

Abstract
Acetaminophen (APAP) hepatotoxicity is protected by S-adenosyl-L-methionine (SAMe) treatment 1 hour (h) after APAP in C57/Bl6 mice. This study examined protein carbonylation as well as mitochondrial and cytosolic protein adduction by 4-hydroxynonenal (4-HNE) using mass spectrometry (MS) analysis. Additional studies investigated the leakage of mitochondrial proteins and 4-HNE adduction of these proteins. Male C57/Bl6 mice (n=5/group) were divided into the following groups and treated as indicated: Veh (15 ml/kg water, ip), SAMe (1.25 mmol/kg, ip), APAP (250 mg/kg), and SAMe given 1 h after APAP (S+A). APAP toxicity was confirmed by an increase (p<0.05) in plasma ALT (U/L) and liver weight/10 g body weight relative to the Veh, SAMe and S+A groups 4 h following APAP treatment. SAMe administered 1 h post APAP partially corrected APAP hepatotoxicity as ALT and liver weight/10 g body weights were lower in the S+A group compared the APAP group. APAP induced leakage of the mitochondrial protein, carbamoyl phosphate synthase-1 (CPS-1) into the cytosol and which was reduced in the S+A group. SAMe further reduced the extent of APAP mediated 4-HNE adduction of CPS-1. MS analysis of hepatic and mitochondrial subcellular fractions identified proteins from APAP treated mice. Site specific 4-HNE adducts were identified on mitochondrial proteins sarcosine dehydrogenase and carbamoyl phosphate synthase-1 (CPS-1). In summary, APAP is associated with 4-HNE adduction of proteins as identified by MS analysis and that CPS-1 leakage was greater in APAP treated mice. SAMe reduced the extent of 4-HNE adduction of proteins as well as leakage of CPS-1.
Keywords: Acetaminophen, Hepatotoxicity, 4-Hydroxynonenal, Protein adduction, Oxidative stress, Mass Spectrometry
INTRODUCTION
Acetaminophen (APAP) overdose induces severe hepatic necrosis in humans (Ameer and Greenblatt, 1977) and in rodent models (Lauterburg et al., 1983). APAP overdose is the major cause of drug induced liver failure and accounts for over 56,000 yearly visits to the hospital emergency room by children and adults (Nourjah et al., 2006). The clinical antidote for APAP toxicity is N-acetylcysteine (Smilkstein et al., 1988) which provides cysteine for glutathione (GSH) synthesis. N-Acetylcysteine (NAC) may be administered 17 times over a 72 h period which increases the likelihood of adverse effects and diminishes compliance.
APAP is biotransformed by cytochrome P450 (Cyp) Cyp 2E1, Cyp 3A4 and Cyp 1A2 to form the toxic metabolite N-acetyl-p-benzoquinoneimine (NAPQI) (Corcoran et al., 1980; Dahlin and Nelson, 1982; Patten et al., 1993). NAPQI is a strong electrophile which covalently binds to proteins as well the cysteine sulfhydryl group in the tripeptide glutathione (GSH) (Streeter et al.,1984). GSH depletion is a critical and early event in APAP hepatotoxicity as NAPQI adduction of proteins occurs in human hepatocytes prior to the appearance of APAP hepatic cytotoxicity (Larrauri et al., 1987). APAP-cysteine adducts appear in plasma presumably due to liver injury and extrusion of hepatocyte cell contents. APAP-cysteine adducts detected in human plasma samples correlated with severity of liver injury (James et al., 2009; Heard et al., 2011). In addition to adducting proteins, NAPQI also induces mitochondrial dysfunction leading to a severe energy debt and the formation of reactive oxygen species (ROS) that induce further damage to hepatocytes (Andersson et al., 1990).
Reactive metabolites can mediate post-translational modifications of proteins in plasma and tissues. Covalent protein binding by reactive metabolites are a consequence of exposure to some drugs including APAP as well as environmental agents such as bromobenzene (Mitchell et al., 1976). APAP is biotransformed by cytochrome P450 isozymes to NAPQI which covalently binds to proteins. The mechanism for APAP toxicity is more complicated than the extent of covalent protein binding by NAPQI. 3-Hydroxyacetanilide (AMAP) is a regioisomer of APAP is not hepatotoxic. AMAP is biotransformed to a metabolite which covalently binds to hepatic proteins but AMAP is not hepatotoxic (Tirmenstein and Nelson, 1989; Holme et al., 1991). Differences in subcellular covalent binding have been suggested to account for some differences in toxicity as AMAP metabolites bind with greater affinity to microsomal proteins while APAP metabolites bind with higher affinity to mitochondrial protein. Covalent binding by NAPQI results in a 70% decline in hepatic GSH in less than 2 h in Swiss Webster mice (Rashed et al., 1990) which initiates a second pathway of damage due to induction of oxidative stress (Hinson et al., 1998; Jaeschke et al., 2003). Oxidative stress induced by APAP overdose increases cellular levels of reactive oxygen (ROS) and nitrogen species (RNS). Peroxynitrite is formed by an interaction of superoxide with nitric oxide and can nitrosylate tyrosine residues in hepatic tissue following APAP overdose (Hinson et al., 1998). Reactive aldehyde products from lipid peroxidation can induce covalent modifications of proteins (Amaranth et al., 1998; LoPachin et al., 2004). Lipid peroxidation products such as 4-hydroxynonenal (4-HNE) and 4-oxononenal (4ONE) can covalently adduct to nucleophilic amino acid side chains of cysteine, lysine and histidine while 4ONE can additionally bind to arginine (Doorn and Petersen, 2002; Sayre et al., 2006). 4-HNE adduction to proteins has the potential to impair enzyme activity and cell function. Very few studies have identified specific proteins and the amino acid residues adducted with 4-HNE using liquid chromatography and tandem mass spectrometry (LC-MS/MS) following exposure to drugs or other chemicals. 4-HNE adducted amino acid residues have been identified in hepatic tissue of rodents fed a high fat and alcohol diet (Carbone et al., 2005; Roede et al., 2008). Alcohol ingestion resulted in 4-HNE adduction of protein disulfide isomerase (PDI) which may impair cellular disulfide exchange as PDI enzyme activity was impaired by incubation of PDI with 4-HNE (Carbone et al., 2005).
Previous studies indicated that S-adenosylmethionine (SAMe) reduced APAP hepatotoxicity (Stramentinoli et al., 1979; Bray et al., 1992; Valentovic et al., 2004; Terneus et al., 2007). SAMe reduced ALT levels and the extent of hepatic centrilobular necrosis based on histopathology following a hepatotoxic dose of APAP (Valentovic et al., 2004). Protection was comparable for SAMe to an equimole/10g body weight dose of NAC administered either just prior or 1 h after APAP (Terneus et al., 2007; Terneus et al., 2008). SAMe administered 1 h after APAP reduced hepatic oxidative stress as SAMe reduced the extent of protein carbonylation and GSH depletion (Terneus et al., 2008). SAMe administered 1 h after a hepatotoxic dose of APAP maintained antioxidant enzyme activity and prevented APAP induced mitochondrial 3-nitrotyrosine (3-NT) formation (Brown et al., 2012) indicating that SAMe reduced APAP mediated oxidative stress.
The purpose of this study was to identify APAP induced hepatic 4-HNE adducted proteins using liquid chromatography and mass spectrometry (LC-MS/MS) as all previous reports of 4-HNE adduction have utilized antibody staining which may contain multiple proteins in one gel spot. This study further characterized the amino acid residues modified by 4-HNE following APAP. The studies also explored the protective effect of SAMe on APAP associated protein carbonylation in whole liver and mitochondrial subcellular fractions to evaluate the specificity of cellular protection by SAMe since the mitochondria are a critical target for APAP. Further studies evaluated APAP induced leakage of mitochondrial carbamoyl phosphate synthetase (CPS-1) into the cytosol and whether CPS-1 was adducted with 4-HNE.
MATERIALS AND METHODS
Materials
SAMe toluene sulfonate salt was used for the experiment and was purchased from Sigma Chemical Company (St. Louis, MO). All other chemicals were purchased from Sigma Chemical Company or Fisher Scientific (Pittsburgh, PA). Alanine aminotransferase (ALT) was assayed using a method based on the discontinued Sigma 505-P kit (Patel et al., 2012).
Animals
Male C57Bl/6 mice were purchased from Hilltop Lab Animals Inc. (Scottsdale, PA). Mice were between 4-8 weeks of age, weighing 18-24 g at the time of the experiment. Mice were housed in animal facilities that have full American Association for Accreditation of Laboratory Animal Care accreditation. All studies were approved by the Institutional Animal Care and Use Committee (IACUC) at Marshall University. Mice were maintained in an environment with controlled temperature (21-23°C), constant humidity (40-55%), and a 12 h light cycle (lights on 6:00 AM to 6:00PM). Mice undergo routine examination and serology to ensure the absence of bacterial or viral infection prior to initiation of any experiments. All mice were given a 7 day minimum acclimation period prior to the beginning of any experiments. The animals were fed Purina rodent chow and water ad libitum. Mice were fasted overnight (approximately 16 h) prior to initiating the experiment.
Treatment Groups
Mice were randomly divided between 4 groups (n=5-10/group): Vehicle (Veh), SAMe, APAP and SAMe+ APAP (S+A). Mice were injected intraperitoneal (ip) at 0900 h with Vehicle (Veh; 15 ml/kg water) or APAP (250 mg/kg in warm water 15 ml/kg) followed at 1000 h by SAMe (1.25 mmol/kg in water 5 ml/kg) or water. Mice were anesthetized with carbon dioxide 2 or 4 h after APAP administration. Blood was collected by cardiac puncture in heparin-rinsed 1 mL syringes for determination of serum ALT activity. Hepatic tissue was collected, homogenized and mitochondria and cytosolic fractions were isolated as described below for Western blot and proteomic analysis.
Mitochondrial Isolation
Hepatic mitochondria were isolated using a modification of a previously published protocol by Gogvadze and colleagues (2006). Briefly, the liver was isolated, blotted, weighed and placed in Mitochondrial Isolation Buffer A (225 mM sucrose, 3 mM KH2PO4, 5 mM MgCl2, 20 mM KCl, 20 mM triethanolamine, 2 mM EGTA; pH 7.4). The liver was minced and homogenized in a Dounce homogenizer on ice. Following homogenization, the liver was centrifuged at 600 × g for 10 min. The resultant pellet was discarded and the supernatant was centrifuged at 15,000 × g for 5 min. After the final centrifugation, the supernatant was retained for analysis of cytosolic SAMe levels. The pellet containing the mitochondria was suspended in Mitochondrial Isolation Buffer B (Same as Buffer A except lacking EGTA) for a final concentration of 1 mg tissue weight/1 ml Buffer B. Samples were stored at -80°C until analysis was completed.
Western Blot
Western blot analysis was conducted to examine expression of protein carbonylation, 4-HNE adduction and CPS-1 expression. Protein carbonylation was evaluated using an OxyBlot Kit (Chemicon International S7150) according to the manufacturer's recommendations. Briefly, the kit utilizes an antibody specific to protein carbonyls derivatized chemically with 2,4-Dinitrophenylhydrazine. Most OxyBlot gels used a 40 ug protein load for all samples on a gel. Mitochondrial and cytosolic samples were examined for CPS-1 and then stripped and reprobed with 4-HNE which would be consistent with 4-HNE adduction. A 100 μg cytosolic protein aliquot was denatured by boiling for 5 min. Samples were separated on a 12.5% polyacrylamide gel and transferred to a nitrocellulose (NC) membrane (Whatman; Dassel, Germany). Transfer efficiency was verified using MemCode® Reversible Protein Stain Kit (Thermo Scientific; Rockford, IL). The membrane was then blocked using a 5% (w/v) milk/TBST solution (10 mM Tris-HCl, 150 mM NaCl, 0.1% Tween-20; pH 8.0) for 1 h. Membranes were next incubated overnight with constant shaking at 4°C in antibody for CPS-1 (sc-10515; Santa Cruz Biotechnology; Santa Cruz, CA) at a 1:200 dilution in 5% (w/v) milk/TBST. The membranes were washed 4 times with TBST. The donkey anti-goat Horseradish Peroxidase (HRP) secondary antibody (sc-2020, Santa Cruz Biotechnology; Santa Cruz, CA) was diluted 1:2000 and incubated with the membrane for 1 h. The membrane was again washed with TBST and developed using Amersham™ ECL™ Western Blotting Detection Reagents (GE Healthcare; Buckinghamnshire, UK). The membranes were subsequently stripped and re-exposed to ECL to confirm successful stripping following which they were blocked and reprobed with 1:1000 dilution of Anti-4-HNE Michael Adducts, reduced rabbit primary antibody (393207; Calbiochem; Merck, Darmstadt, Germany). After appropriate washing, membranes were exposed to goat anti-rabbit IgG HRP for 1 h (DC03L; Calbiochem) washed and developed as stated above.
MS Evaluation of Post-translational modifications in whole liver homogenate
A total of 8 spots were selected from the OxyBlot Western which were more intense for the APAP group and attenuated in the S+A group. The bands were excised from a parallel, protein-stained gel, trypsin digested and analyzed on the LCQ (Deca) LC-MS/MS. The data were analyzed with Turbo SEQEST. Data were also analyzed with X!Tandem. A protein hit required confirmation with SEQEST and X!Tandem. The protein sequence database from the 8 bands was further evaluated using PMOD software (Hansen et al., 2005).
MS Evaluation of Post-translational modifications in mitochondrial protein lysate
Mitochondrial samples from the APAP treated mice were run on a 2D SDS-PAGE gel using a pH 3-10 linear IEF strip from GE Healthcare. Protein was transferred electrophoretically to a NC membrane and probed with 4-HNE antibody (1:1000 dilution). A total of 10 4-HNE-immunopositive spots were identified for MS analysis and excised from a parallel, protein-stained gel. For quality control purposes, the spots were analyzed in the following order: 1, 2, 3, 10, 8, 9, 5, 7, 6, 4. The spots of interest were run at the University of Arizona Proteomics center on a LTQ-Orbitrap mass spectrometer. MS/MS spectra were searched against the ipiMouse v 3.87 protein database downloaded on September 27, 2011 (http://www.ebi.ac.uk/IPI/IPImouse.html) using Thermo Proteome Discoverer 1.3 (Thermo Fisher Scientific).
Statistical analysis
All groups contained 5-10 mice/group with the exception of an n=3 for the CPS-1 Western analysis with 4-HNE adduction. Values are reported as Mean ± SEM. Differences between groups were determined using an Analysis of Variance (ANOVA) followed by a post hoc Tukey or Holm-Sidak test using a statistical software package (SigmaStat; SPSS Inc. Chicago, IL). All tests were conducted using a 95% confidence interval. Groups which are statistically different (p<0.05) from each other are noted by different superscript letters (a, b, c) in all figures.
RESULTS
Hepatic function was not altered by SAMe treatment as ALT values and liver weight per 10 g body weight were comparable between Veh and SAMe groups (Figure 1). APAP administration significantly (p<0.05) elevated plasma ALT levels 77 and 113 fold higher at 4 h when compared to the Veh and SAMe groups, respectively (Figure 1). SAMe administration 1 h after APAP reduced APAP liver injury; the S+A group had ALT levels of 660±93 U/L compared to 14,603±1111 U/L in the APAP group (p<0.05). SAMe administered 1 h after APAP did not entirely reverse APAP hepatic toxicity as ALT values were 3 times higher (p<0.05) in the S+A group compared to the Veh and SAMe groups. The ALT values confirm that APAP was hepatotoxic at 4 h and that SAMe administered 1 h after APAP reduced hepatic damage. Liver weight was highest in the APAP group (p<0.05) and was statistically different (p<0.05) from the Veh, SAMe and S+A groups (Figure 1). SAMe administered 1 h after APAP reduced liver weight to a level lower than the APAP group (p<0.05) but higher (p<0.05) than the Veh or SAMe groups. The increased liver weight and ALT values in the APAP group confirmed the dose of APAP was hepatotoxic and that protection was provided by SAMe administered 1 h after APAP.
Figure 1. Plasma ALT and liver weight/10 g body weight in C57Bl/6 mice 4 h after APAP overdose.
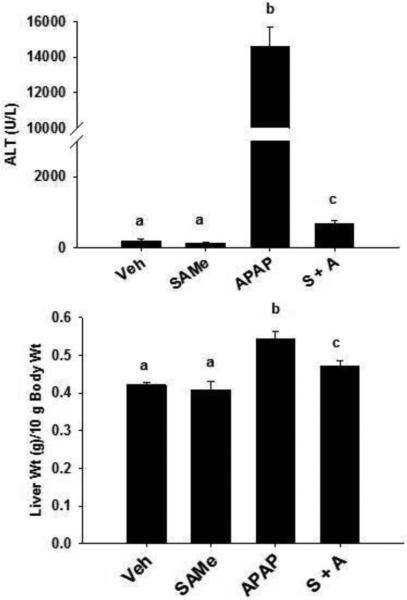
Mice were randomly allocated and treated according to the following groups: Vehicle (Veh, 15 ml/kg water intraperitoneal, ip), SAMe (SAMe, 1.25 mmol/kg in water 5 ml/kg), Acetaminophen (APAP, 250 mg/kg in warm water 15 ml/kg) and SAMe administered 1 h after APAP (S+A). ALT (U/L) was measured using plasma isolated collected 4 h after APAP treatment. Liver weight/10 g body weight is depicted in the bottom Panel. Values represent mean ± S.E.M. with n = 5-10 mice per group. Groups with a different superscript letter (a, b, or c) are statistically different (p<0.05). For example, Veh (a) and SAMe (a) groups were not different from each other for ALT or liver weight/10 g body weight. ALT values were different (p<0.05) between APAP (b) and Veh (a), SAMe (a) and S+A (c) groups.
Protein carbonylation was used as an initial indicator of oxidative stress. Whole liver homogenate was evaluated for protein carbonylation using an OxyBlot kit. Figure 2 (Panel A) confirms that protein loading was comparable between all groups. Protein carbonylation in whole liver homogenate was increased in the APAP group and less pronounced in the S+A group (Figure 2B) indicating that APAP induced protein carbonylation within 4 h and that S+A reduced protein carbonyls. The representative gel was further probed for 4-HNE modifications using LC-MS/MS. A total of 8 bands were excised from the gel for LC-MS/MS analysis. The selected bands are indicated in Figure 2 (Panel C). LC-MS/MS identified 12 proteins (Table 1) in the 8 bands. Further analysis identified selective sites of 4-HNE adduction to sarcosine dehydrogenase and carbamoyl phosphate synthetase-1 (CPS-1) and the MS/MS data are shown in Figure 3. Sarcosine dehydrogenase had an adduction of 4-HNE on lysine 174. CPS-1 was adducted with 4-HNE at arginine 1319.
Figure 2. Protein Carbonylation 4 h after APAP treatment.
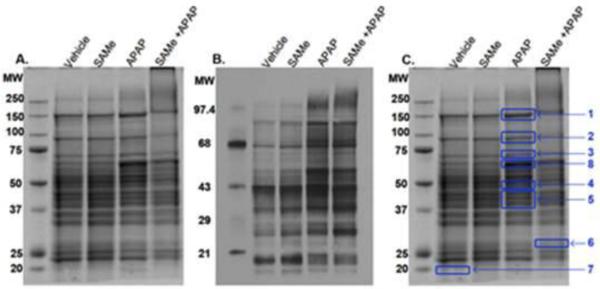
Mice were allocated into the same groups and treated as described in Figure 1. The first left lane in each Panel contains the molecular weight ladder, values are in kD. Panel A indicates proteins (40 μg) stained with coomassie blue. Panel B shows protein carbonylation (18 μg) stained with OxyBlot Kit antibody. Panel C shows a total of 8 bands excised from the same gel in Panel A, for LC-MS/MS. Proteins identified are listed in Table 1.
Table 1.
Proteins identified in OxyBlot of Liver Homogenate 4 h post APAP treatment using Mass Spectrometry.
| Band | Protein | Accession number | Molecular weight (kDa) | Num. of peptides | % Coverage |
|---|---|---|---|---|---|
| 1 | GRP78 (glucose regulated protein precursor) | gi[2506545] | 72.422 | 3 | 7.6 |
| 2 | Aldehyde dehydrogenase | gi[24418394] | 98,709 | 14 | 18.2 |
| Sarcosine dehydrogenase | gi[52000636] | 101,682 | 8 | 10.1 | |
| 3 | Transferrin | gi[20330602] | 76,724 | 14 | 27.0 |
| Heat shock 70 protein | gi[1661134] | 70,837 | 13 | 19.3 | |
| 4 | Tubulin alpha 2 | gi[539933] | 50,036 | 3 | 3.8 |
| Carbamoyl-phosphate synthetase isoform 1 | gi[82879179] | 164,618 | 14 | 35.0 | |
| 5 | Gent1 protein | gi[24657508] | 49,832 | 4 | 3.3 |
| 6 | Dodecenoyl-coenzyme A delta isomerase | gi[12836323] | 32,250 | 4 | 20.4 |
| Carbonic anhydrase III | gi[3198286] | 29,366 | 3 | 11.5 | |
| 7 | ADP-ribosylation factor 2 | gi[14714692] | 20,710 | 3 | 22.1 |
| 8 | Gamma-actin | gi[80956] | 41,793 | 7 | 21.7 |
Bands indicated in Figure 2 were excised, digested, and analyzed via LC/MS-MS. Each protein was required to have a minimum of 3 peptides identified from SEQUEST with reasonable spectra in order to be accepted. The raw data was analyzed using X!Tandem, the table represents proteins fitting the initial criteria as well as being represented in both SEQUEST and X!Tandem.
Figure 3. MS/MS spectra of adducted peptides from Carbamoyl-Phosphate Synthetase 1 (CPS-1) and Sarcosine Dehydrogenase.

TOP: CPS-1's arginine (R) 1319 is adducted with 4-HNE. The dots above or below the amino acids of the peptide above represents ions identified within the spectrum. BOTTOM: The Sarcosine Dehydrogenase lysine (K) 174 residue was adducted with 4-HNE.
Whole liver homogenate showed that APAP increased protein carbonylation and 4-HNE was adducted to sarcosine dehydrogenase and CPS-1. Further studies evaluated the effect of APAP treatment on cytosolic and mitochondrial 4-HNE adduction of proteins using Western blot. Figure 4 depicts representative Western gels for cytosolic (Panel A and mitochondrial (Panel B) fractions for 4-HNE adduction at 4h after APAP treatment. Densitometry analysis (Figure 5) showed that APAP at 4 h induced 4-HNE adduction in the cytosol with the highest intensity (p<0.05) for two bands at 102 and 139 kDa molecular weights in the APAP group when compared to Veh, SAMe and S+A groups. Densitometry of cytosolic 4-HNE gels 2 h after APAP revealed no statistical increase in the APAP group (Figure 5 Panel A) when compared to the Veh, SAMe, APAP and S+A groups (gels not shown). The only difference observed at 2 h was a decrease in intensity at the 109 kDa in the SAMe group compared to the Veh group. Densitometry analysis (Figure 5 Panel C) did reveal that APAP increased (p<0.05) cytosolic protein 4-HNE adduction at the 109 and 139 kDa bands compared to all other groups. S+A diminished 4-HNE adduction at the 109 kDa to a level similar to Veh and SAMe groups. S+A partially reversed 4-HNE adduction at the 139 kDa band as intensity was lower for the S+A group compared to the APAP group but still higher than the Veh and SAMe groups.
Figure 4. Western analysis of cytosolic and mitochondrial fractions 4 h after APAP treatment.

Treatment groups were as described in Figure 1. Liver cytosol and mitochondrial fractions were prepared from liver samples 2 and 4 h post APAP administration. A 100 ug aliquot of protein was loaded in each lane and a 1:1000 primary Anti-4-HNE Michael adduct antibody was used to assess 4-HNE adduction. Representative gels for cytosol (Panel A) and mitochondria (Panel B) are shown for the 4 h post APAP treatment time. Densitometry results are shown in Figure 5.
Figure 5. Densitometry analysis of liver mitochondrial and cytosolic 4-HNE adducted proteins 2 and 4 h following APAP treatment.

Densitometry results for Western blots depicted in Figure 4. Panel A (cytosol) and Panel B (mitochondria) depicts results 2 h following APAP overdose. Panel C and D are the cytosolic (C) and mitochondrial (D) fractions examined 4 h following APAP overdose. Groups with a different superscript letter (a, b or c) are statistically different (p<0.05). Cytosolic bands (Panel C) at 139 and 109 kDa bands were highest (p<0.05) in the APAP group when compared to Veh, SAMe and APAP groups. Mitochondrial 4 h samples (Panel D) had the highest intensity in the 45 kDa band compared to Veh, SAMe and S+A.
More dramatic increases in 4-HNE adduction were evident in the mitochondrial fractions (Figure 4) especially at 4 h after APAP treatment. Densitometry analysis showed that 4-HNE adduction of proteins at the 45 kDa molecular weight were highest (p<0.05) in the APAP group when compared to Veh, SAMe and S+A groups (Figure 5 Panel D). APAP was statistically (p<0.05) higher band intensity at the 91 kDa molecular weight when compared to Veh and SAMe but was not statistically different from the S+A groups. These results indicate that 4-HNE adducts were increased in mitochondrial and cytosolic fractions at 4 h after APAP treatment and that SAMe administered after APAP diminished band intensity at the 45 kDa band (Figure 4 and Figure 5, Panel D). Although 4-HNE adducts were identified by Western analysis, the identity of the specific proteins is not known.
Additional studies investigated the identities of 4-HNE adducted proteins. A 2D gel was run and probed with Anti-4-HNE Michael adduct antibody. The APAP treated group had numerous hits that were positive for 4-HNE (Figure 6). A total of 10 4-HNE positive spots were selected for more rigorous LC-MS/MS analysis. These spots were specifically analyzed for 4-HNE adducts. A total of 13 proteins were identified in the 10 different spots and are listed in Table 2. Proteins identified in the 4-HNE-immunopositive gel spots included: aldehyde dehydrogenase, CPS-1, hypoxia upregulated protein, protein disulfide isomerase, ALT, 78 kDa glucose regulated protein, acyl CoA dehydrogenase family member 9 and plastin.
Figure 6. 2D gel protein probed for 4-HNE and mass spectrometry.
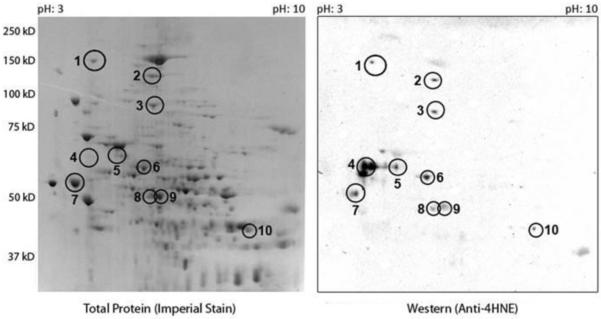
Mitochondrial fraction was isolated 4 h post APAP treatment and a 2D gel was run on the mitochondrial fractions isolated from the APAP group. The left Panel reflects an imperial protein staining. The right Panel depicts 4-HNE adducted proteins. A total of 10 spots were selected for further analysis by mass spectrometry and are denoted by with the numbers 1-10. Proteins identified in each spot are listed in Table 2.
Table 2.
Proteins identified in 2D gel of Mitochondrial 4 h post APAP treatment using Mass Spectrometry.
| Spot | Protein | Accession number | Molecular weight (kDa) | Num. of peptides | % Coverage | Calc. pI |
|---|---|---|---|---|---|---|
| 1 | Hypoxia up-regulated protein 1 | IPI00123342.4 | 111.1 | 46 | 51.2 | 5.19 |
| 2 | Pyruvate carboxylase, mitochondrial | IPI00114710.4 | 129.6 | 40 | 38.3 | 6.71 |
| Carbamoyl-phosphate synthase | IPI00111908.8 | 164.5 | 36 | 26.7 | 6.92 | |
| 3 | Sarcosine dehydrogenase | IPI00136213.5 | 101.6 | 40 | 45.6 | 6.74 |
| Carbamoyl-phosphate synthase | IPI00111908.8 | 164.5 | 49 | 36.0 | 6.92 | |
| 4 | GRP78 (glucose-regulated protein) | IPI00319992.1 | 72.4 | 24 | 38.0 | 5.1 |
| ATP synthase subunit beta | IPI00468481.2 | 56.3 | 13 | 37.6 | 5.3 | |
| 5 | Aldehyde dehydrogenase | IPI00111218.1 | 56.5 | 6 | 21.8 | 7.6 |
| ATP synthase subunit beta | IPI00468481.2 | 56.3 | 8 | 23.8 | 5.3 | |
| 6 | Liver carboxylesterase 31 | IPI00381178.3 | 63.3 | 14 | 34.9 | 6.1 |
| Carbamoyl-phosphate synthase | IPI00111908.8 | 164.5 | 35 | 29.7 | 6.9 | |
| Acyl-CoA dehydrogenase family member 9 | IPI00331710.2 | 68.7 | 13 | 26.2 | 7.4 | |
| 7 | Protein disulfide-isomerase | IPI00133522.2 | 57.0 | 50 | 70.3 | 4.8 |
| 8 | Aldehyde dehydrogenase | IPI00111218.1 | 56.5 | 32 | 55.3 | 7.6 |
| Alanine aminotransferase 1 | IPI00154045.3 | 55.1 | 23 | 55.9 | 6.6 | |
| 9 | Aldehyde dehydrogenase, mitochondrial | IPI00111218.1 | 56.5 | 33 | 55.3 | 7.6 |
| Alanine aminotransferase 1 | IPI00154045.3 | 55.1 | 18 | 50.0 | 6.6 | |
| 10 | 3-ketoacyl-CoA thiolase | IPI00226430.3 | 41.8 | 37 | 85.9 | 8.1 |
| Betaine-homocysteine S-methyltransferase 1 | IPI00130950.1 | 45.0 | 15 | 52.83 | 7.9 |
The 10 spots indicated in Figure 6 were excised, digested, and analyzed via LC/MS-MS. The identified proteins within each spot, their accession number, molecular weight, the number of peptides identified in the MS-MS analysis, the percent protein coverage of the identified peptides and the calculated isoelectric point (pI) are listed in the table.
Studies next investigated whether APAP mediated hepatic toxicity would increase mitochondrial protein leakage of CPS-1. CPS-1 was selected since CPS-1 is a mitochondrial protein identified to be adducted with 4-HNE 4 h after APAP overdose. Studies further examined whether CPS-1 leakage from mitochondria to cytosol was increase with hepatic toxicity. Western blots were performed and stained with CPS-1 antibody. The highest intensity was found in the mitochondria for all groups for CPS-1 (Figure 7 Panels A and B). CPS-1 levels in the mitochondria were not altered by SAMe administration compared to Veh (Figure 7). APAP administration did not alter mitochondrial levels of CPS-1 compared to all other groups. Mitochondrial CPS-1 levels were decreased at 4 h in the S+A mice when compared to Veh but not statistically different from the SAMe or APAP groups. Even though levels of CPS-1 were decreased in the mitochondrial S+A group, there was no increase in cytosolic CPS-1 for the SAMe group.
Figure 7. Mitochondrial and cytosolic fraction CPS-1 expression 4 h following APAP overdose.

Treatment groups were as described in Figure 1. Panel A contains the densitometry for mitochondrial and cytosolic CPS-1 Western blots. Panel B depicts mitochondrial (upper gel) and cytosolic (lower gel) probed for CPS-1. Panel C denotes 4-HNE binding of western blot band which was positive for CPS-1. The upper row depicts mitochondria and the lower bands depict cytosolic. Panel D depicts densitometry analysis for cytosolic 4-HNE adduction. Densitometry was normalized to total protein lane staining. Panel E depicts cytosolic protein gel staining. Values represent mean ± S.E.M. with n=3-5 mice per group. Groups with a different superscript letter (a, b or c) are statistically different (p<0.05).
Some amount of the mitochondrial CPS-1 enzyme was detected in the cytosol of all groups (Figure 7 Panels A and B). CPS-1 leakage into the cytosol correlated with ALT levels as CPS-1 leakage into the cytosol was highest in the APAP group and lowest in the Veh and SAMe groups. CPS-1 positive bands in the cytosol were 800 times higher in the APAP group compared to the Veh and SAMe groups. SAMe treatment (S+A) greatly reduced CPS-1 leakage into the cytosol of APAP treated mice suggesting less mitochondrial damage Cytosolic CPS-1 levels in the S + A group were 50% of the APAP group but still elevated relative to the Veh and SAMe groups indicating SAMe partially corrected APAP hepatic damage but did not totally prevent hepatic damage by APAP.
Our studies next showed that cytosolic CPS-1 when immunostained with 4-HNE antibody had higher 4-HNE adduction in the APAP group than the S+A group (Figure 7 Panels C and D); all lanes were loaded with equal protein (Figure 7 Panel E). Cytosolic CPS-1 was adducted by 4-HNE 4 h following APAP overdose (Figure 7 Panels C and D). Although some CPS-1 leakage was also observed when SAMe was given following APAP, the CPS-1 bands were not positive for 4-HNE adduction after the blots were stripped and re-probed for 4-HNE. No significant adduction of CPS-1 by 4-HNE was observed in either the Veh or SAMe treatment groups. However, APAP increased CPS-1 leakage (Figure 7 Panels C and D).
DISCUSSION
Our studies have shown that 4 h after APAP overdose, protein carbonylation and 4-HNE adduction of hepatic tissue were evident; SAMe administered 1 h after APAP reduced hepatic protein carbonylation and 4-HNE adduction. The liver proteins adducted by 4-HNE following APAP treatment included sarcosine dehydrogenase and CPS-1 as identified by LC-MS/MS analysis of selected spots. The current study is the first to identify by LC-MS/MS the specific proteins adducted by 4-HNE. Sarcosine dehydrogenase was adducted with 4-HNE at lysine residue 174. CPS-1 was adducted at arginine residue 1319. Our results showed that CPS-1 mitochondrial leakage into the cytosol was higher for the APAP treated group and less severe in the S+A group. MS/MS analysis of mitochondrial proteins selected from a 2D gel identified mitochondrial proteins that were adducted with 4-HNE in the APAP group.
APAP is the most common cause of drug induced liver injury in the United States. APAP hepatic toxicity requires cytochrome P450 mediated biotransformation to form NAPQI the toxic APAP metabolite (Corcoran et al., 1980). NAPQI is highly reactive and rapidly depletes intracellular hepatic GSH. Hepatic GSH levels are decreased within 1 h by APAP in mice (Yang et al., 2013) which suggests that within 1 h sufficient NAPQI is formed to deplete GSH. Our experimental model administered SAMe 1 h after a hepatotoxic dose of APAP which is more representative of a clinical overdose since the antidote is administered after the onset of toxicity. Based on our experimental design, it is unlikely that the mechanism for SAMe protection is inhibition of NAPQI formation. CYP 2E1 in rat microsomes can be inhibited by 1.5 mM SAMe (Cado and Cederbaum, 2005) but in our studies we administer 0.0250 mmole to the entire animal 1 h after APAP which is too late to inhibit NAPQI formation.
APAP hepatic toxicity has a second pathway of damage due to an increase in oxidative stress. NAPQI can induce damage by rapid depletion of GSH and by adducting various cellular proteins. Identification NAPQI-APAP adduction of hepatic protein has been a focus of considerable efforts by other investigators (Jollow et al., 1973; Corcoran et al., 1980; Cohen and Khairallah, 1997; Qiu et al., 1998). Jollow and colleagues (1973) showed that radioisotope labeled APAP was converted to a metabolite that was covalently bound to mouse liver. MS analysis of NAPQI-protein adducts identified over 20 hepatic mouse proteins following APAP overdose (Qiu et al., 1998). The proteins adducted with NAPQI included aldehyde dehydrogenase, ATP synthetase, glutathione peroxidase and glutathione S-transferase (Qiu et al., 1998). The effects of these adductions of hepatic proteins by NAPQI are not known and studies are needed to evaluate the long term effects on protein and enzyme function. APAP associated changes in protein levels may also influence response to toxicity as aldehyde dehydrogenase abundance is reduced within 2 h after APAP treatment (Ruepp et al., 2002). Aldehyde dehydrogenase is an important enzyme that can detoxify 4-HNE which is increased by activation of lipid peroxidation. A decrease in aldehyde dehydrogenase activity could increase the levels of cellular 4-HNE resulting in additional cellular dysfunction. Recent studies have also shown that plasma contains APAP-NAPQI adducts which may be a marker of hepatic damage (James et al., 2009). APAP-NAPQI adducts have a half-life over 1 day allowing for assessment of chronic APAP usage (James et al., 2009). It is not clear whether the APAP-NAPQI adducts are the result of leakage from hepatocytes or diffusion of NAPQI into plasma and adduction of proteins (McGill et al., 2013).
Mitochondria impairment has been recognized as an early target for APAP overdose. ROS generation during APAP overdose has been hypothesized as a second hit for hepatic damage following APAP overdose which initiates a second pathway of damage due to induction of oxidative stress (Hinson et al., 1998; Jaeschke et al., 2003). Oxidative stress induced by APAP overdose increases cellular levels of reactive oxygen (ROS), induction of lipid peroxidation and nitrogen species (RNS). Lipid peroxidation as a consequence of ROS generation can increase cellular 4-HNE levels. Increased 4-HNE levels and protein adduction are associated with diseases and cell death (Fritz and Petersen, 2013; Dodson et al., 2013). 4-HNE can lead to post-translational modifications of many proteins. Very little is known regarding the proteins modified by 4-HNE following APAP overdose and even less is known regarding the effects of SAMe on APAP mediated modifications of proteins due to protein carbonylation and 4-HNE adduction. LC-MS/MS analysis of hepatic proteins identified CPS-1 and sarcosine dehydrogenase as proteins adducted with 4-HNE within 4 h of APAP treatment (Figure 2 and 3). Sarcosine dehydrogenase was adducted with 4-HNE at lysine residue 174. CPS-1 was adducted at arginine residue 1319. The arginine 1319 residue is not within the active site or within a functional domain of CPS-1. However arginine 1319 is near the ATP-grasp 2 domain of CPS-1 which is located between residues 1093-1284. Further studies to would be needed to evaluate the impact of 4-HNE adduction of arginine 1319 residue on CPS-1 enzyme activity. Further studies may apply 3-dimensional molecular modeling to predict the potential functional impact of 4-HNE adduction of CPS-1 similar to what has been done with other proteins such as cytochrome c (Monks and Lau, 2012) and peroxiredoxin 6 (Roede et al., 2008).
The results of our study show that 4-HNE adducts mitochondrial proteins which leak from the mitochondria into the cytosol following APAP overdose. It cannot be determined from our study if 4-HNE adduction promotes leakage of proteins from the mitochondria. LC-MS/MS analysis of mitochondrial proteins identified 13 proteins as potential targets of 4-HNE within 4 h of APAP overdose in mice. Alanine aminotransferase (ALT) was one of the identified 4-HNE targets, and this is a protein used as a biomarker of hepatic damage. Future investigations need to evaluate whether alanine aminotransferase adducted proteins are more likely to leak into plasma or occur with other hepatotoxic agents or disease conditions. Other 4-HNE targets included: CPS-1, aldehyde dehydrogenase and protein disulfide isomerase. CPS-1 is a mitochondrial enzyme involved in the urea cycle as well as in the formation of carbamoyl phosphate, an important component in pyrimidine synthesis. CPS-1 enzyme activity was reported to be diminished in mice 4 h after APAP administration (Gupta et al., 1997). CPS-1 enzyme activity was decreased in ICR mice given a 400 mg/kg dose of APAP which was a higher dose than our study. The diminished CPS-1 activity associated with APAP was sufficient to increase plasma ammonia levels within 2 h in mice indicated that the inhibition of CPS-1 was sufficient to induce changes in ammonia balance (Gupta et al., 1997). Our studies did observe some leakage of CPS-1 into the cytosol of all groups. It should be mentioned that Gupta and colleagues (1997) also observed some leakage of CPS-1 into the cytosolic fraction of control and APAP treated tissue which may be due to some leakage from the mitochondria during separation of subcellular fractions. The effect of 4-HNE adduction on CPS-1 and pyrimidine synthesis by APAP is not known. However, our studies show that CPS-1 leakage out of the mitochondria into the cytosol is increased by APAP and that SAMe reduces CPS-1 leakage (Figure 7). SAMe may reduce CPS-1 leakage by maintaining mitochondrial integrity following APAP treatment (Brown et al., 2012).
Previous work in our laboratory showed SAMe provided protection for APAP hepatic toxicity when SAMe was administered concurrently or 1 h after APAP treatment using C57Bl/6 mice (Terneus et al., 2007 and 2008). Our studies further showed that SAMe protections for APAP hepatic toxicity included attenuation of oxidative stress and 3-NT modifications of mitochondrial proteins (Brown et al., 2012). SAMe reduced oxidative stress by preserving antioxidant enzyme activity which was severely impaired by APAP. SAMe administered after APAP maintained manganese superoxide dismutase (Mn SOD), catalase and glutathione peroxidase hepatic enzyme activity which was diminished within 4 h by APAP (Brown et al., 2012). SAMe reduction of APAP mediated oxidative stress would be expected to diminish lipid peroxidation and 4-HNE formation. Our results showed that SAMe did reduce 4-HNE adduction as well as protein carbonylation. 4-HNE is formed due to lipid peroxidation attack of polyunsaturated fatty acids resulting in fragmentation into aldehydes such as 4-HNE and malondialdehyde. 4-HNE is highly reactive and can bind to various amino acids (LoPachin et al., 2009). 4-HNE is metabolized by aldehyde dehydrogenase (Mitchell and Petersen, 1987; Hartley et al., 1995), glutathione-S-transferases (GST) (Sptiz et al., 1991; Fritz and Petersen, 2013) including GST A4 (Hubatsch et al., 1998) and various cytochrome P450 isozymes (Amunom et al., 2011). GST conjugation of 4-HNE is dependent on the intracellular GSH concentration (Spitz et al.,1991). Detoxification of 4-HNE by GST can be diminished by 80% due to depletion of GSH (Spitz et al.,1991) which may result in higher 4-HNE levels following APAP overdose since APAP rapidly induces hepatic GSH depletion (Davis et al., 1974; Yang et al., 2013). Previous work by our laboratory has shown that APAP diminished hepatic GSH level and that SAMe with APAP provided GSH levels that were less markedly diminished compared to APAP alone (Terneus et al., 2007). Our studies indicated that 4 h after APAP overdose, aldehyde dehydrogenase was identified as a target of 4-HNE adduction (Table 2). 4-HNE is highly reactive as studies by other investigators demonstrated that 4-HNE will adduct within 30 min to human recombinant aldehyde dehydrogenase 2 (Doorn et al., 2006). The effect of 4-HNE adduction on aldehyde dehydrogenase enzyme activity remains to be evaluated as to whether 4-HNE adduction induces no change, an increase or a decrease in enzyme activity. Doorn and associates (2006) reported that enzyme activity was diminished by 10% for human recombinant aldehyde dehydrogenase enzyme activity within 5 min of incubation with 4-HNE. These findings would suggest that 4-HNE adduction of aldehyde dehydrogenase may impair aldehyde dehydrogenase detoxification of 4-HNE.
Glucose regulated protein 78 kDa also known as heat shock protein 70 (Hsp70) was identified as a target of 4-HNE. The contribution of HSP70 in APAP toxicity to the liver is not well characterized. One study reported that HSP70 knockout mice showed no difference in susceptibility to APAP hepatic toxicity compared to wild type (Welch et al., 2006). Protein disulfide isomerase was another identified 4-HNE target following APAP overdose (Table 2). Protein disulfide isomerase is located in the endoplasmic reticulum. Protein disulfide isomerase is involved in protein folding and unfolding as it catalyzes the formation and cleaving of disulfide bonds. The effect of APAP overdose on protein disulfide isomerase activity is not known. Protein disulfide isomerase expression was unchanged in HepG2 cells exposed to APAP (Van Summeren et al., 2011). Protein disulfide isomerase can exist in a reduced and oxidized form and the oxidized form is involved in formation of disulfide bridges. A shift toward the oxidized form was reported in CD-1 mouse liver following APAP overdose (Nagy et al., 2007).
In summary, the findings of our study show that SAMe administered 1 h after APAP reduced APAP hepatic toxicity when evaluated 4 h after APAP overdose. Protein carbonylation and 4-HNE adduction of proteins was evident in the mitochondria and cytosol within 4 h of APAP overdose in C57Bl/6 mice. SAMe also prevented 4-HNE adduction of proteins in the mitochondria and cytosol. Further studies indicated that CPS-1 leaked from the mitochondria into the cytosol following APAP administration and that SAMe reduced mitochondrial leakage. APAP induces oxidative stress and 4-HNE adduction of mitochondrial proteins within 4 h of APAP overdose. LC-MS/MS analysis of 4-HNE immunopositive mitochondrial protein gel spots identified: aldehyde dehydrogenase, CPS-1, hypoxia upregulated protein, protein disulfide isomerase, ALT, 78 kDa glucose regulated protein, Acyl CoA dehydrogenase family member 9, sarcosine dehydrogenase, and plastin. Among the identified proteins was aldehyde dehydrogenase, a protein involved in 4-HNE metabolism. These results suggest that APAP mediated protein modifications with 4-HNE may impact hepatic function and that SAMe reduced 4-HNE modification of proteins which have a vital role in protein folding and detoxification of 4-HNE.
Highlights.
Acetaminophen (APAP) Toxicity protected by S-Adenosylmethionine (SAMe)
4-Hydroxynonenal adducted to sarcosine dehydrogenase
4-Hydroxynonenal adducted to carbamoyl phosphate synthetase-1
SAMe reduced APAP mediated CPS-1 mitochondrial leakage
ACKNOWLEDGEMENTS
This work was supported in part by WV-NASA Space consortium (JMB) and NIH grants: 5P20GM103434 (MAV), 5P20RR016477 (MAV), 3P20RR016477-09S4 (MAV), T32ES007091 (CK) and P30ES006694 (SSL). Mass spectrometric data were acquired by the Arizona Proteomic Consortium supported by P30ES06694, P30CA023074 and by the BIO5 Institute of the University of Arizona.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ameer B, Greenblatt DJ. Acetaminophen. Ann Intern Med. 1977;87:202–209. doi: 10.7326/0003-4819-87-2-202. [DOI] [PubMed] [Google Scholar]
- Amarnath V, Valentine WM, Montine TJ, Patterson WH, Amarnath K, Bassett CN, Graham DG. Reactions of 4-Hydroxy-2(E)-nonenal and related aldehydes with proteins studied by carbon-13 nuclear magnetic resonance spectroscopy. Chem. Res. Toxicol. 1998;11:317–328. doi: 10.1021/tx970176n. [DOI] [PubMed] [Google Scholar]
- Amunon I, Dieter LJ, Tamasi V, Cai J, Conklin DJ, Srivastava S, Martin MV, Guengerich FP, Prough RA. Cytochromes P450 catalyze the reduction of α,β-unsaturated aldehydes. Chem Res. Toxicol. 2011;24:1223–1230. doi: 10.1021/tx200080b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson BS, Rundgren M, Nelson SD, Harder S. N-acetyl-p-benzoquinone imine induced changes in the energy metabolism in hepatocytes. Che. Biol. Interact. 1990;75:201–211. doi: 10.1016/0009-2797(90)90118-7. [DOI] [PubMed] [Google Scholar]
- Bray GP, Tredger JM, Williams R. S-adenosylmethionine protects against acetaminophen hepatotoxicity in two mouse models. Hepatology. 1992;15:297–301. doi: 10.1002/hep.1840150220. [DOI] [PubMed] [Google Scholar]
- Brown JM, Ball JG, Wright MS, Van Meter S, Valentovic MA. Novel protective mechanisms for S-adenosyl-L-methionine against acetaminophen hepatotoxicity: improvement of key antioxidant enzymatic function. Toxicol. Lett. 2012;212:320–328. doi: 10.1016/j.toxlet.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SD, Khairallah EA. Selective protein arylation and acetaminophen-induced hepatotoxicity. Drug Metab Rev. 1997;29:59–77. doi: 10.3109/03602539709037573. [DOI] [PubMed] [Google Scholar]
- Corcoran GB, Mitchell JR, Vaishnav YN, Horning EC. Evidence that acetaminophen and N-hydroxy-acetaminophen form a common arylating intermediate, N-acetyl-p-benzoquinoneimine. Mol. Pharmacol. 1980;18:536–542. [PubMed] [Google Scholar]
- Dahlin DC, Nelson SD. Synthesis, decomposition kinetics and preliminary toxicological studies of pure N-acetyl-p-benzoquinoneimine, a proposed toxic metabolite of acetaminophen. J. Med. Chem. 1982;25:885–886. doi: 10.1021/jm00350a001. [DOI] [PubMed] [Google Scholar]
- Davis DC, Potter WZ, Jollow DJ, Mitchell JR. Species differences in hepatic glutathione depletion, covalent binding and hepatic necrosis after acetaminophen. Life Sci. 1974;14:2099–109. doi: 10.1016/0024-3205(74)90092-7. [DOI] [PubMed] [Google Scholar]
- Dodson M, Darley-Usmar V, Zhang J. Cellular metabolic and autophagic pathways: traffic control by redox signaling. Free Radic Biol Med. 2013;63:207–21. doi: 10.1016/j.freeradbiomed.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doorn JA, Petersen DR. Covalent modification of amino acid nucleophiles by the lipid peroxidation products 4-hydroxy-2-nonenal and 4-oxo-2-nonenal. Chem Res. Toxicol. 2002;15:1445–1450. doi: 10.1021/tx025590o. [DOI] [PubMed] [Google Scholar]
- Doorn JA, Hurley TD, Petersen DR. Inhibition of human mitochondrial aldehyde dehydrogenase by 4-hydroxynon-2-enal and 4-oxonon-2-enal. Chem Res. Toxicol. 2006;19:102–110. doi: 10.1021/tx0501839. [DOI] [PubMed] [Google Scholar]
- Fritz KS, Petersen DR. An Overview of the Chemistry and Biology of Reactive AldehydesFree Radic. Biol Med. 2013 Jun;59:85–91. doi: 10.1016/j.freeradbiomed.2012.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogvadze V, Orrenius S, Zhivotovsky B. In: Current Protocols in Toxicology. Maines MD, Costa LG, Reed DJ, Sassa S, Sipes IG, editors. Vol. 1. John Wiley & Sons, Inc.; Hoboken, NJ: 2006. pp. 2.10.11–12.10.27. [Google Scholar]
- Gupta S, Rogers LK, Taylor SK, Smith CV. Inhibition of carbamyl phosphate synthetase-I and glutamine synthetase by hepatotoxic doses of acetaminophen in mice. Toxciol. Appl. Pharmacol. 1997;146:317–327. doi: 10.1006/taap.1997.8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen BT, Davey SW, Ham AJ, Liebler DC. P-Mod: an algorithm and software to map modifications to peptide sequences using tandem MS data. J. Proteome Res. 2005;4:358–368. doi: 10.1021/pr0498234. [DOI] [PubMed] [Google Scholar]
- Hartley DP, Ruth JA, Petersen DR. The hepatocellular metabolism of 4-hydroxynonenal by alcohol dehydrogenase, aldehyde dehydrogenase, and glutathione S-transferase. Arch. Biochem. Biophys. 1995;316:197–205. doi: 10.1006/abbi.1995.1028. [DOI] [PubMed] [Google Scholar]
- Heard KJ, Gree JL, James LP, Judge BS, Zolot L, Rhyee S, Dart RC. Acetaminophen-cysteine adducts during therapeutic dosing and following overdose. BMC Gastroenterology. 2011;11:20. doi: 10.1186/1471-230X-11-20. (March 14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinson JA, Pike SL, Pumford NR, Mayeux PR. Nitrotyrosine-protein adducts in hepatic centrilobular areas following toxic doses of acetaminophen in mice. Chem. Res. Toxicol. 1998;11:604–607. doi: 10.1021/tx9800349. [DOI] [PubMed] [Google Scholar]
- Holme JA, Hongslo JK, Bjørge C, Nelson SD. Comparative cytotoxic effects of acetaminophen (N-acetyl-p-aminophenol), a non-hepatotoxic regioisomer acetyl-m-aminophenol and their postulated reactive hydroquinone and quinone metabolites in monolayer cultures of mouse hepatocytes. Biochem Pharmacol. 1991;42(5):1137–42. doi: 10.1016/0006-2952(91)90299-k. [DOI] [PubMed] [Google Scholar]
- Hubatsch I, Ridderstrom M, Mannervik B. Human glutathione transferase A4-4: an alpha class enzyme with high catalytic efficiency in the conjugation of 4-hydroxynonenal and other genotoxic products of lipid peroxidation. Biochem. J. 1998;330:175–179. doi: 10.1042/bj3300175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H, Knght TR, Bajt ML. The role of oxidant stress and reactive nitrogen species in acetaminophen hepatotoxicity. Toxicol. Lett. 2003;144:279–288. doi: 10.1016/s0378-4274(03)00239-x. [DOI] [PubMed] [Google Scholar]
- James LP, Letzig L, Simpson PM, Capparelli E, Roberts DW, Hinson JA, Davern TJ, Lee WM. Pharmacokinetics of acetaminophen-protein adducts in adults with acetaminophen overdose and acute liver failure. Drug Metab Dispos. 2009;37:1779–84. doi: 10.1124/dmd.108.026195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jollow DJ, Mitchell JR, Potter WZ, Davis DC, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. II. Role of covalent binding in vivo. J Pharmacol Exp Ther. 1973;187:195–202. [PubMed] [Google Scholar]
- Larrauri A, Fabra R, Gomez-Lechon MJ, Trullenque R, Castell JV. Toxicity of paracetamol in human hepatocytes. Comparison of the protective effects of sulfhydryl compounds acting as glutathione precursors. Mol. Toxicol. 1987-1988;1:301–311. [PubMed] [Google Scholar]
- Lauterburg BH, Corcoran GB, Mitchell JR. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J. Clin. Invest. 1983;71:980–91. doi: 10.1172/JCI110853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPachin RM, Gavin T, Petersen DR, Barber DS. Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: nucleophilic targets and adduct formation. Chem Res Toxicol. 2009;22:1499–508. doi: 10.1021/tx900147g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JR, Snodgrass WR, Gillette JR. The role of biotransformation in chemical-induced liver injury. Environ Health Perspect. 1976;15:27–38. doi: 10.1289/ehp.761527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DY, Petersen DR. The oxidation of alpha-beta unsaturated aldehydic products of lipid peroxidation by rat liver aldehyde dehydrogenases. Toxicol. Appl. Pharmacol. 1987;87:403–410. doi: 10.1016/0041-008x(87)90245-6. [DOI] [PubMed] [Google Scholar]
- Monks TJ, Lau SS. Reactive intermediates: molecular and MS-based approaches to assess the functional significance of chemical protein adducts. Toxicol. Pathol. 2013;41:315–321. doi: 10.1177/0192623312467399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nourjah P, Ahmad SR, Karwoski C, Willy M. Estimates of acetaminophen (paracetomal)-associated overdoses in the United States. Pharmacoepidemiol. Drug Saf. 2006;15:398–405. doi: 10.1002/pds.1191. [DOI] [PubMed] [Google Scholar]
- Nagy G, Kardon T, Wunderlich L, Szarka A, Kiss A, Schaff Z, Banhegyi G, Mandl J. Acetaminophen induces ER dependent signaling in mouse liver. Archives Biochem. Biophys. 2007;459:273–279. doi: 10.1016/j.abb.2006.11.021. [DOI] [PubMed] [Google Scholar]
- Patel NN, Crincoli CM, Frederick DM, Tchao R, Harvison PJ. Effect of structural modifications on 3-(3,5-dichlorophenyl)-2,4-thiazolidinedione-induced hepatotoxicity in Fischer 344 rats. J Appl Toxicol. 2012;32:108–117. doi: 10.1002/jat.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patten CJ, Thomas PE, Guy RL, Lee M, Gonzalez FJ, Guengerich FP, Yang CS. Cytochorme P450 Enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol. 1993;6:511–518. doi: 10.1021/tx00034a019. [DOI] [PubMed] [Google Scholar]
- Potter WZ, Davis DC, Mitchell JR, Jollow DJ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. 3. Cytochrome P-450-mediated covalent binding in vitro. J Pharmacol Exp Ther. 1973;187:203–10. [PubMed] [Google Scholar]
- Qiu Y, Benet LZ, Burlingame AL. Identification of the hepatic protein targets of reactive metabolites of acetaminophen in vivo in mice using two-dimensional gel electrophoresis and mass spectrometry. J Biol Chem. 1998;273:17940–17953. doi: 10.1074/jbc.273.28.17940. [DOI] [PubMed] [Google Scholar]
- Rashed MS, Myers TG, Nelson SD. Hepatic protein arylation, glutathione depletion, and metabolite profiles of acetaminophen and a non-hepatotoxic regioisomer, 3’-hydroyacetanilide, in the mouse. Drug Metab. Disp. 1990;18:765–770. [PubMed] [Google Scholar]
- Roede JR, Carbone DL, Doorn JA, Kirichenko OV, Reigan P, Petersen DR. In vitro and in silico characterization of peroxiredoxin 6 modified by 4-hydroxynonenal and 4-oxononenal. Chem. Res. Toxciol. 2008;21:2289–2299. doi: 10.1021/tx800244u. [DOI] [PubMed] [Google Scholar]
- Sayre LM, Lin D, Yuan Q, Zhu X, Tang X. Protein adducts generated from products of lipid oxidation: focus on HNE and ONE. Drug Metab. Rev. 2006;38:651–675. doi: 10.1080/03602530600959508. [DOI] [PubMed] [Google Scholar]
- Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med. 1988;319(24):1557–62. doi: 10.1056/NEJM198812153192401. [DOI] [PubMed] [Google Scholar]
- Spitz DR, Sullivan SJ, Malcolm RR, Roberts RJ. Glutathione dependent metabolism and detoxification of 4-hydroxy-2-nonenal. Free Radic Biol Med. 1991;11:415–23. doi: 10.1016/0891-5849(91)90159-z. [DOI] [PubMed] [Google Scholar]
- Stramentinoli G, Pezzoli C, Galli-Kienle M. Protective role of S-adenosyl-l-methionine against acetaminophen induced mortality and hepatotoxicity in mice. Biochem. Pharmacol. 1979;28:3567–3571. doi: 10.1016/0006-2952(79)90401-5. [DOI] [PubMed] [Google Scholar]
- Streeter AJ, Dahlin DC, Nelson SD, Baillie TA. The covalent binding of acetaminophen to protein. Evidence for cysteine residues as major sites of arylation in vitro. Che. Biol Interact. 1984;48:349–366. doi: 10.1016/0009-2797(84)90145-5. [DOI] [PubMed] [Google Scholar]
- Terneus MV, Kiningham KK, Carpenter AB, Sullivan SB, Valentovic MA. Comparison of S-Adenosyl-L-methionine and N-acetylcysteine protective effects on acetaminophen hepatic toxicity. J Pharmacol Exp Ther. 2007;320:99–107. doi: 10.1124/jpet.106.111872. [DOI] [PubMed] [Google Scholar]
- Terneus MV, Brown JM, Carpenter AB, Valentovic MA. Comparison of S-adenosyl-L-methionine (SAMe) and N-acetylcysteine (NAC) protective effects on hepatic damage when administered after acetaminophen overdose. Toxicology. 2008;244(1):25–34. doi: 10.1016/j.tox.2007.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirmenstein MA, Nelson SD. Subcellular binding and effects on calcium homeostasis produced by acetaminophen and a nonhepatoxic regioisomer, 3’hydroxyacetanilide, in mouse liver. J. Biol. Chem. 1989;264:9814–9819. [PubMed] [Google Scholar]
- Valentovic M, Terneus M, Harmon RC, Carpenter AB. S-Adenosylmethionine (SAMe) attenuates acetaminophen hepatotoxicity in C57BL/6 mice. Toxicol Lett. 2004;154:165–74. doi: 10.1016/j.toxlet.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Van Summeren A, Renes J, Bouwman FG, Noben J-P, van Delft JHM, Kleinjans JCS, Mariman CM. Proteomics investigations of drug-induced hepatotoxicity in HepG2 cells. Toxicol. Sci. 2011;120:109–122. doi: 10.1093/toxsci/kfq380. [DOI] [PubMed] [Google Scholar]
- Welch KD, Reilly TP, Bourdi M, Hays T, Pise-Masison CA, Radonovich MF, Dix DJ, Pohl LR. Genomic identification of potential risk factors during acetaminophen-induced liver disease in susceptible and resistant strains of mice. Chem Res Toxicol. 2006;19:223–33. doi: 10.1021/tx050285z. [DOI] [PubMed] [Google Scholar]
- Yang X, Greenhaw J, Shi Q, Roberts DW, Hinson JA, Muskhelishvili L, Davis K, Salminen WF. Mouse Liver Protein Sulfhydryl Depletion after acetaminophen exposure. J. Pharm. Expt. Ther. 2013;344:286–294. doi: 10.1124/jpet.112.199067. [DOI] [PMC free article] [PubMed] [Google Scholar]