

Abstract
Background & objectives:
Hepatitis A virus usually causes acute viral hepatitis (AVH) in the paediatric age group with a recent shift in age distribution and disease manifestations like acute liver failure (ALF). This has been attributed to mutations in 5’non-translated region (5’NTR) which affects the viral multiplication. The present study was aimed to carry out the molecular detection and phylogenetic analysis of hepatitis A virus strains circulating in north western India.
Methods:
Serum samples from in patients and those attending out patient department of Pediatric Gastroenterology in a tertiary care hospital in north India during 2007-2011 with clinically suspected AVH were tested for anti-hepatitis A virus (HAV) IgM antibodies. Acute phase serum samples were subjected to nested PCR targeting the 5’NTR region followed by sequencing of the representative strains.
Results:
A total of 1334 samples were tested, 290 (21.7%) were positive for anti-HAV IgM antibody. Of these, 78 serum samples (< 7 days old) were subjected to PCR and 47.4% (37/78) samples showed the presence of HAV RNA. Children < 15 yr of age accounted for majority (94%) of cases with highest seropositivity during rainy season. Sequencing of 15 representative strains was carried out and the circulating genotype was found to be III A. The nucleotide sequences showed high homology among the strains with a variation ranging from 0.1-1 per cent over the years. An important substitution of G to A at 324 position was shown by both AVH and ALF strains. The cumulative substitution in AVH strains Vs ALF strains as compared to GBM, Indian and prototype strain in the 200-500 region of 5’ NTR was comparable.
Interpretation & conclusion:
Our results showed hepatitis A still a disease of children with III A as a circulating genotype in this region. The mutations at 5’NTR region warrant further analysis as these affect the structure of internal ribosomal entry site which is important for viral replication.
Keywords: Genotype, hepatitis A virus, mutations, phylogenetic analysis, serology
Hepatitis A virus (HAV) accounts for 60-80 per cent cases of clinical hepatitis in the developing countries1,2. The clinical course may vary from asymptomatic, acute self-limiting viral hepatitis to life threatening acute liver failure (ALF). In 90 per cent of the children, the infection is subclinical followed by seroconversion leading to a life-long immunity. It has been seen that severe manifestations are more common in young adults requiring hospitalization in 22.3 to 52.4 per cent cases with overall case fatality rate of 0.3 per cent3. The changing environmental, socio-economic conditions and the availability of a commercially available vaccine have markedly affected the epidemiology of the disease causing a gradual shift in the age distribution to 2nd and 3rd decade of life.
HAV is the only member of the genus Hepatovirus in the family Picornaviridae causing human infection4. HAV is known to display a high degree of antigenic and genetic conservation contrary to the high frequency of genetic changes seen in RNA viruses5,6. Molecular epidemiology of HAV is important to understand the strains circulating in various geographical regions7 and tracing the source of contamination in an outbreak situation8,9. The HAV strains isolated from various parts of the world constitute a single serotype and are divided into six genotypes (I-VI). Genotypes I-III are most commonly associated with human infections and have a variable geographical distribution. Majority of human strains (80%) belong to genotype I. The predominantly circulating genotype in India is genotype III A8,10,11,12. However, a few studies have reported circulation of genotype IA in New Delhi and also co-circulation of genotypes IIIA and IB has been reported from a day care center in Pune, western India13,14,15. The molecular characterization of the infectious agents is important, as it provides the information about the circulating strains in a particular region, the invasion of new strains from different geographical areas and their role in the pathogenesis and severity of the disease.
The aim of this study was to carry out the molecular characterization of the prevalent strains of HAV over a period of four years. This study was carried out in a tertiary care hospital of north west India which caters Chandigarh and the adjoining States of Haryana, Punjab, Himachal Pradesh, Jammu and Kashmir, parts of Uttar Pradesh and Rajasthan.
Material & Methods
The blood samples were received in the department of Virology from patients with clinically suspected viral hepatitis from March 2007 to August 2011 visiting the in- and out-patients of Pediatric Gastroenterology department of the Postgraduate Institute of Medical Education and Research (PGIMER), Chandigarh. The samples in 2009 could not be tested due to the non-availability of ELISA kits during this time. The blood samples were collected and transported in cold chain system for the detection of anti HAV IgM antibodies. The study protocol was approved by the institute's ethical committee. A total of 1334 clotted blood samples were received, the serum was separated, and the vials were coded and stored at -70 ° C in aliquots till tested.
Case definition: The clinical details were available for some of the patients who were admitted with either acute viral hepatitis (AVH) or acute liver failure (ALF). The AVH was defined as the patients presenting with serum aspartate aminotransferase (AST) or alanine aminotransferase (ALT) elevation of at least five-fold with clinical jaundice and without evidence of any chronic liver disease. ALF was defined as biochemical evidence of liver injury, no history of known chronic liver diseases, coagulopathy not corrected by vitamin K administration, international normalized ratio (INR) >1.5 if the patient had encephalopathy or >2.0 if the patient did not have encephalopathy16.
Serology: The serum samples were tested for anti-HAV IgM antibodies (Immunovision, USA) using commercially available IgM capture ELISA kit with a sensitivity and specificity of >99 per cent as per the manufacturer's instructions.
RNA extraction and reverse transcription-polymerase chain reaction (RT-PCR) for detection of HAV genome: Acute phase serum samples (< 7 days old, n=78) positive for anti-HAV IgM antibodies were subjected to nested PCR targeting the 5’NTR (5’-non translated region)17. Briefly, 140 μl of serum was used for RNA extraction using QIAmp Viral RNA Mini Kit (Qiagen, Germany). The RNA was eluted in 50 μl of the elution buffer and used as a template for cDNA synthesis. On the same day, 10 μl of the dissolved RNA was reverse transcribed to cDNA using external anti-sense primers (Sigma, USA) and RevertAid™ First Strand cDNA Synthesis Kit (MBI Fermentas, USA). Five μl of the cDNA produced was used as a template for the first round of nested PCR. Briefly, the final concentration of various reagents used in a reaction volume of 25 μl was; 1X reaction buffer, 0.2 mM dNTP mix, 1.5 mM MgCl2, 0.5 μM of each outer sense and outer antisense and 0.6 U/μl of Taq polymerase. The thermal profile of PCR amplification cycle was: initial denaturation at 94°C for 3 min followed by 35 cycles of denaturation at 95°C for one min; annealing at 50°C for one min and extension at 72°C for seven min. Five μl of amplicons of first round of PCR were used as template for second round of nested PCR. The inner sense and antisense primers were used in second round of nested PCR keeping the thermal profile unchanged. Positive and negative controls were included with each run and all necessary precautions were taken to prevent cross-contamination. Ten μl of second round amplicons were electrophoresed in two per cent agarose gel containing ethidium bromide and examined for specific band of 563 bp under ultraviolet light. Representative second round amplicons of nested PCR were column purified (QIA gel purification kit, Qiagen, Germany) and both the forward and reverse strands were sequenced for 15 representative strains (10 AVH and all the 5 ALF patients) using Big Dye Terminator cycle sequencing Ready Reaction Kit (Applied Biosystems, USA) and an automated sequencer (ABI PRISM 3100 Genetic Analyzer, Applied Biosystems, USA).
Sequence analysis and phylogenetic tree construction: The phylogenetic status of 15 representative 5’NTR positive specimens covering all years of study was analyzed. ABI chromatogram files were viewed using Finch TV 1.4.0, (Geospiza, Inc., USA) following sequence drafting with bioedit 7.0.9 software. For the confirmation of these sequences, database search was implemented using BLAST program available at NCBI website (http://blast.ncbi.nlm.nih.gov/blast.cgi). For sequence comparison, standard representative strains sequences were retrieved from GenBank (http://www.ncbi.nlm.gov/genbank). Clustal X 2.0.11 programme (http:/www.clustal.org/clustal2/) was used for multiple sequence alignment followed by Molecular Evolutionary Genetics Analysis (MEGA4) (Tempe, USA) for phylogenetic tree construction.
Accession numbers and designations of the complete or partial genome sequence employed for the present study were as follows:
Genotype IA: A B020564, AB020567, AB020566, AB020569, AF485328, AF512536, X75215(GBM/WT), Genotype IB: M 20273, AF268396, M 14707, AF314208, Genotype IIA: AY644670, Genotype IIB: AY644676, Genotype IIIA: AJ299464, AY644337, AB279732, AB279733, AB279734, DQ991029, FJ360734.1, Genotype IIIB: AB258387, AB279735, Genotype VI: D00924.1
Statistical analysis: The statistical analysis was carried out using Statistical Package to Social Sciences (SPSS Inc.Chicago, IL, version 15.0 for windows) and P<0.05 was considered significant.
Results
The mean age group of the study subjects was 8.23 ± 9.45 yr with 95 % confidence interval ranging from 5.98 to 10.47 yr and the male: female ratio was 2.6:1. Majority of patients (94%) were below 15 yr of age. Nearly 70 per cent of patients were hospitalized. The seasonal distribution of anti-HAV IgM positivity over the years is shown in Fig. 1.
Fig. 1.
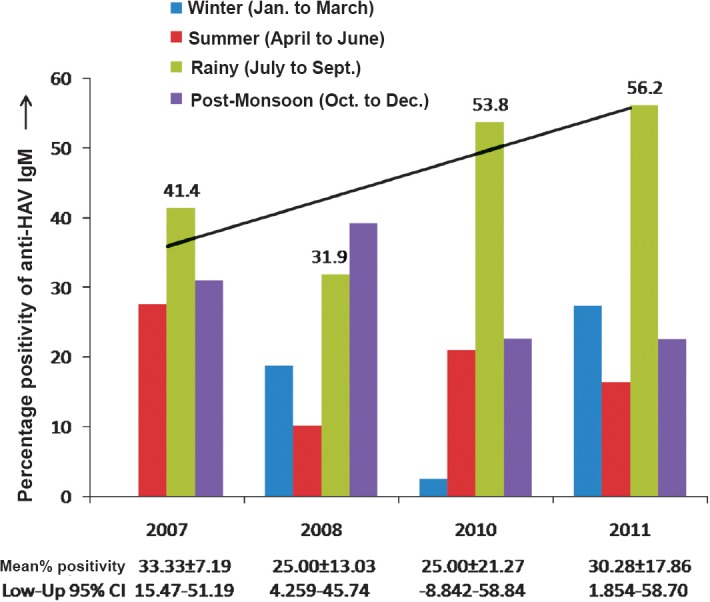
Seasonal distribution of anti-HAV IgM positivity during 2007-2011 (Anti-HAV IgM seropositivity was highest in the rainy season i.e 41.4, 53.8 and 56.2 per cent in the year 2007, 2010 and 2011, respectively, except in the year 2008 where the maximum seropositivity of anti-HAV IgM was observed in the post-monsoon period). The mean ± standard deviation (SD) for anti-HAV IgM seropositivity and lower-upper 95 per cent confidence interval (CI) over the years are represented below the bar graph.
Of the 1334 samples tested, 290 (21.7%) were positive for anti-HAV IgM antibody. Of these, 78 acute serum samples (< 7 days old) were subjected to PCR targeting the 5’NTR of HAV RNA, and 37 (47.4%) samples showed presence of HAV RNA.
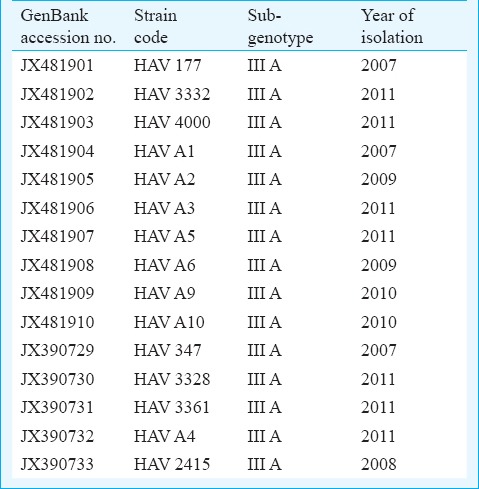
Phylogenetic analysis of HAV isolates: A total of 15 nucleotide sequences from this study have been deposited in the GenBank under the accession numbers from JX390729-JX390733 and JX481901-JX481910 (Table). Phylogenetic analysis was performed using neighbor joining method18 and boot strap values of 1000 replicates were used for statistical verification; >85 per cent similarity has been reported between various genotypes of hepatitis A19. The clustering topology of the phylogram revealed that the HAV strains circulating in our geographical region (2007-2011) belonged to genotype III, subtype IIIA. The sequences obtained over the years showed high level of sequence homology with sequence variability ranging between 0.1-1 per cent. Sequences from AVH and ALF cases showed 97-98 per cent nucleotide sequence identity when compared with the genotype- IIIA prototype strain originally isolated from Panamanian owl monkeys (PA21, accession no. M63026.1).
Table.
GenBank accession numbers, genotype and year of isolation of HAV strains reported from north india
Seven of our strains (JX481901, JX481902, JX481904, JX481906, JX481908, JX390730 and JX390731) clustered along the Indian isolate of HAV (FJ 360734.1) reported from Pune, western India. One of our ALF sequence (JX390732) showed close similarity to the wild type strain of HAV genotype IIIA reported from blood donation camp in Germany AY644337. Three of our sequences (JX481905, JX481910, and JX390729) showed close association with a reported HAV strain from a Guillain-Barre patient with longer duration of viraemia (DQ991029). Another three sequences (JX390733, JX481907, and JX481909) were related to HAV isolate (AJ299464) reported from a HAV outbreak in Norway associated with parental transmission among hemophiliacs and intravenous drug users. One of our strains (JX481903) was related to a strain isolated from Japan AB279732. (Fig. 2).
Fig. 2.

Genetic relatedness of 15 representative HAV strains reported from north India from 2007-2011 with 23 reference HAV strains retrieved from GenBank. Comparison is based on 500 nucleotides from 5’NTR region and analyzed by neighbor-joining method. Bootstrap values of 1000 replicates were used for statistical verification. Our seven strains clustered with isolate from west India, one ALF sequence was closely similar to wild type of HAV IIIA from Germany, three showed close association with HAV strain from a Guillain Barre patient, another three were related to strains isolated from Norway and Japan. Year-wise representation of strains: •, 2007; blank, 2008; □, 2009; ▴, 2010; ○, 2011.
Substitutions between nucleotides 200 and 500 of the 5’NTR region: Interestingly all our strains showed an important substitution of G to A at nucleotide 324 position when compared with wild-type HAV GBM/WT RNA (X75215). As proposed by Fujiwara et al20, this nucleotide is positioned at the beginning of the stem structure of domain IV, and the change increases the free energy resulting in more stable structure. However, no difference was found in distribution of this substitution related to disease severity. On comparison of strains isolated from our region with wild-type HAV GBM/WT RNA (X75215), prototype strain for genotype IIIA (M63026.1) and complete genome sequence of an Indian strain reported from Pune, Maharashtra (FJ360734.1 IND-HAV-99F) nucleotide identities were found to range from 90 to 92 per cent, 97 to 98 per cent and 98 to 99 per cent, respectively. HAV GBM strain was recovered from a human faecal sample collected in the pre-icteric phase of infection in Germany and was named GBM wild type as was adapted for rapid growth and release in HFS (human lung fibroblast cells) and, FRhk-4 cells (Foetal Rhesus monkey kidney-derived cells)21. Genotype circulating in north west India was found to be IIIA. Therefore, the strains were next compared with PA21 which is the prototype strain of HAV genotype IIIA and was originally isolated from Panamanian owl monkey, but has subsequently been found associated with human cases of HAV from Nepal and Northern India during 1989 and 199012. Further, we tried comparing the strain reported in the present study from north west with an indigenous strain. Whole genome sequence of HAV genotype IIIA was available only from western India and, therefore, IND-HAV-99F was selected, which was isolated from stool sample of an icteric patient from western India in 199922. The overall nucleotide substitution in 200-500 nt region as compared to the above mentioned standard strains are represented in Fig. 3a, b and c. Further comparison of our strains with IND-HAV-99F revealed a 161A to G substitution in 5 of 15 (33.3%) cases, 207C to T in 8 of 15 (53.3%) cases and 559A to G in 3 of 15(20%) cases. On comparison with prototype strain of genotype IIIA (PA21) five substitutions were observed in domain III of IRES 105C to T in15 of 15 (100%) cases, 148 T to C in 6 of 15 (40%), 187C to T in 15 of 15(100%), 213G to A in 15 of 15 (100%), 266G to A in 15 of 15 (100%). In domain IV two substitution and one insertion were observed 490G to A in 11 of 15 (73.3%), 517T to C in 15 of 15 (100%) and at position of 431 insertion of G was observed in all the strains.
Fig. 3.
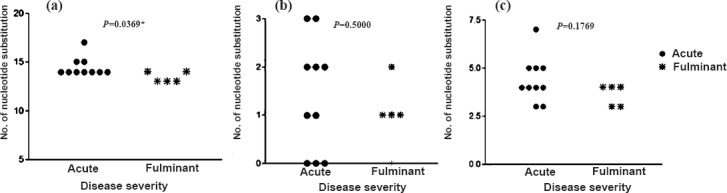
Disease severity and nucleotide substitutions in HAV 5’NTR region (200-500 nt) of standard strains were compared with HAV sequences found circulating in north India reported in the study (a) Wild type strain of HAV genotype IA: GBM/WT (X75215), (b) Whole genome reported from Pune, India genotype IIIA: FJ360734.1, (c) Prototype strain of genotype IIIA: PA21: M63026.1).
On comparison with GBM/WT (X75215) AVH cases had significantly higher number of substitution than ALF cases (14.50 ± 0.97 vs 13.40 ± 0.55; P=0.0369). On comparing with FJ360734.1 Indian strain a higher mean substitution in AVH patients was observed when compared to ALF but the difference was not statistically significant (1.4 ± 1.17 vs 1.0 ± 0.71; P=0.5000). Similar was the observation on comparing AVH and ALF strains with HAV genotype IIIA prototype strain PA21 (M63026.1) (4.4 ± 1.17 vs 3.6 ± 0.55; P=0.1769).
Discussion
Hepatitis A occurs worldwide, however geographic differences exist in its endemicity23. In highly endemic areas like in Asia, Africa, Latin America, and the Middle East, the prevalence of HAV IgG antibodies reaches 90 per cent in adults, and most children have been infected by 10 yr of age2,24. In our study, the predominant age groups affected continued to be children less than 15 yr of age which accounted for 94 per cent of all cases which is in concordance with previous reports from India2,24,25,26. However, a few studies from India have shown a shift in the epidemiology of hepatitis A to older age group, based on the serological surveys1,27. This was because the children belonging to lower socio-economic status excrete HAV in large quantities and a substantial pool of susceptible (anti-HAV negative) adolescents/adults is present among the higher socio-economic class. The molecular detection of HAV RNA ranged from 39.7 per cent in a hospital based study15 to 63.2 per cent in outbreak investigations8. We got a comparable result of 47.4 per cent (37/78).
Using molecular approach two distinct transmission patterns of HAV have been established: (i) infection with indigenous strain, and (ii) infection due to imported strain. Robertson et al7 reported that HAV strains from USA formed a discrete, closely related geographical cluster within genotype IA suggesting existence of circulating endemic population of HAV. In contrast strains collected from Western Europe (Sweden, Germany, Denmark and Switzerland) differed widely in their respective genotypes suggesting little endemic transmission and most cases were related to importation from other endemic regions7. Since, HAV is mostly a self-limiting disease of tropics; data regarding currently circulating strains are limited. In this study, the representative strains collected over the years (2007-2011) from north India were sequenced and found to be genotype IIIA. HAV genotype IIIA has been reported as an aetiological agent of various other water borne outbreaks from northern8, southern10 and western11 India. Hussain et al28 have reported genotype III as the predominant genotype (70%) followed by genotype IA (30%) in Delhi. Co-circulation and co-infections with subgenotypes IIIA and IB have been reported from Pune, western India13. However, none of our isolates belonged to genotype IA and hence we presumed that it was genotype IIIA which was predominantly circulating in our region. In contrast, majority of the circulating strains reported from the adjoining country i.e. China over the year 2003-2008 belonged to genotype I A29.
On evaluating the genetic diversity in the 5’NTR region, it was found that all our strains had 99-99.9 per cent nucleotide identity (PNI) with each other, PNI with an Indian strain reported from western India, Maharashtra (FJ360734.1) was 98-99 per cent and from southern India (DQ004690) was 98-100 per cent, but the query coverage of the sequence was low (41%). When compared with wild-type HAV GBM/WT RNA (X75215) and prototype strain for genotype IIIA (M63026.1) PNI of 90-92 per cent and 97-98 per cent, respectively was observed. This was in concordance with a previous study where the full genome analysis of six Indian isolates obtained from acute sporadic hepatitis A patients from western India showed highly conserved pattern at the nucleotide level which differed from each other by 2.3-3.7 per cent over the entire genome length22.
The variation in the severity of hepatitis A may be attributed to various viral and host factors. Among the viral factors, the proposed factors are duration of viraemia, viral load and unique amino acid substitutions in hepatitis A virus strains, leading to alterations in the surface charge distribution on the VP1 protein. This contributes to neutralization escape of virus leading to a longer duration of viraemia30. Further, association between severity of hepatitis A and nucleotide substitution in 5’ NTR region has also been postulated. Fujiwara et al17 reported whole genome analysis of hepatitis A virus in three cases with AVH and showed possible association between the severity of hepatitis A and nucleotide substitution in 5’NTR region. They also reported that the mutations in 5’NTR were associated with cytopathic variants in cultured cell, and virulence in tamarin17. The 5’NTR region forms a highly ordered secondary structure and contains elements necessary for RNA translation and viral replication. Schultz and colleagues31 have reported that mutations within 5’NTR of cell adapted HAV enhance cap-independent translation directed by HAV internal ribosomal entry site (IRES) in a cell type specific fashion.
Both the AVH and ALF strains reported from our region showed an important substitution of G to A at nucleotide 324 at the beginning of stem structure of domain IV, this change increases the free energy resulting in more stable structure as proposed by Fujiwara et al17. These strains share high nucleotide homology with each other as well as with strains reported from western (FJ360734.1) and southern India (DQ004690). Hence, the 5’NTR region of hepatitis A virus is conserved and may represent a viable antiviral agent. On comparing the mutations in the IRES region with respect to disease severity the strains from AVH patients had higher substitution compared to strains from ALF patients, though the difference was not significant. However, a limitation of our study was that sequences from only five ALF patients were analyzed.
The present findings showed the predominance of HAV genotype IIIA circulating in north west India from 2007-2011. However, other genotypes with limited circulation might be missed because of the restricted number of samples sequenced. Therefore, continuous surveillance is required to get a comprehensive overview of the circulating genotypes. On evaluating the per cent nucleotide identity it was observed that the 5’NTR region of the strains circulating in north west India was highly conserved. Evaluation of the nucleotide substitutions of the strains depending on disease severity i.e AVH versus ALF showed higher number of substitutions in AVH strains than ALF strains but the difference was non significant. Therefore, further pathogenicity studies involving larger number of samples need to be carried out to understand the functional aspect of these mutations. In the present study we were not able to perform the viral load, hence future studies are required to see correlation of viral load with mutation and the disease severity.
Acknowledgment
The authors thank Shriyut Om Prabhash, Junior laboratory technician and B.M. Dhawan, Senior laboratory technician of the Department of Virology for performing the serological tests.
References
- 1.Poddar U, Thapa BR, Prasad A, Singh K. Changing spectrum of sporadic acute viral hepatitis in Indian children. J Trop Pediatr. 2002;48:210–3. doi: 10.1093/tropej/48.4.210. [DOI] [PubMed] [Google Scholar]
- 2.Thapa BR, Singh K, Singh V, Broor S, Singh V, Nain CK. Pattern of hepatitis A and hepatitis B virus markers in cases of acute sporadic hepatitis and in healthy school children from north west India. J Trop Pediatr. 1995;41:328–9. doi: 10.1093/tropej/41.6.328. [DOI] [PubMed] [Google Scholar]
- 3.Wasley A, Grytdal S, Gallagher K. Centers for Disease Control and Prevention (CDC). Surveillance for acute viral hepatitis - United States, 2006. MMWR Surveill Summ. 2008;57:1–24. [PubMed] [Google Scholar]
- 4.Marvil P, Knowles NJ, Mockett AP, Britton P, Brown TD, Cavanagh D. Avian encephalomyelitis virus is a picornavirus and is most closely related to hepatitis A virus. J Gen Virol. 1999;80:653–62. doi: 10.1099/0022-1317-80-3-653. [DOI] [PubMed] [Google Scholar]
- 5.Costa-Mattioli M, Di Napoli A, Ferre V, Billaudel S, Perez-Bercoff R, Cristina J. Genetic variability of hepatitis A virus. J Gen Virol. 2003;84:3191–201. doi: 10.1099/vir.0.19532-0. [DOI] [PubMed] [Google Scholar]
- 6.Nainan OV, Xia G, Vaughan G, Margolis HS. Diagnosis of hepatitis a virus infection: a molecular approach. Clin Microbiol Rev. 2006;19:63–79. doi: 10.1128/CMR.19.1.63-79.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robertson BH, Jansen RW, Khanna B, Totsuka A, Nainan OV, Siegl G, et al. Genetic relatedness of hepatitis A virus strains recovered from different geographical regions. J Gen Virol. 1992;73:1365–77. doi: 10.1099/0022-1317-73-6-1365. [DOI] [PubMed] [Google Scholar]
- 8.Chobe LP, Arankalle VA. Investigation of a hepatitis A outbreak from Shimla Himachal Pradesh. Indian J Med Res. 2009;130:179–84. [PubMed] [Google Scholar]
- 9.Hutin YJ, Pool V, Cramer EH, Nainan OV, Weth J, Williams IT, et al. A multistate, foodborne outbreak of hepatitis A. National Hepatitis A Investigation Team. N Engl J Med. 1999;340:595–602. doi: 10.1056/NEJM199902253400802. [DOI] [PubMed] [Google Scholar]
- 10.Arankalle VA, Sarada Devi KL, Lole KS, Shenoy KT, Verma V, Haneephabi M. Molecular characterization of hepatitis A virus from a large outbreak from Kerala, India. Indian J Med Res. 2006;123:760–9. [PubMed] [Google Scholar]
- 11.Chadha MS, Lole KS, Bora MH, Arankalle VA. Outbreaks of hepatitis A among children in western India. Trans R Soc Trop Med Hyg. 2009;103:911–6. doi: 10.1016/j.trstmh.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 12.Khanna B, Spelbring JE, Innis BL, Robertson BH. Characterization of a genetic variant of human hepatitis A virus. J Med Virol. 1992;36:118–24. doi: 10.1002/jmv.1890360208. [DOI] [PubMed] [Google Scholar]
- 13.Chitambar S, Joshi M, Lole K, Walimbe A, Vaidya S. Cocirculation of and coinfections with hepatitis A virus subgenotypes IIIA and IB in patients from Pune, western India. Hepatol Res. 2007;37:85–93. doi: 10.1111/j.1872-034X.2007.00025.x. [DOI] [PubMed] [Google Scholar]
- 14.Chitambar SD, Chadha MS, Yeolekar LR, Arankalle VA. Hepatitis A in day care centre. Indian J Pediatr. 1996;6:781–3. doi: 10.1007/BF02730929. [DOI] [PubMed] [Google Scholar]
- 15.Hussain Z, Das BC, Husain SA, Asim M, Chattopadhyay S, Malik A, et al. Hepatitis A viral genotypes and clinical relevance: clinical and molecular characterization of hepatitis A virus isolates from northern India. Hepatol Res. 2005;32:16–24. doi: 10.1016/j.hepres.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 16.Bucuvalas J, Yazigi N, Squires RH., Jr Acute liver failure in children. Clin Liver Dis. 2006;10:149–68. doi: 10.1016/j.cld.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 17.Fujiwara K, Yokosuka O, Ehata T, Saisho H, Saotome N, Suzuki K, et al. Association between severity of type A hepatitis and nucleotide variations in the 5’ non-translated region of hepatitis A virus RNA: strains from fulminant hepatitis have fewer nucleotide substitutions. Gut. 2002;51:82–8. doi: 10.1136/gut.51.1.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamura K, Dudlay J, Nei M, Kumar S. MEGA 4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 19.Stene-Johansen K, Jonassen TØ, Skaug K. Characterization and genetic variability of hepatitis A virus genotype IIIA. J Gen Virol. 2005;86:2739–45. doi: 10.1099/vir.0.81155-0. [DOI] [PubMed] [Google Scholar]
- 20.Fujiwara K, Yokosuka O, Imazeki F, Saisho H, Miki M, Omata M. Do high levels of viral replication contribute to fulminant hepatitis A? Liver Int. 2005;25:194–5. doi: 10.1111/j.1478-3231.2004.0981.x. [DOI] [PubMed] [Google Scholar]
- 21.Graff J, Normann A, Feinstone SM, Flehmig B. Nucleotide sequence of wild-type hepatitis A virus GBM in comparison with two cell culture-adapted variants. J Virol. 1994;68:548–54. doi: 10.1128/jvi.68.1.548-554.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kulkarni MA, Walimbe AM, Cherian S, Arankalle VA. Full length genomes of genotype IIIA hepatitis A virus strains (1995-2008) from India and estimates of the evolutionary rates and ages. Infect Genet Evol. 2009;9:1287–94. doi: 10.1016/j.meegid.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 23.World Health Organization. Hepatitis A: Fact Sheet No. 328. [accessed on June 12, 2013]. Available from: http://www.who.int/mediacentre/ factsheets/fs328/en/
- 24.Batra Y, Bhatkal B, Ojha B, Kaur K, Saraya A, Panda SK, et al. Vaccination against hepatitis A virus may not be required for schoolchildren in northern India: results of a seroepidemiological survey. Bull World Health Organ. 2002;80:728–31. [PMC free article] [PubMed] [Google Scholar]
- 25.Khanna S, Vohra P, Jyoti R, Vij JC, Kumar A, Singal D, et al. Changing epidemiology of acute hepatitis in a tertiary care hospital in Northern India. Indian J Gastroenterol. 2006;25:101–2. [PubMed] [Google Scholar]
- 26.Mohanavalli B, Dhevahi E, Menon T, Malathi S, Thyagarajan SP. Prevalence of antibodies to hepatitis A and hepatitis E virus in urban school children in Chennai. Indian Pediatr. 2003;40:328–31. [PubMed] [Google Scholar]
- 27.Arankalle VA, Chadha MS, Chitambar SD, Walimbe AM, Chobe LP, Gandhe SS. Changing epidemiology of hepatitis A and hepatitis E in urban and rural India (1982-98) J Viral Hepat. 2001;8:293–303. doi: 10.1046/j.1365-2893.2001.00279.x. [DOI] [PubMed] [Google Scholar]
- 28.Hussain Z, Husain SA, Almajhdi FN, Kar P. Immunological and molecular epidemiological characteristics of acute and fulminant viral hepatitis A. Virol J. 2011;8:254. doi: 10.1186/1743-422X-8-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao J, Wang Y, Song H, Meng Q, Sheng L, Bian T, et al. Hepatitis A outbreaks in China during 2006: application of molecular epidemiology. Hepatol Int. 2009;3:356–63. doi: 10.1007/s12072-008-9116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joshi MS, Cherian SS, Bhalla S, Chitambar SD. Longer duration of viremia and unique amino acid substitutions in a hepatitis A virus strain [corrected] associated with Guillain-Barre syndrome (GBS) J Med Virol. 2010;82:913–9. doi: 10.1002/jmv.21757. [DOI] [PubMed] [Google Scholar]
- 31.Schultz DE, Honda M, Whetter LE, McKnight KL, Lemon SM. Mutations within the 5’ nontranslated RNA of cell culture-adapted hepatitis A virus which enhance cap-independent translation in cultured African green monkey kidney cells. J Virol. 1996;70:1041–9. doi: 10.1128/jvi.70.2.1041-1049.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]