

Abstract
The protein tyrosine phosphatase 1B (PTP1B), a non-transmembrane protein tyrosine phosphatase, has been implicated in gastric pathogenesis. Several lines of recent evidences have shown that PTP1B is highly amplified in breast and prostate cancers. The aim of this study was to investigate PTP1B amplification in gastric cancer and its association with poor prognosis of gastric cancer patients, and further determine the role of PTP1B in gastric tumorigenesis. Our data demonstrated that PTP1B was significantly up-regulated in gastric cancer tissues as compared with matched normal gastric tissues by using quantitative RT-PCR (qRT-PCR) assay. In addition, copy number analysis showed that PTP1B was amplified in 68/131 (51.9%) gastric cancer cases, whereas no amplification was found in the control subjects. Notably, PTP1B amplification was positively associated with its protein expression, and was significantly related to poor survival of gastric cancer patients. Knocking down PTP1B expression in gastric cancer cells significantly inhibited cell proliferation, colony formation, migration and invasion, and induced cell cycle arrested and apoptosis. Mechanically, PTP1B promotes gastric cancer cell proliferation, survival and invasiveness through modulating Src-related signaling pathways, such as Src/Ras/MAPK and Src/phosphatidylinositol-3-kinase (PI3K)/Akt pathways. Collectively, our data demonstrated frequent overexpression and amplification PTP1B in gastric cancer, and further determined the oncogenic role of PTP1B in gastric carcinogenesis. Importantly, PTP1B amplification predicts poor survival of gastric cancer patients.
Keywords: Gastric cancer, PTP1B, genomic amplification, poor prognosis, signaling pathways
Abbreviations: Akt, serine/threonine protein kinase; DMEM, Dulbecco's modified Eagles medium; DMSO, dimethyl sulfoxide; EDTA, Ethylenediaminetetraacetic acid; EMT, epithelial-to-mesenchymal transition; Erk, elk-related tyrosine kinase; FAK, focal adhesion kinase; FITC, fluoresceine isothiocyanate; FOXO3a, Forkhead class O transcription factor 3a; H&E, hematoxylin and eosin; HR, hazard ratio; HRP, horseradish peroxidase; IHC, immunohistochemistry; MAPK, mitogen-activated protein kinase; Mdm2, mouse double minute 2; MMPs, metalloproteinases; MTT, 3-[4, 5-dimethylthiazol-2-yl]-2, 5-diphenyl tetrazolium bromide; PBS, phosphate buffered saline; PI3K, phosphatidylinositol 3-kinase; PTP1B, protein tyrosine phosphatase 1B; PVDF, polyvinylidene fluoride; RPMI 1640, Roswell Park Memorial Institute 1640; RT-PCR, Reverse-transcription polymerase chain reaction; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; siRNA, short interfering RNA.
Introduction
Gastric cancer is one of the most common malignancies in Asia, particularly in China, and remains the second leading cause of cancer-related death.1 Although the recent advances in diagnostic and effective therapeutic strategies have significantly improved the survival of patients with early gastric cancer, gastric cancer is usually diagnosed at an advanced stage and clinical outcomes of patients remain dismal.2 Thus, a better understanding of molecular events in gastric carcinogenesis will hopefully provide new insights in mechanisms responsible for the development of gastric cancer and potentially guide the design of new diagnostic and therapeutic strategies to this disease.
Protein phosphorylation and dephosphorylation, which are fundamental cellular events mediated by kinases and phosphatases, govern a host of physiological processes, such as cell growth, differentiation, intercellular communication, cell adhesion and migration.3,4 The protein tyrosine phosphatase 1B (PTP1B) is a classical non-transmembrane protein tyrosine phosphatase that is located on the cytoplasmic face of the endoplasmic reticulum and plays an important role in regulating the activity of major signaling pathways involved in human diseases such as diabetes, obesity and cancer.5 Aberrant expression of PTP1B has been frequently reported in diverse human cancers and accumulating evidences suggest that it exerts both tumor suppressing and tumor promoting effects.5 On one hand, PTP1B can induce tumor cell apoptosis as a putative tumor suppressor.6-8 On the other hand, PTP1B plays an oncogenic role in certain cancers. For example, PTP1B is frequently overexpressed in various cancers, including breast cancer, colon cancer, prostate cancer and gastric cancer,9-12 and has been demonstrated to promote cancer cell growth, migration and invasion both in vitro and in vivo.10-12
In this study, we demonstrated frequent PTP1B amplification in a cohort of gastric cancers by using real-time PCR, and revealed an association of PTP1B amplification with poor survival of gastric cancer patients. Functional studies showed that PTP1B promoted gastric cancer cell growth and invasiveness by modulating major signaling pathways.
Results
Frequent overexpression and amplification of PTP1B in gastric cancer
As the first step to determine the role of PTP1B gene in gastric carcinogenesis, its mRNA expression was assessed in 29 pairs of gastric cancer tissues and matched normal gastric tissues using quantitative RT-PCR (qRT-PCR) assay. As shown in Figure 1A, PTP1B expression was significantly upregulated in gastric cancer tissues as compared with matched normal tissues (P = 0.002). Given that increased gene dosage by gene amplification is a common mechanism for oncogene overexpression during tumorigenesis,13 including gastric cancer,14-16 real-time quantitative PCR method was performed to analyze copy number of PTP1B gene in 131 gastric cancers and 37 control subjects. Copy number of PTP1B gene corresponding to each individual case was shown in Figure 1B. Further analysis showed that copy number of PTP1B gene in gastric cancer cases was significantly higher than control subjects (P =0.0001). With a gene copy number of 4 or more defined as gene amplification, PTP1B amplification was found in 68/131 (51.9%) gastric cancers, whereas no PTP1B amplification was found in the control subjects.
Figure 1.
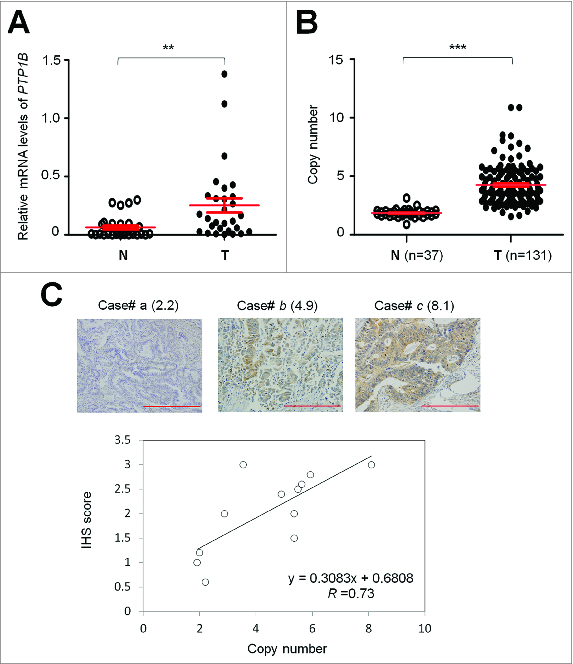
Overexpression and amplification of PTP1B in gastric cancer. (A) PTP1B mRNA expression was significantly up-regulated in primary gastric cancers (T) as compared with matched normal gastric tissues (N) as determined by qRT-PCR assay (n = 29). PTP1B expression level was normalized with 18S rRNA level. Horizontal lines indicate mean ± SE. **, P < 0.01. (B) Real-time quantitative PCR was performed to analyze PTP1B copy number in a cohort of gastric cancers (T) and control subjects (N). Horizontal lines indicate mean ± SE. ***, P < 0.001. (C) Increasing extent of specific staining (brown color) was associated with increasing PTP1B copy number (number inside brackets). Shown are representative cases of immunohistostainging on gastric cancer histologic slides using anti-PTP1B antibody (upper panel). Linear regression analysis was performed to assess the relationship between immunohistostaining score and PTP1B copies on 12 randomly selected gastric cancer cases (lower panel, R = 0.73).
To investigate the association of copy number of PTP1B with its protein expression, we randomly selected 12 gastric cancer cases with different PTP1B copies and did immunohistostaining for PTP1B. As shown in Figure 1C (upper panel), increased staining of PTP1B was seen with increased PTP1B copies. Linear regression analysis on the 12 cases revealed a positive correlation between the PTP1B immunohistostaining score and PTP1B copy number (Fig. 1C, lower panel; R = 0.73).
Association of PTP1B amplification with poor prognosis of gastric cancer patients
Given highly frequent PTP1B amplification in gastric cancer, the association of PTP1B amplification with clinicopathological characteristics was investigated in a large cohort of clinically well-characterized gastric cancers. As shown in Table 1, PTP1B amplification was significantly associated with age (P = 0.03), tumor invasion (P = 0.01), tumor stage (P = 0.04) and cancer-related death (P = 0.009). Although no statistical significance was noted, there was a positive association of PTP1B amplification with tumor size (P =0.06). In order to assess the independent association of PTP1B amplification with age, differentiation, tumor invasion, tumor stage, lymph node metastasis and survival status, we conducted a multivariable logistic regression (Table 2). Similarly, PTP1B amplification was still significantly associated with age (OR = 1.51, 95% CI = 1.03–2.21; P = 0.018), and remained positively associated with tumor invasion (OR = 2.85, 95% CI = 0.52–15.79) and cancer-related death (OR = 2.26, 95% CI = 0.94–5.45), although these associations did not reach statistical difference (Table 2).
Table 1.
Association of PTP1B amplification with clinicopathologic variables in gastric cancer
| Variables | PTP1B amplification (n =131) | ||
|---|---|---|---|
| Yes | No | P | |
| No. of patients | 68 | 63 | |
| Gender | |||
| Male | 53 | 49 | 0.98 |
| Female | 15 | 14 | |
| Age, years | |||
| Mean ± SD | 61.09 ± 13.11 | 57.83 ± 12.66 | 0.03 |
| ≤50 | 12 | 21 | |
| 50–60 | 12 | 17 | |
| 60–70 | 31 | 15 | |
| >70 | 13 | 10 | |
| Tumor localization | |||
| gastric cardia | 21 | 14 | 0.44 |
| gastric body | 15 | 19 | |
| gastric antrum | 32 | 30 | |
| Tumor size (cm3) | |||
| ≤3 | 16 | 27 | 0.06 |
| 3–5 | 28 | 19 | |
| >5 | 24 | 17 | |
| Differentiation | |||
| well/moderate | 26 | 30 | 0.29 |
| poor/undifferentiation | 42 | 33 | |
| Tumor invasion | |||
| T1/T2 | 12 | 24 | 0.01 |
| T3/T4 | 56 | 39 | |
| Tumor stage | |||
| I | 9 | 21 | 0.04 |
| II | 13 | 8 | |
| III | 42 | 32 | |
| IV | 4 | 2 | |
| Lymph node metastasis | |||
| Yes | 46 | 35 | 0.21 |
| No | 22 | 28 | |
| Survival status | |||
| Dead | 42 | 24 | 0.009 |
| Alive | 26 | 39 |
Table 2.
PTP1B amplification in gastric cancer―multivariable models assessing age, differentiation, tumor invasion, tumor stage, lymph node metastasis and survival status
| Factors | PTP1B amplification [OR† (95% CI)] |
|---|---|
| Age1 | 1.51 (1.03–2.21)* |
| Differentiation2 | 1.93 (0.87–4.27) |
| Tumor invasion3 | 2.85 (0.52–15.79) |
| Tumor stage4 | 0.85 (0.29–2.47) |
| Lymph node metastasis | 0.67 (0.20–2.22) |
| Survival status5 | 2.26 (0.94–5.45) |
†OR: odds ratio with 95% confidence interval;1Age (per 10 years);2Differentiation (well or moderate; poor or no differentiation);3Tumor invasion (T1+T2; T3+T4);4Tumor stage (I; II; III; IV);5Survival status (Alive vs. Dead); *Significant at P < 0.05; **Significant at P < 0.01.
To investigate the effect of PTP1B amplification on the survival of gastric cancer patients, univariate survival analysis was performed in this study. As shown in Table 3, PTP1B amplification was significantly associated with poor survival with a hazard ratio (HR) of 2.12 (95% CI, 1.28–3.51; P = 0.003). Next, Kaplan-Meier survival curves were used to further determine the effect of PTP1B amplification on the survival of patients. As shown in Figure 2A, the patients with PTP1B amplification had significantly shorter median survival times than the patients without PTP1B amplification (55.9 months vs. 84.8 months; P =0.003). Given that residual tumor after surgery is an independent risk factor for gastric cancer patients,14 we excluded the patients with residual tumor after surgery to explore the effect of PTP1B amplification on the survival of gastric cancer patients. Similarly, PTP1B amplification still significantly affected clinical outcomes of gastric cancer patients. The patients with PTP1B amplification had significantly shorter survival times than the patients without PTP1B amplification (59.8 months vs. 88.1 months; P = 0.005) (Fig. 2B). Next, we attempted to determine whether the prognostic value of PTP1B amplification as found in univariate analysis is attributable to its association with other factors or whether PTP1B amplification itself contributes independently to prognosis. Thus, Cox multivariate regression analysis was performed in this study. The results suggested that PTP1B amplification is a predictor of poor survival in gastric cancer (HR = 1.72; 95% CI, 1.03–2.86; P = 0.04) as an independently variable with respect to age, differentiation, tumor invasion, tumor stage and lymph node metastasis (Table 3).
Table 3.
Prognostic value of clinicopathologic factors and PTP1B amplification in univariate and multivariate Cox regression analysis (HR† and 95% CI)
| Factors | Univariate analysis | Multivariable analysis |
|---|---|---|
| PTP1B amplification | 2.12 (1.28–3.51)** | 1.72 (1.03–2.86)* |
| Age1 | 1.33 (1.05–1.69)* | 1.19 (0.93–1.54) |
| Differentiation2 | 1.45 (0.88–2.38) | 1.32 (0.78–2.23) |
| Tumor invasion3 | 4.40 (2.01–9.65)*** | 1.17 (0.39–3.54) |
| Tumor stage4 | 2.81 (1.94–4.07)*** | 1.53 (0.77–3.03) |
| Lymph node metastasis5 | 7.46 (3.55–15.71)*** | 3.95 (1.54–10.12)** |
†HR: Hazard Ratio;1Age (per 10 years);2Differentiation (well or moderate; poor or no differentiation);3Tumor invasion (T1+T2; T3+T4);4Tumor stage (I; II; III; IV);5Lymph node metastasis (Yes vs. No); *Significant at P < 0.05; **Significant at P < 0.01; ***Significant at P < 0.001.
Figure 2.
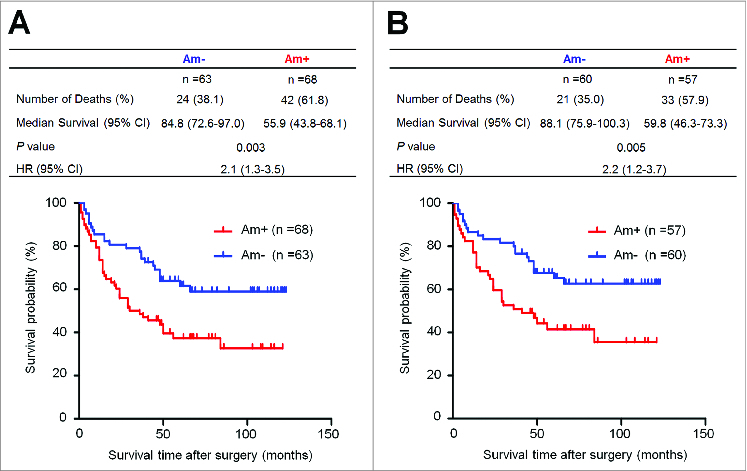
Association of PTP1B amplification with poor survival of gastric cancer patients. Kaplan–Meier survival curves were used to assess the survival of primary gastric cancer patients. (A) The patients with PTP1B amplification (n = 68) had significantly shorter survival times than the patients without PTP1B amplification (n = 63). Median survival was 55.9 months among the patients with PTP1B amplification compared with 84.8 months among the patients without PTP1B amplification. The hazard ratio was 2.1 (95% confidence interval = 1.3, 3.5; P = 0.003, log rank test). (B) When the patients with residual cancers were excluded, the patients with PTP1B amplification (n = 57) still had significantly poor survival compared with the patients without PTP1B amplification (n = 60). Median survival was 59.8 months in the former compared with 88.1 months in the latter. The hazard ratio was 2.2 (95% confidence interval = 1.2, 3.7; P = 0.005, log rank test). Am+, PTP1B amplification; Am-, no amplification.
PTP1B knockdown inhibits gastric cancer cell growth
Frequent overexpression and amplification of PTP1B in primary gastric cancers but not in matched normal tissues suggested that PTP1B may be a putative oncogene in gastric cancer. We thus examined the growth-suppressive effect through knocking down PTP1B expression in SGC7901 and MGC803 cells using siRNA approach. Down-regulation of PTP1B by using 2 different siRNA sequences (si-PTP1B #1 and #2) was confirmed by qRT-PCR and western blot assays (Fig. 3A). These two specific PTP1B siRNAs strongly inhibited cell proliferation in these cells as compared with control siRNA (si-NC), particularly si-PTPN1 #1 (Fig. 3B). In addition, to exclude off-target effect of the si-PTP1B, siRNA rescue assay was also performed in this study. As shown in Figure S1A, introduction of 3 silent mutations into the si-PTP1B #1 binding sequence was sufficient to abolish downregulation of exogenous PTP1B protein by si-PTPN1 #1, indicating that si-PTP1B #1 is highly selective for PTP1B gene. Moreover, our data demonstrated that mutant PTP1B construct was resistant to si-PTP1B in gastric cancer cells as compared with wild-type PTP1B construct, further supporting high specificity of si-PTP1B #1 and suggesting that this phenotype is directly linked to oncogenic function of PTP1B (Fig. S1B). The inhibitory effect on cell growth was also confirmed by colony formation assay. As shown in Figure 3C, the colonies formed in si-PTP1B-transfected cells were fewer than those formed in si-NC transfected cells. Collectively, PTP1B exhibits a growth-promoting activity in gastric cancer cells.
Figure 3 (See previous page).
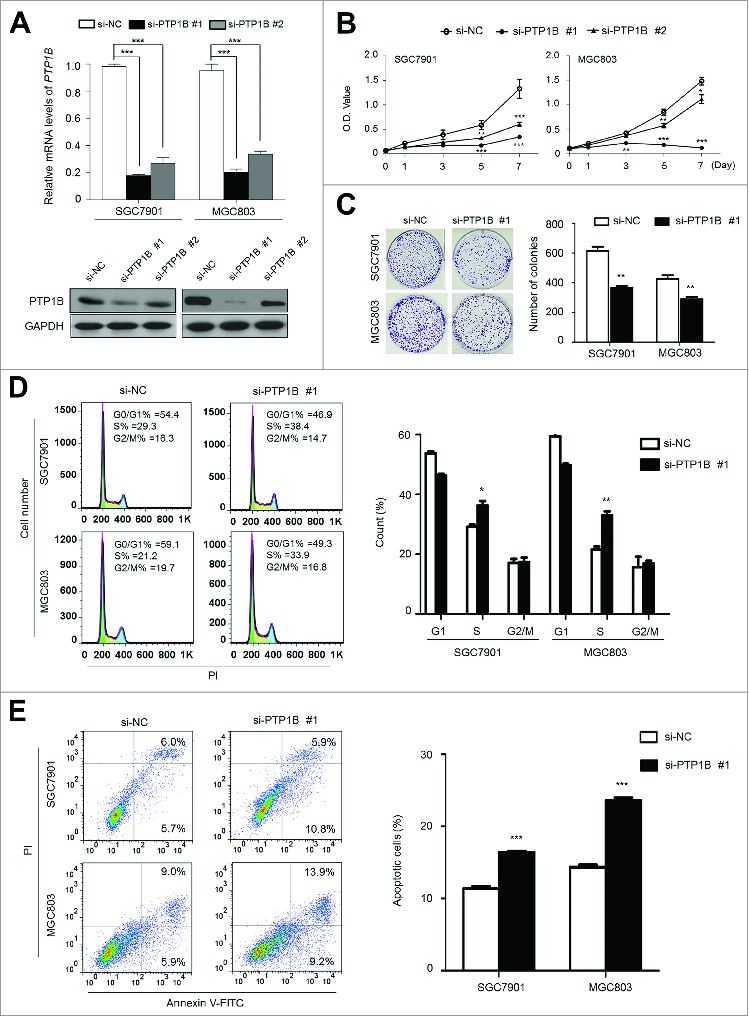
Inhibition of cell growth and induction of cell cycle arrest and apoptosis by knocking down PTP1B expression in gastric cancer cells. (A) Knockdown of PTP1B mRNA (upper panel) and protein (lower panel) by using 2 different siRNAs (si-PTP1B #1 and #2) in gastric cancer cell lines SGC7901 and MGC803 was evidenced by qRT-PCR and western blot, respectively. 18S rRNA was used as a normalized control for qRT-PCR assay. GAPDH was used as loading control in western blot analysis. ***, P < 0.001. (B) PTP1B downregulation significantly inhibited cell proliferation in gastric cancer cells. **, P < 0.01; ***, P < 0.001. (C) The effect of PTP1B knockdown on cell growth was further confirmed by colony formation assay. Left panel shows the representative images of colony formation in cells transfected with si-PTP1B #1 or si-NC. Quantitative analysis of colony numbers is shown in right panel. Data were presented as mean ± SE of values from 3 different assays. **, P < 0.01. (D) Cells were transiently transfected with si-PTP1B #1 or si-NC. After 72 h post-transfection, DNA content was measured by flow cytometry to determine cell cycle fractions. Representative flow cytometric histograms of cell transfected with si-PTP1B #1 and si-NC were shown in left panel. The fraction of cells in each cell cycle phase was indicated in right panel. Data were presented as mean ± SE of values from 3 independent experiments. *, P < 0.05; **, P < 0.01. (E) Cells transiently transfected with si-PTP1B #1 or si-NC. Cell apoptosis was then measured 72 h after transfection by flow cytometry analysis of Annexin V-FITC/PI double-labeled cells. Flow cytometry profiles represent SGC7901 and MGC803 cells with Annexin V-FITC staining in x axis and Pl in y axis, respectively (left panel). The percentages of early apoptotic (bottom right quarter) and late apoptotic (top right) cells were presented in the figures. The percentage of apoptotic cells was presented in right panel. The experiment was repeated 3 times and data were presented as mean ± SE. ***, P < 0.001.
PTP1B knockdown induces cell cycle arrest and apoptosis in gastric cancer cells
We also examined the contribution of cell cycle arrest and apoptosis to the observed growth-inhibitory of si-PTP1B transfected cells. As shown in Figure 3D, as compared with si-NC transfected cells, cell cycle was arrested at the S phase in si-PTP1B transfected cells. The percentage of S phase was increased from 29.2 ± 1.2% to 36.3 ± 2.6% in SGC7901 cells (P = 0.013) and from 21.6 ± 1.7% to 33.1 ± 2.2% in MGC803 cells (P = 0.002), respectively. Next, the effect of PTP1B knockdown on apoptosis was investigated in these 2 cell lines. As shown in Figure 3E, si-PTP1B transfection showed an increase in cell apoptosis as compared with si-NC transfection (16.4 ± 0.3% vs. 11.4 ± 0.4% in SGC7901 cells, P = 0.0001; 23.6 ± 1.6% vs. 14.4 ± 0.5% in MGC803 cells, P = 0.0001).
PTP1B knockdown inhibits gastric cancer cell migration and invasion
Given that cancer cell invasion and metastasis is the main cause of gastric cancer-related death,1,17 we attempted to explore the effect of PTP1B knockdown on the migration and invasion abilities of gastric cancer cells. As expected, there was a significantly lower number of migrated cells in siPTP1B-transfected cells than siNC-transfected cells (Fig. 4A, upper panel), suggesting that PTP1B silencing inhibited cancer cell migration. Moreover, the Matrigel invasion assays showed that the number of cells passed through Matrigel-coated membrane into the lower chamber was significantly lower in siPTP1B-transfected cells than in siNC-transfected cells (Fig. 4A, lower panel), indicating that PTP1B knockdown inhibited the invasive potential of gastric cancer cells. To determine whether the effect of PTP1B down-regulation on cell metastasis was associated with matrix metalloproteinases (MMPs), qRT-PCR was used to assess the expression of MMP-2,-9 and -14 genes, 3 representative MMPs involved in cancer cell invasion.18 As shown in Figure 4B, PTP1B knockdown significantly inhibited the expression of these 3 genes in these cells, suggesting that the decrease in the metastasis-associated phenotypes may be link to the inhibition of MMP -2, -9 and -14.
Figure 4.
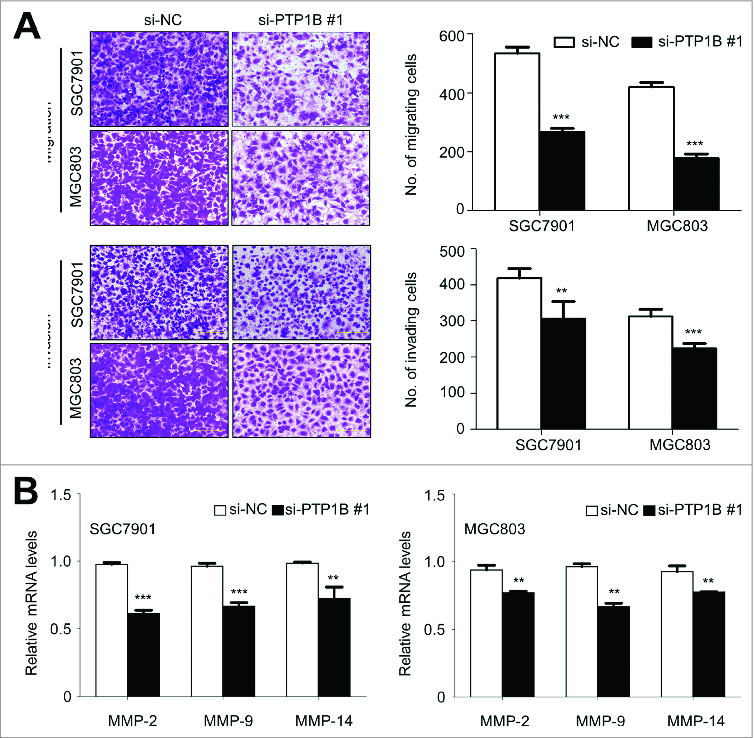
Inhibition of gastric cancer cell migration and invasion by down-regulating PTP1B expression. (A) Cells transfected with si-PTP1B #1 or si-NC were starved overnight and then seeded in the Transwell chambers without Matrigel for migration assay, and coated with Matrigel for invasion assay, respectively. After a 12 h or 24 h-culture, non-migrating (or non-invading) cells in the upper chamber were removed and migrating (or invading) cells were stained and calculated in 5 microscopic fields per sample. Shown are representative images of migrating (or invading) cells (left panels). The bar graphs (right panels), corresponding to left panels, show means ± SE of the numbers of migrating (or invading) cells from 3 independent assays. **, P < 0.01; ***, P < 0.001. (B) qRT-PCR assay was performed to assess the effect of PTP1B knockdown on the expression of metastasis-related genes MMP-2,-9 and -14 in gastric cancer cells. Expression levels of these genes were normalized with 18S rRNA levels. Data were presented as mean ± SE. *, P < 0.05;**, P < 0.01.
To further clarify the mechanism of PTP1B contributing to cell migration and invasion, we investigated the effect of PTP1B on the process of epithelial-mesenchymal transition (EMT), which is one of the critical steps during tumor metastasis and plays a central role in the invasion and metastasis of various cancers, including gastric cancer.19 As shown in Figure 5A, immunofluorescence assay showed that PTP1B knockdown substantially promoted the expression of epithelial cell marker E-cadherin and reduced the expression of 2 well-known mesenchymal markers N-cadherin and Vimentin in these 2 gastric cancer cells. As compared with si-NC transfection, the number of E-cadherin positive cells was increased in si-PTP1B transfected cells (20.2 ±1 .1% vs. 2.9 ± 0.5% in SGC7901 cells, P = 0.0001; 22.9 ± 1.1% vs. 2.5 ± 0.6% in MGC803 cells, P = 0.0001), whereas the number of N-cadherin and Vimentin positive cells was significantly reduced in si-PTP1B transfected cells (N-cadherin: 6.5 ± 0.9% vs. 34.0 ±2 .3% in SGC7901 cells, P = 0.0001; 22.7 ± 1.8% vs. 54.3 ± 1.3% in MGC803 cells, P = 0.0001; Vimentin: 7.4 ± 0.4% vs. 23.4 ±1 .3% in SGC7901 cells, P = 0.0001; 9.8 ± 1.3% vs. 36.5 ± 0.4% in MGC803 cells, P = 0.0001) (Fig. 5B). These findings suggest that inhibition of the EMT process by PTP1B downregulation may contribute to suppression of gastric cancer cell migration and invasion.
Figure 5 (See previous page).
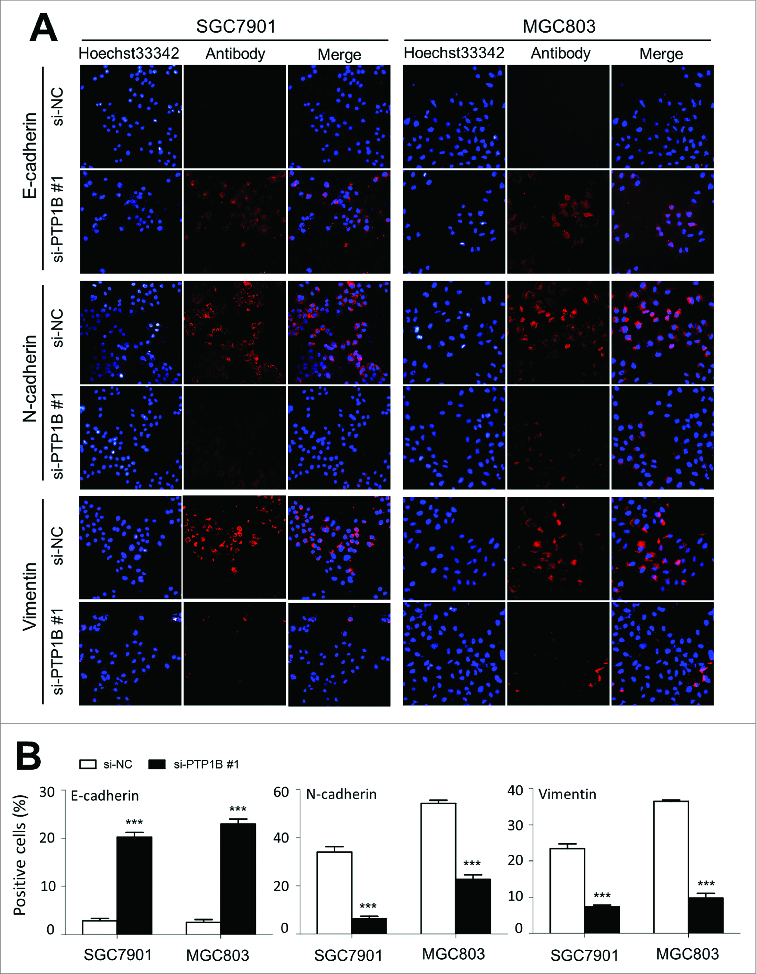
Effect of PTP1B down-regulation on the process of EMT in gastric cancer cells. Cells transfected with si-PTP1B #1 or si-NC were seeded on the coverslips in 6-well plates. After a 24 h-culture, immunofluorescence staining was performed to assess the expression of an epithelial cell marker E-cadherin and 2 mesenchymal markers N-cadherin and Vimentin in SGC7901 and MGC803 cells. Representative immunofluorescence stainings of target protein in cells transfected with si-PTP1B #1 and si-NC were shown in panel (A). Red color represents target protein fluorescence and blue color represents Hoechst33342 staining for nuclei. The percentage of cells showing positive E-cadherin, N-cadherin and Vimentin expression was indicated in panel (B). The experiment was repeated 3 times and data were presented as mean ± SE. ***, P < 0.001.
PTP1B regulates major signaling pathways in gastric cancer
To gain insights into molecular mechanisms underlying oncogenic role of PTP1B in gastric cancer, we investigated the effect of PTP1B on the activities of Ras/MAPK and phosphatidylinositol-3-kinase (PI3K)/Akt pathways, which play a crucial role in cell proliferation and survival in gastric cancer.20 As expected, PTP1B knockdown significantly inhibited the activities of both pathways in gastric cancer cells, characterized by reduced phosphorylation of Erk and Akt (Fig. 6A). It is well known that FOXO3a, a primary transcription factor and downstream effector of MAPK and PI3K/Akt pathways,21 can transcriptionally regulate a number of cell cycle- and apoptosis-related genes, contributing to carcinogenesis.22,23 Thus, we investigated the effect of PTP1B on FOXO3a expression in these cells. As shown in Figure 6A, PTP1B down-regulation did not affect FOXO3a expression in these cell lines. However, we found that phosphorylation of FOXO3a was dramatically inhibited upon PTP1B knockdown in these 2 cell lines (Fig. 6A), as supported by the previous studies that FOXO3a can be post-transcriptionally regulated by these 2 pathways.21,24 Taken together, our data implicate that PTP1B promotes gastric cancer cell growth and invasiveness through activating the MAPK and PI3K/Akt signaling pathways.
Figure 6.
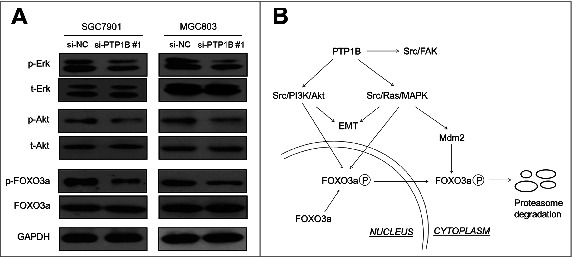
PTP1B acts as a potential oncogene by activating major signaling pathways. (A) Cells transfected with si-PTP1B #1 or si-NC were lysed and lysates were subjected to protein gel blot assays. The antibodies against phospho-Erk (p-Erk), total Erk (t-Erk), phospho-Akt (p-Akt) and total Akt (t-Akt) were used to determine the effect of PTP1B downregulation on the activities of the MAPK and PI3K/Akt pathways. The antibodies against FOXO3a and phosphorylated FOXO3a (p-FOXO3a) were used to test the effect of knocking down PTP1B expression on the transcriptional activity of FOXO3a. GAPDH was used as a loading control. (B) Schematic model of molecular mechanisms underlying oncogenic role of PTP1B in gastric cancer. PTP1B overexpression or amplification in gastric cancer cells increases Src activity by reducing phosphorylation at tyrosine 530 of Src and sequentially activates Src-related signaling pathways, such as Src/FAK, Src/Ras/MAPK and Src/PI3K/Akt. Moreover, knocking down PTP1B expression inhibits phosphorylation of FOXO3a, nuclear-cytoplasmic translocation and proteasome degradation through blockade of the MAPK and PI3K/Akt pathways, further contributing to the enhancement of FOXO3a transcriptional activity in gastric cancer cells. Taken together, PTP1B overexpression or amplification promotes gastric cancer cell growth and invasiveness through activating Src-related signaling pathways, ultimately contributing to poor prognosis of gastric cancer patients.
Discussion
In this study, we found that PTP1B was frequently overexpressed in gastric cancer tissues as compared with matched normal gastric tissues, suggesting that PTP1B may be a potential oncogene in gastric cancer. The increased expression is bound up with genomic amplification, as demonstrated by qRT-PCR analysis, implicating that gene amplification is one of major mechanisms for PTP1B up-regulation in gastric cancer. Given frequent PTP1B amplification in gastric cancer cases, we investigated its clinical significances and prognostic values in a cohort of clinically well-characterized gastric cancers. Our data showed that PTP1B amplification was associated with a significantly increased risk of cancer-related death. More noteworthy, its amplification dramatically affected the overall survival of gastric cancer patients. Collectively, PTP1B amplification may be used as a potential prognostic marker for gastric cancer patients.
Considering that frequent overexpression and amplification of PTP1B may contribute to gastric tumorigenesis, oncogenic effect of PTP1B was tested in gastric cancer cells. PTP1B knockdown in the overexpressed gastric cancer cell lines showed significant growth-inhibitory effect by suppression of cell proliferation and colony formation as well as induction of cell cycle arrest and apoptosis, further confirming its potential oncogenic function. Notably, down-regulating PTP1B strongly inhibited gastric cancer cell migration and invasion, as supported by our findings that PTP1B knockdown inhibited the EMT process in gastric cancer cells. In addition, given that matrix metalloproteinases (MMPs) play a key role in tumor metastasis,25 the effect of PTP1B on expression of MMP-2,-9 and -14 was tested in gastric cancer cells. Our data showed that PTP1B knockdown significantly inhibited expression of these genes, implicating that the decrease in the metastasis-associated phenotypes may be mediated by suppressing expression of some MMPs genes.
To better understand oncogenic role of PTP1B in gastric cancer, we assessed the impact of PTP1B on aberrant signaling of MAPK and PI3K/Akt pathways in gastric cancer cells. These two pathways play a fundamental role in gastric tumorigenesis, and are therefore used as therapeutic targets for this cancer.20,26,27 As expected, our data demonstrated that down-regulating PTP1B expression significantly inhibited phosphorylation of Erk and Akt in gastric cancer cells, suggesting that PTP1B plays its oncogenic role through activating the MAPK and PI3K/Akt pathways. We also found that, although protein expression level of FOXO3a, a downstream effector of MAPK and PI3K/Akt pathways, was not influenced by down-regulating PTP1B expression, phosphorylation of FOXO3a was strongly inhibited in gastric cancer cells. This is supported by a previous study that FOXO3a can be post-transcriptionally regulated by multiple major pathways among which MAPK and PI3K/Akt cascades are the 2 primary ones.21 FOXO3a has been demonstrated to be phosphorylated at Thr32 and Ser253 by the PI3K/Akt pathway, which can not only disrupt the interaction of FOXO3a with DNA, but also cause its subsequent nuclear export and proteasome degradation.21,28 Accumulating evidences have shown that FOXO3a is frequently inactivated in various human cancers due to the overactivation of PI3K/Akt pathway, leading to loss of its tumor suppressive function.23,28 It has also been reported that decreased expression of FOXO3a is found in gastric cancer and is associated with poor prognosis of gastric cancer patients.29 Moreover, the constitutive activation of MAPK pathway also can phosphorylate FOXO3a at multiple residues, leading to Mdm2-mediated ubiquitination and proteasome degradation.24 Therefore, the development of new and effective therapeutic drugs to multitarget the MAPK and PI3K/Akt pathways is expected to boost anti-tumor activity by maximal activation FOXO3a.21,30
It is well documented that PTP1B overexpression increases Src specific activity in cancer cells by reducing phosphorylation at tyrosine 530 of Src.10,12,31 Accordingly, suppression of PTP1B activity by PTP1B inhibitor or knocking down PTP1B expression using siRNA reduces Src kinase activity. Src tyrosine kinase is the first characterized proto-oncogene, specifically c-Src acts at points of integration, relaying signals from cell surface receptors to the nucleus.32,33 Src activation has been associated with proliferation, survival, differentiation and motility in cancer cells through modulating multiple signaling pathways, such as Src/FAK, Src/PI3K/Akt and Src/Ras/MAPK,33 as supported by the findings in the present study that PTP1B knockdown strongly inhibits phosphorylation of Erk and Akt in gastric cancer cells. These observations suggest that decreasing Src activity through inhibition of PTP1B may even provide a means to treat a subset of cancers. Therefore, small molecular inhibitors of PTP1B can be promising drug candidates in recent years. However, targeting PTP1B for drug discovery is still challenging because of the highly conserved and positively charged active-site pocket, although some of PTP1B inhibitors have been developed and used in the treatment of cancers.34-36
In summary, we found frequent overexpression and amplification of PTP1B in a cohort of gastric cancers, and demonstrated a strong association of PTP1B amplification with poor prognosis of gastric cancer patients. Our data are consistent with a model (Fig. 6B) in which PTP1B contributes to gastric carcinogenesis by promoting cell growth and invasiveness through modulating Src-related signaling pathways.
Materials and Methods
Clinical samples
With the institutional review board approval, a total of 29 matched pairs of normal and resected primary gastric cancer tissues were obtained from the First Affiliated Hospital of Xi’an Jiaotong University School of Medicine, and were used for mRNA expression analysis. For copy number analysis, formalin-fixed and paraffin-embedded tissue sections from 131 gastric cancer patients were randomly obtained from the First Affiliated Hospital of Xi’an Jiaotong University School of Medicine. The normal controls from 37 patients with chronic gastritis who underwent endoscopic biopsy, were also obtained from the First Affiliated Hospital of Xi’an Jiaotong University School of Medicine. Informed consent was obtained from each patient before the surgery. All patients did not receive chemotherapy and radiotherapy before the surgery, and all sections were histologically examined by a senior pathologist at Department of Pathology of the Hospital based on World Health Organization (WHO) criteria. Clinicopathological data were obtained from the patients’ files or by interview with the patients or their relatives, and were summarized inTable 1.
Cell culture and short interfering RNA (siRNA) transfection
Human gastric cancer cell lines SGC7901 and MGC803 were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum. For transient siRNA transfection, cells were transfected at 70% confluence using Lipofectamine 2000 (Invitrogen, Grand Island, NY), with a final siRNA concentration of 50 nM for SGC7901 and 100 nM for MGC803. Oligonucleotides were obtained from GenePharma (Shanghai, P.R. China) and the sequences of siRNA targeting PTP1B (si-PTP1B) and control siRNA (si-NC) were presented in Table S1. For the siRNA rescue assay, cells were co-transfected with 1 μg/ml wild-type or mutant PTP1B expression plasmid (Biofeng Inc., Shanghai, P.R. China) as well as the indicated concentrations of si-PTP1B #1 and si-NC. In the mutant PTP1B construct, 3 silent mutations were introduced into si-PTP1B #1 binding sequence (GTCGGATaAAgCTtCATCA) by using QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). Lowercase letters indicate altered nucleotides in the PTP1B gene at positions 171, 174 and 177.
RNA extraction and qRT-PCR
According to the instructions of manufacturer, total RNA was extracted from tissues and cell lines using TRIzol reagent (Takara Inc., Dalian, P.R. China) and cDNA was prepared using PrimeScript RT reagent Kit (Takara Inc., Dalian, P.R. China). Quantitative RT-PCR (qRT-PCR) was performed on a CFX96 Thermal Cycler DiceTM real-time PCR system (Bio-Rad Laboratories, Inc., CA) using SYBR Premix Ex TaqTM (Takara Inc.., Dalian, P.R. China). The mRNA expression of each gene was normalized to 18S rRNA cDNA. Each sample was run in triplicate. The primer sequences were presented in Table S2.
Tissues and DNA preparation
Serial sections from each tumor tissue were cut at 5 μm. One of sections was stained by hematoxylin and eosin (H&E) and was marked as a tumor representative tissue by a senior pathologist for gastric cancer. Other tumor tissues were then isolated by manual microdissection under an inverted microscope using the marked H&E section as target tissue identification. DNA was extracted from isolated tissues as previously described.14 Briefly, after a treatment for overnight at room temperature with xylene to remove pareffin, tissues were digested with sodium dodecyl sulfate (SDS)/proteinase K solution at 48°C for 48 h. DNA was then isolated using standard phenol/chloroform protocol, and was dissolved in distilled water and stored at −80°C until use. Subsequent sections were mounted on 3-aminopropyltriethoxysilane-coated slides for immunohistochemical assay.
Copy number analysis
Real-time quantitative PCR technique was used to analyze the copy number of PTP1B gene in 131 gastric cancer sample and 37 control subjects on a CFX384 Thermal Cycler DiceTM real-time PCR system (Bio-Rad Laboratories, Inc., CA) as described previously.15 This method was well established and widely used in diverse human cancers.14,15,37-39 Specific primers and TaqMan probes were designed using Primer Express 3.0 (Applied Biosystems) to amplify both the PTP1B and β-actin genes. For the PTP1B gene, the TaqMan probe used was 5′-6FAM-TAA CCC ATC TCT GCC CTC TGA TTC CTC AG-TAMRA-3′, and the primers were 5′-GCC ATT CAT TTT CTC CAA AGT GA-3′ (forward) and 5′-CGA CCC GAC TTC TAA CTT CAG TGT-3′ (reverse). The TaqMan probe and primers for β-actin gene were described previously.14 Using a PCR protocol described previously,14,15 the samples were run in triplicate, and β-actin was run in parallel to normalize input DNA. Standard curves were established using serial dilutions of normal leukocyte DNA. PTP1B amplification was defined by a copy number ≥ 4.
Immunohistochemistry (IHC)
This procedure was pursued to investigate the association of PTP1B protein expression with its amplification in the tumor tissues. Briefly, paraffin-embedded sections (5 μm) were deparaffinized and rehydrated in a graded series of ethanol, and washed in distilled water. The sections were then incubated with anti-PTP1B antibody (Epitomics, Inc..) overnight at 4°C after antigen retrieval and blocking. Immunodetection was performed with the Streptavidin-Peroxidase system (ZSGB-bio, Beijing China) according the manufacture's protocol, followed by reaction with diaminobenzidine and counterstaining with hematoxylin. To insure the comparability of immunohistochemical staining, a common reference standard was included to serve as an internal or intra-assay control in each batch. PTP1B protein expression was scored in double-blinding way (i.e., without knowing the PTP1B copy number of the case), and 0, 1, 2, 3 reprents negative, weak positive, positive, and strong positive, respectively.
Western blot analysis
Cells were lysed in prechilled RIPA buffer containing protease inhibitors. Supernatants were collected and subjected to 10% SDS-PAGE, and transferred onto PVDF membranes (Roche Diagnostics, Mannheim, Germany). The membranes were then incubated with primary antibodies. Anti-PTP1B was purchased from Epitomics, Inc.. Anti-phospho-AktSer473, anti-phospho-Erk1/2 and anti-total-Akt (t-Akt) were purchased from Bioworld Technology, co, Ltd. Anti-total-Erk1/2 (t-Erk) was purchased from Abcam Inc. Anti-FOXO3a, anti-phospho-FOXO3aThr32 were purchased from Cell Signaling Technology, Inc.. Anti-GAPDH were purchased from Abgent, Inc. This was followed by incubation with species-specific HRP-conjugated secondary antibodies from ZSGB-BIO, and immunoblotting signals were visualized using the Western Bright ECL detection system (Advansta, CA).
Cell proliferation and colony formation assays
Cell proliferation was determined by the MTT assay. In brief, cells transfected with siPTP1B or siNC (200/well) were seeded and cultured in 96-well plates for 1, 3, 5 and 7 d At the indicated times, 20 μl of 0.5 mg/ml MTT (Sigma, Saint Louis, MO) was added into the medium and incubated for 4 h, followed by adding 150 μl of DMSO for additional 15 min. The plates were then read on a microplate reader using a test wavelength of 570 nm and a reference wavelength of 670 nm. Three triplicates were done for each datapoint.
Colony formation assay was performed using monolayer culture. Cells (1000/well) transfected with different siRNAs were seeded in 6-well plates. The medium was refreshed every 3 d After 14 d of culture, surviving colonies (≥50 cells per colony) were fixed with methanol and stained with 0.5% crystal violet, and the colonies were then counted. The experiments were performed in triplicate.
Cell cycle and apoptosis assays
For cell cycle assay, cells transfected with different siRNAs were harvested at 72 h when the confluence reached ∼80%, and washed twice with PBS. Cells were then fixed in ice-cold 70% ethanol for 30 min, and stained with propidium iodide solution (50 μg/mL propidium iodide, 50 μg/mL RNase A, 0.1% Triton-X, 0.1 mM EDTA). Cell cycle phase distributions were determined on a Flow Cytometer (BD Biosciences, NJ). For apoptosis analysis, the indicated cells were harvested, washed with PBS, suspended in binding buffer, and sequentially stained with Annexin V-FITC Detection Kit (Roche Applied Science, Penzberg, Germany) by flow cytometer according to the manufacturer's protocol. Each experiment was performed in triplicate.
Cell migration and invasion assays
Cell migration and invasion assays were assessed by transwell chambers (8.0 μm pore size; Millipore, MA). For cell invasion assay, chambers were coated with Matrigel (4 × dilution; 15 μl/well; BD Bioscience, NJ). Cells transfected with different siRNAs were starved overnight and then seeded in the upper chamber at a density of 2 × 104 cells/ml for migration assay and 4 × 104 cells/ml for invasion assay in 200 μl of medium containing 0.5% FBS. Medium with 10% FBS (1 ml) was added to the lower chamber. After a 12- or 24-h incubation, non-migrating/non-invading cells in the upper chamber were removed using a cotton swab, and migrating/invading cells were then fixed in 100% methanol and stained with crystal violet solution (0.5% crystal violet in 2% ethanol). Photographs were taken randomly for 5 fields of each membrane. The number of migrating/invading cells was expressed as the average number of cells per microscopic field over 5 fields.
Immunofluorescence staining
Cells transfected with different siRNAs were seeded on the coverslips in 6-well plates and cultured for 24 h. Next, cells were fixed with 4.0% formaldehyde in phosphate-buffered saline for 15 min, and blocked with 5% goat serum for 30 min. The coverslips were then incubated at 4°C with primary antibodies overnight in a moist chamber. Anti-E-cadherin and anti-Vimentin were purchased from Epitomics, Inc.. Anti-N-cadherin was purchased from Abcam Inc.. The coverslips were incubated with Cy3-conjugated goat anti-rabbit secondary antibody (Bioss, Beijing, P.R. China) at room temperature for 90 min. After three washes with PBS, the coverslips were then dried, dyed with Hoechst33342, and fixed in glycerol. The images were obtained with an Olympus IX71 microscope (Olympus, Tokyo, Japan), and color mergence was performed using ImageJ image software (ImageJ version 1.44p, NIH, MD). Photographs were taken randomly for 5 fields of each coverslip. The percentage of positive cells was expressed as the average percent of cells per microscopic field over 5 fields.
Statistical analysis
The Mann-Whitney U test was used to compare copy number of PTP1B gene between gastric cancer tissues and control subjects. Correlation between PTP1B amplification and clinicopathological characteristics was analyzed by Pearson's Chi square test. Using the SPSS statistical package (11.5, Chicago, IL, USA), multivariate models were developed that adjusted for the most important covariates, including age, differentiation, tumor invasion, tumor stage, lymph node metastasis and survival status. Survival length was determined from the day of primary tumor surgery to the day of death or last clinical follow-up. Kaplan–Meier method was used for survival analysis grouping with PTP1B amplification. Differences between curves were analyzed using the log-rank test. Multivariate Cox regression analysis was used to evaluate the effect of PTP1B amplification on survival of independently of age, differentiation, tumor invasion, tumor stage and lymph node metastasis. All statistical analyses were performed using the SPSS statistical package (11.5, Chicago, IL, USA). P values < 0.05 were considered significant.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81171969, 81272933 and 81372217), and the Fundamental Research Funds for the Central Universities.
Author Contributions
PH conceived, guided and supervised the research project. PH and NW designed the experiments. NW, JS, WL, JS, and QY performed the experiments. BS and PH provided tumor samples and research materials. NW, JS and PH analyzed the data. PH wrote the manuscript. All authors contributed to and approved the final version of the manuscript.
References
- 1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61:69-90; PMID:21296855; http://dx.doi.org/ 10.3322/caac.20107 [DOI] [PubMed] [Google Scholar]
- 2. Shi Y, Zhou Y. The role of surgery in the treatment of gastric cancer. J Surg Oncol 2010; 101:687-92; PMID:20512944; http://dx.doi.org/ 10.1002/jso.21455 [DOI] [PubMed] [Google Scholar]
- 3. Ostman A, Hellberg C, Bohmer FD. Protein-tyrosine phosphatases and cancer. Nat Rev Cancer 2006; 6:307-20; PMID:16557282; http://dx.doi.org/ 10.1038/nrc1837 [DOI] [PubMed] [Google Scholar]
- 4.Soulsby M, Bennett AM. Physiological signaling specificity by protein tyrosine phosphatases. Physiology (Bethesda) 2009; 24:281-9; PMID:19815854; http://dx.doi.org/ 10.1152/physiol.00017.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lessard L, Stuible M, Tremblay ML. The two faces of PTP1B in cancer. Biochim Biophys Acta. 2010; 1804:613-9; PMID:19782770; http://dx.doi.org/ 10.1016/j.bbapap.2009.09.018 [DOI] [PubMed] [Google Scholar]
- 6. Brown-Shimer S, Johnson KA, Hill DE, Bruskin AM. Effect of protein tyrosine phosphatase 1B expression on transformation by the human neu oncogene. Cancer Res 1992; 52:478-82; PMID:1345814 [PubMed] [Google Scholar]
- 7. Sangwan V, Paliouras GN, Cheng A, Dube N, Tremblay ML, Park M. Protein-tyrosine phosphatase 1B deficiency protects against fas-induced hepatic failure. J Biol Chem 2006; 281:221-8; PMID:16234234; http://dx.doi.org/ 10.1074/jbc.M507858200 [DOI] [PubMed] [Google Scholar]
- 8. Zheng LY, Zhou DX, Lu J, Zhang WJ, Zou DJ. Down-regulated expression of the protein-tyrosine phosphatase 1B (PTP1B) is associated with aggressive clinicopathologic features and poor prognosis in hepatocellular carcinoma. Biochem Biophys Res Commun 2012; 420:680-4; PMID:22450318; http://dx.doi.org/ 10.1016/j.bbrc.2012.03.066 [DOI] [PubMed] [Google Scholar]
- 9. Wiener JR, Kerns BJ, Harvey EL, Conaway MR, Iglehart JD, Berchuck A, Bast RC., Jr. Overexpression of the protein tyrosine phosphatase PTP1B in human breast cancer: association with p185c-erbB-2 protein expression. J Natl Cancer Inst 1994; 86:372-8; PMID:7905928; http://dx.doi.org/ 10.1093/jnci/86.5.372 [DOI] [PubMed] [Google Scholar]
- 10. Zhu S, Bjorge JD, Fujita DJ. PTP1B contributes to the oncogenic properties of colon cancer cells through src activation. Cancer Res 2007; 67:10129-37; PMID:17974954; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-4338 [DOI] [PubMed] [Google Scholar]
- 11. Lessard L, Labbé DP, Deblois G, Bégin LR, Hardy S, Mes-Masson AM, Saad F, Trotman LC, Giguère V, Tremblay ML. PTP1B is an androgen receptor-regulated phosphatase that promotes the progression of prostate cancer. Cancer Res 2012; 72:1529-37; PMID:22282656; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-2602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang J, Liu B, Chen X, Su L, Wu P, Wu J, Zhu Z. PTP1B expression contributes to gastric cancer progression. Med Oncol 2012; 29:948-56; PMID:21442314; http://dx.doi.org/ 10.1007/s12032-011-9911-2 [DOI] [PubMed] [Google Scholar]
- 13. Schwab M. Amplification of oncogenes in human cancer cells. Bioessays 1998; 20:473-9; PMID:9699459; http://dx.doi.org/ 10.1002/(SICI)1521-1878(199806)20:6%3c473::AID-BIES5%3e3.0.CO;2-N [DOI] [PubMed] [Google Scholar]
- 14. Shi J, Yao D, Liu W, Wang N, Lv H, Zhang G, Ji M, Xu L, He N, Shi B, et al. Highly frequent PIK3CA amplification is associated with poor prognosis in gastric cancer. BMC Cancer 2012; 12:50; PMID:22292935; http://dx.doi.org/ 10.1186/1471-2407-12-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shi J, Yao D, Liu W, Wang N, Lv H, He N, Shi B, Hou P, Ji M. Frequent gene amplification predicts poor prognosis in gastric cancer. Int J Mol Sci 2012; 13:4714-26; PMID:22606006; http://dx.doi.org/ 10.3390/ijms13044714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang D, Wang Z, Luo Y, Xu Y, Liu Y, Yang W, Zhang X. Analysis of DNA copy number aberrants by multiple ligation-dependent probe amplification on 50 intestinal typegastric cancers. J Surg Oncol 2011; 103:124-32; PMID:21259245; http://dx.doi.org/ 10.1002/jso.21792 [DOI] [PubMed] [Google Scholar]
- 17. Yasui W, Oue N, Aung PP, Matsumura S, Shutoh M, Nakayama H. Molecular-pathological prognostic factors of gastric cancer: a review. Gastric Cancer 2005; 8:86-94; PMID:15864715; http://dx.doi.org/ 10.1007/s10120-005-0320-0 [DOI] [PubMed] [Google Scholar]
- 18. Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer 2002; 2:161-74; PMID:11990853; http://dx.doi.org/ 10.1038/nrc745 [DOI] [PubMed] [Google Scholar]
- 19. Peng Z, Wang CX, Fang EH, Wang GB, Tong Q. Role of epithelial-mesenchymal transition in gastric cancer initiation and progression. World J Gastroenterol 2014; 20:5403-10; PMID:24833870; http://dx.doi.org/ 10.3748/wjg.v20.i18.5403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Qu Y, Dang S, Hou P. Gene methylation in gastric cancer. Clin Chim Acta 2013; 424:53-65; PMID:23669186; http://dx.doi.org/ 10.1016/j.cca.2013.05.002 [DOI] [PubMed] [Google Scholar]
- 21. Yang JY, Hung MC. A new fork for clinical application: targeting forkhead transcription factors in cancer. Clin Cancer Res 2009; 15:752-7; PMID:19188143; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-0124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005; 24:7410-25; PMID:16288288; http://dx.doi.org/ 10.1038/sj.onc.1209086 [DOI] [PubMed] [Google Scholar]
- 23. Zhang X, Tang N, Hadden TJ, Rishi AK. 2011 Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta 2011; 1813:1978-86; PMID:21440011; http://dx.doi.org/ 10.1016/j.bbamcr.2011.03.010 [DOI] [PubMed] [Google Scholar]
- 24. Yang JY, Zong CS, Xia W, Yamaguchi H, Ding Q, Xie X, Lang JY, Lai CC, Chang CJ, Huang WC, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol 2008; 10:138-48; PMID:18204439; http://dx.doi.org/ 10.1038/ncb1676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stetler-Stevenson WG. The role of matrix metalloproteinases in tumor invasion, metastasis, and angiogenesis. Surg Oncol Clin N Am 2001; 10:383-92; PMID:11382593 [PubMed] [Google Scholar]
- 26. Sutter AP, Zeitz M, Scherübl H. Recent results in understanding molecular pathways in the medical treatment of esophageal and gastric cancer. Onkologie 2004; 27:17-21; PMID:15007244; http://dx.doi.org/ 10.1159/000075600 [DOI] [PubMed] [Google Scholar]
- 27. Wadhwa R, Song S, Lee JS, Yao Y, Wei Q, Ajani JA. Gastric cancer-molecular and clinical dimensions. Nat Rev Clin Oncol 2013; 10:643-55; PMID:24061039; http://dx.doi.org/ 10.1038/nrclinonc.2013.170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weidinger C, Krause K, Klagge A, Karger S, Fuhrer D. Forkhead box-O. transcription factor: critical conductors of cancer's fate. Endocr Relat Cancer 2008; 15:917-29; PMID:18775975; http://dx.doi.org/ 10.1677/ERC-08-0153 [DOI] [PubMed] [Google Scholar]
- 29. Yang XB, Zhao JJ, Huang CY, Wang QJ, Pan K, Wang DD, Pan QZ, Jiang SS, Lv L, Gao X, et al. Decreased expression of the FOXO3a gene is associated with poor prognosis in primary gastricadenocarcinoma patients. PLoS One 2013; 8:e78158; PMID:24194912; http://dx.doi.org/ 10.1371/journal.pone.0078158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang JY, Hung MC. Deciphering the role of forkhead transcription factors in cancer therapy. Curr Drug Targets 2011; 12:1284-90; PMID:21443462; http://dx.doi.org/ 10.2174/138945011796150299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Egan C, Pang A, Durda D, Cheng HC, Wang JH, Fujita DJ. Activation of Src in human breast tumor cell lines: elevated levels of phosphotyrosine phosphatase activity that preferentially recognizes the Src carboxy terminal negative regulatory tyrosine 530. Oncogene 1999; 18:1227-37; PMID:10022129; http://dx.doi.org/ 10.1038/sj.onc.1202233 [DOI] [PubMed] [Google Scholar]
- 32. Martin GS. The road to Src. Oncogene 2004; 23:7910-7; PMID:15489909; http://dx.doi.org/ 10.1038/sj.onc.1208077 [DOI] [PubMed] [Google Scholar]
- 33. Chen T, George JA, Taylor CC. Src tyrosine kinase as a chemotherapeutic target: is there a clinical case? Anticancer Drugs 2006; 17:123-31; PMID:16428929; http://dx.doi.org/ 10.1097/00001813-200602000-00002 [DOI] [PubMed] [Google Scholar]
- 34. Zhang S, Zhang ZY. PTP1B as a drug target: recent developments in PTP1B inhibitor discovery. Drug Discov Today 2007; 12:373-81; PMID:17467573; http://dx.doi.org/ 10.1016/j.drudis.2007.03.011 [DOI] [PubMed] [Google Scholar]
- 35. Lee S, Wang Q. Recent development of small molecular specific inhibitor of protein tyrosine phosphatase 1B. Med Res Rev 2007; 27:553-73; PMID:17039461; http://dx.doi.org/ 10.1002/med.20079 [DOI] [PubMed] [Google Scholar]
- 36. Krishnan N, Koveal D, Miller DH, Xue B, Akshinthala SD, Kragelj J,Jensen MR, Gauss CM, Page R, Blackledge M, et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat Chem Biol 2014; 10:558-66; PMID:24845231; http://dx.doi.org/ 10.1038/nchembio.1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316:103-43; PMID:17463250 [DOI] [PubMed] [Google Scholar]
- 38. Hou P, Liu D, Shan Y, Hu S, Studeman K, Condouris S, Wang Y, Trink A, El-Naggar AK, Tallini G, et al. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/Akt pathway in thyroid cancer. Clin Cancer Res 2007; 13:1161-70; PMID:17317825; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-1125 [DOI] [PubMed] [Google Scholar]
- 39. Ji M, Guan H, Gao C, Shi B, Hou P. Highly frequent promoter methylation and PIK3CA amplification in non-small cell lung cancer (NSCLC). BMC Cancer 2011; 11:147; PMID:21507233; http://dx.doi.org/ 10.1186/1471-2407-11-147 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.