

Abstract
Clival chordoma is a rare primary bone tumour that arises from the remnant of the notochord and typically occurs in older adults. Upon imaging, the tumour can be seen arising from the clivus and causes clival destruction. This usually provides insight for a diagnosis. Here we present a case of a non-enhancing, pre-pontine mass that was hypointense on T1W and hyperintense on T2W in an adolescent. No clival bone erosion was observed. Based on the age group, imaging findings, and lack of clival erosion, a provisional diagnosis of epidermoid cyst was made and the tumour was resected. This patient was eventually diagnosed with a clival chordoma based on histopathological examination.
Keywords: magnetic resonance imaging, adolescent, clivus, chordoma, atypical
Introduction
Chordomas are rare primary bone tumours that arise from embryonic remnants of the notochord and typically occur in the clivus (spheno-occipital) region or the sacrum. Chordomas are locally aggressive with a high risk of local recurrence (1,2). The mean age at diagnosis is 58.5 years (3), with cases rarely reported in children and adolescents (4). Imaging by computed tomography (CT) or magnetic resonance imaging (MRI) can identify clival chordoma based on its midline location and destruction of the clivus (5). This case report describes our diagnosis of clival chordoma in an adolescent with no clival destruction seen on MRI or CT.
Case Report
A previously healthy 15-year-old girl was referred to our neurosurgical center with a seven-month history of a headache that had worsened in the previous week and was accompanied by intermittent nausea. She also felt weakness on the left side of her face and complained of double vision for the previous five days.
Examination showed left lower motor neuron facial nerve palsy with partial ptosis of the left eyelid. The rest of the cranial nerves were intact as well as the musculoskeletal systems bilaterally. Eye examination showed bilateral papilloedema, and left eye keratitis secondary to facial nerve palsy was detected. Her blood pressure was low at 103/76 mmHg and pulse rate was high at 106 beats/min. Biochemical analysis showed normal levels of prolactin, growth hormone (GH), thyroid stimulating hormone (TSH), follicle-stimulating hormone (FSH), luteinising hormone (LH), and early morning cortisol.
A contrast-enhanced computed tomography (CT) of the patient’s brain performed by the referring doctor demonstrated a well-defined, non-enhancing hypodense mass at the prepontine region. The mass compressed the pons and fourth ventricle, resulting in acute obstructive hydrocephalus with cerebrospinal fluid seepage (Figure 1).
Figure 1:
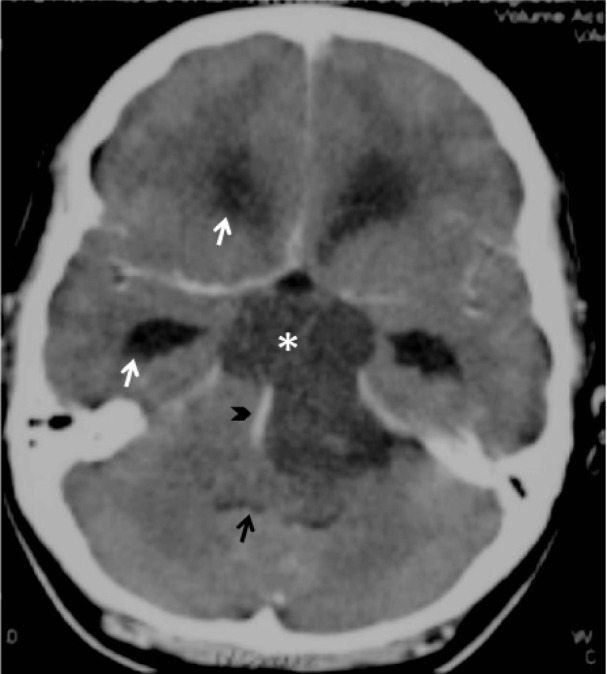
Post-contrast computed tomography (CT) brain shows a well-defined, nonenhancing hypodense mass (*) at the prepontine region compressing the pons and fourth ventricle (black arrow), resulting in obstructive hydrocephalus evidenced by the dilated temporal and frontal horn of lateral ventricle (white arrow). The basilar artery is displaced laterally but remained patent (black arrowhead).
An MRI performed at our center showed a well-defined, lobulated prepontine mass that was predominantly hypointense on T1W and Fluid attenuation inversion recovery (FLAIR) sequences and hyperintense on T2W, suggesting a lesion with fluid content. Small scattered areas of hyperintensity were evident on T1W and FLAIR, possibly representing foci of haemorrhage. No enhancement was seen post-contrast. In addition, no tumour calcification was observed on CT or MRI. The mass extended from the sellar region superiorly and inferiorly towards the craniocervical junction. No suprasellar extension was seen. The pituitary was intact. The clivus was also intact with no destruction or expansion seen. The clival marrow demonstrated normal mixed signal intensity on T1 pre-contrast sequence, which corresponded to mixed red and yellow marrow expected in this age group. There was also mild enhancement of the clivus post-contrast, which was normal (6,7). The basilar artery was patent and displaced posteromedially. The effect of the mass on the brainstem and fourth ventricle resulting in acute hydrocephalus was in agreement with that observed on the previous CT scan (Figure 2). Although we noted atypical findings, a differential diagnosis of epidermoid cyst was made.
Figure 2:

Magnetic resonance imaging (MRI) (a) sagittal T1W, (b) axial T2W and (c) sagittal T1W. Fat suppressed post-contrast shows a well defined, lobulated prepontine mass (*), which is predominantly hypointense on T1W (a) and hyperintense on T2W (b), suggesting a lesion with fluid content. No enhancement is seen post contrast (c). Small scattered areas of hyperintensity on T1W may represent foci of haemorrhage (black arrowhead). The pituitary (white arrow) and clivus (black arrow) are intact.
A craniotomy was performed and excision of the tumour was conducted using a subfrontal approach. The surgery was limited by space, making it difficult to properly evaluate the clivus. The tumour was retroclival in location and occupied the entire pre-pontine, pre-medullary, and crural cistern. It extended from the foramen magnum up to the level of the upper pons. The tumour was greyish in colour, and soft and semisolid in texture. The cranial nerves were stretched and encased by the tumour. The surgical team removed as much of the tumour as possible.
Histopathological examination (HPE) showed that the entire specimen consisted of tumour with no accompanying normal tissue. The tumour was homogenous with large vacuolated cells and some myxoid background (Figure 3). Vacuolated cells, also known as physaliferous cells, characterise a chordoma. There are an admixture of macrophages, lymphocytes, plasma cells, and eosinophils within the interlobules septae. Varying amounts of myxoid matrix were also present in the background (Figure 4). Thick fibrous tissue was not observed, suggesting the absence of a dural component. In addition, no loose fibroconnective tissue was observed that would suggest periosteum. The tumour cells were arranged in sheets and lobules separated by vascularised fibrous septae, which is best highlighted by the immunohistochemical staining shown in Figure 5. The staining shows positivity for pan cytokeratin, epithelial membrane antigen (EMA), and S100 protein. It was negative for glial fibrillary acidic protein (GFAP). Taking into account the retroclival location of the tumour, a diagnosis of clival chordoma was made.
Figure 3:

(a) Hematoxylin and eosin stain (×20 magnification) and (b) The tumour is homogenous and made up of vacuolated cells.
Figure 4:

(a, b) Hematoxylin and eosin stain (×200 magnification): Vacuolated cells (physaliferous cells) (black arrow) on a myxoid background (*) with adjacent fibrovascular septa (arrowhead) filled with mixed inflammatory cells.
Figure 5:

(a) Pan-cytokeratin immunohistochemistry (×40 magnification) and (b) (×200 magnification) Cytokeratin positive tumour cells (stained brown) are arranged in lobules and separated by fibrous septa (arrows). Some tumour cells are arranged in cords within some of the lobules (*).
This patient was subsequently referred to an oncology center for further clinical management. She required a second excision 13 months later as the residual tumour was increasing in size. The patient also completed 3 months of radiotherapy. She is currently doing well and attending school again while being closely followed-up. The most recent MRI performed two years post-surgery showed no progression of the residual tumour.
Discussion
Chordoma is a rare primary bone tumour that accounts for 2–5% of all primary bone tumours (8). It arises from embryonic remnants of the primitive notochord, which are entrapped within the cranial and caudal ends of the vertebral column (1,2). Chordomas have an incidence rate of 0.08 per 100,000, with a median age at diagnosis of 58.5 years and prevalence in males (3). Previous reports have described cases of clival chordoma in a younger population. In a study of cases over a 15-year span in his center, Menezes et al. reported eight cases of intracranial chordoma in children (4). McMaster’s analysis of chordoma incidence in the United States over an 18-year period found that 33/400 (8.3%) cases occur in patients less than 25 years of age, with propensity towards cranial presentation (3). Therefore, based on demographics alone, it was difficult to diagnose clival chordoma in this 15-year-old female patient.
Imaging is useful not only for diagnostic purposes, but also for selection of operative approaches. However, this case proved to be diagnostically challenging. On CT scan, the typical description of a clival chordoma is a well-defined, hyperdense soft-tissue mass with moderate to marked enhancement post-contrast. Destruction of the clivus is detectable, which may also give rise to the appearance of intratumoural calcification. On MRI, the tumour will be lobulated, intermediate to hypointense on T1W, and very hyperintense on T2W, secondary to high fluid content of vacuolated cellular components. Involvement of the clivus will be clear with a “thumb” of tissue indenting the pons posteriorly. Similar to a CT scan, the tumours show moderate to marked enhancement post-contrast on MRI (2,8).
As described above, our patient’s imaging results did not conform to the typical description. The pre-contrast appearance was suggestive of chordoma; however, there was lack of enhancement post-contrast. Although the tumour was in close contact with the clivus, the lack of bony destruction, cortical erosion, and marrow involvement suggested the lack of clivus involvement. In addition, the imaging results made clival chordoma even less likely in our patient given the typical demographics of these tumours. A CT viewed in a bone window would best demonstrate the lack of clival destruction; however, the bone window images were not included in the printed films sent to us from the other center where the procedure was performed. Nevertheless, the reporting radiologist did note the absence of clival destruction in the report.
In an analysis of 42 skull base-chordoma and chondrosarcoma cases by Pamir et al., only two (4.8%) of the lesions did not enhance on MRI and two (4.8%) did not have bone destruction on CT, one being an intradural lesion and another a chondrosarcoma in the cavernous sinus (9). Moreover, Niida et al. reported a case of a 5-year-old boy with clival chordoma without bone involvement, which was similar to our patient (10).
We acknowledge that this lesion does not conform to the typical appearance of an epidermoid cyst, which was a point factored into the differential diagnosis. However, this case was diagnosed as such because epidermoid cysts have a similar appearance to this lesion on MRI. Other differential diagnoses of a prepontine mass include a hypothalamus hamartoma and a midline lipoma. However, hypothalamus hamartoma is unlikely, as this lesion clearly did not arise from the hypothalamic region. In contrast to our imaging findings, a midline lipoma would be hyperintense on T1W, T2W, and FLAIR due to the fat content.
We postulate that this lesion may have been intradural, thus explaining why no bone involvement was observed. This was not apparent in the pre-operative imaging or in the surgical findings. The pathology findings also did not identify any dural component. However, the absence of a dural component does not exclude the possibility of an intradural lesion (11). Intradural chordoma is very rare and has been previously reported (12–14). Another hypothesis is that this may be an extraosseus chordoma that developed from ectopic notochord remnants, which have been found intradurally anterior to the pons in 2% of autopsies (15).
Vacuolated cells, also known as physaliferous cells, characterise a chordoma. Intracranial tumours that may resemble chordomas include a rare variant of meningioma, chordoid meningioma, and third ventricular chordoid glioma (16). No region of the tissuespecimen exhibited recognisable features of meningioma. Moreover, a diffuse and strong positivity for pan cytokeratin allowed us to differentiate chordoma from chordoid meningioma. Regarding a diagnosis of third ventricular chordoid glioma, this tumor is typically negative for Glial fibrillary acidic protein (GFAP) and positive for Epithelial Membrane Antigen (EMA) and pan cytokeratin. Thus, we could exclude a diagnosis of chordoid glioma. The microscopic features described above together with the immunohistochemical (IHC) studies are consistent with a chordoma. We note that microscopically, there was no loose fibroconnective tissue to suggest the presence of periosteum. Although the presence of fibroconnective tissue may be helpful, it does not unequivocally indicate a periosteal origin (11).
Surgery continues to be the primary modality in the management of chordoma. Lanzino et al. proposed cytoreductive resection to reduce tumour bulk and minimise complications. Aggressive resection may not be possible, particularly when important structures such as cavernous sinuses and cranial nerves are involved (17). Radiotherapy has been increasingly used as an adjunct to gain local control, as chordoma is known to have a high local recurrence rate.
Conclusion
In summary, this case report describes our challenge in diagnosing a prepontine mass in an adolescent without imaging evidence of clival involvement. Our findings emphasise that even without clival erosion, clival chordoma should be considered as a differential diagnosis when a lesion is seen in close relation to the clivus, particularly when the precontrast images are suggestive.
Acknowledgments
The authors would like to thank the Director General of Health Malaysia for his permission to publish this paper. We would also like to thank all other staffs of the Department of Neurosurgery and Department of Radiology, Sungai Buloh Hospital, Malaysia for their support. We also thank Dr Fauziah Kassim, neuropathologist from Hospital Kuala Lumpur for her help in facilitating the pathology diagnosis.
Footnotes
Conflict of interest
None.
Funds
None.
Authors’ contributions
Conception and design, drafting of the article: HH, AKR
Critical revision of the article for the important intellectual content: HH, AKR, AAA, AKR, NSB
Final approval of the article: HH, AKR, AAA, AKR, NSB
Provision of study materials or patient: AKR, AAA, NSB
References
- 1.Chugh R, Tawbi H, Lucas DR, Biermann JS, Schuetze SM, Baker LH. Chordoma: The Nonsarcoma Primary Bone Tumor. Oncologist. 2007;12(11):1344–1350. doi: 10.1634/theoncologist.12-11-1344. [DOI] [PubMed] [Google Scholar]
- 2.Erdem E, Angtuaco EC, Van Hemert R, Park JS, Al-Mefty O. Comprehensive Review of Intracranial Chordoma1. Radiographics. 2003;23(4):995–1009. doi: 10.1148/rg.234025176. [DOI] [PubMed] [Google Scholar]
- 3.McMaster M, Goldstein A, Bromley C, Ishibe N, Parry D. Chordoma: incidence and survival patterns in the United States, 1973–1995. Cancer Causes Control. 2001;12(1):1–11. doi: 10.1023/a:1008947301735. doi: 10.1023/A:1008947301735 . [DOI] [PubMed] [Google Scholar]
- 4.Menezes A. Craniovertebral junction neoplasms in the pediatric population. Childs Nerv Syst. 2008;24(10):1173–1186. doi: 10.1007/s00381-008-0598-4. [DOI] [PubMed] [Google Scholar]
- 5.Handa J, Suzuki F, Nioka H, Koyama T. Clivus chordoma in childhood. Surg Neurol. 1987;28(1):58–62. doi: 10.1016/0090-3019(87)90207-2. doi: 10.1023/A:1008947301735 . [DOI] [PubMed] [Google Scholar]
- 6.Kimura F, Kim KS, Friedman H, Russell EJ, Breit R. MR imaging of the normal and abnormal clivus. AJR Am J Roentgenol. 1990;155(6):1285–1291. doi: 10.2214/ajr.155.6.2122682. doi: 10.1023/A:1008947301735 . [DOI] [PubMed] [Google Scholar]
- 7.Oyar O, Govsa F, Sener RN, Kayalioglu G. Assessment of normal clivus related to age with magnetic resonance imaging. Surg Radiol Anat. 1996;18(1):47–49. doi: 10.1007/BF03207762. doi: 10.1023/A:1008947301735 . [DOI] [PubMed] [Google Scholar]
- 8.Osborn AG. Philadelphia (US): Lippincott Williams & Wilkins Amirsys; 2013. Chordoma. Osborn's Brain: Imaging, Pathology and Anatomy; pp. 736–737. [Google Scholar]
- 9.Pamir MN, Özduman K. Analysis of radiological features relative to histopathology in 42 skull-base chordomas and chondrosarcomas. Euro J Radiol. 2006;58(3):461–470. doi: 10.1016/j.ejrad.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 10.Niida H, Tanaka R, Tamura T, Takeuchi S. Clival chordoma in early childhood without bone involvement. Childs Nerv Syst. 1994;10(8):533–535. doi: 10.1007/BF00335078. doi: 10.1007/BF00334959 . [DOI] [PubMed] [Google Scholar]
- 11.Burkitt HG, Young B, Heath JW. 3rd ed. Edinburgh (UK): Churchill Livingstone; 1995. Wheater’s Functional Histology. A Text and Colour Atlas. [Google Scholar]
- 12.Choo YS, Joo SW, Noh SJ, Lee SI. Intradural Retroclival Chordoma. J Korean Neurosurg Soc. 2009;46(2):152–155. doi: 10.3340/jkns.2009.46.2.152. doi: 10.3340/jkns.2009.46.2.152 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ito E, Saito K, Nagatani T, Ishiyama J, Terada K, Yoshida M, et al. Intradural cranial chordoma. World Neurosurg. 2010;73(3):194–197;e31. doi: 10.1016/j.surneu.2009.01.003. doi: 10.1016/j.surneu.2009.01.003 . [DOI] [PubMed] [Google Scholar]
- 14.Tashiro T, Fukuda T, Inoue Y, Nemoto Y, Shakudo M, Katsuyama J. Intradural chordoma: case report and review of the literature. Neuroradiology. 1994;36(4):313–315. doi: 10.1007/BF00593269. [DOI] [PubMed] [Google Scholar]
- 15.Ribbert H, Steiner H. [Place of publication unknown]: Zentralbl Allg Pathol Anat; 1894. Uber die Ecchondrosis Physalifora Sphenooccipitalis; p. 5. [Google Scholar]
- 16.Prayson RA, Cohen ML. Totowa (NJ): Humana Press Inc; 2000. Practical Differential Diagnosis in Surgical Neuropathology. [Google Scholar]
- 17.Lanzino G, Dumont AS, Lopes MBS, Laws ER. Skull base chordomas: overview of disease, management options, and outcome. Neurosurg Focus. 2001;10(3):1–9. doi: 10.3171/foc.2001.10.3.13. [DOI] [PubMed] [Google Scholar]