P values obtained by unpaired t-tests assuming unequal sample sizes and unequal variance s. In most cases the differences are significant at very low P, thus it is unlikely the deviations from normality would change the outcome.
Table S4. Since we compare samples with equal sizes, we again used the Mann-Whitney U (rank) test to calculate the P values shown in the table.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by NIH grants GM094551 to AW, SV and JAP, GM064700 to S.V., and NIH diversity supplement GM094551-01S1 to EAV.
Footnotes
AUTHOR CONTRIBUTIONS: AW, SV and DK conceived of and directed the study; EAV performed the calculations and analysis, with advice from DB; SC under the supervision of JAP analyzed the physicochemical properties of the MC drugs; AW and EAV wrote the manuscript with input from SV, DK and SC.
COMPETING FINANCIAL INTERESTS: The authors declare no competing financial interests.
ADDITIONAL INFORMATION: Supplementary information is available in the online version of the paper. Reprints and permissions information is available online at http//:www.nature.com/reprints/index.html. Correspondence and requests for reprints should be directed to AW, SV or DK.
References
- 1.Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 2.Berg T. Small-molecule inhibitors of protein-protein interactions. Curr Opin Drug Discov Devel. 2008;11:666–674. [PubMed] [Google Scholar]
- 3.Buchwald P. Small-molecule protein-protein interaction inhibitors: therapeutic potential in light of molecular size, chemical space, and ligand binding efficiency considerations. IUBMB life. 2010;62:724–731. doi: 10.1002/iub.383. [DOI] [PubMed] [Google Scholar]
- 4.Whitty A, Kumaravel G. Between a rock and a hard place? Nat Chem Biol. 2006;2:112–118. doi: 10.1038/nchembio0306-112. [DOI] [PubMed] [Google Scholar]
- 5.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 6.Hajduk PJ, Greer J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat Rev Drug Discov. 2007;6:211–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 7.Scott DE, et al. Using a Fragment-Based Approach To Target Protein-Protein Interactions. Chembiochem : a European journal of chemical biology. 2013;14:332–342. doi: 10.1002/cbic.201200521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morelli X, Bourgeas R, Roche P. Chemical and structural lessons from recent successes in protein-protein interaction inhibition (2P2I) Curr Opin Chem Biol. 2011;15:475–481. doi: 10.1016/j.cbpa.2011.05.024. [DOI] [PubMed] [Google Scholar]
- 9.Basse MJ, et al. 2P2Idb: a structural database dedicated to orthosteric modulation of protein-protein interactions. Nucleic acids research. 2013;41:D824–827. doi: 10.1093/nar/gks1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 11.Walensky LD, et al. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murray JK, Gellman SH. Targeting protein-protein interactions: lessons from p53/MDM2. Biopolymers. 2007;88:657–686. doi: 10.1002/bip.20741. [DOI] [PubMed] [Google Scholar]
- 13.Raj M, Bullock BN, Arora PS. Plucking the high hanging fruit: A systematic approach for targeting protein-protein interactions. Bioorganic & medicinal chemistry. 2013;21:4051–4057. doi: 10.1016/j.bmc.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Driggers EM, Hale SP, Lee J, Terrett NK. The exploration of macrocycles for drug discovery--an underexploited structural class. Nat Rev Drug Discov. 2008;7:608–624. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- 15.Wessjohann LA, Ruijter E, Garcia-Rivera D, Brandt W. What can a chemist learn from nature’s macrocycles?--a brief, conceptual view. Mol Divers. 2005;9:171–186. doi: 10.1007/s11030-005-1314-x. [DOI] [PubMed] [Google Scholar]
- 16.Marsault E, Peterson ML. Macrocycles are great cycles: applications, opportunities, and challenges of synthetic macrocycles in drug discovery. J Med Chem. 2011;54:1961–2004. doi: 10.1021/jm1012374. [DOI] [PubMed] [Google Scholar]
- 17.Mallinson J, Collins I. Macrocycles in new drug discovery. Future medicinal chemistry. 2012;4:1409–1438. doi: 10.4155/fmc.12.93. [DOI] [PubMed] [Google Scholar]
- 18.Yu X, Sun D. Macrocyclic drugs and synthetic methodologies toward macrocycles. Molecules. 2013;18:6230–6268. doi: 10.3390/molecules18066230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giordanetto F, Kihlberg J. Macrocyclic drugs and clinical candidates: what can medicinal chemists learn from their properties? J Med Chem. 2014;57:278–295. doi: 10.1021/jm400887j. [DOI] [PubMed] [Google Scholar]
- 20.Ganesan A. The impact of natural products upon modern drug discovery. Curr Opin Chem Biol. 2008;12:306–317. doi: 10.1016/j.cbpa.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 21.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44:235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 22.Vistoli G, Pedretti A, Testa B. Assessing drug-likeness--what are we missing? Drug discovery today. 2008;13:285–294. doi: 10.1016/j.drudis.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 23.Brandt W, Haupt VJ, Wessjohann LA. Chemoinformatic analysis of biologically active macrocycles. Current topics in medicinal chemistry. 2010;10:1361–1379. doi: 10.2174/156802610792232060. [DOI] [PubMed] [Google Scholar]
- 24.Bockus AT, McEwen CM, Lokey RS. Form and function in cyclic peptide natural products: a pharmacokinetic perspective. Current topics in medicinal chemistry. 2013;13:821–836. doi: 10.2174/1568026611313070005. [DOI] [PubMed] [Google Scholar]
- 25.White TR, et al. On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds. Nat Chem Biol. 2011;7:810–817. doi: 10.1038/nchembio.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brenke R, et al. Fragment-based identification of druggable ‘hot spots’ of proteins using Fourier domain correlation techniques. Bioinformatics. 2009;25:621–627. doi: 10.1093/bioinformatics/btp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landon MR, Lancia DR, Jr., Yu J, Thiel SC, Vajda S. Identification of hot spots within druggable binding regions by computational solvent mapping of proteins. Journal of Medicinal Chemistry. 2007;50:1231–1240. doi: 10.1021/jm061134b. [DOI] [PubMed] [Google Scholar]
- 28.Kozakov D, et al. Structural conservation of druggable hot spots in protein-protein interfaces. Proc Natl Acad Sci U S A. 2011;108:13528–13533. doi: 10.1073/pnas.1101835108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gaulton A, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic acids research. 2012;40:D1100–1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jorgensen WL. The many roles of computation in drug discovery. Science. 2004;303:1813–1818. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- 31.Lo Conte L, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. J Mol Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 32.Nayal M, Honig B. On the nature of cavities on protein surfaces: application to the identification of drug-binding sites. Proteins. 2006;63:892–906. doi: 10.1002/prot.20897. [DOI] [PubMed] [Google Scholar]
- 33.Ciulli A, Williams G, Smith AG, Blundell TL, Abell C. Probing hot spots at protein-ligand binding sites: a fragment-based approach using biophysical methods. J Med Chem. 2006;49:4992–5000. doi: 10.1021/jm060490r. [DOI] [PubMed] [Google Scholar]
- 34.DeLano WL. Unraveling hot spots in binding interfaces: progress and challenges. Curr Opin Struct Biol. 2002;12:14–20. doi: 10.1016/s0959-440x(02)00283-x. [DOI] [PubMed] [Google Scholar]
- 35.Landon MR, Lancia DR, Jr., Yu J, Thiel SC, Vajda S. Identification of hot spots within druggable binding regions by computational solvent mapping of proteins. J Med Chem. 2007;50:1231–1240. doi: 10.1021/jm061134b. [DOI] [PubMed] [Google Scholar]
- 36.Chuang GY, et al. Binding hot spots and amantadine orientation in the influenza a virus M2 proton channel. Biophys J. 2009;97:2846–2853. doi: 10.1016/j.bpj.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hall DH, et al. Robust identification of binding hot spots using continuum electrostatics: application to hen egg-white lysozyme. J Am Chem Soc. 2011;133:20668–20671. doi: 10.1021/ja207914y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buhrman G, et al. Analysis of binding site hot spots on the surface of Ras GTPase. Journal of molecular biology. 2011;413:773–789. doi: 10.1016/j.jmb.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zerbe BS, Hall DR, Vajda S, Whitty A, Kozakov D. Relationship between hot spot residues and ligand binding hot spots in protein-protein interfaces. Journal of chemical information and modeling. 2012;52:2236–2244. doi: 10.1021/ci300175u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hall DR, Ngan CH, Zerbe BS, Kozakov D, Vajda S. Hot spot analysis for driving the development of hits into leads in fragment-based drug discovery. Journal of chemical information and modeling. 2012;52:199–209. doi: 10.1021/ci200468p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Golden MS, et al. Comprehensive Experimental and Computational Analysis of Binding Energy Hot Spots at the NF - κ B Essential Modulator/IKKβ Protein-Protein Interface. J Am Chem Soc. 2013;135:6242–6256. doi: 10.1021/ja400914z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roe SM, et al. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- 43.Hajduk PJ, Huth JR, Fesik SW. Druggability indices for protein targets derived from NMR-based screening data. J Med Chem. 2005;48:2518–2525. doi: 10.1021/jm049131r. [DOI] [PubMed] [Google Scholar]
- 44.Landon MR, et al. Novel druggable hot spots in avian influenza neuraminidase H5N1 revealed by computational solvent mapping of a reduced and representative receptor ensemble. Chemical biology & drug design. 2008;71:106–116. doi: 10.1111/j.1747-0285.2007.00614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rand AC, et al. Optimizing PK properties of cyclic peptides: the effect of side chain substitutions on permeability and clearance() MedChemComm. 2012;3:1282–1289. doi: 10.1039/C2MD20203D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug discovery today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 47.Hajduk PJ. Fragment-based drug design: how big is too big? J Med Chem. 2006;49:6972–6976. doi: 10.1021/jm060511h. [DOI] [PubMed] [Google Scholar]
- 48.Veber DF, et al. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 49.Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/ elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22:11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 50.Vieth M, et al. Characteristic physical properties and structural fragments of marketed oral drugs. J Med Chem. 2004;47:224–232. doi: 10.1021/jm030267j. [DOI] [PubMed] [Google Scholar]
- 51.Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 52.Hartshorn MJ, et al. Diverse, high-quality test set for the validation of protein-ligand docking performance. J Med Chem. 2007;50:726–741. doi: 10.1021/jm061277y. [DOI] [PubMed] [Google Scholar]
- 53.Kozakov D, et al. Structural conservation of druggable hot spots in protein-protein interfaces. Proc Natl Acad Sci U S A. 2011;108:13528–13533. doi: 10.1073/pnas.1101835108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hall DR, Ngan CH, Zerbe BS, Kozakov D, Vajda S. Hot spot analysis for driving the development of hits into leads in fragment-based drug discovery. J Chem Inf Model. 2012;52:199–209. doi: 10.1021/ci200468p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mann H, Whitney D. On a test of whether one of two random variables is stochastically larger than the other. Annals of Mathematical Statistics. 1947;18:50–60. [Google Scholar]
- 56.Anderson T, Darling D. Asymptotic theory of certain “goodness-of-fit” criteria based on stochastic processes. Annals of Mathematical Statistics. 1952;23:193–212. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.