

Abstract
Purpose
Interleukin-2 inducible T-cell kinase (ITK) promoter CpG sites are hypomethylated in melanomas compared to nevi. The expression of ITK in melanomas, however, has not been established and requires elucidation.
Experimental Design
An ITK specific monoclonal antibody was used to probe sections from de-identified, formalin-fixed paraffin-embedded tumor blocks or cell line arrays and ITK was visualized by immunohistochemistry. Levels of ITK protein differed among melanoma cell lines and representative lines were transduced with four different lentiviral constructs that each contained an shRNA designed to knockdown ITK mRNA levels. The effects of the selective ITK inhibitor BI 10N on cell lines and mouse models were also determined.
Results
ITK protein expression increased with nevus to metastatic melanoma progression. In melanoma cell lines, genetic or pharmacological inhibition of ITK decreased proliferation and migration and increased the percentage of cells in the G0/G1 phase. Treatment of melanoma-bearing mice with BI 10N reduced growth of ITK-expressing xenografts or established autochthonous (Tyr-Cre/Pten null/Braf V600E) melanomas.
Conclusions
We conclude that ITK, formerly considered an immune cell-specific protein, is aberrantly expressed in melanoma and promotes tumor development and progression. Our finding that ITK is aberrantly expressed in most metastatic melanomas suggests that inhibitors of ITK may be efficacious for melanoma treatment. The efficacy of a small molecule ITK inhibitor in the Tyr-Cre/Ptennull/BrafV600E mouse melanoma model supports this possibility.
Keywords: melanoma, ITK (interleukin-2 inducible T-cell kinase), genetically engineered animals, tumor microenvironment, TEC kinase
Introduction
Advances in understanding the genetic aberrations associated with melanoma initiation and progression have led to the recent U.S. Food and Drug Administration approval of three small molecule inhibitors against components in the BRAF-MEK pathway (1-3). However, these systemic therapies rarely lead to melanoma cures, even if used in combination (4). Next generation sequencing analyses of melanoma specimens identified a handful of genetic aberrations that may have “driver” roles in melanoma development and progression but some distinct melanoma subtypes bear none of the identified “driver” genetic aberrations (5). Current immunotherapies have admittedly led to cures, albeit in a small group of patients (6-8). While early clinical trials testing combinations of antibodies against immune checkpoint proteins have shown great promise, the toxicities and long-term efficacy are unknown and under investigation (9).
Using DNA-methylation profiling to discriminate primary cutaneous melanomas from benign moles, we determined that the promoter for IL-2 inducible T-cell kinase (ITK) was significantly hypomethylated in primary melanomas compared to nevi (10). This finding was unexpected because the expression of ITK, a member of the TEC family of tyrosine kinases that includes the Bruton's tyrosine kinase (BTK), is normally restricted to distinct immune cell subsets. In T cells, ITK functions downstream of the T-cell receptor and is important for T-cell activation, development, differentiation, and production of many pro-inflammatory cytokines (11). Given previous reports about the potential for lineage reprogramming of melanoma cells with acquisition of neural or angiogenic properties (12, 13), we hypothesized that aberrant ITK expression in melanoma cells might foster tumor growth
Based on the finding that ITK CpG sites are hypomethylated in melanoma compared to nevi, we conducted experiments to determine if ITK was expressed in nevi and melanomas. The availability of potent ITK-selective inhibitors makes ITK an attractive target, and the TEC kinase BTK has been successfully targeted in hematologic malignancies (14). We therefore examined ITK expression in primary and metastatic melanomas and tested the effects of genetic and pharmacologic inhibition of ITK in cultured cells and murine melanoma models.
Materials and Methods
Tissue procurement
Human tissues analyzed (Supplementary Table S1) were obtained as de-identified, formalin-fixed paraffin-embedded tumor blocks from the UNC Health Care archives under Institutional Review Board (IRB) approved protocols.
Tissue sections and tissue microarray (TMA) construction
Four micron-thick whole tissue sections (WTS) of nevi (n = 30) and primary invasive melanomas (n = 20) were used for tumor tissue analyses. TMAs of metastatic melanomas (n = 82) containing triplicate cores (0.6 mm) from each specimen were also constructed. A pathologist (P.A.G.) reviewed hematoxylin and eosin (H&E; Hematoxylin 7211, Eosin 7111, Richard-Allan)-stained tissue sections to confirm the specimens' diagnoses and marked representative tumor areas on the H&Es for cores to be included in TMAs. TMA blocks were cut into 4 micron-thick sections. A pathologist examined H&E slides of the TMAs to confirm tumor presence.
Cell culture and western blots
Cells were grown in 10% FBS (S12450, Atlanta Biologicals) in base media without antibiotic unless noted. Human melanoma cell lines A375, VMM 39, SKMEL 103, SKMEL 131, SKMEL 153, SKMEL 147, RPMI 8322, and PMWK were used for the cell biology experiments. A375, VMM 39, SKMEL 103, and SKMEL 147 were passaged in Dulbecco's Modified Eagle Medium (DMEM) (Gibco). SKMEL 131, SKMEL 153, and RPMI 8322 were grown in RPMI 1640 media. PMWK cells were passaged in Alpha Minimum Essential Medium (MEM) media (Gibco) with 0.5x minimal essential amino acids. The sources and base media for the cell lines have been reported (15) except for SKMEL 235 and WM 1232 (obtained from Wistar, M. Herlyn) and grown in RPMI 1640 (11875-093, Gibco) and TU 2% [80% MCDB 153 (M-7403, Sigma), 20% L-15 media (41300-070, Gibco)], 2% fetal bovine serum (FBS) (S12450, Atlanta Biologicals), 5 μg/ml Bovine insulin (I-5500, Sigma), 1.68 mM Calcium Chloride (C-5670, Sigma). Cell lines have been tested and authenticated and verified as mycoplasma-free. Normal human melanocytes (NHMs) (Clonetech) were cultured and western blots were performed as described previously (16).
Melanocyte-melanoma cell line array (CLA) construction
CLAs were constructed from 38 melanoma cell line and 3 NHM pellets that were fixed in 10% buffered formalin (SF98-4, Fisher) for 16-24h, washed twice in 70% ethanol, clotted in 2% low-melting agarose (BP165-25, Fisher), processed, and embedded in paraffin wax (23-021-400, Fisher). Cell pellet blocks were sectioned and H&E-stained to verify the quality of the cell clots and guide CLA construction. Three 1-mm diameter cores were removed from each cell pellet block and randomly embedded into recipient CLA blocks, which were cut into 5 micron-thick sections.
Specimen pathology review
Melanomas were reviewed by a dermatopathologist. Demographic characteristics were extracted from the clinical record.
Antibodies, tissue staining protocols, imaging, and analysis
ITK (rabbit monoclonal Y401; Abcam ab32039) was utilized to probe human ITK. GAPDH (Abcam ab9484 mouse monoclonal), cyclin D2 (Santa Cruz Biotechnology sc-563056), and LC3 (Novus Biologicals NB 100-2220) were used for western Blots. Other antibodies and tissue staining protocols for the immunohistochemistry experiments are summarized in Supplementary Table S2. Single or dual IF on CLAs, melanoma TMAs and WTS of nevi and melanomas were performed in the Bond fully-automated slide staining system as previously described (17).
H&E stained WTS, CLA, and melanoma TMA slides were digitally imaged at 20x magnification using the Aperio ScanScope XT (Aperio Technologies). High-resolution acquisition (20x objective) of fluorescently stained slides in the DAPI, Cy3 and Cy5 channels was performed in the Aperio-FL (Aperio Technologies) and fluorescently stained CLAs in PM2000 (HistoRx). Fluorescently stained WTS and TMAs (ITK-S100) were submitted for analysis through Spectrum using HistoRx automated quantitative analysis (AQUA) software version 2.2. Expression of ITK protein labeled by Cy5 (red) was measured in an S100-specific tumor mask labeled by Alexa555 (green) (6). ITK expression in CLAs was measured in the autofluorescent (Cy3) mask. Same specimen array cores were averaged. AQUA scores were normalized across the TMAs and WTS using cell line standards run in every batch. The Aperio Area Quantification FL algorithm was used to quantify ITK (Cy5) co-localization with CD3, CD19, CD56, CD57, CD68, MPO, and MCT markers (Cy3) in 3 primary melanomas and 2 melanomas metastatic to lymph nodes from 5 different patients.
Tyr-Cre/Ptennull/BrafV600Emouse melanomas were stained as described (18) (Supplementary Table S2). Slides were blocked with DakoCytomation, X0909 (KEY 2006) and incubated with Abcam ab32113 for 1 hour at room temperature or 4°C overnight. The slides were blocked with Dako X0590 biotin blocking solution (X059030-2, Agilent). Chromogenic and fluorescently stained images were stored within the Aperio Spectrum Database.
Melanoma mutational status
The BRAF (exons 11 and 15) and NRAS (exons 2 and 3) mutational status of primary melanomas and cell lines was determined as reported (19). Metastatic melanoma TMA cores were stained with BRAF VE1 antibody (20) and scored for VE1 cytoplasmic staining by a pathologist as 0 (no staining), 1+ (weak background), 2+ (moderate staining), or 3+ (strong staining). VE1 scores of 2+ and 3+ were considered positive for BRAFV600E.
shRNA lentivirus production and use
Lentiviral small hairpin RNA (shRNA) constructs TRCN0000010020 (ITK4), TRCN0000010021 (ITK5), TRCN0000010022 (ITK6), and TRCN0000010023 (ITK7) from the Thermo Scientific TRC shRNA library TRC-Hs1.0 (Human) were supplied by the UNC-CH's Lenti-shRNA Core Facility. Lentivirus was produced according to the ViraPower™ Lentiviral Packaging Mix instructions (#44-2050, Invitrogen). Approximately 1×106 lentiviral particles were added to transduce approximately 50% of the cells in a 7.5 cm dish. On day 2, media was removed and fresh complete media added then on day 3 media was replaced with fresh complete media containing puromycin (final concentration 10 μg/ml). Cells were allowed to grow for 4 days before use.
BI 10N, a small molecule ITK inhibitor
BI 10N (21) was from Changchun Discovery Sciences Ltd (Changchun, Jilin, China). Aliquots of a 1,000x stock solution in dimethylsulphoxide (DMSO) were prepared and stored at -20° C. Carna Biosciences (Kobe, Japan) performed selectivity assays.
Two-dimensional gel electrophoresis
Two-dimensional gel electrophoresis was performed using the Immobiline DryStrip 7 cm, pH 6-11 gel system according to the manufacturer's specifications (17-6001-94, GE Healthcare). Gels were then transferred and western blots were performed using the Y401 antibody.
Proliferation and migration assays
Human melanoma cell lines were added to 10 cm2 6-well dishes at a density of 50,000 cells per well. BI 10N was added in DMSO; DMSO was used as a drug vehicle control. Cells were harvested using Trypsin (0.025%) in PBS solution (R-001-100, Gibco) containing 0.01% EDTA for approximately 5 minutes. Cells were counted using the Countess® Automated Cell Counter (C10227, Life Technologies). Graphs were generated using GraphPad Prism version 5 (GraphPad Software, San Diego, CA).
Single-cell tracking was performed to calculate the average motility rate, as described previously (22). Cells were incubated for 24 hours with BI 10N prior to tracking. At least 50 cells were tracked at each BI 10N concentration.
EdU – FxCycle violet staining of melanoma cells
Melanoma cells were grown to approximately 60% confluence in T25 tissue culture flasks (Corning Product #430639). Cells were labeled with Click-iT EdU Alexa fluor 488 (C10425, Invitrogen) followed by detection using FxCycle Violet (F-10347, Invitrogen), per manufacturer's recommendations. Data acquisition was accomplished using CyanADP from Bechman Coulter, and cell cycle analysis accomplished using Summit (version 4.3) software (DAKO).
Caspase glo 3/7 assay
The Promega Caspase-GLO 3/7 Assay Kit (G8090, Promega) was utilized per the manufacture's protocols. 10,000 melanoma cells were plated in 96 well dishes in quadruplicate with the indicated drug concentrations. 100 nM staurosporine was used as the positive control.
Reverse Phase Protein Array (RPPA)
PMWKs and RPMI 8322 cells were treated with increasing BI 10 concentrations, lysates were produced, and the MD Anderson core facility performed RPPA analyses (23).
In vivo studies
All mice were housed and followed in the UNC LCCC mouse phase 1 unit (MP1U) under UNC-CH Institute for Animal Care and Use Committee approved protocols. 10 week old male nude athymic mice (Jackson Labs 000819) were subcutaneously injected into the flank with 500,000 cells, which were previously suspended in a 50:50 mixture of Matrigel Basement Membrane Matrix (356234, BD Biosciences) and Hank's Solution with 2% FBS. The Tyr-Cre/Ptennull/BrafV600E GEMM was induced as described (24). Treatment began when tumors reached an approximate size of 60mm3. BI 10N was administered orally via medicated diet (25) at 15mg/kg/d. Body mass and body condition score (26) were monitored weekly via caliper measurements for the duration of the experiment as a marker for toxicity. Tumor volume was calculated using the formula: (Width2) × (Length)/2.
Statistical methods
Statistical analyses of IF data were accomplished using R software (The R project for Statistical Computing). Mann-Whitney and Kruskal-Wallis tests were used to compare ITK expression with clinical attributes in subsets of nevi, primary melanoma, and metastatic melanoma samples. Mann-Whitney tests were used to compare ITK expression with mutational status in melanoma metastases (mutant versus wild type) and genetic alterations (negative versus positive) in cell lines.
Linear regression analyses were performed for shRNA cell proliferation data using logarithmically transformed baseline normalized cell counts (y) against days (x). Similar regression analyses were performed for mouse tumor growth data using the square root-transformed tumor volume (y) against weeks (x). Likelihood ratio tests were conducted to assess if slopes between control and treatment groups were statistically different from each other. Differences in melanoma cell motility among different treatment groups were assessed using Mann-Whitney U tests. P-values were Bonferroni corrected to account for multiple comparisons of different treatment groups for each cell line.
For RPPA data, a linear regression line was fitted using protein expression as the dependent variable and BI 10N concentration as the independent variable to analyze RPPA data. T-tests were used to assess if the estimated slopes significantly differed from zero. The P-values of the t-tests for slope were adjusted using the False Discovery Rate control to account for multiple testings. Data were analyzed through the use of QIAGEN's Ingenuity® Pathway Analysis (IPA®, QIAGEN Redwood City, CA).
Microarray analysis
Analysis of microarray data (GEO accession number GSE54623) was performed using Biometric Research Branch (BRB, National Cancer Institute) array tools software (27). The quantitative trait analysis (QTA) tool used the Spearman correlation coefficient to identify genes whose expression was correlated with ITK expression in melanoma cell lines. The correlated (P-value <0.005) genes were investigated to determine if they were overrepresented in Biologic Biochemical Image Database (BBID) (28), Kyoto Encyclopedia of Genes and Genomes (KEGG) (29), BioCarta (30), Panther (31), or Reactome (30) pathways.
Study approval
Human tissues studies were IRB-approved under UNC protocols 07-0450 and 09-0737. Animal experiments were approved by the UNC Institutional Animal Care and Use Committee.
Results
ITK protein expression in melanocytic tissues
Sections of normal skin, benign nevus, primary melanoma, and metastatic melanomas were dually probed with antibodies against ITK (red) and S100 (green) (a marker for melanocytic lineage cells) and visualized using immunofluorescence (IF) (Example shown in Fig. 1A). ITK in melanocytic lineage cells of benign nevi (n = 30), primary melanomas (n = 20), and metastatic melanomas (n = 70) was quantified in the S100-positive cells using AQUA (32, 33) (Fig. 1B and Supplementary Table S1). Mean ITK expression was higher in primary melanomas than benign nevi, and even higher in metastatic melanomas (P < 0.001). The mean ITK protein levels of nevi and melanomas were each not significantly associated (all P > 0.05) with demographic or tumor characteristics, and the melanomas' ITK protein levels were not associated with BRAFV600E status (Supplementary Table S1).
Figure 1.

ITK expression in melanocytic tissues. (A) Representative tissue sections from normal skin (a), benign nevus (b), primary melanoma (c), and metastatic melanoma (d) were stained with H&E (top row) or probed with ITK-Cy5 (red) and S100-Alexa 555 (green) primary and secondary antibodies and counterstained with DAPI (blue). Orange indicates colocation of ITK with S100, a melanocytic lineage marker. The clinical and histologic characteristics of the melanocytic tissues stained and their ITK levels are in Supplementary Table S1. Staining protocols are in Supplementary Table S2. (B) Box-and-Whisker plots (median with the 25-75th percentiles and outliers) of ITK expression in S100+ cells from nevi (n = 30), primary melanomas (n = 20), and metastatic melanomas (n = 70). Of the metastatic melanomas, 91% (64/70) had ITK expression above that of the range of ITK expression found in nevi.
ITK protein expression in immune cell subsets
Since ITK has been shown to have a role in Th2-mediated responses, which have been previously associated with ineffective antitumor responses (34), we investigated whether ITK was expressed in immune subsets infiltrating the tumors. To accomplish this, we performed dual color IF analysis of ITK and markers of immune cell subsets on 3 primary melanomas and 2 melanomas metastatic to lymph nodes from 5 patients. ITK expression was observed in a few small mononuclear cells in the melanoma microenvironment, but did not colocalize with CD3+, CD19+, CD68+, myeloperoxidase (MPO)+, mast cell tryptase (MCT)+, CD56+, or CD57+ markers for T-cells, B-cells, macrophages, neutrophilic granulocytes, mast cells, and subsets of natural killer cells (Supplementary Fig. S1A). We therefore could not confirm ITK expression in the most abundant tumor-infiltrating immune cell subsets.
Melanoma cell line characterization and ITK levels
To investigate whether ITK expression is associated with a particular gene expression or mutation signature, we first probed a melanocyte-melanoma cell line array (CLA) with the ITK antibody (Supplementary Table S2). These melanoma cell lines had previously undergone next generation sequencing (35) and gene expression profiling (16). The three different NHM cultures in the CLA had a mean ITK signal of 38.5 (range 26.7-52.3) in arbitrary IF units while 38 melanoma cell lines had a mean ITK signal of 278 (range 30.4-4,941). To group the melanoma cell lines in the CLA by ITK levels, we used the ITK IF arbitrary units obtained for each cell line to classify them as ITK low (ITKLO) (< 38.5, the mean NHM IHC value), intermediate (ITKINT) (38.5 to <77.0, twice the mean NHM IHC ITK signal), or high (ITKHI) (≥ 77.0) (Supplementary Table S3). In comparison, western blots of 5 of these cell lines give an order from lowest to highest ITK expression (PMWK, SKMEL 147, A375, RPMI 8322, to VMM 39) indistinguishable from the IF determined order (Fig. 2A).
Figure 2.
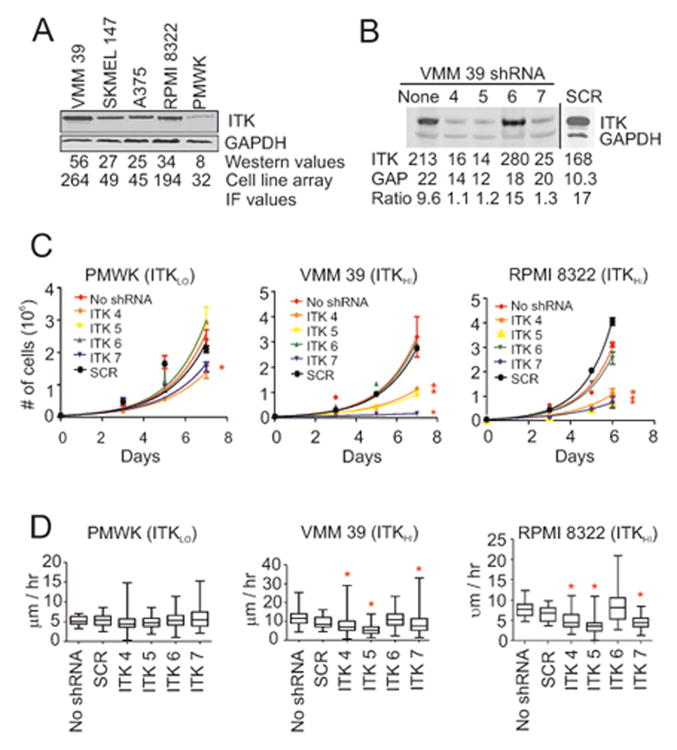
ITK Expression in melanoma cell lines and effects of ITK depletion on their proliferation and motility. (A) Western blot analysis for total ITK in whole cell protein lysates obtained from five melanoma cell lines. Signal intensities of the protein bands were normalized to GAPDH. Cell array IF values from Supplementary Table S3 are also shown for comparison. (B) Western blot analysis for total ITK in protein lysates obtained from VMM 39 melanoma cells that were transduced with shRNA sequences designed to target ITK mRNA (ITK 4, 5, 6, and 7) and with scrambled (SCR) shRNA. The integrated values of ITK containing bands shown in the figure were normalized against the GAPDH values to produce the ratio shown in the figure. (C) Effects of the five shRNAs on the proliferation rates of PMWK, VMM 39 and RPMI 8322 melanoma cells. Asterisks denote melanoma cell lines whose proliferation was significantly reduced compared to SCR shRNA (P < 0.05, Bonferroni corrected; Supplementary Table S4A). (D) Single-cell motility analysis of PMWK, VMM 39 and RPMI 8322 cells transduced with various shRNAs. Asterisks denote significant changes in motility compared to SCR shRNA (P < 0.05, Bonferroni corrected; Supplementary Table S4B).
There were no significant associations (all P > 0.05) of ITK protein levels with NRAS and BRAF mutational status or CDKN2A, PTEN, or TP53 genetic alterations in the cell lines (data not shown). Quantitative Trait Analysis (QTA) was performed to correlate ITK expression with gene expression changes identified by the next generation sequencing and gene expression profiling. However, no (P < 0.05, Bonferroni corrected) gene or pathway alterations were associated with ITK expression (data not shown).
ITK protein depletion using small hairpin RNA (shRNA)
To confirm ITK's presence in melanoma cells and elucidate its role in melanoma, we stably transduced melanoma cell lines with lentiviral particles that contained a single shRNA designed to knockdown ITK mRNA levels (ITK 4, 5, 6, or 7). For negative controls, we transduced the same melanoma cell lines with lentiviral vectors containing a scrambled (SCR) shRNA sequence. As shown in Fig. 2B, western blot analysis of whole cell lysates showed a >85% decrease in the ITK protein level of VMM 39 (ITKHI) cells stably transduced with shRNAs ITK 4, 5, and 7, but ITK 6 and SCR shRNAs appeared inactive. No increase in dead cells or other morphological changes were noted in the lentivirally transduced melanoma cells. Comparable results were noted with the RPMI 8322 (ITKHI) cell line after lentiviral transduction (data not shown), whereas ITK protein levels in the PMWK (ITKLO) cell line were too low to reliably measure significant changes in ITK protein levels (Fig. 2A).
The shRNAs were utilized to study the effects of decreased ITK protein expression on melanoma cell proliferation, cell cycle profile, apoptosis, and motility. Melanoma cell lines VMM 39 and RPMI 8322 (both ITKHI) transduced with shRNAs ITK 4, 5, or 7 proliferated at a decreased rate (P < 0.05, Bonferroni corrected) compared to those transduced with SCR (Fig. 2C-D and Table S4). shRNAs ITK 5 and 7 had no affect (P > 0.05) on PMWK (ITKLO) proliferation, although ITK 4 slightly decreased the proliferation of PMWK compared to SCR.
To assess whether ITK suppression induced changes in the cell cycle, incorporation of 5-ethynyl-2″-deoxyuridine (EdU) was measured to determine de novo DNA synthesis (S-phase). In addition, each cell line was stained using FxCycle violet to measure DNA content. Inhibition of ITK expression in the VMM 39 (ITKHI) and RPMI 8322 (ITKHI) cell lines increased the proportion of cells in G0/G1 (FxCyclelow EdUlow) and decreased the number of cells in S phase (FxCyclemedium/lowEdUhigh). There were no significant cell cycle changes in PMWK (ITKLO) melanoma cells transduced with any of the shRNAs (data not shown).
Since ITK regulates migration in T cells (36, 37), we investigated the effect of ITK expression on melanoma cell line motility quantified by single-cell tracking analysis (38). Migration rates of the ITK 4, 5, or 7- transduced VMM 39 (ITKHI) and RPMI 8322 (ITKHI) cell lines were significantly decreased (P < 0.05, Bonferroni corrected) compared to the corresponding SCR-transduced cell lines (Fig. 2E and 2F and Supplementary Table S4). No significant changes in the migration rate of the PMWK (ITKLO) cells were observed following transduction with any of the lentiviral clones.
BI 10N decreases ITK activity in melanoma cells
To assess the effect of pharmacologic inhibition of ITK activity in melanoma cell lines, we used BI 10N, a small molecule inhibitor of ITK (21, 39). We investigated the potency and selectivity of BI 10N at a concentration of 200 nM against 56 kinases using a cell-free assay (Supplementary Table S5). IC50s were measured for the kinases most potently inhibited by BI 10N in the initial screen, and tight binding studies were performed as needed. BI 10N is a highly selective inhibitor of ITK, and this affinity is described by an apparent dissociation constant (KI,apparent) of 8.6 pM. Of the 56 protein kinases tested, BI 10N is at least 1,000-fold more selective for ITK than for 51 of the protein kinases and is about 10-fold selective over three members of the neurotrophic tyrosine receptor kinase family (TRKA, TRKB, TRKC) and the SRC family member non-receptor tyrosine kinase YES. BI 10N is about 1,000-fold more selective for ITK than for the seven other SRC family tyrosine kinases tested including SRC and FYN.
To assess whether ITK expressed in melanoma cells is active, we probed phosphorylation at Tyr-180 and 511 residues (40) in whole cell lysates from high ITK-expressing melanoma cell lines with the commercially available antibodies 4F10 and 8D11, but could not identify a correctly migrating band. To quantitate ITK phosphorylation in the melanoma cells, we therefore performed two-dimensional electrophoresis on whole cell lysates obtained from RPMI 8322 cells followed by western blot analysis using the ITK Y401 antibody. Lysates profiled included those from untreated cells, phosphatase treated cells, and from cells treated with different concentrations of BI 10N for 3 days prior to harvest (Fig. 3A). In untreated RPMI 8322 cells, two differentially charged immune reactive bands that migrated with the appropriate weight for ITK were identified as ITK by western blot analysis. However, one of the immune reactive bands was absent in extracts treated with lambda phosphatase and from extracts from cells treated with concentrations of BI 10N ≥ 25 nM. In untreated cells, about 14% of ITK is phosphorylated.
Figure 3.
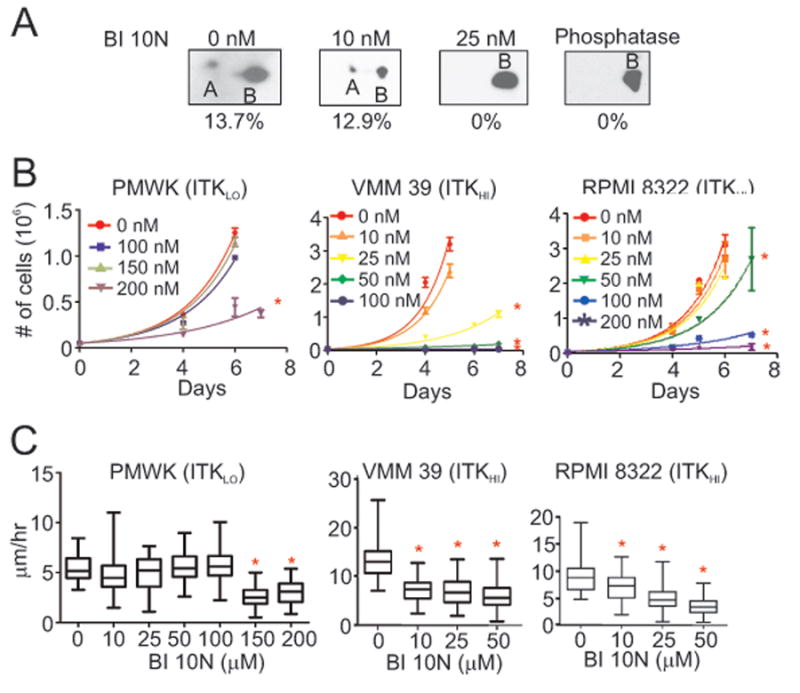
Effects of BI 10N on the phosphorylation of ITK and on the proliferation and motility of melanoma cells. (A) Western blot analysis on whole cell lysates obtained from RPMI 8322 cells. Cells were treated with BI 10N for 3 days at the indicated concentrations prior to the analysis. Extracts of cells that did not receive BI 10N were treated with phosphatase prior to analysis. Proteins from lysates were separated by two-dimensional polyacrylamide gel electrophoresis prior to Western blot analysis using an antibody against total ITK. The percentages indicate the area of the smaller band as a percent of the whole signal. (B) Effects of increasing BI 10N concentrations on the proliferation rates of PMWK, VMM 39 and RPMI 8322 melanoma cells. Asterisks denote significant decreases in proliferation compared to untreated cells (P < 0.05, Bonferroni corrected; Supplementary Table S4C). (C) Effects of increasing BI 10N concentrations on the motility of PMWK, VMM 39 and RPMI 8322 melanoma cells treated for 24 hours prior to the assay. Asterisks denote significant decreases in motility in BI 10N-treated compared to untreated melanoma cells (P < 0.05, Bonferroni corrected; Supplementary Table S4D).
To determine whether the ITK's catalytic activity stimulates the proliferation and migration of melanoma cell lines, we inhibited ITK using BI 10N. Proliferation assays were conducted on PMWK (ITKLO), VMM 39 (ITKHI), RPMI 8322 (ITKHI), and other melanoma cell lines (Fig. 3B, 3C, data not shown, and Supplementary Table S4). In these studies, BI 10N was added at the inception of the assay at the indicated concentrations when the cells were plated. Similar to the equivalent shRNA studies, proliferation of the ITKHI cell lines (VMM 39, RPMI 8322, and SKMEL 153) and an ITKINT cell line (A375) decreased by about 50% in the presence of 50 nM BI 10N, whereas the effects on the proliferation of PMWK (ITKLO) cells was weaker and required 200 nM BI 10N. ITK catalytic activity inhibition by BI 10N reduced ITKHI cell proliferation and migration similar to shRNA suppression of ITK expression. Isogenic overexpression of ITK was not performed to confirm these results.
Cell cycle analysis of VMM 39 (ITKHI) and RPMI 8322 (ITKHI) cell lines using EdU labeling and FxCycle Violet staining indicated that, similar to the shRNA studies, treatment of cells with BI 10N increased the percentage of cells in the G0/G1 phase and decreased the percentage of cells in the S phase in a dose-dependent manner (Supplementary Fig. S1B). No cell cycle-specific changes were observed between the treated and untreated PMWK (ITKLO) cells. BI 10N did not affect the percentage of cells in pre-G0/G1 in PMWK, RPMI 8322, or VMM 39 cells (data not shown). To further assess the effects of ITK activity on apoptosis, caspase 3/7 activities in PMWK, RPMI, and VMM 39 were assessed; no consistent effects by BI 10N on the caspase activity levels were noted (data not shown).
The effects of BI 10N on melanoma cell migration was measured for PMWK (ITKLO), VMM 39 (ITKHI), RPMI 8322 (ITKHI), and additional melanoma cell lines (Fig. 3D, 3E, data not shown, and Supplementary Table S4), by single-cell tracking analysis. Similar to the results with the shRNA clones targeting ITK, the migration by the ITKHI cell lines (VMM39, RPMI 8322, and SKMEL 153) decreased about 50% in the presence of 10 – 25 nM BI 10N while an ITKINT cell line (A375) and the ITKLO cell line PMWK required BI 10N concentrations of 100 nM, or greater, to significantly decrease migration.
Effects of ITK activity on specific cellular proteins
Westerns were performed on RPMI 8322 cells treated with BI 10N administered at concentrations as high as 50nM for 3 days. Inhibition of ITK resulted in cyclin D2 (CCND2) decrease by about one-third but no change in LC3 (MAP13LC3A) level (related to autophagy) (Supplementary Fig. 2A). AKT and ERK protein levels and phosphorylation were unchanged by western analyses (data not shown).
To further investigate the effects of ITK activity on cells, RPPAs (282 proteins or phosphorylated species) were performed on triplicate extracts of PMWK and RPMI 8322 cells treated with up to 50nM BI 10N. The RPPA results confirmed the phosphor western analyses we performed for AKT and ERK showing no change in the phoshphorylation state of those proteins (data not shown). Ingenuity Pathway Analysis (IPA) using a cutoff FDR value of 0.15 indicated that the most affected pathway was p53 signaling with cell cycle arrest being influenced but not apoptosis or cell survival (Supplementary Table S6 and Fig. S2B). RPPA-determined levels of protein analyzed in the p53 pathway are plotted in Supplementary Fig. S2C.
Efficacy of BI 10N in murine melanoma models
To investigate whether BI 10N reduces melanoma growth in vivo, we treated mouse melanoma models with medicated chow containing BI 10N (21). Dose-finding studies based on the published literature were performed to establish an effective dose of 15 mg per kg (mpk)/day. No signs of toxicity (e.g. weight loss, lethargy) were noted in immunodeficient or genetically engineered mice at this dose. Human melanoma xenograft mice were established from two melanoma cell lines with high ITK expression (VMM 39 and RPMI 8322) and from a lower ITK expressing line, A375. Treatment of each xenograft mouse model with BI 10N at 15 mpk significantly slowed tumor growth in the VMM 39 and RPMI 8322 mice, whereas no such effect was seen in the melanoma xenograft mice established from A375 cells (Fig. 4A).
Figure 4.

Effects of BI 10N on tumor growth in vivo. (A) Effects of BI 10N (15mpk) on the growth of human melanoma xenografts (mean and standard error of mean). Asterisks indicate significant tumor growth inhibition by BI 10N-treatment compared to untreated mice (P < 0.05, Bonferroni corrected). AZD6244 (37 mpk), an active agent against melanoma, is shown for comparison. Samples sizes are indicated in parentheses. (B) Representative sections from an untreated melanoma tumor tissue that was induced within the CRE-ERT2 B-RafL/+PtenL/L genetically engineered mouse model (GEMM). The left panel is stained with H&E, The center panel is stained with the biotin linked secondary antibody, while the right panel is stained both with antibodies against total ITK (red) and biotin linked secondary antibody then counterstained with hematoxylin. The lower panels are magnified areas of each of these images. (C) Effect of BI 10N on tumor growth (mean and standard error of mean) in the PTEN/BRAF GEMM. Asterisks indicate significant tumor growth inhibition of BI 10N-treated compared to untreated mice (P < 0.05, Bonferonni corrected). The effects of AZD6244 and PLX4032 on tumor growth are also shown. The activity of PLX4032 shown in C is typical for the GEMM model. Samples sizes are indicated in parentheses. (D) Melanoma bearing mice survive longer when orally dosed with 15 milligrams/kilogram BI 10N. According to the protocol governing the use of mice, the animals have to be sacrificed when the tumors reach a specific size. Of the 9 BI 10N treated animals, 7 were culled due to a multiple masses developing at the end of therapy while the primaries were still responding or had not reached a terminal burden. 1 animal had a primary that achieved a complete response and was culled at 133 days due to age and IACUC time limits. 1 animal was culled due to a progressing primary with no secondary mass.
Given ITK's possible effect on immune system function, we tested BI 10N in immunocompetent mice with autochthonous melanoma. For this purpose, we employed the Tyr-CRE-ERT2 B-RafCA PtenL/L genetically engineered murine model (GEMM) (24). In this system, endogenous tumors featuring V600E B-Raf mutation and Pten loss are generated by tissue-specific CRE recombinase activation in melanocytes. Melanomas from this model expressed high levels of ITK in all 5 excised tumors tested (Fig. 4B). Mice were dosed with BI 10N after the tumors reached a size of about 60mm3. Treatment of mice with BI 10N at 15 mpk resulted in arrest of tumor growth compared to the untreated mice (Fig. 4C). These results suggest that ITK kinase activity is a driver of tumor growth in a GEMM of melanoma. GEMM mice survive significantly longer when treated with BI 10N (Fig. 4D). Similar to the treated cell lines, cyclin D2 was decreased by at least one-third in BI 10N treated versus untreated tumors, whereas LC3 showed no change (data not shown).
Discussion
Our work provides several lines of evidence that ITK, a gene whose promoter CpG sites are hypomethylated in primary melanomas compared to benign nevi, is expressed in a majority of metastatic and primary melanoma tumors. Specifically, we show that ITK protein expression increased with tumor progression from nevus to primary melanoma and even more so to metastatic melanoma. Furthermore, suppression of ITK expression or kinase function in melanoma cells reduced cell attributes associated with tumorigenesis, including proliferation and motility. Finally, inhibition of ITK activity using BI 10N reduced tumor growth in both animal models and in addition extended the survival of the Tyr-Cre/Ptennull/BrafV600E mice.
To our knowledge, this is the first report that ITK is highly expressed in non-hematopoietic tissues and even more so by a solid tumor malignancy, such as melanoma. One group reported low level expression of ITK mRNA in colon cancer tumor tissues, but commented that its expression was probably derived from contaminating host cells (41). Previous studies about the putative oncogenic role of ITK have been limited to its increased or aberrant expression in T-cell malignancies (42-44). It has also been reported that an ITK inhibitor had cytotoxic effects on malignant T cells (43). We are not aware of previous studies reporting ITK inhibition of solid tumor malignancies.
Our review of the 226 melanoma samples that have undergone next generation sequencing and putative copy-number analysis as part of The Cancer Genome Atlas (TCGA) Project (45, 46) using the GISTIC tool revealed no evidence of copy number alterations or somatic mutations that could account for the ITK expression in the cells or clinical samples reported here. The lack of ITK gene copy number alterations in the TCGA melanomas together with our finding that melanomas are hypomethylated in the ITK promoter relative to nevi (10) and express ITK protein suggests that epigenetic rather than genetic mechanisms, at least in part, account for the high ITK expression in melanoma.
Therapeutic targeting of ITK in melanoma should critically consider its bystander effect on the host immune response given the overwhelming evidence that a functioning immune system is fundamental for antitumor responses against this disease. Of note, our investigations failed to demonstrate ITK expression in several tumor-infiltrating immune cell subsets such as T cells, polymorphonuclear cells, mast cells, natural killer cells and macrophages. Importantly, the antitumor effect of BI 10N remained significant in immunocompetent mice with melanoma, suggesting at least the lack of a significant negative effect of ITK inhibition on the host anti-tumor response.
In this work, we characterized the kinase inhibitor profile of BI 10N, a small molecule selective inhibitor of ITK previously used in animal studies (21). BI 10N was highly selective for ITK over a large panel of kinases, but not for 3 TRK family members and the SFK member YES. Although TRKA and TRKB are highly expressed in melanoma (47), the effects of BI 10N are most likely mediated through the inhibition of ITK as the effects of BI 10N on melanoma cells are similar to those achieved with ITK depletion using shRNAs.
A limitation to our work is that the mechanism by which ITK increases cell proliferation and migration remains unknown. BI 10N treatment did not change AKT and ERK phosphorylation levels in RPMI 8322 cells. However, evidence presented indicates that the mechanism by which ITK activity drives the cell cycle may involve changes in the p53 pathway, and in particular modulation of cyclin levels, but further studies are needed to clarify these issues.
In summary, ITK appears to be a driver of melanoma as demonstrated by its expression in most of the metastatic melanomas examined and by the significant effects of BI 10N on the proliferation and migration of melanoma cells and its efficacy in mouse melanoma models. We are currently investigating rational treatment combinations between ITK inhibitors and standard treatments for metastatic melanoma in various preclinical melanoma models that may serve as the basis for future clinical trials in metastatic melanoma. Besides BI 10N, several other ITK inhibitors previously have been developed to target Th2 dominant autoimmune, inflammatory, and infectious diseases (48, 49). Notably, ibrutinib (IMBRUVICA, Pharmacyclics, Inc.), an irreversible inhibitor of BTK and ITK (50), has been granted approval by the U.S. Food and Drug Administration. We conclude that ITK is a novel target for the treatment of melanoma that, at least in animal models, is amenable to pharmacologic intervention.
Supplementary Material
Translational Relevance.
Emerging systemic therapies for metastatic melanoma extend life but are rarely curative for the majority of patients. Our finding that interleukin-2 inducible T-cell kinase (ITK), a TEC family tyrosine kinase, is aberrantly expressed in most metastatic melanomas suggests that inhibitors of ITK might be useful for melanoma treatment. The efficacy of a small molecule ITK inhibitor in the Tyr-Cre/Ptennull/BrafV600E mouse melanoma model suggests that ITK is a driver of melanoma progression and a potential therapeutic target. Ibrutinib, an inhibitor of the TEC family Bruton's tyrosine kinase, also inhibits ITK and was recently FDA-approved for treatment of hematologic malignancies. Therefore, considering the clinical success of targeting tyrosine kinases for treatment of other malignancies, trials using ITK inhibitors for treatment of human melanoma, alone or in combination with existing therapies, are warranted.
Acknowledgments
The authors wish to thank M. Olorvida and B. Midkiff (UNC TPL) for assistance with staining and tissue imaging analysis of the immunofluorescence experiments. We thank the UNC MP1U personnel for assistance with animal handling, therapeutic studies and tumor serial assessment. We thank UNC Dermatopathology (director: Dr. Daniel Zedek) for immunostaining of the metastatic tumor tissues with the VE1 BRAFV600E mutation specific antibody. We thank Dr. Marcus W. Bosenberg M.D., Ph.D. for kindly providing the 4-hydroxytamoxifen Tyr::CreER; BrafCA; Ptenlox/lox mouse for our in vivo studies. UNC-CH's Lenti-shRNA Core Facility supplied lentiviral small hairpin RNA. We would like to thank Dr. Alan Houghton and Memorial Sloan-Kettering Cancer Center for providing the SKMEL cell lines used in this research.
Funding sources: This work was supported by National Cancer Institute (NCI) grants R33CA10704339 (N.E.T; K.C.), R21CA134368 (N.E.T; K.C.), R01CA112243 (N.E.T), and P30CA016086; National Institute of Environmental Health Sciences (NIEHS) grant P30ES010126; University Cancer Research Fund (UCRF) at University of North Carolina (UNC) Chapel Hill's Lineberger Comprehensive Cancer Center (LCCC) (N.E.T); Harry J. Lloyd Charitable Trust (N.E.T.; S.J.M.); UNC LCCC Developmental Grant; The V Foundation (N.E.T; D.W.O.); Doris Duke Charitable Foundation Clinical Research Fellowship (S.P); National Center for Advancing Translational Sciences award ULTR000083 (N.E.T.); and The Irene and Robert Alan Briggaman Distinguished Professorship (N.E.T.). UCRF supported, in part, work in the Mouse Phase 1 Unit (MP1U) and the UNC Translational Pathology Laboratory (TPL).
Footnotes
Disclosure of Potential Conflicts of Interest. The authors have no conflict of interest to disclose.
References
- 1.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–65. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 3.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–14. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 4.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–63. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105–16. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 7.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–44. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conway K, Edmiston SN, Khondker ZS, Groben PA, Zhou X, Chu H, et al. DNA-methylation profiling distinguishes malignant melanomas from benign nevi. Pigment Cell Melanoma Res. 2011;24:352–60. doi: 10.1111/j.1755-148X.2011.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andreotti AH, Schwartzberg PL, Joseph RE, Berg LJ. T-cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harb Perspect Biol. 2010;2:a002287. doi: 10.1101/cshperspect.a002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mourad-Zeidan AA, Melnikova VO, Wang H, Raz A, Bar-Eli M. Expression profiling of Galectin-3-depleted melanoma cells reveals its major role in melanoma cell plasticity and vasculogenic mimicry. Am J Pathol. 2008;173:1839–52. doi: 10.2353/ajpath.2008.080380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zabierowski SE, Baubet V, Himes B, Li L, Fukunaga-Kalabis M, Patel S, et al. Direct reprogramming of melanocytes to neural crest stem-like cells by one defined factor. Stem Cells. 2011;29:1752–62. doi: 10.1002/stem.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sambade MJ, Peters EC, Thomas NE, Kaufmann WK, Kimple RJ, Shields JM. Melanoma cells show a heterogeneous range of sensitivity to ionizing radiation and are radiosensitized by inhibition of B-RAF with PLX-4032. Radiother Oncol. 2011;98:394–9. doi: 10.1016/j.radonc.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carson C, Omolo B, Chu H, Zhou Y, Sambade MJ, Peters EC, et al. A prognostic signature of defective p53-dependent G1 checkpoint function in melanoma cell lines. Pigment Cell Melanoma Res. 2012;25:514–26. doi: 10.1111/j.1755-148X.2012.01010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nikolaishvilli-Feinberg N, Cohen SM, Midkiff B, Zhou Y, Olorvida M, Ibrahim JG, et al. Development of DNA damage response signaling biomarkers using automated, quantitative image analysis. J Histochem Cytochem. 2014;62:185–96. doi: 10.1369/0022155413516469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi SR, Imam SA, Young L, Cote RJ, Taylor CR. Antigen retrieval immunohistochemistry under the influence of pH using monoclonal antibodies. J Histochem Cytochem. 1995;43:193–201. doi: 10.1177/43.2.7822775. [DOI] [PubMed] [Google Scholar]
- 19.Thomas NE, Edmiston SN, Alexander A, Millikan RC, Groben PA, Hao H, et al. Number of nevi and early-life ambient UV exposure are associated with BRAF-mutant melanoma. Cancer Epidemiol Biomarkers Prev. 2007;16:991–7. doi: 10.1158/1055-9965.EPI-06-1038. [DOI] [PubMed] [Google Scholar]
- 20.Pearlstein MV, Zedek DC, Ollila DW, Treece A, Gulley ML, Groben PA, et al. Validation of the VE1 immunostain for the BRAF V600E mutation in melanoma. J Cutan Pathol. 2014;41:724–32. doi: 10.1111/cup.12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riether D, Zindell R, Kowalski JA, Cook BN, Bentzien J, Lombaert SD, et al. 5-Aminomethylbenzimidazoles as potent ITK antagonists. Bioorg Med Chem Lett. 2009;19:1588–91. doi: 10.1016/j.bmcl.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 22.Williams HC, San Martin A, Adamo CM, Seidel-Rogol B, Pounkova L, Datla SR, et al. Role of coronin 1B in PDGF-induced migration of vascular smooth muscle cells. Circulation research. 2012;111:56–65. doi: 10.1161/CIRCRESAHA.111.255745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu W, Ju Z, Lu Y, Mills GB, Akbani R. A comprehensive comparison of normalization methods for loading control and variance stabilization of reverse-phase protein array data. Cancer Inform. 2014;13:109–17. doi: 10.4137/CIN.S13329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, Jr, et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41:544–52. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roberts PJ, Usary JE, Darr DB, Dillon PM, Pfefferle AD, Whittle MC, et al. Combined PI3K/mTOR and MEK inhibition provides broad antitumor activity in faithful murine cancer models. Clin Cancer Res. 2012;18:5290–303. doi: 10.1158/1078-0432.CCR-12-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ullman-Cullere MH, Foltz CJ. Body condition scoring: a rapid and accurate method for assessing health status in mice. Laboratory animal science. 1999;49:319–23. [PubMed] [Google Scholar]
- 27.Simon R, Lam A, Li MC, Ngan M, Menenzes S, Zhao Y. Analysis of gene expression data using BRB-ArrayTools. Cancer Inform. 2007;3:11–7. [PMC free article] [PubMed] [Google Scholar]
- 28.Becker KG, White SL, Muller J, Engel J. BBID: the biological biochemical image database. Bioinformatics. 2000;16:745–6. doi: 10.1093/bioinformatics/16.8.745. [DOI] [PubMed] [Google Scholar]
- 29.Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–80. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schaefer CF, Anthony K, Krupa S, Buchoff J, Day M, Hannay T, et al. PID: the Pathway Interaction Database. Nucleic Acids Res. 2009;37:D674–9. doi: 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mi H, Guo N, Kejariwal A, Thomas PD. PANTHER version 6: protein sequence and function evolution data with expanded representation of biological pathways. Nucleic Acids Res. 2007;35:D247–52. doi: 10.1093/nar/gkl869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berger AJ, Davis DW, Tellez C, Prieto VG, Gershenwald JE, Johnson MM, et al. Automated quantitative analysis of activator protein-2alpha subcellular expression in melanoma tissue microarrays correlates with survival prediction. Cancer Res. 2005;65:11185–92. doi: 10.1158/0008-5472.CAN-05-2300. [DOI] [PubMed] [Google Scholar]
- 33.Camp RL, Chung GG, Rimm DL. Automated subcellular localization and quantification of protein expression in tissue microarrays. Nat Med. 2002;8:1323–7. doi: 10.1038/nm791. [DOI] [PubMed] [Google Scholar]
- 34.Dai M, Wei H, Yip YY, Feng Q, He K, Popov V, et al. Long-lasting complete regression of established mouse tumors by counteracting Th2 inflammation. J Immunother. 2013;36:248–57. doi: 10.1097/CJI.0b013e3182943549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeck WR, Parker J, Carson CC, Shields JM, Sambade MJ, Peters EC, et al. Targeted next generation sequencing identifies clinically actionable mutations in patients with melanoma. Pigment Cell Melanoma Res. 2014 Mar 15; doi: 10.1111/pcmr.12238. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gomez-Rodriguez J, Kraus ZJ, Schwartzberg PL. Tec family kinases Itk and Rlk / Txk in T lymphocytes: cross-regulation of cytokine production and T-cell fates. The FEBS journal. 2011;278:1980–9. doi: 10.1111/j.1742-4658.2011.08072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fischer AM, Mercer JC, Iyer A, Ragin MJ, August A. Regulation of CXC chemokine receptor 4-mediated migration by the Tec family tyrosine kinase ITK. J Biol Chem. 2004;279:29816–20. doi: 10.1074/jbc.M312848200. [DOI] [PubMed] [Google Scholar]
- 38.Wu C, Asokan SB, Berginski ME, Haynes EM, Sharpless NE, Griffith JD, et al. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell. 2012;148:973–87. doi: 10.1016/j.cell.2011.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kashem MA, Nelson RM, Yingling JD, Pullen SS, Prokopowicz AS, 3rd, Jones JW, et al. Three mechanistically distinct kinase assays compared: Measurement of intrinsic ATPase activity identified the most comprehensive set of ITK inhibitors. Journal of biomolecular screening. 2007;12:70–83. doi: 10.1177/1087057106296047. [DOI] [PubMed] [Google Scholar]
- 40.Wilcox HM, Berg LJ. Itk phosphorylation sites are required for functional activity in primary T cells. J Biol Chem. 2003;278:37112–21. doi: 10.1074/jbc.M304811200. [DOI] [PubMed] [Google Scholar]
- 41.Chen WS, Kung HJ, Yang WK, Lin W. Comparative tyrosine-kinase profiles in colorectal cancers: enhanced arg expression in carcinoma as compared with adenoma and normal mucosa. Int J Cancer. 1999;83:579–84. doi: 10.1002/(sici)1097-0215(19991126)83:5<579::aid-ijc1>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 42.Kaukonen J, Savolainen ER, Palotie A. Human Emt tyrosine kinase is specifically expressed both in mature T-lymphocytes and T-cell associated hematopoietic malignancies. Leukemia & lymphoma. 1999;32:513–22. doi: 10.3109/10428199909058409. [DOI] [PubMed] [Google Scholar]
- 43.Guo W, Liu R, Ono Y, Ma AH, Martinez A, Sanchez E, et al. Molecular characteristics of CTA056, a novel interleukin-2-inducible T-cell kinase inhibitor that selectively targets malignant T cells and modulates oncomirs. Molecular pharmacology. 2012;82:938–47. doi: 10.1124/mol.112.079889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shin J, Monti S, Aires DJ, Duvic M, Golub T, Jones DA, et al. Lesional gene expression profiling in cutaneous T-cell lymphoma reveals natural clusters associated with disease outcome. Blood. 2007;110:3015–27. doi: 10.1182/blood-2006-12-061507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.The Human Protein Atlas.org [Internet] NAlbaNova and SciLifeLab, KTH - Royal Institute of Technology; Stockholm, Sweden: the Rudbeck Laboratory, Uppsala University; Uppsala, Sweden: Lab Surgpath; Mumbai, India: Version: 13 [updated 2014 Nov 06 cited 2014 Dec 19]. Available from: http://www.proteinatlas.org/ [Google Scholar]
- 48.Charrier JD, Knegtel RM. Advances in the design of ITK inhibitors. Expert opinion on drug discovery. 2013;8:369–81. doi: 10.1517/17460441.2013.769520. [DOI] [PubMed] [Google Scholar]
- 49.Lo HY. Itk inhibitors: a patent review. Expert opinion on therapeutic patents. 2010;20:459–69. doi: 10.1517/13543771003674409. [DOI] [PubMed] [Google Scholar]
- 50.Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122:2539–49. doi: 10.1182/blood-2013-06-507947. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.