

Abstract
Syt1 (synaptotagmin 1), a major Ca2+ sensor for fast neurotransmitter release, contains tandem Ca2+-binding C2 domains (C2AB), a single transmembrane α-helix and a highly charged 60-residue-long linker in between. Using single-vesicle-docking and content-mixing assays we found that the linker region of Syt1 is essential for its two signature functions: Ca2+-independent vesicle docking and Ca2+-dependent fusion pore opening. The linker contains the basic-amino-acid-rich N-terminal region and the acidic-amino-acid-rich C-terminal region. When the charge segregation was disrupted, fusion pore opening was slowed, whereas docking was unchanged. Intramolecular disulfide cross-linking between N- and C-terminal regions of the linker or deletion of 40 residues from the linker reduced docking while enhancing pore opening, although the changes were subtle. EPR analysis showed Ca2+-induced line broadening reflecting a conformational change in the linker region. Thus the results of the present study suggest that the electrostatically bipartite linker region may extend for docking and fold to facilitate pore opening.
Keywords: bipolar charge distribution, Ca2+-dependent content mixing, Ca2+-independent docking, linker region, soluble N-ethylmaleimide-sensitive fusion protein-attachment protein receptor (SNARE), synaptotagmin 1 (Syt1)
INTRODUCTION
Neurotransmitter release at the synapse constitutes the fundamental basis for major brain functions including cognition, memory and motor control. The precise temporal control of the release is essential for healthy brain activities. SNAREs (soluble N-ethylmaleimide-sensitive fusion protein-attachment protein receptors) are known to be the core fusion machinery in neuro-exocytotic pathways [1–3]. SNARE complex formation between the synaptic vesicle and the plasma membrane is mediated by the cognate coiled-coil motifs on v (vesicle)- and plasma membrane t (target)-SNAREs: one such motif from t-SNARE syntaxin 1A, two from t-SNARE SNAP-25 (25 kDa synaptosome-associated protein) and another from v-SNARE VAMP2 (vesicle-associated membrane protein 2) form a parallel four-stranded coiled-coil, which brings about the apposition of two membranes [4–6]. SNARE complex formation might proceed in sequential steps. Two or three distinct stages have been observed with optical or magnetic tweezer set-ups [7,8]. It is thought, however, that SNARE proteins themselves do not have the required regulatory function that controls the timing of SNARE complex formation [9,10], which confers the temporal on/off switching capability for vesicle fusion. A vesicular protein, Syt1 (synaptotagmin 1), is instead believed to be the key regulator, which senses the spike of the Ca2+ level in response to the action potential and helps trigger fast vesicle fusion [10–12].
At the molecular level, Syt1 contains a transmembrane domain, a putatively unstructured linker region (~60 amino acids) and two cytoplasm tandem C2 domains (C2AB) that bind Ca2+ [13]. Syt1 is thought to be involved both in docking and fusion pore opening [14–16]. In early steps, Syt1 binding to the binary t-SNARE (syntaxin 1A/SNAP-25) on the plasma membrane may mediate vesicle docking [14,16–18]. Recent in vitro experiments indicate that the negatively charged lipid PIP2 [PtdIns(4,5)P2] plays a role in docking via the t-SNARE–Syt1 interaction [17]. In response to the Ca2+ influx, Syt1 inserts itself into the target plasma membrane [19,20], which triggers membrane fusion [19,21].
Mechanistically, the C2AB domain is considered as the functional domain and has been widely used as a soluble substitute for Syt1 [22–25]; however, studies have indicated that the C2AB domain is not an adequate model to recapitulate important Syt1 functions [26,27]. The C2AB domain promotes fusion by aggregating vesicles in response to Ca2+ and thereby enhancing v- and t-SNARE pairing [22,23,27,28], whereas Syt1 is supposed to stimulate membrane fusion by the trans-interaction with the plasma membrane [14,15,26,29]. For the trans-interaction the 60 residue-long linker region appears to be essential as an in vitro study suggests that Syt1 is a distance regulator reaching out to the plasma membrane using this long linker [30].
In the present study, using single-vesicle-docking and content-mixing assays, which has the capacity to detect the docking and the fusion pore opening steps separately [31–33], we found that the Syt1’s linker region is essential for both docking and fusion pore opening. The linker region is featured with the basic-residue-rich N-terminal half and the acidic-residue-rich C-terminal region. Interestingly, this feature is well conserved in many species from Caenorhabditis elegans to humans. When the asymmetric charge distribution was disrupted by double or triple mutations, it hardly affected the docking step, but impaired fusion pore opening. However, disulfide cross-linking between the basic and the acidic regions reduced docking while enhancing Ca2+-triggered fusion pore opening. Our SDSL (site-directed spin labelling) and EPR analysis of the linker showed that upon Syt1 binding to t-SNARE-reconstituted (t-)vesicles in the presence of Ca2+ it became motionally restricted; however, this conformational change was not seen when the charge segregation in the linker was disrupted. Thus the results of the present study suggest that the flexible linker region of Syt1 undergoes conformational changes during vesicle fusion: it stretches out to mediate vesicle docking, but folds to assist the C2AB domain for fusion pore opening.
MATERIALS AND METHODS
Plasmid constructs and site-directed mutagenesis
DNA sequences encoding rat syntaxin 1A (amino acids 1–288 with three native cysteine residues, Cys145, Cys271 and Cys272, replaced by alanines), rat VAMP2 (amino acids 1–116 with Cys103 replaced by an alanine residue), rat SNAP-25 (amino acids 1–206 with four native cysteine residues, Cys85, Cys88, Cys90 and Cys92, replaced by alanines), the C2AB domain (amino acids 140–421), soluble syntaxin 1A (amino acid 191–266) and soluble VAMP2 (amino acids 1–96) were inserted into the pGEX-KG vector as N-terminal GST (glutathione transferase)-fusion proteins. SNAP-25 was also inserted into pET-28b vector as an N-terminal His6 tag fusion protein. Full-length synaptotagmin 1 (Syt1, amino acids 50–421 with four native cysteine residues, Cys74, Cys75, Cys77 and Cys79, replaced by alanines and another, Cys82, replaced by serine) was inserted into the pET-28b vector as a C-terminal His-tagged protein. We used the QuikChange® site-directed mutagenesis kit (Stratagene) to generate all of the Syt1 mutants including Syt1 K86E, K86E/K90E, K86E/K90E/K95E, E131K, E131K/E135K, E131K/E135K/E139K, C277A/G92C/G130C and the Syt1 linker deletion mutant [Δ(99–140)aa]. DNA sequences were confirmed by the Iowa State University DNA Sequencing Facility.
Protein expression and purification
The GST-tagged proteins were expressed in Escherichia coli Rosetta (DE3) pLysS cells (Novagen). Details can be found in our previous work [14]. The His-tagged proteins were expressed in E. coli BL21 (DE3) cells (Novagen) and purified with the same protocol as described previously [14].
Membrane reconstitution
The lipid molecules used in this study are DOPS (1,2-dioleoyl-sn-glycero-3-phospho-L-serine), POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine), PIP2 (from porcine brain), cholesterol and biotin-DPPE [1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(biotinyl)]. All lipids were obtained from Avanti Polar Lipids. DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate), DiD (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate) and sulforhodamine B were obtained from Invitrogen.
For the bulk lipid-mixing assay, the molar ratios of lipids were 15:61:20:2:2 (DOPS/POPC/cholesterol/PIP2/DiI) for the t-SNARE-reconstituted (t-) vesicles and 5:73:20:2 (DOPS/POPC/cholesterol/DiD) for the v-SNARE-reconstituted (v-) vesicles. For the single-vesicle-docking assays, 0.1% biotin-DPPE was added in v-vesicles. The lipid mixture was first completely dried and then hydrated by dialysis buffer [25 mM Hepes (pH 7.4) and 100 mM KCl]. After five freeze–thaw cycles, protein-free large unilamellar vesicles (~100 nm in diameter) were prepared by extrusion through a 100 nm polycarbonate filter (Whatman). For membrane reconstitution, SNARE proteins and Syt1 were mixed with protein-free vesicles at the protein to lipid molar ratio of 1:200 for each protein component (this ratio was kept for all experiments including the single-vesicle content-mixing assay) with ~0.8% OG (N-octyl-β-D-glucopyranoside) in the dialysis buffer at 4°C for 15 min. The mixture was diluted twice times with dialysis buffer and this diluted mixture was then dialysed in 2 litres of dialysis buffer at 4°C overnight. Details for reconstitution were discussed in our previous work [14].
For the single-vesicle content-mixing assay with the small sulforhodamine B content indicator, the lipid compositions were the same as those used in the single-vesicle-docking assay except that the fluorescent lipid dyes (DiI and DiD) were replaced by the equal amount of POPC. The lipid mixture was first completely dried and then hydrated by dialysis buffer, but a population of vesicles to make v-vesicles was hydrated in the presence of 50 mM sulforhodamine B. The overall vesicle preparation and protein reconstitution process was the same as above except that v-vesicles were always kept in the 50 mM sulforhodamine B prior to dialysis overnight. Free sulforhodamine B was removed using a PD-10 desalting column (GE Healthcare) after dialysis.
Bulk lipid-mixing assay
Reconstituted t- and v-vesicles were mixed at a ratio of 1:1. The final lipid concentration was 0.1 mM. The fluorescence intensity was monitored in two channels with the excitation wavelength of 530 nm and emission wavelengths of 570 and 670 nm for donor DiI and acceptor DiD respectively. Fluorescence changes were recorded with the Varian Cary Eclipse model fluorescence spectrophotometer using a quartz cell of 100 μl with a 2 mm path length. All measurements were performed at 35°C.
Single-vesicle-docking and content-mixing assays
After coating the quartz surface with a solution of mPEG [methoxypoly(ethylene glycol)] and biotin-PEG molecules to eliminate non-specific binding of vesicles, the quartz slide was assembled into a flow chamber and coated with neutravidin (0.2 mg/ml). Following a 30 min incubation at room temperature (~25°C), the v-vesicles were immobilized on the PEG-coated surface. After two rounds of washing with 200 μl of buffer, the t-vesicles (100~200 nM) were injected into the flow chamber for 30 min pre-docking at room temperature. After washing out the free t-vesicles, the docking probability was calculated by the ratio of docked t-vesicles and total anchored v-vesicles in the imaging area (45×90 μm2). The details of the single-vesicle-docking assay have been reported in our previous work [14].
For real-time imaging of the small sulforhodamine B content release, the sulforhodamine-B-containing v-vesicles were immobilized on the PEG-coated surface. After two rounds of washing with 1 ml of dialysis buffer, empty t-vesicles were injected into the channel to make them bind to v-vesicles. After 30 min incubation at room temperature, the dialysis buffer with or without 500 μM Ca2+ was injected into the flow chamber at a speed of 33 μl/s by a motorized syringe pump. The details of the single-vesicle content-mixing assay were reported previously [29,31,33].
GST pull-down assay
To form the binary complex, His-tagged SNAP-25 and GST-tagged soluble syntaxin 1A (amino acids 191–266) cell lysates were mixed and loaded on to Ni-NTA (Ni2+-nitrilotriacetate) beads. The binary complex was purified following the same procedure described for His-tagged SNAP-25. The purified binary complex was then loaded on to glutathione–agarose beads. After washing thoroughly with cleavage buffer [50 mM Tris/HCl (pH 8.0), 150 mM NaCl and 0.8% OG] to remove unbound His-tagged SNAP-25, the beads immobilized with the binary complex were separated equally into 1.7 ml Eppendorf tubes. An approximately equal amount of wild-type Syt1 or its mutants and an equal volume of cleavage buffer were added to the immobilized binary complex and the mixture was incubated at 4°C for 1 h. The beads were then washed thoroughly with the cleavage buffer. 5× SDS loading buffer [0.313 M Tris/HCl (pH 6.8), 10% SDS, 0.05% Bromophenol Blue, 50% glycerol and 0.5 M DTT] was added to the samples and boiled for 10 min. Proteins were resolved by precast SDS/PAGE (12% gels) and visualized by Coomassie Blue staining. To test binding of wild-type Syt1 and its mutants to the ternary SNARE complex, purified soluble syntaxin 1A (amino acids 191–266) was mixed with His-tagged SNAP-25 and GST-tagged soluble VAMP2 (amino acids 1–96) cell lysates before loading on to Ni-NTA beads. The ternary complex was purified following the same procedure described for His-tagged SNAP-25. Then the purified ternary complex was loaded on to glutathione–agarose beads. After washing thoroughly with cleavage buffer to remove unbound His-tagged SNAP-25 and the binary complex, binding of wild-type Syt1 and its mutants was carried out following the same procedure as the immobilized binary complex, except that 500 μM Ca2+ was added to the cleavage buffer.
Cross-linking and gel-filtration assays
A native cysteine residue at position 277 in wild-type Syt1 was mutated to an alanine, followed by mutation of Gly92 and Gly130 to cysteine. H2O2 (1 mM) was added to the cell lysate before loading on to Ni-NTA beads, and then the protein was purified following the same procedure described for wild-type Syt1. To separate the intermolecular disulfide-bonded oligomers from the intramolecular disulfide-bond monomer the protein was concentrated and loaded on to a 10/300 GL Superdex 200 column (GE Healthcare) using the Bio-Rad Laboratories biologic Duoflow system. Multi-peak Gaussian curves were fitted to the elution profile obtained from the gel-filtration experiment using Origin 8.0 (Figure 4A). The sample was collected with the elution volume from 15 to 20 ml as a monomer. The samples were analysed using precast SDS/PAGE (12% gels) and visualized by Coomassie Blue staining (Figure 4A, inset).
Figure 4. Disulfide cross-linking or deletion in the Syt1 linker region reduces docking, but enhances pore opening.

(A) FPLC profile showing the separation of the Syt1 monomer. Inset, SDS/PAGE showing purification and separation of Syt1 cross-linked mutants with or without DTT. Lane 1, protein markers; lane 2, wild-type Syt1; lane 3, Syt1 cross-linked mutants eluted from Ni-NTA beads; and lane 4, Syt1 cross-linked monomer. AU, arbitrary units. (B) Normalized docking probability for Syt1 cross-linked monomer mutant with or without DTT. Results are means±S.D. from three independent experiments. Summary of immobilized v-vesicles and docked t-vesicles are shown in Supplementary Table S2 at http://www.biochemj.org/bj/456/bj4560025add.htm. (C) Real-time content mixing with the cross-linked monomer mutant. The solid symbol lines represent wild-type Syt1 in the presence of 500 μM Ca2+ without (●) or with (▼) 1 mM DTT. The open symbol lines represent Syt1 cross-linked monomer mutant in the present of 500 μM Ca2+ without (○) or with (△) 1 mM DTT. (D) The histograms represent content-mixing events within 1 min. Black and grey bars represent content mixing in the presence of 500 μM Ca2+ without and with 1 mM DTT respectively. Results are means±S.D. from three independent experiments (*P < 0.05). Histograms representing content-mixing events in the absence of Ca2+ within 1 min are shown in Supplementary Figure S4 at http://www.biochemj.org/bj/456/bj4560025add.htm. Summary of fusion events among docked vesicles are shown in Supplementary Table S1 at http://www.biochemj.org/bj/456/bj4560025add.htm. (E) Histograms representing the normalized docking probability (left-hand bars) and content-mixing events within 1 min (right-hand bars) with the Syt1 linker deletion mutant. The black bars represent wild-type Syt1 and white bars represent the Syt1 linker deletion mutant. Results are means±S.D. from three independent experiments (*P < 0.05). A summary of fusion events among docked vesicles are shown in Supplementary Table S1 and a summary of immobilized v-vesicles and docked t-vesicles are shown in Supplementary Table S2. WT, wild-type.
SDSL and CW-EPR (continuous wave EPR) data collection
A single cysteine residue mutant, G130C, of wild-type Syt1 and its triple lysine-to-glutamic acid and glutamic acid-to-lysine mutants were labelled using MTSSL [(1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl) methanethiosulfonate], and reconstituted on to v-vesicles with VAMP2. CW-EPR spectra were collected by using a Bruker ESP 300 spectrometer with a loop-gap resonator (Medical Advances) and a low-noise microwave amplifier (Miteq) at room temperature. To examine the conformational changes derived from t-vesicle binding and Ca2+ binding, t-vesicles were added to v-vesicles three times with or without 1 mM Ca2+.
RESULTS
Membrane-anchored Syt1 is essential for fusion pore opening
The C2AB domain is often used as an alternative model for Syt1 in a variety of in vitro studies [22–25]. The C2AB domain has been shown to recapitulate some features of Syt1 functions such as Ca2+-triggered enhancement of SNARE-driven lipid mixing, which has been later found to be via aggregating t- and v-vesicles in the presence of Ca2+ [22,23,27,28]. In the present study, we revisited the Ca2+ control of SNARE-dependent lipid mixing by the C2AB domain and that by membrane-anchored Syt1. As demonstrated previously [14,15,22,23,27], both the C2AB domain and Syt1 show strong Ca2+-dependent stimulation of lipid mixing (Figure 1A and Supplementary Figure S1 at http://www.biochemj.org/bj/456/bj4560025add.htm). Without SNAP-25 no lipid mixing was observed (Figure 1A), indicating that both lipid mixing cases are SNARE-dependent. In the absence of Ca2+, however, the C2AB domain had no effect on lipid mixing, whereas Syt1 still had a substantial stimulatory effect (Figure 1A). This Ca2+-independent stimulation of lipid mixing by Syt1 may be mainly derived from the enhancement of vesicle docking (Supplementary Figure S3A at http://www.biochemj.org/bj/456/bj4560025add.htm), similar to previously published work [14,17,26,27], owing to the t-SNARE–Syt1–lipid interaction, which is absent for the recombinant soluble C2AB domain.
Figure 1. Syt1 is a superior fusion pore opener to the C2AB domain.

(A) Bulk lipid-mixing assays with the C2AB domain (2 μM) or with reconstituted Syt1 (~0.5 μM). Initial rate of changes of the FRET efficiency (Supplementary Figure S1 at http://www.biochemj.org/bj/456/bj4560025add.htm) was calculated (ΔE s−1). The black bar is the control with SNAREs only without Syt1. The white and grey bars represent lipid mixing in the presence of 2 μM C2AB domain with 100 μM Ca2+ (grey bar) or without Ca2+ (white bar), whereas the pattern-filled bars represent the reaction in the presence of Syt1 with 100 μM Ca2+ (pattern-filled and grey bar) or without Ca2+ (pattern-filled and white bar). The light grey bar is the negative control without SNAP-25. Results are means±S.D. from three independent experiments (*P < 0.05). n.s., not significant. (B) The single-vesicle content-mixing assay with 2 μM C2AB domain at different Ca2+ concentrations (grey bars) or reconstituted Syt1 (black bar). White bars represent SNAREs with or without Ca2+. Pattern-filled bar represents reconstituted Syt1 without VAMP2 in the presence of 1 mM Ca2+. Results are means± S.D. from three independent experiments (**P < 0.01). An exemplary image of acceptor and donor channels for content mixing and a typical time trace of dequenching of small sulforhodamine B are shown in Supplementary Figure S2 at http://www.biochemj.org/bj/456/bj4560025add.htm. A summary of fusion events among docked vesicles are shown in Supplementary Table S1 at http://www.biochemj.org/bj/456/bj4560025add.htm.
Next, we compared the capacity of the soluble C2AB domain to drive fusion pore opening with that of membrane-anchored Syt1. As shown previously [31], SNAREs alone without Syt1 cannot efficiently open the fusion pore even in the presence of 1 mM Ca2+ (Figure 1B, lower panel). Pore opening happens in less than 2% of docked vesicles. In the presence of 500 μM Ca2+ Syt1 stimulates fusion pore opening significantly, leading to pore opening for 15% of the docked vesicles (Figure 1B, lower panel). Surprisingly, however, when 2 μM C2AB was used instead of membrane-anchored Syt1, we did not observe efficient content mixing in the Ca2+ range up to 2 mM (Figure 1B, lower panel). When we reconstituted Syt1 into vesicles in the absence of VAMP2, little content mixing was observed in the presence of 1 mM Ca2+ (Figure 1B, lower panel), indicating that the stimulation of fusion pore opening by Syt1 and Ca2+ is strictly SNARE-dependent. On the other hand, when we reconstituted Syt1 into t-vesicles, we observed only very mild enhancement of content mixing (Supplementary Figure S3B). Thus the results of the present study show that anchoring of Syt1 to the vesicular membrane is essential for efficient fusion pore opening.
The linker region of Syt1 is highly conserved, but does not contribute to the binding to SNARE complexes
The fact that the C2AB domain does not reproduce two important functions of Syt1, Ca2+-independent enhancement of docking and Ca2+-dependent stimulation of fusion pore opening, shows that the linker region is indispensable for Ca2+-triggered vesicle fusion. We now take a close look at its amino acid sequence. Strikingly, within the linker region the N-terminal half is enriched with positively charged amino acids, whereas the C-terminal half is abundant in negatively charged amino acids (Figure 2A). Moreover, this feature of the bipolar charge distribution is highly conserved from C. elegans to humans and all of them have almost equal net charge, although the amino acids in linker regions are not as highly conserved as those in the C2 domains in different species.
Figure 2. Asymmetric distribution of charged amino acids on the linker region does not contribute to SNARE binding.

(A) Multiple alignment of the Syt1 linker amino acid sequences from C. elegans to humans shows that the Syt1 linker is highly conserved. Residue numbers of the rat Syt1 sequence are labelled. The positive-charged residues lysine and arginine are shown in blue and the negative charged residues aspartic acid and glutamic acid are shown in red. Net charges are shown on the right-hand side. The highly asymmetric distribution of charged residues is simply emphasized in the upper bar. All amino acid sequences are obtained from the UniProtKB database: Syt1_RAT (P21707), Syt1_HUMAN (P21579), Syt1_CHICK (P47179), Syt1_APLCA (P41823), Syt1_DROME (P21521) and Syt1_CAEEL (P34693). The mutiple alignment was carried out using MutiAlin [35]. (B) The single, double and triple K-to-E (K86E, K86E/K90E and K86E/K90E/K95E) mutants and those of E-to-K (E131K, E131K/E135K and E131K/E135K/E139K) are made to disrupt the asymmetric distribution of charged residues. (C) The charge-disrupting mutations do not alter Syt1’s binding abilities to SNARE complexes as shown by the GST pull-down assay for the binary complex and the ternary complex. The binary complex is formed between GST-tagged soluble Syt1A (amino acids 191–266) and His-tagged SNAP-25. The ternary complex is formed among GST-tagged soluble VAMP2 (amino acids 1–96), Syt1A (amino acids 191–266) and His-tagged SNAP-25. WT, wild-type.
To test if this bipolar charge distribution in the linker region is important for the Syt1’s function, we made several mutants on the linker region to disrupt this asymmetric charge distribution by changing lysine to glutamic acid (K-to-E) or glutamic acid to lysine (E-to-K). We made single, double and triple K-toE mutants (K86E, K86E/K90E and K86E/K90E/K95E mutants respectively; Figure 2B). We also made single, double and triple E-to-K mutants (E131K, E131K/E135K and E131K/E135K/E139K mutants respectively; Figure 2B). We tested if the bipolar charge distribution is important for the SNARE-binding affinity of Syt1. As expected, in our pull-down assay all mutants showed similar binding abilities to t-SNAREs without Ca2+ and to the ternary SNARE complex in the presence of Ca2+ (Figure 2C), indicating that the linker region of Syt1 does not directly contribute to the interaction with SNARE complexes.
The bipolar charge distribution in the Syt1 linker plays a role in fusion pore opening
To test if the asymmetric charge distribution in the Syt1 linker region influences vesicle fusion, we investigated the effect of those Syt1 mutants we made (K86E, K86E/K90E and K86E/K90E/K95E and E131K, E131K/E135K and E131K/E135K/E139K) on vesicle docking and content mixing. The single-vesicle-pairing analysis shows that all mutations have a negligible effect on vesicle docking (Figure 3A). In the content-mixing assay with 500 μM Ca2+, we did not observe any change in content mixing for single E-to-K or K-to-E mutants (Figure 3B, left-hand panel).
Figure 3. Asymmetric distribution of charged amino acids in the Syt1 linker region is important for fusion pore opening.

(A) Normalized single-vesicle-docking probabilities for wild-type (WT) Syt1 and its mutants. Results are means± S.D. from three independent experiments. Summary of immobilized v-vesicles and docked t-vesicles are shown in Supplementary Table S2 at http://www.biochemj.org/bj/456/bj4560025add.htm. (B) Real-time single-vesicle content mixing with Syt1 single (left-hand panel), double (middle panel) and triple (right-hand panel) E-to-K or K-to-E mutants. ●, wild-type Syt1 with Ca2+ ; ○, wild-type Syt1 without Ca2+, ▲ and △ Syt1 K-to-E mutants; and ■ and □, Syt1 E-to-K mutants. (C) Single-vesicle content-mixing events within 1 min. Black bars represent mixing without Ca2+, whereas the grey bars represent mixing in the presence of 500 μM Ca2+. Results are means±S.D. from three independent experiments (*P < 0.05). Summary of fusion events among docked vesicles are shown in Supplementary Table S1 at http://www.biochemj.org/bj/456/bj4560025add.htm.
However, for the double or triple mutants we clearly observed the impairment of fusion pore opening (Figure 3B, middle and right-hand panels) in the presence of 500 μM Ca2+. The quantitative analysis revealed that after 1 min, content mixing for the double mutants was reduced by 20~30% when compared with that of wild-type Syt1, whereas the triple mutants had as much as a 50% reduction (Figure 3C). Thus the results of the present study suggest that the bipolar charge distribution contributes to fusion pore opening in a positive way.
Disulfide cross-linking in the linker reduces docking while enhancing fusion pore opening
By what mechanism does the bipartite linker region contribute to fusion pore opening? One can envision that an electrostatic interaction between the basic N-terminal region and the acidic C-terminal region drives some folding of the linker region, which would effectively shorten the tether between the C2AB and the transmembrane domain.
To further verify the hypothesis that the folding and unfolding state of the linker region of Syt1 regulates synaptic vesicle fusion we designed a double cysteine residue mutant of Syt1, G92C/G130C, which can lock the folded conformation by cross-linking the N-terminal to C-terminal regions by the intramolecular disulfide bond. The double cysteine residue mutant was cross-linked with 1 mM H2O2. Both intermolecular and intramolecular disulfide bonds formed in this process, in which intermolecular dimers or higher oligomers had larger molecular masses than the monomer with the intramolecular disulfide bond. Thus we were able to further purify the monomer component via gel filtration using a 10/300 GL Superdex 200 column (Figure 4A), which was confirmed by SDS/PAGE (Figure 4A, inset). The Syt1 mutant with the disulfide bond in its linker region ran slightly faster than the wide-type monomer, making it easy to confirm the intramolecular disulfide bond formation. We then reconstituted the purified intramolecular cross-linked Syt1 into v-vesicles.
We found that the cross-linked Syt1 mutants could reduce vesicle docking by approximately 40%. Although the changes appear to be statistically insignificant, the inhibition of vesicle docking could be recovered when the sample was treated with DTT within experimental errors (Figure 4B). Thus the results suggest that the flexibility of the linker region is favourable for Syt1 in assisting vesicle docking.
In contrast, in the content-mixing assay, cross-linked Syt1 enhanced fusion pore opening compared with wild-type Syt1 by approximately 50% (Figures 4C and 4D), which also could be negated by DTT. Thus the results show that the folded linker region is preferable for Syt1/Ca2+ to drive opening of the fusion pore for content mixing.
To further substantiate this observation, we made the linker of Syt1 shorter by deleting 40 amino acids [Δ(99–140)aa]. As expected, the truncated mutant of Syt1 reduced the vesicle docking by ~50%, but enhanced fusion pore opening by more than 40% (Figure 4E). Thus the results of the present study support the notion that an extended and flexible linker region is preferred for Syt1 to induce vesicle docking to the plasma membrane, whereas a shorter linker region is favoured for Syt1 to drive fusion pore opening.
Syt1’s binding to t-vesicles in the presence of Ca2+ induces conformation change in the linker
To examine whether t-vesicle binding in the absence or presence of Ca2+ could induce conformation changes in the linker of Syt1, we used SDSL and the EPR analysis of the spin-labelled Syt1 G130C mutant. We found that the linker was in a flexible conformation when reconstituted on v-vesicles (Figure 5A, □). Upon binding to t-vesicles in the absence of Ca2+, the linker region became slightly more flexible as the EPR lines got somewhat narrower (Figure 5A, ○). However, an opposite conformation change happened when Syt1 bound to t-vesicles with Ca2+ (Figure 5A, △), which indicated a conformation change from flexible to restricted. Interestingly, this conformational change was lost when the bipolar charge distribution of linker was disrupted by triple K-to-E or E-to-K mutants (Figures 5B and 5C), consistent with our result from the fusion assay.
Figure 5. Asymmetric distribution of charged amino acids in the Syt1 linker region is important for the Ca2+-dependent conformational change.

First-derivative EPR spectra of spin-labelled wild-type Syt1 (A) and its triple K-to-E (B) and E-to-K (C) mutants. □, EPR spectra of v-vesicles reconstituted with wild-type Syt1 or its triple K-to-E and E-to-K mutants; ○, EPR spectra of v-vesicles reconstituted wild-type Syt1 or its triple K-to-E and E-to-K mutants bound to t-vesicles; and △, EPR spectra of v-vesicles reconstituted wild-type Syt1 or its triple K-to-E and E-to-K mutants bound to t-vesicles in the presence of 1 mM Ca2+.
DISCUSSION
In the present study our single-vesicle-docking and content-mixing assays helped reconstitute two signature Syt1 functions in synaptic vesicle fusion successfully: Ca2+-independent docking and Ca2+-dependent content mixing [14,31]. When the recombinant soluble C2AB domain, for which the linker and the transmembrane regions were deleted, was used instead, these two important Syt1 functions were both lost, consistent with the prediction by Chapman and co-workers [27].
The linker region is featured with the distinct charge segregation: positively charged amino acids are enriched in the N-terminal region, whereas the negatively charged amino acids are clustered in the C-terminal region. Therefore one might wonder if the linker region is capable of folding as an electrostatic zipper, which could regulate the distance between the functional C2AB domain and the membrane anchor. Indeed, the molecular dynamics simulation for the wild-type sequence reveals that the linker region can collapse owing to electrostatic interactions (Supplementary Figure S5 at http://www.biochemj.org/bj/456/bj4560025add.htm). When the charge segregation was disrupted by triple E-to-K or K-to-E mutations in the simulation, the linker region instead prefers to be in an extended conformation (Supplementary Figure S5).
Our single-vesicle experiments showed that the double and triple E-to-K and K-to-E mutations impair content mixing to some extent. Thus the results of the present study suggest that the folded conformation is favourable for content mixing. For the double and triple mutants, the electrostatic interactions are weakened and the tendency for the linker to fold is reduced and, consequently, content mixing is reduced. In contrast, the disulfide cross-linked mutant and the mutant with shortened linker both showed enhanced content mixing, which corroborated the argument.
For both the disulfide cross-linked mutant and the mutant with the shortened linker, vesicle docking was reduced, although the changes were subtle. Thus the data suggest that the extended linker region might be favoured for docking. It is, however, intriguing that for the E-to-K or K-to-E mutants, docking was unchanged when compared with the wild-type Syt1. The results might suggest that the linker region of the wild-type Syt1 stays in an extended conformation most of the time only spending a small portion of time in the folded conformation.
Our EPR experiments provided some structural basis for the proposed mechanistic model for the linker region. The results showed that the Syt1 linker region prefers a flexible conformation during docking, but a folded conformation during content mixing. However, for the triple K-to-E and E-to-K mutants, the ability to flex was lost.
For Syt1-mediated docking the longer linker would be much more effective. The 60-residue-long linker can theoretically extend as much as 30 nm, which will allow the C2AB domain to reach out to bind t-SNARE on the target membrane [16,17]. In contrast, for Ca2+-triggered vesicle fusion the short linker would be the better because it would allow more efficient SNARE complex formation in a confined environment. Thus to make the linker be effective for both docking and fusion it has perhaps evolved to become an electrostatic zipper that can extend to facilitate docking, but fold to help SNARE assembly.
The conformation of the linker region may be in equilibrium between extended and folded conformation. Hypothetically, it suggests an extended conformation in the absence of Ca2+, thereby assisting Syt1-mediated docking (Figure 6A) [33]; however, the equilibrium shifts to the folded conformation in the presence of Ca2+ to reduce the gap between two membranes (Figures 6B and 6C). Conceptually, the hypothetical model presented here is similar to the distance regulation model proposed by Jahn and co-workers for Ca2+-triggered stimulation of SNARE-dependent membrane fusion by Syt1 [30,34]. In the present study we propose that the linker region plays a role in regulating the distance between two opposing membranes.
Figure 6. A hypothetical model summarizing the mechanism by which the Syt1 linker assists docking and fusion.
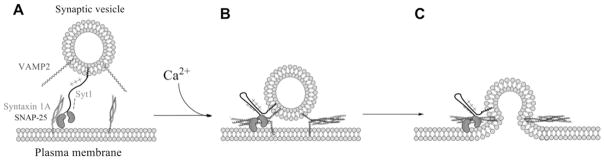
(A) For docking, the Syt1 linker region extends the C2AB domain to reach out to t-SNAREs residing on the plasma membrane surface. (B) Upon Ca2+ arrival the linker region folds, reducing the gap between the vesicle membrane and the plasma membrane, which may facilitate SNARE complex formation. (C) The SNARE complex and Syt1/Ca2+ drive fusion pore opening.
In summary, we show using a single-vesicle-fusion assay that the linker region of Syt1 plays a subtle role in Ca2+-independent docking as well as in Ca2+-dependent content mixing. Such dual function might exist due to the capacity of the electrostatically bipartite linker region to extend and fold to some extent in response to Ca2+ signals.
Supplementary Material
Acknowledgments
We thank Dr Taekjip Ha (University of Illinois at Urbana-Champaign, Urbana, IL, U.S.A.) for providing the analysis program for the single-vesicle assay.
FUNDING
This work was supported by the National Institutes of Health [grant number R01 GM051290 (to Y.-K.S.)].
Abbreviations used
- biotin-DPPE
1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(biotinyl)
- CW-EPR
continuous wave EPR
- DiD
1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate
- DiI
1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate
- DOPS
1,2-dioleoyl-sn-glycero-3-phospho-L-serine
- GST
glutathione transferase
- Ni-NTA
Ni2+-nitrilotriacetate
- OG
N-octyl-β-D-glucopyranoside
- PIP2
PtdIns(4,5)P2
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- SDSL
site-directed spin labelling
- SNAP-25
25 kDa synaptosome-associated protein
- SNARE
soluble N-ethylmaleimide-sensitive fusion protein-attachment protein receptor
- Syt1
synaptotagmin 1
- t-SNARE
target SNARE
- v-SNARE
vesicle SNARE
- VAMP2
vesicle-associated membrane protein 2
Footnotes
AUTHOR CONTRIBUTION
Ying Lai, Xiaochu Lou and Yeon-Kyun Shin designed the experiments; Ying Lai and Xiaochu Lou carried out the experiments; Yongseok Jho carried out the molecular dynamics simulation; and Ying Lai, Xiaochu Lou, Tae-Young Yoon and Yeon-Kyun Shin wrote the paper.
References
- 1.Jahn R, Scheller RH. SNAREs: engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 2.Brunger AT. Structure and function of SNARE and SNARE-interacting proteins. Q Rev Biophys. 2005;38:1–47. doi: 10.1017/S0033583505004051. [DOI] [PubMed] [Google Scholar]
- 3.Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 4.Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 5.Poirier MA, Xiao W, Macosko JC, Chan C, Shin YK, Bennett MK. The synaptic SNARE complex is a parallel four-stranded helical bundle. Nat Struct Biol. 1998;5:765–769. doi: 10.1038/1799. [DOI] [PubMed] [Google Scholar]
- 6.Stein A, Weber G, Wahl MC, Jahn R. Helical extension of the neuronal SNARE complex into the membrane. Nature. 2009;460:525–528. doi: 10.1038/nature08156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao Y, Zorman S, Gundersen G, Xi Z, Ma L, Sirinakis G, Rothman JE, Zhang Y. Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science. 2012;337:1340–1343. doi: 10.1126/science.1224492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Min D, Kim K, Hyeon C, Cho H, Shin YK, Yoon TY. Mechanical unzipping and rezipping of a single SNARE complex reveals hysteresis as a force-generating mechanism. Nat Commun. 2013;4:1705. doi: 10.1038/ncomms2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wickner W, Schekman R. Membrane fusion. Nat Struct Mol Biol. 2008;15:658–664. doi: 10.1038/nsmb.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rizo J, Rosenmund C. Synaptic vesicle fusion. Nat Struct Mol Biol. 2008;15:665–674. doi: 10.1038/nsmb.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- 12.Chapman ER. Synaptotagmin: a Ca2+ sensor that triggers exocytosis? Nat Rev Mol Cell Biol. 2002;3:498–508. doi: 10.1038/nrm855. [DOI] [PubMed] [Google Scholar]
- 13.Perin MS, Brose N, Jahn R, Sudhof TC. Domain structure of synaptotagmin (p65) J Biol Chem. 1991;266:623–629. [PubMed] [Google Scholar]
- 14.Lai Y, Shin YK. The importance of an asymmetric distribution of acidic lipids for synaptotagmin 1 function as a Ca2+ sensor. Biochem J. 2012;443:223–229. doi: 10.1042/BJ20112044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee HK, Yang Y, Su Z, Hyeon C, Lee TS, Lee HW, Kweon DH, Shin YK, Yoon TY. Dynamic Ca2+-dependent stimulation of vesicle fusion by membrane-anchored synaptotagmin 1. Science. 2010;328:760–763. doi: 10.1126/science.1187722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Wit H, Walter AM, Milosevic I, Gulyas-Kovacs A, Riedel D, Sorensen JB, Verhage M. Synaptotagmin-1 docks secretory vesicles to syntaxin-1/SNAP-25 acceptor complexes. Cell. 2009;138:935–946. doi: 10.1016/j.cell.2009.07.027. [DOI] [PubMed] [Google Scholar]
- 17.Kim JY, Choi BK, Choi MG, Kim SA, Lai Y, Shin YK, Lee NK. Solution single-vesicle assay reveals PIP2-mediated sequential actions of synaptotagmin-1 on SNAREs. EMBO J. 2012;31:2144–2155. doi: 10.1038/emboj.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vennekate W, Schroder S, Lin CC, van den Bogaart G, Grunwald M, Jahn R, Walla PJ. Cis- and trans-membrane interactions of synaptotagmin-1. Proc Natl Acad Sci USA. 2012;109:11037–11042. doi: 10.1073/pnas.1116326109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hui E, Johnson CP, Yao J, Dunning FM, Chapman ER. Synaptotagmin-mediated bending of the target membrane is a critical step in Ca2+-regulated fusion. Cell. 2009;138:709–721. doi: 10.1016/j.cell.2009.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arac D, Chen X, Khant HA, Ubach J, Ludtke SJ, Kikkawa M, Johnson AE, Chiu W, Sudhof TC, Rizo J. Close membrane-membrane proximity induced by Ca2+-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat Struct Mol Biol. 2006;13:209–217. doi: 10.1038/nsmb1056. [DOI] [PubMed] [Google Scholar]
- 21.Martens S, Kozlov MM, McMahon HT. How synaptotagmin promotes membrane fusion. Science. 2007;316:1205–1208. doi: 10.1126/science.1142614. [DOI] [PubMed] [Google Scholar]
- 22.Hui E, Gaffaney JD, Wang Z, Johnson CP, Evans CS, Chapman ER. Mechanism and function of synaptotagmin-mediated membrane apposition. Nat Struct Mol Biol. 2011;18:813–821. doi: 10.1038/nsmb.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xue M, Ma C, Craig TK, Rosenmund C, Rizo J. The Janus-faced nature of the C2B domain is fundamental for synaptotagmin-1 function. Nat Struct Mol Biol. 2008;15:1160–1168. doi: 10.1038/nsmb.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schaub JR, Lu X, Doneske B, Shin YK, McNew JA. Hemifusion arrest by complexin is relieved by Ca2+-synaptotagmin I. Nat. Struct Mol Biol. 2006;13:748–750. doi: 10.1038/nsmb1124. [DOI] [PubMed] [Google Scholar]
- 25.Tucker WC, Weber T, Chapman ER. Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science. 2004;304:435–438. doi: 10.1126/science.1097196. [DOI] [PubMed] [Google Scholar]
- 26.Stein A, Radhakrishnan A, Riedel D, Fasshauer D, Jahn R. Synaptotagmin activates membrane fusion through a Ca2+-dependent trans interaction with phospholipids. Nat Struct Mol Biol. 2007;14:904–911. doi: 10.1038/nsmb1305. [DOI] [PubMed] [Google Scholar]
- 27.Wang Z, Liu H, Gu Y, Chapman ER. Reconstituted synaptotagmin I mediates vesicle docking, priming, and fusion. J Cell Biol. 2011;195:1159–1170. doi: 10.1083/jcb.201104079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diao J, Yoon TY, Su Z, Shin YK, Ha T. C2AB: a molecular glue for lipid vesicles with a negatively charged surface. Langmuir. 2009;25:7177–7180. doi: 10.1021/la901676e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kyoung M, Srivastava A, Zhang Y, Diao J, Vrljic M, Grob P, Nogales E, Chu S, Brunger AT. In vitro system capable of differentiating fast Ca2+-triggered content mixing from lipid exchange for mechanistic studies of neurotransmitter release. Proc Natl Acad Sci USA. 2011;108:E304–E313. doi: 10.1073/pnas.1107900108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van den Bogaart G, Thutupalli S, Risselada JH, Meyenberg K, Holt M, Riedel D, Diederichsen U, Herminghaus S, Grubmuller H, Jahn R. Synaptotagmin-1 may be a distance regulator acting upstream of SNARE nucleation. Nat Struct Mol Biol. 2011;18:805–812. doi: 10.1038/nsmb.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lai Y, Diao J, Liu Y, Ishitsuka Y, Su Z, Schulten K, Ha T, Shin YK. Fusion pore formation and expansion induced by Ca2+ and synaptotagmin 1. Proc Natl Acad Sci USA. 2013;110:1333–1338. doi: 10.1073/pnas.1218818110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kyoung M, Zhang Y, Diao J, Chu S, Brunger AT. Studying calcium-triggered vesicle fusion in a single vesicle-vesicle content and lipid-mixing system. Nat Protoc. 2013;8:1–16. doi: 10.1038/nprot.2012.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Diao J, Grob P, Cipriano DJ, Kyoung M, Zhang Y, Shah S, Nguyen A, Padolina M, Srivastava A, Vrljic M, et al. Synaptic proteins promote calcium-triggered fast transition from point contact to full fusion. eLife. 2012;1:e00109. doi: 10.7554/eLife.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park Y, Hernandez JM, van den Bogaart G, Ahmed S, Holt M, Riedel D, Jahn R. Controlling synaptotagmin activity by electrostatic screening. Nat Struct Mol Biol. 2012;19:991–997. doi: 10.1038/nsmb.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988;16:10881–10890. doi: 10.1093/nar/16.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.