

Abstract
Tandem mass spectrometry (MS/MS) has enabled researchers to analyze complex biological samples since the original concept inception. It facilitates the identification and quantification of modifications within tens of thousands of proteins in a single large-scale proteomic experiment. Phosphorylation analysis, as one of the most common and important post-translational modifications, has particularly benefited from such progress in the field. Here we showcase the technique through in-depth analyses of B cell signaling based on quantitative phosphoproteomics. As a complement to the previously described PolyMAC-Ti (polymer-based metal ion affinity capture using titanium) reagent, we introduce here PolyMAC-Fe, which utilizes a different metal ion, Fe(III). An extensive comparison using the different available MS/MS fragmentations techniques was made between PolyMAC-Fe, PolyMAC-Ti and IMAC (immobilized metal ion affinity chromatography) reagents in terms of specificity, reproducibility and type of phosphopeptides being enriched. PolyMAC-Fe based chelation demonstrated good selectivity and unique specificity toward phosphopeptides, making it useful in specialized applications. We have combined PolyMAC-Ti and PolyMAC-Fe, along with SILAC-based quantitation and large-scale fractionation, for quantitative B cell phosphoproteomic analyses. The complementary approach allowed us to identify a larger percentage of multiply phosphorylated peptides than with PolyMAC-Ti alone. Overall, out of 13,794 unique phosphorylation sites identified, close to 20% were dependent on BCR signaling. These sites were further mapped to a variety of major signaling networks, offering more detailed information about the biochemistry of B cell receptor engagement.
Keywords: Proteomics, Tandem mass spectrometry, kinase, protein phosphorylation
Graphical Abstract
INTRODUCTION
The seminal paper by Cooks and co-workers in 1978 described the concept of direct mixture analysis through the separation of individual components in a mass spectrometer [1]. The experiments were performed with the mass-analyzed ion kinetic energy spectrometer (MIKES), one of the major instruments that demonstrated the unique capability of tandem mass spectrometry (MS/MS). Since then, MS/MS has become the essential tool for shotgun-based proteomics where tens hundreds of peptides are typically introduced into mass spectrometer at a given time and each peptide is isolated, activated and then fragmented for sequencing information [2–4].
MS/MS has also become the method of choice to analyze important protein modifications such as phosphorylation [5, 6]. It allows not only for the identification of individual phosphorylation sites, but also for comprehensive phosphoproteome analyses. Phosphorylated proteins typically exist in sub-stoichiometric ratios when compared to unmodified proteins. Additionally, phosphorylated proteins and, in particular, the dynamically phosphorylated forms of signaling proteins, are often of low abundance. With these additional complications, therefore, an efficient enrichment of actual phosphopeptides is typically employed before MS/MS analyses. Several approaches have been explored to date for the selective isolation of phosphopeptides, such as immobilized metal ion affinity chromatography (IMAC) [7, 8], TiO2 [9, 10], ZrO2 [11, 12], and Al(OH)3 [13] beads and chromatography columns. Our group has recently introduced a new concept for phosphopeptide enrichment to facilitate phosphorylation analyses by MS. The new reagent and novel chemical strategy, termed Polymer-based Metal-ion Affinity Capture (PolyMAC), is based on a metal ion-functionalized soluble nanopolymer to chelate phosphopeptides in a homogeneous aqueous environment [14]. The technology is based on water soluble, globular nanopolymers (i.e., dendrimers) that are multi-functionalized with reactive groups (metal ions) for the site-specific recognition of phosphate groups, and with ‘handle’ groups that facilitate the isolation. PolyMAC replaces commonly used solid-phase extraction methods with soluble functionalized dendrimers capable of selective and strong binding to low-abundant phosphopeptides by creating a homogeneous reaction condition that allows for excellent reproducibility, robustness, high yield and fast procedure. The first PolyMAC was functionalized with titanium ions, demonstrating exceptional selectivity and binding efficiency toward phosphorylated residues [14–16].
We present here a MS/MS-based large scale phosphoproteomic study by expanding the PolyMAC technology with other metal ions to increase the coverage of phosphoproteome. We introduce a novel PolyMAC reagent functionalized with iron metal ions (PolyMAC-Fe), capable of enriching a unique set of phosphopeptides complementary to those isolated by PolyMAC-Ti. We performed a thorough comparison between PolyMAC functionalized with Ti or Fe using a variety of MS/MS fragmentation methods, including collision-induced dissociation (CID), high-energy collisional dissociation (HCD) and electron-transfer dissociation (ETD), as well as using different proteolytic enzymes (trypsin and Lys-C). Finally, we combined the two PolyMAC variations for an in-depth quantitative analysis of the phosphoproteome after B-cell receptor (BCR) engagement in human B cells.
As signaling through the BCR is critical for the development and activation of B cells [17, 18], understanding the role of phosphorylation would provide a vast amount of information about B cell activation and development, as well as offer insights into the signaling irregularities of lymphomas and leukemia where tonic BCR signaling is thought to occur [19, 20]. To date, only limited large-scale studies have been performed on the effects of BCR engagement on downstream signaling, offering some information on the changes in transcriptional responses [21] or in tyrosine phosphorylation [22]. However, aside from receptor-proximal tyrosine kinase signaling, the majority of affected downstream molecules are likely phosphorylated on serine or threonine residues. Thus, an examination of the overall changes in the phosphoproteome would offer much more detail about the role of the BCR in regulating various pathways. Our current approach allowed the identification of 2,737 peptides with significant changes in phosphorylation after BCR stimulation, including numerous previously unanticipated phosphoserine and phosphothreonine residues. This, to our knowledge, is the first large-scale phosphoproteomic analysis in such a system.
METHODS
Materials
All reagents for PolyMAC synthesis, cell lysis, protein digestion and Phos-Select IMAC enrichment were obtained from Sigma-Aldrich. Affi-Gel Hydrazide gel was purchased from Bio-Rad, and empty spin columns from Boca Scientific. AminoLink aldehyde beads, SnakeSkin dialysis tubing, dialyzed FBS and SILAC media were bought from Thermo-Pierce. Stably labeled 13C-Arg and 13C-Lys isotopes were obtained from IsoTec. Cell culture reagents were acquired from Invitrogen. RapiGest denaturing agent, C18 Sep-Pak and Xbridge C18 BEH130 RPLC columns were from Waters. Amicon Ultra centrifugal filter devices (5 kDa MW cutoff) were obtained from Millipore.
Synthesis of PolyMAC-Fe
A total of 200 μL of PAMAM (polyamidoamine) dendrimer generation 4 solution (provided as 10% (w/v) in methanol) was dried in a microfuge tube, resolubilized in 2 mL of anhydrous DMSO and transferred into a 10-mL round-bottom flask with a magnetic stir bar. In a microfuge tube, 5.5 mg of Boc-amino-oxyacetic acid, 10 mg of HOBt (hydroxybenzotriazole) and 10 μL of DIPCI (1,3-diisopropylcarbodiimide) were dissolved in 1 mL of DMSO and incubated for 30 min at room temperature. The mixture was then added into the round bottom flask containing dendrimer and stirred overnight to add the “handle” group. The solution was further dialyzed against water for 7–8 h using Snakeskin® pleated dialysis tubing (3,500 MWCO, 22 mm dry diameter, Pierce) to remove any remaining unreacted reagents. After transferring the solution into an Amicon Ultra centrifugal filter device (5 kDa MW cutoff; Millipore), it was concentrated to a volume under 2 mL. The solution was then transferred into a clean 10-mL round-bottom flask with a stir bar and 1.5 mL of 150 mM MES buffer in water (2-(N-morpholino)ethanesulfonic acid; pH 5.5) was added along with 16 mg of 2-carboxyethyl-phosphonic acid, 16 mg of N-hydroxysuccinimide (dissolved in 100 μL of water), and 160 mg of EDC (1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride) and stirred overnight to functionalize the dendrimer with phosphonic acid. For the next step, the mixture was dialyzed against water for 7–8 hours to overnight to remove any remaining unreacted reagents. At this point, the mixture was collected and stored at 4°C. One-third of the reagent solution was dried in a glass tube under vacuum using a SpeedVac concentrator (Savant). The reagent was dissolved in 100 μL of water and 400 μL of 100% TFA (trifluoroacetic acid) were added, followed by 1 h incubation at room temperature to remove the Boc protecting group. The mixture was dried using nitrogen positive flow and resolubilized in 750 μL of DMSO. After resuspending the reagent in 250 μL of the 4:1 mixture of 0.1% HCl in water and DMSO, it was dialyzed against the 4:1 mixture of 0.1% HCl in water and DMSO, followed by dialysis in 0.01% HCl. After dialysis, the solution was transferred into a microfuge tube, iron chloride was added into the solution to a final concentration of 100 mM and incubated for 1 h with agitation at room temperature to chelate iron with the phosphonic acid groups on the dendrimer. The solution was finally dialyzed against 0.01% HCl overnight to remove any unbound iron.
Capture of phosphopeptides using PolyMAC-Fe
To prepare the capturing gel, 50 μL of the AminoLink aldehyde resin were transferred into a frit-based spin column (Boca Scientific). The resin was washed twice with 200 μL water by centrifuging down the column at 2,300 × g for 30 s. The peptide mixture was resuspended in 100 μL of loading buffer (100 mM glycolic acid, 1% trifluroacetic acid, 50% acetonitrile), to which 10 μL of the PolyMAC-Fe reagent was added. The mixture was incubated for 5 min, and 200 μL of the capture buffer (300 mM HEPES, pH 7.7) was added to bring the pH to above 6.3. The mixture was transferred into the spin column containing the washed resin. The column was incubated for 10 min with agitation, and then centrifuged at 2,300 × g for 30 s to collect the unbound flowthrough. The resin with the captured dendrimer was washed once with 200 μL of the loading buffer by incubating the mixture for 5 min with agitation and centrifuging the column at 2,300 × g for 30 s. The resin was further washed twice with washing buffer (100 mM acetic acid, 1% trifluoroacetic acid, 80% acetonitrile) and once with water. The phosphopeptides were eluted off the dendrimer by incubating the resin twice with 100 μL of 400 mM ammonium hydroxide using a 5 min agitation, and centrifuging the column at 2,300 × g for 30 s. Both 100 μL elutions were collected into the same low-binding microfuge tube and dried down completely using a SpeedVac concentrator.
Phosphopeptide enrichment using PolyMAC-Ti and IMAC
PolyMAC-Ti based phosphopeptide enrichment was done essentially as described before [14] using a similar protocol and solutions as above (except PolyMAC-Fe was replaced with PolyMAC-Ti and the aldehyde beads were replaced with Hydrazide Affi-Gel Hydrazine gel).
IMAC phosphopeptide capture was done using the Phos-Select IMAC Fe beads according to the previously published protocol with some modifications [23]. Briefly, 50 μL of the Phos-Select resin slurry was transferred into a spin column and washed twice with water. The peptide mixture was resuspended in 200 μL IMAC loading buffer (25 mM formic acid, 40% acetonitrile), added to the spin column with the resin, incubated for 1 h and then centrifuged at 2,300 × g for 30 s to collect the unbound flowthrough. The resin was washed twice with 200 μL of the loading buffer for 5 min and one last time with water. The phosphopeptides were eluted twice with 100 μL of 400 mM of ammonium hydroxide and dried completely in a SpeedVac concentrator.
Preparation of DG75 cell lysate samples
Burkitt’s Lymphoma DG75 human B cells (ATCC) were cultured in RPMI-1640 media supplemented with 10% heat-inactivated FBS, 1% sodium pyruvate, 0.5% streptomycin/penicillin, and 0.05% 2-mercaptoethanol. Before collection, the cells were washed once with PBS and stimulated with 10 μM of sodium pervanate solution for 30 min. After stimulation, the cells were washed again with PBS, collected, and frozen at −80 °C. 1 × 108 cells were lysed in 1 mL of lysis solution (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 1 mM sodium orthovanadate, 1x phosphatase inhibitor cocktail (Sigma), 10 mM sodium fluoride) for 20 min on ice. The cell debris was cleared by centrifugation at 16,100 × g for 10 min. The supernatant containing soluble proteins was collected. The concentration of the cell lysate was determined using the BCA assay (Bio-Rad). Proteins were denatured and reduced in 50 mM trimethyl ammounium bicarbonate containing 0.1% RapiGest (Waters) and 5 mM dithiothreitol for 30 min at 50 °C. The proteins were further alkylated in 15 mM iodoacetamide for 1 h in the dark at room temperature and digested with proteomics grade trypsin or endoproteinase Lys-C at 1:100 ratio overnight at 37 °C. 100 mM HCl was added to the peptide mix to lower the pH below 3 and the sample was incubated for 40 min at 37 °C. The sample was centrifuged at 16,100 × g to remove RapiGest and the supernatant collected. The sample was mixed in equal amounts with 1% TFA and loaded onto a 100-mg Sep-Pak C18 column (Waters) to remove buffer and small molecules. The peptides were eluted with 0.1% trifluoroacetic acid in 80% acetonitrile and dried completely using a SpeedVac.
Preparation of SILAC −/+ IgM stimulation samples
Two sets of DG75 cells were prepared as described above, except SILAC RPMI-1640 media (containing no Lys and Arg) and dialyzed FBS were used. For “light” labeled cells, regular Lys and Arg were added to the media, while 13C6-Lys and 13C6-Arg were added to make the “heavy” media. The cells were grown over 4–5 generations to incorporate the labeled amino acids (MS analysis showed over 99% incorporation with less that 2% 13C6-Arg to Pro conversion; data not shown). After counting and normalizing the cells to 4 × 107 cell for each set, cells were collected by centrifugation and resuspended in 4 mL of PBS. The “light”-labeled cells were incubated with the standard protocol of 4 μL/mL of Fab fragment of goat anti-human IgM (from 1 mg/mL stock) for 10 min at 37 °C (“heavy” cells were incubated with PBS as a control). Ten min stimulation time point was chosen from the time course experiment (Supplementary Fig. 1) in order to ensure stimulation of more down-stream signaling events. The cells were washed again and combined together (previous experiments showed that the ratio was well-preserved when the samples were combined at the cell stage; data not shown). The cells were then lysed, digested and RapiGest removed as described above. The resulting peptides were then diluted to 5 mL with 10 mM trimethylamminium bicarbonate (pH 8.0) and loaded onto a 15-cm XBridge C18 BEH130 reverse-phase liquid chromatography (RPLC) column. 20 fractions were collected using a 60-min 5% to 40% 10 mM TMAB/acetonitrile gradient at pH 8.0 (0.5 mL/min flowrate). At this point, each fraction was split in two, lyophilized and enriched for phosphopeptides using either PolyMAC-Fe or PolyMAC-Ti.
Mass spectrometry analyses
Peptide samples were re-dissolved in 8 μL of 0.1% formic acid and injected into an Eksigent nanoflow 2D HPLC system. The separation was performed using an in-house C18 capillary column packed in-house with 5 μm C18 Magic beads resin (Michrom; 75 μm i.d. and 12 cm of bed length) on an Eksigent nanoflow 2D HPLC.[24] The mobile phase buffer consisted of 0.1% formic acid in ultra-pure water with the eluting buffer of 0.1% formic acid in acetonitrile, run over a shallow linear gradient over 90 min (2% to 30% acetonitrile) with a flow rate of 0.3 μl/min. The electrospray ionization emitter tip was generated on the prepacked column with a laser puller (Model P-2000, Sutter Instrument Co.). The Eksigent nanoflow 2D HPLC system was coupled online with an LTQ Orbitrap Velos hybrid mass spectrometer equipped with ETD capability (Thermo Fisher, San Jose, CA, USA). The mass spectrometer was either set to operate in CID only mode, in decision-tree mode where either CID or ETD is done depending on the charge and the size of each ion, or in HCD mode for MS and MS/MS. The mass spectrometer was operated in the data-dependent mode in which a full-scan MS was followed by 20 MS/MS scans of the most abundant ions for CID and decision-tree, and by 10 MS/MS for HCD [25]. Charge state of +1 and unassigned charge states were excluded. The mass exclusion time was 90 s. For SILAC samples, to improve the MS peak shapes for quantitation, CID and decision-tree modes were carried out using 6 MS/MS for each scan, while 3 most abundant MS/MS scans were done for HCD method.
Data Acquisition and Analysis
Data analysis was performed with SEQUEST, Mascot and Z-core (for ETD data) algorithms using Proteome Discoverer software V1.3 (ThermoFisher Scientific, San Jose, CA, USA). RAW files were searched against Homo sapiens database with no redundant entries. Peptide mass tolerance was set at 10 ppm, and MS/MS tolerance was set at 0.8 Da (0.05 Da for HCD data). Search criteria included a static modification of cysteine residues of +57.0214 Da, variable modifications of +15.9949 Da to include potential oxidation of methionines, and a modification of +79.996 Da on serines, threonines and tyrosines for identification of phosphorylation. Searches were performed with full tryptic or Lys-C digestion and allowed a maximum of two missed cleavages on the peptides analyzed from the sequence database. The parameters for FDR were set for 1% for each analysis. Proteome Discoverer software generated a reverse “decoy” database from the chosen database, and any peptides passing the initial filtering parameters that were derived from this decoy database are defined as false positive identifications. The minimum cross-correlation factor (Xcorr) filter was then re-adjusted for each individual charge state separately in order to optimally meet the predetermined target FDR of 1% based on the number of random false-positive matches from the reversed “decoy” database. Thus, each dataset had its own passing parameters. The most likely phosphorylation site localization from CID mass spectra was determined by PhosphoRS algorithm within the Proteome Discoverer 1.3 software. The number of unique phosphopeptides and nonphosphopeptides identified were then counted and compared. SILAC quantitation was carried out using Proteome Discoverer software V1.3, which uses the MS peak areas of the “light” and “heavy” peptides and reports “light/heavy” (L/H) ratios. The quantitation significance threshold was based on −/+ 3x standard deviation of variance calculated from the identified nonphosphopeptides data, resulting in the quantitative significance limits of 1.04 to 3.04 for L/H ratio after the removal of outliers.
IPA pathways analysis
For pathway analyses, high confidence peptides (<1% FDR) with significantly changed levels of phosphorylation from −/+ IgM stimulated cells were used along with their quantitative ratios. To identify canonical and metabolic pathways and their protein partners, Ingenuity Pathway Analysis (IPA) (Ingenuity Systems Inc., Redwood City, CA; http://www.ingenuity.com/), software version 7.6, was used for categorizing these proteins and grouping then according to processes, pathways, and networking. The IPA analyses criteria were set to include only known human cellular proteins and their interactions. Top identified networks and pathways (those with most significant changes in phosphorylation) were further inspected using NCBI and Swissprot websites, as well as the peer-reviewed literature.
RESULTS AND DISCUSSION
Rationale for development of PolyMAC-Fe
The design of the PolyMAC technology takes advantage of not only the multi-functionalized nanoparticles, but more importantly, the soluble nature of molecules, allowing for the recognition of a limited sample of phosphopeptides in the solution phase for optimum efficiency and maximum yield. In general, PolyMAC is functionalized with high density metal ions on the surface to capture phosphopeptides in a homogeneous aqueous environment, eliminating the heterogeneous binding conditions that limit the reproducibility of solid–phase isolation techniques [14]. In the second step, the PolyMAC-bound phosphopeptides are pulled out of solution via a second functional group (“handle” group) on the dendrimer that binds to modified agarose beads. A high concentration ratio of the reactive groups to the “handle” groups facilitates the complete recovery of the bound phosphopeptides while eliminating the need for extra steps to remove excess reagents. To utilize the PolyMAC reagent, a typical protocol starts with a peptide mixture from a single protein, protein complex, whole cell or tissue extract, which is incubated with the PolyMAC reagents for a few minutes. Subsequently, “handle” group-specific functionalized agarose beads are added to capture PolyMAC-bound phosphopeptides. Extensive washing steps can be applied to remove nonphosphopeptides. Finally, phosphopeptides are recovered in the solution under the basic conditions, followed by mass spectrometric analysis for the identification of peptide sequences and phosphorylation sites.
The first PolyMAC product was functionalized with titanium ions (PolyMAC-Ti) and has been examined extensively with multiple systems [14, 26]. We reason that PolyMAC reagents functionalized with different metal ions are capable of capturing different and complementary segments of the phosphoproteome. In this study, we introduce the use of iron metal ions for PolyMAC-based enrichment (a schematic of the PolyMAC-Fe and its proposed chemical structure are shown in Fig. 1A and Fig. 1B respectively). Although the first PolyMAC technology utilizes an aldehyde “handle” group and hydrazide capture beads, surprizingly, in the case of PolyMAC-Fe, we found that more efficient enrichment was achieved when the groups were reversed (hydroxylamine “handle” groups and aldehyde capture beads).
Figure 1.

A) Schematic representation of the PolyMAC-Fe strategy. In the first step, PolyMAC-Fe binds to phosphopeptides under homogeneous conditions. In the second step, the PolyMAC-Fe-phosphopeptide complexes are covalently linked via hydroxylamine “handle” groups to aldehyde beads. B) Proposed chemical structure of the PolyMAC-Fe functional groups.
Using the same starting material (50 μg of lysate), a comparison was carried out between PolyMAC-Ti, PolyMAC-Fe and IMAC-Fe (selectivity and phosphopeptide yield are demonstrated in Fig. 2; lists of identified peptides in Supplementary Tables 1–3). As in the previous report investigating PolyMAC-Ti, PolyMAC-Fe has demonstrated more efficient enrichment than its metal ion counterpart IMAC, again likely due to the homogeneous nature and other advantageous properties of the soluble nanopolymer foundation. In our hands for this particular sample, an improvement was observed in the parameter of phosphopeptide enrichment selectivity, reaching close to 60% for PolyMAC-Fe, while less than 30% for IMAC (Fig. 2B). Although PolyMAC-Ti did exhibit much higher selectivity and yield than either of the other two methods, PolyMAC-Fe was still capable of capturing close to 400 unique phosphopeptides not identified by PolyMAC-Ti, from a single enrichment (Fig. 2C). This reveals only a 44% unique phosphopeptides overlap of iron compared to titanium. The observation is consistent with previous reports about TiO2 and IMAC complementarity [27]. While there is a certain difference between the configuration of PolyMAC-Ti and PolyMAC-Fe (specifically regarding the “handle” groups), this change did not affect the complementarity of the two reagents; with only metal ion substitution producing such an effect. We reason that the primary difference in the types of phosphopeptides isolated comes from the inherent coordination variations between the two metals. It has been previously suggested that iron metal ions are typically bound in a heterocyclic chelation format [28, 29], while titanium is immobilized through bidentate bridging coordination [10, 30]. This has also been recently confirmed for the bioactive form of Ti(IV) [31]. Such differences in chelation formats, as well as changes in redox potential and Lewis acid stregths, are likely the source of differential binding of these metals to phosphopeptide subsets.
Figure 2.

Comparison of the enrichment efficiency among PolyMAC-Ti, PolyMAC-Fe and IMAC, including A) number of unique phosphopeptides identified and B) percent enrichment selectivity (# unique phosphopeptides/total # of peptides). C) Overlap of the unique phosphopeptides identified by PolyMAC-Ti and PolyMAC-Fe. A 50 μg lysate from pervanadate-treated DG-75 cells was used for the comparison; error bars represent 3 separate enrichment experiments.
Therefore, we decided to utilize this outcome to further examine the novel iron-functionalized PolyMAC nanomolecule as a tool for enrichment of a unique subset of the phosphoproteome alongside PolyMAC-Ti. The complete isolation of phosphopeptides with multiple polymer-based reagents has the following advantages: first, each reagent has higher efficiency and selectivity than its corresponding existing counterpart; second, the use of complementary methods allows for the individual analysis of different pools of phosphorylated peptides using mass spectrometry parameters differentially optimized for their unique properties; and third, separations on similar soluble polymers will minimize variations due to differences in the support, pore size, particle size and surface area, as typically is the case with IMAC and TiO2/ZrO2.
Detailed comparison between PolyMAC-Fe and PolyMAC-Ti
For a comparison between PolyMAC-Ti and PolyMAC-Fe, 50 μg of the digested DG75 lysate was used as the starting material for each enrichment procedure. Additionally, we examined three MS fragmentation methods for the phosphoproteomic analysis, CID, HCD, and a decision-tree approach that combines CID and ETD for optimal phosphorylation detection. Though each case has its reported advantages for the phosphopeptide identification [23, 32], our aim was to identify which fragmentation approach is best suited for our enrichment techniques. As shown in Figure 3, most methods resulted in very similar data with a large overlap among the samples, demonstrating that each technique is suitable and can detect comparable subsets of phosphopeptides (lists of identified peptides in Supplementary Tables 4–9). HCD after PolyMAC-Ti enrichment was the only method that demonstrated a significant (~20%) improvement in the number of phosphopeptides identified (Fig. 3A; selectivity was very similar in all cases, data not shown). Therefore, the combination of PolyMAC-Ti and HCD fragmentation appears optimal for this type of sample. This is likely due to a number of advantages associated with HCD [33], including better fragmentation of the phosphopeptide backbone due to higher collision energy, ability to fragement peptides of any standard size, and highly accurate MS/MS carried out in Orbitrap, enabling more precise peptide identification. However, the latter point can also be considered a negative aspect, depending on the complexity of a sample, because MS/MS analysis in the Orbitrap is relatively time-consuming, potentially resulting in lower identification. This could be particularly true for quantitative analyses performed on the MS stage, where it is desirable to obtain a maximum number of MS scans to achieve optimal peak shape.
Figure 3.

Analysis of differences in phosphopeptide identification using three separate fragmentation methods. A) Number of unique phosphopeptides identified after enrichment with PolyMAC-Ti or PolyMAC-Fe using different fragmentation methods. Overlap of unique phosphopeptides identified by different fragmentation methods using B) PolyMAC-Ti or C) PolyMAC-Fe. A 50 μg lysate from DG-75 cells was used for the comparison; error bars represent 3 separate enrichment experiments.
To further examine the influence of different fragmentation techniques on the differentially enriched samples, we utilized SILAC labeled samples, which would represent our large-scale peptides pool in the future analysis. Additionally, to investigate the preference of PolyMAC-Ti or PolyMAC-Fe toward different types of peptides, we also utilized endoproteinase Lys-C as an alternative digestion method. In each case, 50 μg of “light” and “heavy” SILAC samples were processed as before, enriched with either PolyMAC-Fe (“light”) or PolyMAC-Ti (“heavy”), and analyzed by MS using various fragmentation techniques. Direct quantitative comparison of each phosphopeptide identified provided a wealth of information about the type of phosphorylation preferred by each enrichment approach (quantitative histograms of the identified phosphopeptides are provided in Fig. 4; lists of identified peptides in Supplementary Tables 10–15). As expected, the reduction in the number of MS/MS done after each scan from 10 to 3 during the HCD fragmentation (in favor of enhanced quantitation) has reduced the overall number of phosphopeptides identified. Here, both CID and decision-tree approaches appeared more suitable for large-scale complex quantitative analyses. As predicted from previous analyses, in every case after trypsin digestion, PolyMAC-Ti demonstrated a much better enrichment efficiency than PolyMAC-Fe. However, a sufficiently large number of unique phosphopeptides captured only by iron warrant its complemetary use in future studies. Interestingly, Lys-C based digestion resulted in the overwhelming majority of the phosphopeptides being identified after PolyMAC-Fe enrichment, demonstrating that indeed PolyMAC-Fe exhibits a preference for larger peptides. Nonetheless, the sheer number of phosphopeptides identified after PolyMAC-Fe enrichment was still much larger after digestion with trypsin than Lys-C. Finally, a further examination of the data demonstrated yet again the preference of PolyMAC-Fe for multiply phosphorylated and longer peptides (Fig. 5), confirming previous reports [8, 34]. Therefore, based on these results, to acquire a more complete set of phosphoproteins from a large-scale quantitative proteomics experiment, we have selected to use trypsin digestion, followed by a Ti/Fe side-by-side PolyMAC enrichment of phosphopeptides and CID MS/MS fragmentation method.
Figure 4.

Quantitative histograms of the unique phosphopeptides identified using different proteases, enrichment techniques and fragmentation methods. The x-axis represents the log(2) values of L/H ratios (Light – PolyMAC-Fe; Heavy – PolyMAC-Ti), and the values were normalized to the non-phosphopeptides data. The black dotted lines represent the quantitative significance threshold of 0.5 and 2.0 L/H ratios, an internal statistical algorithm setting of Proteome Discoverer software. The colors categorize groups of quantified phosphopeptides, where red = phosphopeptides overrepresented in PolyMAC-Ti sample, blue = phosphopeptides overrepresented in PolyMAC-Fe sample, and green = similar representation.
Figure 5.

Quantitative comparison of the types of phosphopeptides enriched by PolyMAC-Ti and PolyMAC-Fe, including A) percent of unique multiply-phosphorylated peptides identified and B) average length of the phosphopeptides identified.
Large-scale quantitative analysis of the BCR-dependant phosphoproteome
The system to which we applied our novel enrichment approach is B cell receptor (BCR)-dependent signaling in human B cells. B cell activation through BCR stimulation is a very important starting point for numerous signaling cascades that can lead to various cellular fates, including proliferation, cell death, differentiation and controlled immune responses [17, 18, 35]. In the primary level of BCR engagement, a number of tyrosine kinases are involved, which furher relay the signals to other networks. Though some limited analyses of these signaling pathways have been reported [22], a comprehensive quantitative examination of changes in the phosphoproteome after BCR activation has not been carried out.
In this study, we utilized the human Burkitt’s lymphoma DG75 cell line. To observe the changes in phosphorylation quantitatively, we labeled the cells using the SILAC approach [36]. After normalizing the cells based on cell density and treating them with either control PBS (“heavy”) or anti-IgM antibody fragment (“light”) to induce BCR engagement (time-course in Supplementary Fig. 1), we collected the cells, lysed them and normalized again based on protein concentration to ensure proper quantitation. The cell lysates were then combined, digested, and fractionated using a revese phase column-based liquid chromatography (RPLC) under slightly basic conditions (pH 8.0). The good orthogonality of this chromatography to acidic RPLC in LC-MS and elimitation of the need for prior sample desalting make this fractionation method a very practical approach [37]. A total of 20 fractions were collected from the RPLC column and each equally split in two for phosphopeptide enrichment using either PolyMAC-Ti or PolyMAC-Fe technology for every fraction. The resulting phosphopeptide eluents were then analyzed by an Orbitrap Velos mass spectrometer coupled to an Eksigent nano-LC system (the schematic flowchart of the protocol is depicted in Fig. 6). The overall data analysis resulted in 13,794 phosphorylation sites being identified, with 2,737 unique peptides showing a significant change in phosphorylation after BCR engagement (the list of identified phosphopeptides is in Supplementary Table 16). Majority of these phosphopeptides exhibited increased phosphorylation, with only 607 having decreased phosphorylation levels, consistent with basic roles of the BCR activation.
Figure 6.
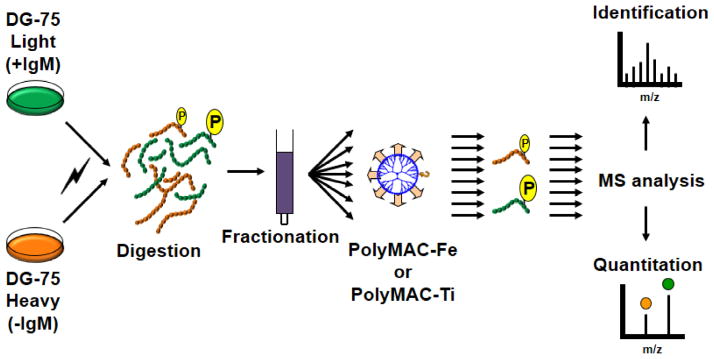
Schematic illustration of the experimental flowchart for the large-scale phosphoproteomic analysis. SILAC-labeled DG-75 B cells were either not treated (H) or treated with Fab fragment of anti-IgM (L) to stimulate BCR signaling. After lysing and combining the cells, the proteins were digested, fractionated by RPLC under slightly basic conditions, and 20 fractions collected. Each fraction was then split in two and the phosphopeptides enriched either with PolyMAC-Ti or PolyMAC-Fe, followed by MS analysis for identification and quantitation.
Analysis of the networks affected through the changes in phosphorylation
The proteins with significant changes in phosphorylation levels were input into the Ingenuity Pathway Analysis (IPA) software, which allows a closer assessment of the affected networks. The depth of color gradient for each pathway node represents the degree in phosphorylation change for that protein, with red representing increased phosphorylation and green – decreased after BCR engagement. As expected, the B cell receptor signaling pathway received one of the highest significance scores, with its many nodes being affected by phosphorylation (Supplementary Fig. 2). We have found not only phosphopeptides belonging to Syk, Lyn, Vav1, Cbl, CD19, CD22 and other direct signaling nodes [17, 38–43], but the changes in phosphorylation have also been directly linked to dowstream pathways of MEKK/MAPK/JNK, Ras/ERK, NF-AT, PKC and NF-κB, all of which have been previously reported [44–49]. These findings resulted in identification of cell death/survival, gene expression, cell proliferation, differentiation and assembly as some of the most over-represented networks in our phosphoproteome analysis. Moreover, a number of other attractive signaling networks found to be significantly affected by BCR-induced changes in phosphorylation are molecular transport and protein trafficking (Supplementary Fig. 3), post-transcriptional regulation (Supplementary Fig. 4), and Peroxisome proliferator-activated receptor network (PPAR/RXR), (Supplementary Fig. 5).
Overall, we believe that the large scale of this phosphoproteome analysis could provide insights regarding the regulation of the proximal and distant signaling pathways and offer numerous potential leads for future biological studies. It provides not only information about the B cell activation itself, but also suggests some putative targets for treatment of B cell-generated diseases. As an example, a number of important regulating proteins involved in Chronic Myeloid Leukemia (CML) signaling have beed identified as differentially phosphorylated (Supplementary Fig. 6), including Ras, NF-κB, p53, c-Myc, Cdk4/6 complex and Rb. This is particularly interesting because BCR signaling has been shown to play a vital role in both early and late stages of leukemia pathogenesis [19, 20, 50]. These proteins have been previously linked to the anti-apoptotic signals in the CML and CLL cells. Consequently, a further, more in-depth investigation of the phosphorylation-dependant regulation of this network would be enlightening. Therefore, it is our belief that such large-scale functional phosphoproteomic analyses of biologically relevant networks could serve as an outstanding explanatory and discovery tool for any system.
CONCLUSION
The present study provides a general tool needed for investigators in the molecular signaling and proteomics fields that will allow them to examine important roles of kinases and phosphorylation in multiple signaling pathways. Although for a single type of enrichment, PolyMAC-Ti is still the obvious choice, its combination with another metal-based PolyMAC (such as PolyMAC-Fe) would offer a particularly valuable approach for comprehensive phosphoproteome analyses. Such explorations could help provide detailed knowledge at the molecular level on precisely how kinases and phosphatases are involved in cellular, particularly disease, signaling through dynamic changes in protein phosphorylation.
Supplementary Material
Highlights.
Developed a novel iron-based PolyMAC phosphopeptide enrichment method
Carefully examined the new PolyMAC-Fe approach
PolyMAC-Fe demonstrated unique enrichment capabilities compared to Ti
Applied both PolyMAC methods for global B cell phosphoproteomics
Uncovered new downstream pathways and networks activated by BCR stimulation
Acknowledgments
This project has been funded in part by National Institutes of Health grants GM088317 (WAT), AI098132 (RLG), and 1R43CA162767 (AI).
Footnotes
Supporting Information (SI_PolyMAC-Fe.pdf which includes Supplementary Figures S1–6) is available free of charge at the journal website.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kondrat RW, Cooks RG. Direct Analysis of Mixtures by Mass-Spectrometry. Anal Chem. 1978;50:A81–A92. [Google Scholar]
- 2.Kruger M, Moser M, Ussar S, Thievessen I, Luber CA, Forner F, Schmidt S, Zanivan S, Fassler R, Mann M. SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell. 2008;134:353–364. doi: 10.1016/j.cell.2008.05.033. [DOI] [PubMed] [Google Scholar]
- 3.Dong MQ, Venable JD, Au N, Xu T, Park SK, Cociorva D, Johnson JR, Dillin A, Yates JR., 3rd Quantitative mass spectrometry identifies insulin signaling targets in C. elegans. Science. 2007;317:660–663. doi: 10.1126/science.1139952. [DOI] [PubMed] [Google Scholar]
- 4.Wolters DA, Washburn MP, Yates JR., 3rd An automated multidimensional protein identification technology for shotgun proteomics. Anal Chem. 2001;73:5683–5690. doi: 10.1021/ac010617e. [DOI] [PubMed] [Google Scholar]
- 5.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 6.Tan CS, Bodenmiller B, Pasculescu A, Jovanovic M, Hengartner MO, Jorgensen C, Bader GD, Aebersold R, Pawson T, Linding R. Comparative analysis reveals conserved protein phosphorylation networks implicated in multiple diseases. Sci Signal. 2009;2:ra39. doi: 10.1126/scisignal.2000316. [DOI] [PubMed] [Google Scholar]
- 7.Porath J. High-performance immobilized-metal-ion affinity chromatography of peptides and proteins. J Chromatogr. 1988;443:3–11. doi: 10.1016/s0021-9673(00)94778-2. [DOI] [PubMed] [Google Scholar]
- 8.Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J, Hunt DF, White FM. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat Biotechnol. 2002;20:301–305. doi: 10.1038/nbt0302-301. [DOI] [PubMed] [Google Scholar]
- 9.Pinkse MW, Uitto PM, Hilhorst MJ, Ooms B, Heck AJ. Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D-NanoLC-ESI-MS/MS and titanium oxide precolumns. Anal Chem. 2004;76:3935–3943. doi: 10.1021/ac0498617. [DOI] [PubMed] [Google Scholar]
- 10.Larsen MR, Thingholm TE, Jensen ON, Roepstorff P, Jorgensen TJ. Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol Cell Proteomics. 2005;4:873–886. doi: 10.1074/mcp.T500007-MCP200. [DOI] [PubMed] [Google Scholar]
- 11.Zhou H, Xu S, Ye M, Feng S, Pan C, Jiang X, Li X, Han G, Fu Y, Zou H. Zirconium phosphonate-modified porous silicon for highly specific capture of phosphopeptides and MALDI-TOF MS analysis. J Proteome Res. 2006;5:2431–2437. doi: 10.1021/pr060162f. [DOI] [PubMed] [Google Scholar]
- 12.Kweon HK, Hakansson K. Selective zirconium dioxide-based enrichment of phosphorylated peptides for mass spectrometric analysis. Anal Chem. 2006;78:1743–1749. doi: 10.1021/ac0522355. [DOI] [PubMed] [Google Scholar]
- 13.Wolschin F, Wienkoop S, Weckwerth W. Enrichment of phosphorylated proteins and peptides from complex mixtures using metal oxide/hydroxide affinity chromatography (MOAC) Proteomics. 2005;5:4389–4397. doi: 10.1002/pmic.200402049. [DOI] [PubMed] [Google Scholar]
- 14.Iliuk AB, Martin VA, Alicie BM, Geahlen RL, Tao WA. In-depth analyses of kinase-dependent tyrosine phosphoproteomes based on metal ion functionalized soluble nanopolymers. Mol Cell Proteomics. 2010;9:2162–2172. doi: 10.1074/mcp.M110.000091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iliuk A, Jayasundera K, Schluttenhofer R, Tao WA. Functionalized soluble nanopolymers for phosphoproteome analysis. Methods Mol Biol. 2011;790:277–285. doi: 10.1007/978-1-61779-319-6_21. [DOI] [PubMed] [Google Scholar]
- 16.Munagala N, Nguyen S, Lam W, Lee J, Joly A, McMillan K, Zhang W. Identification of small molecule ceramide kinase inhibitors using a homogeneous chemiluminescence high throughput assay. Assay Drug Dev Technol. 2007;5:65–73. doi: 10.1089/adt.2006.046. [DOI] [PubMed] [Google Scholar]
- 17.Kurosaki T. Regulation of BCR signaling. Mol Immunol. doi: 10.1016/j.molimm.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 18.Brezski RJ, Monroe JG. B-cell receptor. Adv Exp Med Biol. 2008;640:12–21. doi: 10.1007/978-0-387-09789-3_2. [DOI] [PubMed] [Google Scholar]
- 19.Gobessi S, Laurenti L, Longo PG, Carsetti L, Berno V, Sica S, Leone G, Efremov DG. Inhibition of constitutive and BCR-induced Syk activation downregulates Mcl-1 and induces apoptosis in chronic lymphocytic leukemia B cells. Leukemia. 2009;23:686–697. doi: 10.1038/leu.2008.346. [DOI] [PubMed] [Google Scholar]
- 20.Longo PG, Laurenti L, Gobessi S, Sica S, Leone G, Efremov DG. The Akt/Mcl-1 pathway plays a prominent role in mediating antiapoptotic signals downstream of the B-cell receptor in chronic lymphocytic leukemia B cells. Blood. 2008;111:846–855. doi: 10.1182/blood-2007-05-089037. [DOI] [PubMed] [Google Scholar]
- 21.Franke A, Niederfellner GJ, Klein C, Burtscher H. Antibodies against CD20 or B-Cell Receptor Induce Similar Transcription Patterns in Human Lymphoma Cell Lines. PLoS One. 6:e16596. doi: 10.1371/journal.pone.0016596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsumoto M, Oyamada K, Takahashi H, Sato T, Hatakeyama S, Nakayama KI. Large-scale proteomic analysis of tyrosine-phosphorylation induced by T-cell receptor or B-cell receptor activation reveals new signaling pathways. Proteomics. 2009;9:3549–3563. doi: 10.1002/pmic.200900011. [DOI] [PubMed] [Google Scholar]
- 23.Xiao K, Sun J, Kim J, Rajagopal S, Zhai B, Villen J, Haas W, Kovacs JJ, Shukla AK, Hara MR, Hernandez M, Lachmann A, Zhao S, Lin Y, Cheng Y, Mizuno K, Ma’ayan A, Gygi SP, Lefkowitz RJ. Global phosphorylation analysis of beta-arrestin-mediated signaling downstream of a seven transmembrane receptor (7TMR) Proc Natl Acad Sci U S A. 107:15299–15304. doi: 10.1073/pnas.1008461107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ficarro SB, Zhang Y, Lu Y, Moghimi AR, Askenazi M, Hyatt E, Smith ED, Boyer L, Schlaeger TM, Luckey CJ, Marto JA. Improved electrospray ionization efficiency compensates for diminished chromatographic resolution and enables proteomics analysis of tyrosine signaling in embryonic stem cells. Anal Chem. 2009;81:3440–3447. doi: 10.1021/ac802720e. [DOI] [PubMed] [Google Scholar]
- 25.Nagaraj N, D’Souza RC, Cox J, Olsen JV, Mann M. Feasibility of large-scale phosphoproteomics with higher energy collisional dissociation fragmentation. J Proteome Res. 9:6786–6794. doi: 10.1021/pr100637q. [DOI] [PubMed] [Google Scholar]
- 26.Xue L, Wang WH, Iliuk A, Hu L, Galan JA, Yu S, Hans M, Geahlen RL, Tao WA. Sensitive kinase assay linked with phosphoproteomics for identifying direct kinase substrates. Proc Natl Acad Sci U S A. 2012;109:5615–5620. doi: 10.1073/pnas.1119418109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang X, Fonnum G, Hajivandi M, Stene T, Kjus NH, Ragnhildstveit E, Amshey JW, Predki P, Pope RM. Quantitative comparison of IMAC and TiO2 surfaces used in the study of regulated, dynamic protein phosphorylation. J Am Soc Mass Spectrom. 2007;18:1932–1944. doi: 10.1016/j.jasms.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 28.Kalinowski DS, Richardson DR. The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacological reviews. 2005;57:547–583. doi: 10.1124/pr.57.4.2. [DOI] [PubMed] [Google Scholar]
- 29.du d’Hardemare AM, Torelli S, Serratrice G, Pierre JL. Design of iron chelators: syntheses and iron (III) complexing abilities of tripodal tris-bidentate ligands. Biometals : an international journal on the role of metal ions in biology, biochemistry, and medicine. 2006;19:349–366. doi: 10.1007/s10534-005-2997-2. [DOI] [PubMed] [Google Scholar]
- 30.Connor PAMAJ. Phosphate Adsorption onto TiO2 from Aqueous Solutions: An in Situ Internal Reflection Infrared Spectroscopic Study. Langmuir. 1999;15:2916–2921. [Google Scholar]
- 31.Buettner KM, Collins JM, Valentine AM. Titanium(IV) and vitamin C: aqueous complexes of a bioactive form of Ti(IV) Inorganic chemistry. 2012;51:11030–11039. doi: 10.1021/ic301545m. [DOI] [PubMed] [Google Scholar]
- 32.Swaney DL, McAlister GC, Coon JJ. Decision tree-driven tandem mass spectrometry for shotgun proteomics. Nat Methods. 2008;5:959–964. doi: 10.1038/nmeth.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nat Methods. 2007;4:709–712. doi: 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- 34.Thingholm TE, Jensen ON, Robinson PJ, Larsen MR. SIMAC (sequential elution from IMAC), a phosphoproteomics strategy for the rapid separation of monophosphorylated from multiply phosphorylated peptides. Mol Cell Proteomics. 2008;7:661–671. doi: 10.1074/mcp.M700362-MCP200. [DOI] [PubMed] [Google Scholar]
- 35.Harwood NE, Batista FD. Early events in B cell activation. Annu Rev Immunol. 28:185–210. doi: 10.1146/annurev-immunol-030409-101216. [DOI] [PubMed] [Google Scholar]
- 36.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 37.Gilar M, Olivova P, Daly AE, Gebler JC. Two-dimensional separation of peptides using RP-RP-HPLC system with different pH in first and second separation dimensions. J Sep Sci. 2005;28:1694–1703. doi: 10.1002/jssc.200500116. [DOI] [PubMed] [Google Scholar]
- 38.Geahlen RL. Syk and pTyr’d: Signaling through the B cell antigen receptor. Biochim Biophys Acta. 2009;1793:1115–1127. doi: 10.1016/j.bbamcr.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richards S, Watanabe C, Santos L, Craxton A, Clark EA. Regulation of B-cell entry into the cell cycle. Immunol Rev. 2008;224:183–200. doi: 10.1111/j.1600-065X.2008.00652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang F, Gu H. Negative regulation of lymphocyte development and function by the Cbl family of proteins. Immunol Rev. 2008;224:229–238. doi: 10.1111/j.1600-065X.2008.00655.x. [DOI] [PubMed] [Google Scholar]
- 41.Mocsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. 10:387–402. doi: 10.1038/nri2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pierce SK, Liu W. The tipping points in the initiation of B cell signalling: how small changes make big differences. Nat Rev Immunol. 10:767–777. doi: 10.1038/nri2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Batista FD, Treanor B, Harwood NE. Visualizing a role for the actin cytoskeleton in the regulation of B-cell activation. Immunol Rev. 237:191–204. doi: 10.1111/j.1600-065X.2010.00943.x. [DOI] [PubMed] [Google Scholar]
- 44.Mackay F, Figgett WA, Saulep D, Lepage M, Hibbs ML. B-cell stage and context-dependent requirements for survival signals from BAFF and the B-cell receptor. Immunol Rev. 237:205–225. doi: 10.1111/j.1600-065X.2010.00944.x. [DOI] [PubMed] [Google Scholar]
- 45.Takada E, Hata K, Mizuguchi J. Requirement for JNK-dependent upregulation of BimL in anti-IgM-induced apoptosis in murine B lymphoma cell lines WEHI-231 and CH31. Exp Cell Res. 2006;312:3728–3738. doi: 10.1016/j.yexcr.2006.06.036. [DOI] [PubMed] [Google Scholar]
- 46.Jiang A, Craxton A, Kurosaki T, Clark EA. Different protein tyrosine kinases are required for B cell antigen receptor-mediated activation of extracellular signal-regulated kinase, c-Jun NH2-terminal kinase 1, and p38 mitogen-activated protein kinase. J Exp Med. 1998;188:1297–1306. doi: 10.1084/jem.188.7.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herzog S, Reth M, Jumaa H. Regulation of B-cell proliferation and differentiation by pre-B-cell receptor signalling. Nat Rev Immunol. 2009;9:195–205. doi: 10.1038/nri2491. [DOI] [PubMed] [Google Scholar]
- 48.Shinohara H, Kurosaki T. Comprehending the complex connection between PKCbeta, TAK1, and IKK in BCR signaling. Immunol Rev. 2009;232:300–318. doi: 10.1111/j.1600-065X.2009.00836.x. [DOI] [PubMed] [Google Scholar]
- 49.Yasuda T, Sanjo H, Pages G, Kawano Y, Karasuyama H, Pouyssegur J, Ogata M, Kurosaki T. Erk kinases link pre-B cell receptor signaling to transcriptional events required for early B cell expansion. Immunity. 2008;28:499–508. doi: 10.1016/j.immuni.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 50.Baudot AD, Jeandel PY, Mouska X, Maurer U, Tartare-Deckert S, Raynaud SD, Cassuto JP, Ticchioni M, Deckert M. The tyrosine kinase Syk regulates the survival of chronic lymphocytic leukemia B cells through PKCdelta and proteasome-dependent regulation of Mcl-1 expression. Oncogene. 2009;28:3261–3273. doi: 10.1038/onc.2009.179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.