

Abstract
Here we report the discovery of oncogenic mutations in the Hedgehog and mitogen-activated protein kinase (MAPK) pathways in over 80% of ameloblastomas, locally destructive odontogenic tumors of the jaw, by genomic analysis of archival material. Mutations in SMO (encoding Smoothened, SMO) are common in ameloblastomas of the maxilla, whereas BRAF mutations are predominant in tumors of the mandible. We show that a frequently occurring SMO alteration encoding p.Leu412Phe is an activating mutation and that its effect on Hedgehog-pathway activity can be inhibited by arsenic trioxide (ATO), an anti-leukemia drug approved by the US Food and Drug Administration (FDA) that is currently in clinical trials for its Hedgehog-inhibitory activity. In a similar manner, ameloblastoma cells harboring an activating BRAF mutation encoding p.Val600Glu are sensitive to the BRAF inhibitor vemurafenib. Our findings establish a new paradigm for the diagnostic classification and treatment of ameloblastomas.
Ameloblastoma, a locally destructive tumor, is thought to exhibit characteristics of ameloblastic differentiation1. Tumor cells resemble ameloblasts, cells in the developing tooth responsible for depositing enamel during tooth development (odontogenesis). Therapeutic options are few, and these tumors often require disfiguring wide local excision with high rates of recurrence. Research into the pathogenesis of ameloblastoma has largely been driven by clues derived from histological appearance and from normal tooth development. Rare tumor types such as ameloblastoma are not only understudied but are typically only accessible as formalin-fixed, paraffin-embedded (rather than freshly frozen) specimens that have been thought to be suboptimal for genomic analysis. Thus, relatively little genomic data have been generated on this tumor type. We have recently shown that transcriptome sequencing of formalin-fixed, paraffin-embedded specimens can effectively identify gene transcript fusions, suggesting that it might represent a more generally useful approach to study rare tumor genetics2.
In a survey of rare neoplasia to discover driver mutations, we performed whole-transcriptome sequencing on formalin-fixed, paraffin-embedded material from two cases of ameloblastoma. This is an approach that may be efficient for the screening of rare neoplasia for clinically targetable, activating mutations, as these mutations are typically in well-expressed genes and thus easily detected in full-transcriptome libraries. Libraries of total RNA were prepared from rRNA-depleted RNA isolated from formalin-fixed, paraffin-embedded specimens. A custom analytical pipeline (Online Methods) identified high-confidence single-nucleotide variations (SNVs) but no gene fusions. Candidate SNVs were prioritized for further validation on the basis of their presence in both tumor samples and/or on the basis of previously known involvement of the identified gene or pathway in tooth bud development3. Candidate mutations were validated in an independent cohort consisting of 26 cases from 4 institutions (Supplementary Table 1), using targeted-capture deep sequencing and/or PCR with Sanger sequencing. Analysis of paired tumor-normal tissue in a subset of the validation cohort confirmed that the mutations were somatic.
From this analysis, we identified highly recurrent somatic mutations in two key developmental or growth factor signaling pathways—the Hedgehog and MAPK pathways. In all, 39% (11/28) of the tumors had mutations in SMO (an essential seven-transmembrane Hedgehog signal transduction component; 10 encoding p.Leu412Phe and 1 encoding p.Trp535Leu) and 46% (13/28) had BRAF mutations (12 encoding p.Val600Glu and 1 encoding p.Leu597Arg) (Fig. 1a and Supplementary Fig. 1). SMO and BRAF mutations tended to be mutually exclusive (P = 0.02, two-sided Fisher’s exact test), suggesting that these alterations might define two independent genetic etiologies for ameloblastoma. There was some correlation between mutation status and previously established morphological subtypes, as most (8/10) plexiform variants had a SMO mutation (P < 0.02), whereas most follicular and desmoplastic variants carried either SMO or BRAF mutation. Strikingly, SMO mutations exhibited a marked preponderance in maxillary ameloblastomas (9/11 cases) compared to mandibular cases (1/13) (P < 0.001), whereas BRAF mutations exhibited the reverse pattern, with a higher frequency in mandibular (9/13) compared to maxillary (1/11; encoding p.Leu597Arg) cases (P = 0.01) (Fig. 1b). Using available information on clinical outcome, we observed a trend toward earlier recurrence for tumors with SMO mutations (three of five SMO mutants versus one of six BRAF mutants recurred within 3 years after initial treatment; P = 0.24; Supplementary Table 1); analysis of a larger cohort is needed to substantiate this finding. Additional mutations in the MAPK pathway were also identified, including four cases (15%) with mutation of KRAS (encoding p.Gly12Arg) and five cases (19%) with mutation of FGFR2 (four encoding p.Cys382Arg and one encoding p.Asn549Lys), the presumptive upstream receptor tyrosine kinase. In all but one case, mutation of BRAF was mutually exclusive with mutations in KRAS and FGFR2 (P < 0.05). Expression of mutant BRAF protein, evaluated by immunohistochemistry for BRAF Val600Glu, was only seen in cases with confirmed presence of the corresponding mutation in BRAF, with some qualitative increase in expression at the leading edge of the epithelial cell component, adjacent to the stroma (Supplementary Fig. 2).
Figure 1.
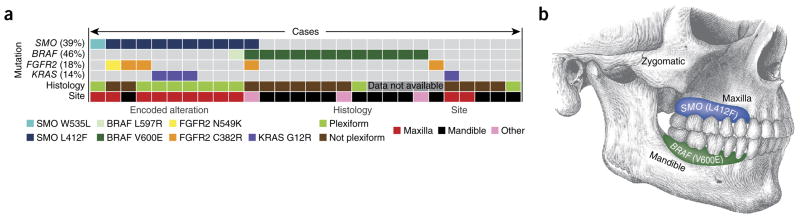
Mutation frequency, distribution and relationship with pathological features. (a) Mutation status for four genes is indicated, and the overall percentage of mutant cases is given in parentheses. Information on histology and anatomical site is included below for each case. Each column represents a single case. Colors correspond to a specific mutation, histology or anatomical site. Details on additional clinical parameters are included in supplementary table 1. (b) Illustration of the distribution of tumors with the identified mutations.
Previous studies have demonstrated that the recurring BRAF, KRAS and FGFR2 mutations identified in this ameloblastoma cohort are activating mutations present in other cancers4–6. The SMO mutation encoding p.Trp535Leu, found in one case, is also known to be a frequent activating mutation in sporadic basal cell carcinoma7. The SMO mutation encoding p.Leu412Phe, the ‘hotspot’ SMO mutation in our study, was only recently reported in a subset of meningiomas8. To evaluate the functional consequences of the p.Leu412Phe alteration, we measured Hedgehog-pathway activation mediated by wild-type or mutant forms of SMO using a previously established Gli-driven luciferase reporter assay in Smo−/− mouse embryonic fibroblasts (MEFs) (Fig. 2)9. Of note, these fibroblasts also express basal levels of Ptch1, the 12-pass Hedgehog receptor that, in the absence of Hedgehog ligand, inhibits Smo. As expected, wild-type human SMO was essentially devoid of basal activity in this assay (resulting in <1% of the maximal Sonic Hedgehog (Shh)-induced response). In contrast, the Leu412Phe mutant showed substantially elevated, constitutive activity (34 ± 8% of the maximal Shh-driven response; P < 0.01), although activation was at a lower level than with the Trp535Leu variant (54 ± 12%) (Fig. 2a). Notably, overexpression of SMO Leu412Phe in immortalized mouse ameloblast-lineage cells (the ALC line10) enhanced cell proliferation in comparison to overexpression of wild-type SMO or empty vector control (Fig. 3), demonstrating a relevant phenotype in a germane cell type.
Figure 2.

SMO Leu412Phe activity and inhibition. (a) Relative expression of a Hedgehog-sensitive Gli-driven luciferase reporter in Smo−/− MEFs expressing GFP (negative control), wild-type human SMO (WT) or the Leu412Phe or Trp535Leu SMO mutants following stimulation with control medium or medium containing the Shh N-terminal domain (ShhN). Note the significant basal induction of Hedgehog-pathway activity by Leu412Phe and Trp535Leu. **P < 0.01, two-sided Student’s t test. Results are representative of five independent trials.
(b) Effect of treatment with vismodegib (200 nM), itraconazole (2 μM), KAAD-cyclopamine (300 nM) and ATO (8 μM), showing significant reduction in Hedgehog activity in cells expressing SMO Leu412Phe and SMO Trp535Leu with KAAD-cyclopamine and ATO treatment. ***P < 1 × 10−5, ****P < 1 × 10−6, two-sided Student’s t test. Results are representative of two independent trials. Data in a,b are from three independent transfections (three biological replicates), and error bars represent s.d. (c) Crystal structure of human SMO (Protein Data Bank (PDB) 4JKV) bound to the LY2940680 inhibitor10 (yellow) with amino acids 412 and 535 highlighted (red) to show transmembrane domain positioning.
Figure 3.

SMO Leu412Phe enhances ameloblast-lineage cell proliferation. (a) Overexpression of wild-type SMO, SMO Trp535Leu, SMO Leu412Phe or empty vector control in mouse ameloblast-lineage (ALC) cells, shown by protein blot (antibody to Myc tag). GAPDH serves as a loading control. (b) Relative cell proliferation (in optical density (OD) units) evaluated by WST-1 assay (Roche) 24, 48 and 72 h after plating equal numbers of cells. Overexpression of SMO constructs significantly enhances cell proliferation compared to empty vector control; SMO Leu412Phe also enhances proliferation in comparison to wild-type SMO (two-sided Student’s t test, P values indicated). SMO expression was engineered by lentiviral transduction, and stable cell pools (with approximately 1 × 105 independent integrations) were assayed. Cell proliferation was evaluated by three independent cell platings; error bars, s.d. Results presented are representative of three independent trials.
Next, we evaluated the response of the SMO Leu412Phe mutant to various pharmacological Hedgehog-pathway inhibitors (Fig. 2b), including the SMO antagonists KAAD-cyclopamine and vismodegib (Erivedge, Genentech), which bind the cyclopamine pocket of SMO, and itraconazole, which acts at the level of SMO but does not bind the cyclopamine pocket. KAAD-cyclopamine effectively inhibited the Leu412Phe mutant (P < 1 × 10−6), comparable to the Trp535Leu mutant, whereas an inhibitory effect was not observed for vismodegib. This difference in inhibition is likely due to a combination of the effect of the mutation on the binding pocket and the divergent chemical structures of the two compounds. Itraconazole was also found to be ineffective at inhibition; however, both SMO mutants were sensitive to ATO (also known as Trisenox, Teva) (P < 1 × 10−5), an inhibitor of downstream GLI effectors. For both SMO mutants, constitutive activity was also suppressed by supraphysiological overexpression of mouse Ptch1 in these Smo−/− MEFs (Supplementary Fig. 3). Interestingly, the Leu412 and Trp535 residues are both located within the SMO seven-transmembrane domain but map outside the crystallographically resolved binding pocket for small molecule cyclopamine mimics (Fig. 2c)11. These observations suggest that the p.Leu412Phe substitution, as with p.Trp535Leu, leads to constitutive SMO activation via similar allosteric effects on SMO conformation.
To further investigate the activity of the inhibitors, we sought to evaluate their efficacy in human ameloblastoma cell lines. Few such cell lines have been reported, but we found that the AM-1 line12, derived from a mandibular tumor, harbored the BRAF mutation encoding p.Val600Glu but had no SMO mutation (Fig. 4a,b). Notably, AM-1 cells were sensitive to the BRAF inhibitor vemurafenib, with a half-maximal inhibitory concentration (IC50) of 0.19 μM (Fig. 4c), within the range of IC50 values reported for vemurafenib-sensitive cell lines for melanoma (0.1–0.8 μM)13 and colorectal cancer (0.025–0.35 μM)14 harboring the BRAF p.Val600Gu alteration. These data support the potential efficacy of BRAF inhibitors in treating BRAF-mutant ameloblastomas.
Figure 4.
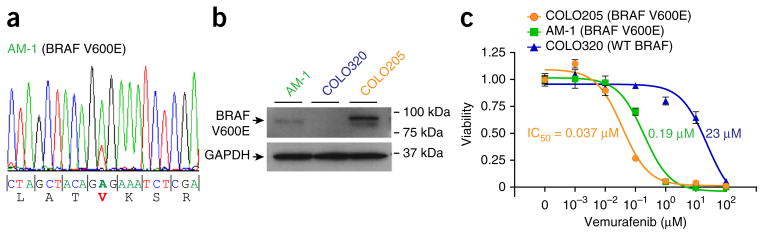
An ameloblastoma cell line harboring BRAF p.Val600Glu is sensitive to the BRAF inhibitor vemurafenib. (a) PCR amplification with Sanger sequencing identifies a BRAF mutation encoding p.Val600Glu in the AM-1 ameloblastoma cell line. (b) Expression of RAF Val600Glu in AM-1 cells was confirmed by protein blot using Val600Glu-specific antibody. COLO320 (wild-type BRAF) and COLO205 (BRAF Val600Glu) colorectal cancer cell lines served as controls.
(c) Vemurafenib inhibits AM-1 cell proliferation/viability (fractional viability normalized to vehicle control) with an IC50 of 0.19 μM. Respective IC50 values for the control BRAF-mutant COLO205 and BRAF–wild type COLO320 cell lines are indicated. Data are representative of three independent cell platings; error bars, s.d. Results presented are representative of two independent trials.
Hedgehog and FGFR-MAPK pathway components are known to be expressed during tooth development and in ameloblastoma15–17. In particular, analyses using gene expression microarrays, immunohistochemistry and quantitative RT-PCR (qRT-PCR) have demonstrated differential expression of Hedgehog pathway genes in ameloblastoma16,17. While this manuscript was under preparation, Kurppa et al.18, using targeted Sanger sequencing, reported BRAF mutations encoding p.Val600Glu in 63% (15/24) of ameloblastomas (all mandibular), consistent with our findings. Nonetheless, to our knowledge, our study is the first to identify a common mutation of the Hedgehog pathway component SMO and to functionally characterize the mutant SMO Leu412Phe protein. Our study is also the first, to our knowledge, to distinguish two molecular subclasses of ameloblastoma (SMO versus BRAF mutated) with different histological and odontological features that are potentially responsive to different molecularly targeted therapies, either established or in clinical trials.
Our findings highlight the relationship between ontogenesis and oncogenesis, in particular, with respect to the biology of epidermal placodes, which are miniorgans that generate both teeth and hair11,15,16. The Hedgehog and FGFR-MAPK pathways are essential for both tooth and hair genesis, and their expression patterns are quite similar. Both SHH and FGFR are expressed at the tip of the invaginating hair bud19,20 and at the tip of the tooth invagination15. In both structures, loss of SHH signaling leads to stunted growth and morphogenesis but does not prevent differentiation: enamel and dentin secretion occur in the tooth, and hair keratins are synthesized in the hair follicle. As with the genesis of their normal counterparts, both ameloblastoma and basal cell carcinoma have mutations in the Hedgehog pathway. We found that nearly half of our ameloblastomas had activating SMO mutations. Likewise, basal cell carcinomas, which are derived from the hair follicle, harbor mutations in PTCH1 (30–40%) and SMO (6–13%), and nevoid basal cell carcinoma syndrome (or Gorlin syndrome) is defined by germline inactivating PTCH1 mutations21. Interestingly, some individuals with Gorlin syndrome also develop keratocystic odontogenic tumors, distinct from ameloblastomas but underscoring the role of Hedgehog signaling in odontogenic neoplasms.
Also notable was the observed relationship between anatomical site and driver mutation, with ameloblastomas arising in the maxilla predominantly carrying SMO mutations and those occurring in the mandible mainly harboring BRAF mutations. This finding may reflect distinctive odontogenic pathways in the upper and lower dentition3. More broadly, this result underscores an emerging appreciation of the anatomical specificity of driver mutations, with this specificity presumably reflecting distinctive developmental pathways based on spatial, temporal and/or cell type–specific cues. Other recently identified examples include meningiomas, in which NF2-mutant tumors originate from lateral and posterior regions along the skull base, whereas tumors with wild-type NF2 (including those with SMO mutation) originate from anterior and medial regions8, and high-grade astrocytomas, in which H3F3A mutations encoding p.Lys27Met (in histone H3.3) characterize brainstem and thalamic tumors, whereas H3F3A mutations encoding p.Gly34Arg or p.Gly34Val and IDH1 or IDH2 mutations characterize cortical tumors22.
From a clinical perspective, tumors (for example, melanomas) with BRAF mutation encoding p.Val600Glu are already being treated with FDA-approved targeted therapies (vemurafenib). Our data suggest that such therapies may be immediately relevant for patients with ameloblastomas positive for BRAF mutation encoding p.Val600Glu. Drug discovery for Hedgehog-pathway inhibitors is also an active area of research; however, current Hedgehog-pathway inhibitors are effective only with inactivating mutations of PTCH as demonstrated by the resistance of SMO Trp535Leu to FDA-approved SMO inhibitors. Experimental trials are using ATO to treat advanced basal cell carcinomas and medulloblastomas with SMO-activating mutations23,24. Our findings suggest that determining the molecular subtype, characterized either by activating SMO or MAPK–pathway mutations, might provide an accurate biomarker test to guide molecularly targeted therapy in ameloblastoma.
In summary, this study demonstrates an emerging approach of transcriptome sequencing in formalin-fixed, paraffin-embedded samples to identify clinically actionable mutations in rare cancers and molecularly defines the majority of the instances of a tumor type that previously had no effective medical therapy. Our findings suggest an immediately actionable drug target (BRAF Val600Glu) and targets of experimental therapies (SMO) for the majority of ameloblastoma cases.
URLs
SIFT, http://sift.jcvi.org/www/SIFT_enst_submit.html; Sequence Read Archive (SRA), http://www.ncbi.nlm.nih.gov/sra/; deFuse, http://compbio.bccrc.ca/; Chimerascan, https://code.google.com/p/chimerascan/; Primer3web, http://primer3.wi.mit.edu/.
ONLINE METHODS
Samples
Paraffin blocks from 28 cases of ameloblastoma were collected from the Departments of Pathology at Stanford University Hospital, the Cleveland Clinic, Oregon Health and Sciences University and the University of British Columbia, with Health Insurance Portability and Accountability Act (HIPAA)-compliant Stanford University Medical Center Institutional Review Board approval. Tissue sections stained with hematoxylin and eosin were reviewed by pathologists R.B.W. and R.T.S. Tumors were morphologically classified by R.B.W. and R.T.S. as plexiform, follicular or desmoplastic25. If a tumor of mixed morphology had a plexiform component, it was counted as plexiform. BRAF Val600Glu expression was evaluated by immunohistochemistry, using a Val600Glu-specific antibody (VE1, Ventana; 12 μg/ml) and peroxidase-based chromogenic staining (EnVision, Dako).
RNA sequencing library preparation and sequencing
Paired-end transcriptome sequencing (RNA-seq) was performed using sequencing libraries prepared from rRNA-depleted RNA isolated from archival formalin-fixed, paraffin-embedded ameloblastoma samples. In brief, tumor cores were sectioned (thickness of 10 μm), RNA was isolated using the AllPrep RNA/DNA FFPE kit (Qiagen) and RNA quality was verified by Bioanalyzer (Agilent Technologies). Sequencing libraries (insert size of 150 bp) were then prepared from 100 ng of rRNA-depleted RNA26 using TruSeq RNA Sample Preparation Kit v2 (Illumina), with four indexed libraries loaded per flow-cell lane. Paired-end 75-bp sequencing was carried out on a HiSeq 2000 instrument (Illumina). The two ameloblastoma libraries yielded 101 and 277 million uniquely mapped reads.
Sequence analysis
For SNV analysis, FASTQ reads were uniquely mapped to hg19 using Bowtie 2 and TopHat 2 (refs. 27,28), and duplicate mapping reads were removed with Picard. SNVs were called with SNVMix2 (ref. 29) and further filtered and annotated with ANNOVAR (SIFT < 0.05)30. High-interest mutations had high driver prediction scores in Cravat and Chasm31 and/or were confirmed to be somatic and reported more than once in the Catalogue of Somatic Mutations in Cancer (COSMIC). Gene mutations selected for validation included the high-interest mutations that were present in both samples and/or involved a gene or pathway that had been implicated in tissue-specific proliferation, differentiation or neoplasia. Analysis for fusion transcripts was performed with SnowShoes32, deFuse33 and Chimerascan34.
Targeted-capture deep sequencing
Genomic DNA was extracted from paraffin-embedded tumor sections with the QIAamp DNA FFPE Tissue kit (Qiagen). DNA (500 ng) was then sequenced using a multiplexed targeted resequencing assay including 48 genes in relevant cancer-associated loci (TruSeq Amplicon Cancer panel, Illumina). Sequencing was carried out to an average depth of 1,000-fold on an Illumina MiSeq next-generation DNA sequencer. Variants were identified by the Illumina variant caller and further analyzed by filtering out common variants and polymorphisms. All mutations were confirmed bidirectionally. The assay had the sensitivity to detect a 1% mutation allele frequency in a wild-type background.
PCR and Sanger sequencing
Genomic DNA was isolated using the QIAamp DNA FFPE Tissue kit (Qiagen). Hot-start PCR using AmpliTaq Gold polymerase (Applied Biosystems) was performed in two rounds (with either the same or nested primer pairs), respectively, for 30 and 20 cycles (94 °C for 30 s, 54 °C for 30 s and 72 °C for 45 s). Sequencing primers were designed using Primer3 (ref. 35) and were vetted using SNPCheck 3. See Supplementary Table 2 for primer sequences. PCR products were verified by gel electrophoresis and purified with the QIAquick PCR Purification kit (Qiagen), and Sanger sequencing (Quintara Biosciences) was then performed. Sequence reads were examined by BLAST alignment to RefSeq transcripts and by manual review of the sequence traces.
SMO functional assays
Human SMO cDNA was obtained from Origene Technologies (clone SC122724). Constructs encoding wild-type, Leu412Phe and Trp535Leu SMO were PCR amplified and inserted into the pGEN expression vector using one-step isothermal DNA assembly as previously described36; all constructs were verified by automated DNA sequencing. 4C20 Smo−/− MEFs were used for signaling assays as previously described12. Briefly, Smo−/− cells were transfected with a plasmid DNA mixture composed of 2% (w/w) SMO (with or without 10% (w/w) Ptch1 cDNA) along with a mixture of 8×Gli-driven luciferase and SV40-driven Renilla luciferase reporter plasmids; a GFP expression plasmid was included to normalize the amount of transfected DNA in each well. Upon reaching confluence, cells were shifted to DMEM with 0.5% serum containing ShhN-conditioned medium, agonists or antagonists (or appropriate vehicle controls) where indicated and incubated for 48 h, at which point, luciferase activity was measured. ShhN-conditioned medium was collected from HEK293-ShhN cells as previously described12 and diluted 20-fold (Fig. 2a,b) or 4-fold (Supplementary Fig. 1) for cell treatment. Cells were tested and confirmed to be negative for mycoplasma. Vismodegib was purchased from LC Laboratories. KAAD-cyclopamine was purchased from Toronto Research. Itraconazole was purchased from Sigma. Clinical-grade, pH-buffered ATO solution was a generous gift from M. Monje (Stanford University School of Medicine). All data points represent means ± s.d. (n = 3 wells per condition) and are representative of multiple independent experiments. For ameloblast-lineage cell experiments, ALC cells10 were grown in DMEM with 10% FBS. The above constructs encoding Myc-tagged SMO were subcloned into pLentiCMV-Blast (Addgene), and the viruses generated were transduced into ALC cells; stably transduced cell pools (with approximately 1 × 105 independent integrations) were selected in 40 μg/ml blasticidin for 7 d. SMO expression was verified by protein blotting using an antibody to the Myc tag (Cell Signaling Technology, 2276; 1:1,000 dilution). Cells were then plated (30,000 cells per well of a 6-well plate; in triplicate), and relative cell proliferation was determined by WST-1 assay after 24, 48 and 72 h. Data shown are representative of multiple independent experiments.
BRAF inhibitor studies
AM-1 cells12 were grown in KSFM complete medium (Life Technologies), and COLO205 and COLO320 cells were grown in RPMI medium with 10% FBS. Cells were not tested for mycoplasma. BRAF Val600Glu protein expression was evaluated using a Val600Glu-specific antibody (NewEast Biosciences, 26039; 1:1,000 dilution). We plated 30,000–50,000 cells per well of a 6-well plate in triplicate, and vemurafenib (Santa Cruz Biotechnology) was added at the indicated concentrations or cells received vehicle control (1% DMSO). Cell viability was measured after 3–5 d by WST-1 assay. IC50 values were determined by fitting a nonlinear log (inhibitor) versus response curve using GraphPad Prism. Data shown are representative of multiple independent experiments.
Supplementary Material
Acknowledgments
We would like to thank E. Epstein for assistance. We would like to thank C. Millward, H. Kaplan and M. Labusch for histology and pathology support.
We are also grateful to H. Harada (Iwate Medical University) for sharing the AM-1 cell line.
Footnotes
AUTHOR CONTRIBUTIONS
R.T.S., A.C.M., J.R.P. and R.B.W. designed the study and wrote the manuscript. A.C.M., B.R.M., J.R.P. and R.B.W. designed the figures. R.T.S., X.G., J.R.P. and R.B.W. analyzed raw sequence data. J.B. and J.R.P. performed mutation validation (PCR and Sanger sequencing). B.R.M., L.N., J.B., J.R.P. and P.A.B. designed and implemented functional studies. C.D.J., J.I.O. and J.L.Z. performed targeted sequencing (TruSeq). K.A.K., K.Q. and R.J.P. performed transcriptome sequencing. S.V. performed immunohistochemistry. T.N., B.P.R. and M.L.T. provided cases for evaluation. T.S. and S.K. provided key cell line reagents.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Accession codes. Raw sequence reads are available in the database of Genotypes and Phenotypes (dbGaP) under accession phs000739.v1.p1.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
References
- 1.Gorlin RJ, et al. Odontogenic tumors. Classification, histopathology, and clinical behavior in man and domesticated animals. Cancer. 1961;14:73–101. doi: 10.1002/1097-0142(196101/02)14:1<73::aid-cncr2820140111>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 2.Sweeney RT, et al. Desktop transcriptome sequencing from archival tissue to identify clinically relevant translocations. Am J Surg Pathol. 2013;37:796–803. doi: 10.1097/PAS.0b013e31827ad9b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tucker A, et al. The cutting-edge of mammalian development; how the embryo makes teeth. Nat Rev Genet. 2004;5:499–508. doi: 10.1038/nrg1380. [DOI] [PubMed] [Google Scholar]
- 4.Davies H, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 5.Parada LF, et al. Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature. 1984;312:649–651. doi: 10.1038/312649a0. [DOI] [PubMed] [Google Scholar]
- 6.Li Y, et al. Activation of FGF receptors by mutations in the transmembrane domain. Oncogene. 1997;14:1397–1406. doi: 10.1038/sj.onc.1200983. [DOI] [PubMed] [Google Scholar]
- 7.Xie J, et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391:90–92. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- 8.Clark VE, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science. 2013;339:1077–1080. doi: 10.1126/science.1233009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taipale J, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005–1009. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- 10.Nakata A, et al. Establishment and characterization of a spontaneously immortalized mouse ameloblast-lineage cell line. Biochem Biophys Res Commun. 2003;308:834–839. doi: 10.1016/s0006-291x(03)01467-0. [DOI] [PubMed] [Google Scholar]
- 11.Wang C, et al. Structure of the human smoothened receptor bound to an antitumour agent. Nature. 2013;497:338–343. doi: 10.1038/nature12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harada H, et al. Establishment of ameloblastoma cell line, AM-1. J Oral Pathol Med. 1998;27:207–212. doi: 10.1111/j.1600-0714.1998.tb01943.x. [DOI] [PubMed] [Google Scholar]
- 13.Søndergaard JN, et al. Differential sensitivity of melanoma cell lines with BRAFV600E mutation to the specific Raf inhibitor PLX4032. J Transl Med. 2010;8:39. doi: 10.1186/1479-5876-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang H, et al. Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res. 2012;72:779–789. doi: 10.1158/0008-5472.CAN-11-2941. [DOI] [PubMed] [Google Scholar]
- 15.Dassule HR, et al. Sonic hedgehog regulates growth and morphogenesis of the tooth. Development. 2000;127:4775–4785. doi: 10.1242/dev.127.22.4775. [DOI] [PubMed] [Google Scholar]
- 16.Kumamoto H, et al. Expression of Sonic hedgehog (SHH) signaling molecules in ameloblastomas. J Oral Pathol Med. 2004;33:185–190. doi: 10.1111/j.0904-2512.2004.00070.x. [DOI] [PubMed] [Google Scholar]
- 17.Heikinheimo K, et al. Gene expression profiling of ameloblastoma and human tooth germ by means of a cDNA microarray. J Dent Res. 2002;81:525–530. doi: 10.1177/154405910208100805. [DOI] [PubMed] [Google Scholar]
- 18.Kurppa KJ, et al. High frequency of BRAF V600E mutations in ameloblastoma. J Pathol. 2014;232:492–498. doi: 10.1002/path.4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiang C, et al. Essential role for Sonic hedgehog during hair follicle morphogenesis. Dev Biol. 1999;205:1–9. doi: 10.1006/dbio.1998.9103. [DOI] [PubMed] [Google Scholar]
- 20.Schneider MR, et al. The hair follicle as a dynamic miniorgan. Curr Biol. 2009;19:R132–R142. doi: 10.1016/j.cub.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 21.Iwasaki JK, et al. The molecular genetics underlying basal cell carcinoma pathogenesis and links to targeted therapeutics. J Am Acad Dermatol. 2012;66:e167–e178. doi: 10.1016/j.jaad.2010.06.054. [DOI] [PubMed] [Google Scholar]
- 22.Fontebasso AM, et al. Chromatin remodeling defects in pediatric and young adult glioblastoma: a tale of a variant histone 3 tail. Brain Pathol. 2013;23:210–216. doi: 10.1111/bpa.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J, et al. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc Natl Acad Sci USA. 2010;107:13432–13437. doi: 10.1073/pnas.1006822107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim J, et al. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell. 2013;23:23–34. doi: 10.1016/j.ccr.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sciubba JJ, et al. Atlas of Tumor Pathology. Vol. 29. Armed Forces Institute of Pathology; Washington, DC: 2001. pp. 71–84. [Google Scholar]
- 26.Morlan JD, et al. Selective depletion of rRNA enables whole transcriptome profiling of archival fixed tissue. PLoS ONE. 2012;7:e42882. doi: 10.1371/journal.pone.0042882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langmead B, et al. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim D, et al. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12:R72. doi: 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goya R, et al. SNVMix: predicting single nucleotide variants from next-generation sequencing of tumors. Bioinformatics. 2010;26:730–736. doi: 10.1093/bioinformatics/btq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang K, et al. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Douville C, et al. CRAVAT: cancer-related analysis of variants toolkit. Bioinformatics. 2013;29:647–648. doi: 10.1093/bioinformatics/btt017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asmann YW, et al. A novel bioinformatics pipeline for identification and characterization of fusion transcripts in breast cancer and normal cell lines. Nucleic Acids Res. 2011;39:e100. doi: 10.1093/nar/gkr362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McPherson A, et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput Biol. 2011;7:e1001138. doi: 10.1371/journal.pcbi.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iyer MK, et al. ChimeraScan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics. 2011;27:2903–2904. doi: 10.1093/bioinformatics/btr467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Untergasser A, et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Myers BR, et al. Hedgehog pathway modulation by multiple lipid binding sites on the smoothened effector of signal response. Dev Cell. 2013;26:346–357. doi: 10.1016/j.devcel.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.